An integrated gene annotation and transcriptional profiling approach towards the full gene content of the Drosophila genome (original) (raw)
- Research
- Published: 22 December 2003
- B Beckmann2,
- SA Haas3,
- B Koch1,
- V Solovyev4,
- C Busold2,
- K Fellenberg2,
- M Boutros5,
- M Vingron3,
- F Sauer1,6,
- JD Hoheisel2 &
- …
- R Paro1
Genome Biology volume 5, Article number: R3 (2003)Cite this article
- 21k Accesses
- 82 Citations
- Metrics details
Abstract
Background
While the genome sequences for a variety of organisms are now available, the precise number of the genes encoded is still a matter of debate. For the human genome several stringent annotation approaches have resulted in the same number of potential genes, but a careful comparison revealed only limited overlap. This indicates that only the combination of different computational prediction methods and experimental evaluation of such in silico data will provide more complete genome annotations. In order to get a more complete gene content of the Drosophila melanogaster genome, we based our new D. melanogaster whole-transcriptome microarray, the Heidelberg FlyArray, on the combination of the Berkeley Drosophila Genome Project (BDGP) annotation and a novel ab initio gene prediction of lower stringency using the Fgenesh software.
Results
Here we provide evidence for the transcription of approximately 2,600 additional genes predicted by Fgenesh. Validation of the developmental profiling data by RT-PCR and in situ hybridization indicates a lower limit of 2,000 novel annotations, thus substantially raising the number of genes that make a fly.
Conclusions
The successful design and application of this novel Drosophila microarray on the basis of our integrated in silico/wet biology approach confirms our expectation that in silico approaches alone will always tend to be incomplete. The identification of at least 2,000 novel genes highlights the importance of gathering experimental evidence to discover all genes within a genome. Moreover, as such an approach is independent of homology criteria, it will allow the discovery of novel genes unrelated to known protein families or those that have not been strictly conserved between species.
Background
Knowledge of the complete gene set of a genome is a prerequisite to an integrated view of the network of encoded functions. One major obstacle to this goal is the reliable identification of genes within the vast excess of non-coding sequences within eukaryotic genomes. While bioinformatics methods have substantially evolved in their prediction capabilities, they still compromise on sensitivity versus specificity. Most genome annotations performed so far have concentrated on maximizing the number of 'real' genes by requiring multiple evidence before accepting an annotation, such as: the presence of expressed sequence tags (ESTs); homologies to known proteins or the conservation of genomic sequences between related organisms; or by raising the thresholds of the prediction software in order to keep the number of false positives to a minimum. Another problem of such mere in silico approaches is that even if different approaches result in the same number of annotated genes, the overlap of such predictions can be limited depending on the extent to which the criteria for predicting a gene differ [1, 2].
For D. melanogaster, the initially published genome annotation proposed the existence of about 14,000 genes [3]. A first comparison of this annotation with known protein sequences from SwissProt showed that the predictions must be treated with caution [4]. Moreover, based on in silico data [5], novel ESTs [6, 7] and a protein trap approach [8], the gene numbers in the Drosophila genome annotation (Berkeley Drosophila Genome Project (BDGP) Release 1 and 2) were challenged rapidly after publication. Although, in the meantime, the annotation has seen substantial changes in gene models, the absolute gene number has only changed marginally in the latest release (now FlyBase Release 3.1) [9]. The FlyBase annotation process relies to a large extent on EST evidence and homology criteria in addition to the ab initio gene prediction based on Genie [10] and GENSCAN [11] and, as a result, only 6% of the gene models in Release 3 stem from gene prediction data only [9]. While such an approach may reliably detect already known genes or genes that at least show substantial similarity to known proteins, it will most likely omit genes encoding proteins or functions that are currently not undescribed. One way out of this dilemma may be the incorporation of information gathered by genome-wide comparisons of related species, like Anopheles gambiae [12] and Drosophila pseudoobscura [[13](/articles/10.1186/gb-2003-5-1-r3#ref-CR13 "Drosophila pseudoobscura Genome Project. [ http://www.hgsc.bcm.tmc.edu/projects/drosophila/
]")\], again assuming that all genes will be sufficiently conserved to allow their unequivocal detection.Most whole-transcriptome microarrays will therefore remain incomplete, as the only safe way to include all potential genes (and even their splice forms) is the construction of arrays based on a whole genome tiling path. Whilst the feasibility and superiority of such an approach has been shown for parts of the human chromosome 22 [14], its application to complete genomes is limited due to the technical restrictions of microarray fabrication. A practicable alternative will be to concentrate on genomic regions that, even with only low confidence, are predicted to be protein coding.
We decided to combine the published, conservative BDGP genome annotation Release 2 with an alternative, less stringent gene prediction for the design of a new PCR-fragment based whole-transcriptome microarray for D. melanogaster (Heidelberg FlyArray, HD FlyArray). Although this approach will unavoidably overestimate the number of genes by including many false-positives, subsequent experimental validation by expression profiling enables true and false positives to be distinguished. By focusing on the developmental life-cycle of Drosophila and relying on multiple experimental validations our data demonstrate the existence of at least 2,000 novel genes.
Results and discussion
Combined annotation
To overcome the known limitations in gene prediction, we constructed our Drosophila transcriptome microarray by first combining the BDGP Drosophila genome annotation Release 2 and the BDGP cDNA collection Release 1 [15] and then we also included an ab initio prediction based on the Fgenesh software [16]. We merged the combined BDGP set with the 20,622 Fgenesh predicted genes (Heidelberg Prediction, Heidelberg Collection (HDC)), based on the assumption that predictions showing an overlap of more than 30% of their exon sequences represent the same gene, resulting in a set of 21,396 potential genes (Figure 1). While the fact that nearly 97% of the BDGP genes were also predicted by Fgenesh validates our overlap criterion, we still found a further 7,464 predicted genes (36.2%; HDC unique) not represented in the BDGP annotation.
Figure 1
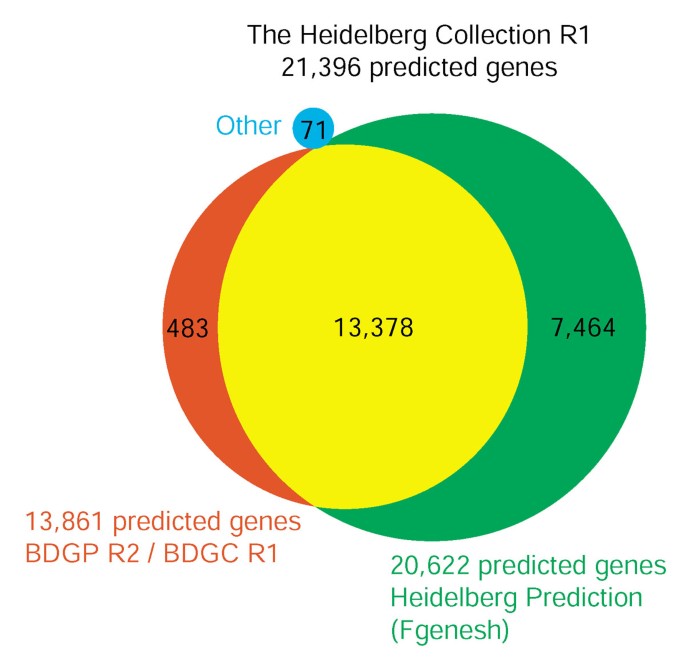
The Heidelberg Collection R1. The combination of the BDGP cDNA Collection (BDGC) R1 with the BDGP genome annotation Release 2 contained 13,861 genes. The Heidelberg Prediction based on the Fgenesh ab initio gene prediction software contains 20,622 predictions. Assuming that genes that overlap by more than 30% of their exon sequence represent the same gene, we combined these two annotation sets. In addition we included 71 genes from different databases that were not present in either annotation. The resulting Heidelberg Collection consists of 21,396 potential genes and is the basis for the Heidelberg FlyArray.
Computational analysis of the combined annotation
The simplest explanation for the high number of HDC unique predictions may be the relaxed stringency criterion applied. Consequently, a careful inspection of the two sets (BDGP/FlyBase versus HDC) showed a high degree of similarity for most common predictions; differences were largely confined to the 5' and 3' ends of the predictions as may be expected. This is not only because ab initio gene prediction algorithms have most difficulties in locating the precise ends of a gene, but also because the HDC predictions contain only coding regions - while the BDGP/FlyBase annotation may also include untranslated regions (UTRs). Next, we compared the median open reading frame (ORF) size of predictions unique for either BDGP/FlyBase or the HDC as this might reflect a tendency to arbitrarily split a single gene into multiple annotations. For the BDGP/FlyBase unique predictions we found a median ORF size of 113 amino acids for Release 2 which increases to 163 amino acids in Release 3.1. The median ORF size for the HDC unique predictions (139 amino acids) falls well within this range. In addition, no significant difference in the median ORF size was found between the predictions that were expressed or unexpressed according to our developmental expression profiling (see below). In summary, these results suggest that the HDC unique predictions are not simply additional exons of BDGP/FlyBase genes and, in addition, argue that most of the differences between the two annotations are based solely on stringency.
The assumption that most of the HDC unique predictions have been omitted from Release 3.1 due to the lack of significant homologies is further substantiated by homology searches performed against the SwissProt database, which were positive for only 1.5% of the HDC unique predictions. Likewise, only 1.8% scored a hit in an InterPro protein domain search and only 10.2% showed an overlap with EST sequences [17]. This low degree of conservation was expected as most of the information was already available to FlyBase. In contrast, the sequence of D. pseudoobscura [[13](/articles/10.1186/gb-2003-5-1-r3#ref-CR13 "Drosophila pseudoobscura Genome Project. [ http://www.hgsc.bcm.tmc.edu/projects/drosophila/
]")\] became available only recently and therefore might validate more of the HDC unique predictions, fulfilling the expectation that interspecies comparisons will fill the gaps in _in silico_ gene predictions. While DNA sequence comparison revealed that, independent of the overlap requested, about 50% of the common predictions showed conservation between _D. melanogaster_ and _D. pseudoobscura_ (Table [1](/articles/10.1186/gb-2003-5-1-r3#Tab1)), the HDC unique predictions were less well conserved. Only 31.3-32.5% showed an overlap of at least 30% of their length with conserved regions, and only 13.4-13.7% extended this overlap to more than 50%. As exemplified by our analysis, the definition of meaningful cut-off values for the sequence comparisons, as well as for the overlap criterion, is arbitrary and will remain the main weakness of this approach. Even for more sophisticated bioinformatics approaches this constraint will limit the success in complementing genome annotations.Table 1 Conservation between D. melanogaster and D. pseudoobscura
In summary, we find that most of the HDC unique predictions cannot be confirmed by in silico approaches based on homology criteria such as conservation of protein motifs or DNA sequence between species and therefore have been omitted from the BDGP/FlyBase annotations. Reversing this argument, the lack of conservation argues that the HDC unique predictions may code for novel proteins, representatives of hitherto undescribed protein families, and will only be accessible to experimental validation.
Amplicon selection and primer design
Using GenomePride [18], we automatically designed primer pairs for 21,306 (99.6%) of the 21,396 potential genes in the final Heidelberg Collection R1 (Figure 1). Based on an all-against-all comparison of the transcripts of the Heidelberg Collection, we detected regions of homology as well as repetitive sequences common to some of the transcripts. These regions of similarity were penalized in the subsequent selection of optimal amplicons such that each amplicon is likely to be unique. In order to minimize the potential ill effects of false gene models for our microarray, we aimed to exclude most 5' and 3' regions from the amplicon design, as ab initio gene prediction programs tend to have most problems in finding the correct start and end. Consequently, we observed that shorter Fgenesh predictions and the BDGP annotations often disagree on the gene borders, while the central regions that were used for amplicon design are in good agreement. As a homogenous amplicon size will help to ensure comparable hybridization conditions for all genes analyzed we aimed for fragments of about 500 bp in length. In addition, we avoided introns, thus targeting a single exon whenever possible. Using a two-step PCR protocol we produced amplicons for 97.9% of all predicted genes from genomic DNA. Besides the advantage of limiting the amount of contaminating genomic DNA present in the samples which will be spotted, as well as an increased sensitivity, the two-step PCR approach opens the way for a very efficient re-amplification of the amplicon set. This requires only a limited set of primers (we used a combination of one unique tag-primer in combination with nine different tag-primers to limit cross-well contaminations) and thereby also facilitates the re-use of this set for other purposes, such as the production of a template set for genome-wide dsRNA production (see below).
Expression profiling I - quality of the novel array design
Together with a number of controls the complete set was spotted in duplicates, resulting in a high density (47,616 spots) transcriptome microarray. Initial hybridizations already indicated on visual inspection that a high percentage of the novel genes are expressed (Figure 2a, positive spots within green frame). To assess the overall quality of our array design as well as to validate the novel predictions, we performed developmental profiling of the Drosophila life-cycle using nine different stages and analyzed the data by correspondence analysis (CA) [19]. This is an explorative computational method of studying the associations between variables. Similar to other projection methods, CA represents variables such as gene expression as vectors in a multi-dimensional space. Like principal component analysis (PCA), CA reveals the principal axes of this n-dimensional space that account for the main variance. However, CA is distinguished by its ability to account for genes in hybridization-dimensional space and for the hybridizations in gene-dimensional space at the same time. Projection of both representations of the data matrix into the same low-dimensional sub-space, for example a plane, reveals the associations both within and between these variables. Moreover, CA does not require any prior choice of parameters and thus allows an unbiased view of the structures within data. As a consequence, the quality of hybridizations in multiconditional experiments may be validated by a clustering of repeated hybridizations from the same condition. In our case, CA showed the validity of our array design and the quality of the hybridizations by the clear distinction between the clusters of different developmental stages hybridized to the arrays (Figure 2b). Only the larval stage was not completely resolved, due to the fact that early larval stages and adult stages, as well as the late larval stages and pupal and embryonic stages, are related [20]. Therefore, such overlap is expected as all larval stages were pooled in our experiments. A detailed analysis (B.B., M.H., S.A.H., B.K., C.B., K.F., M.V., F.S., J.D.H. and R.P., unpublished observations) demonstrated that our gene expression results successfully reproduced most of the temporal regulation reported previously [20–22].
Figure 2
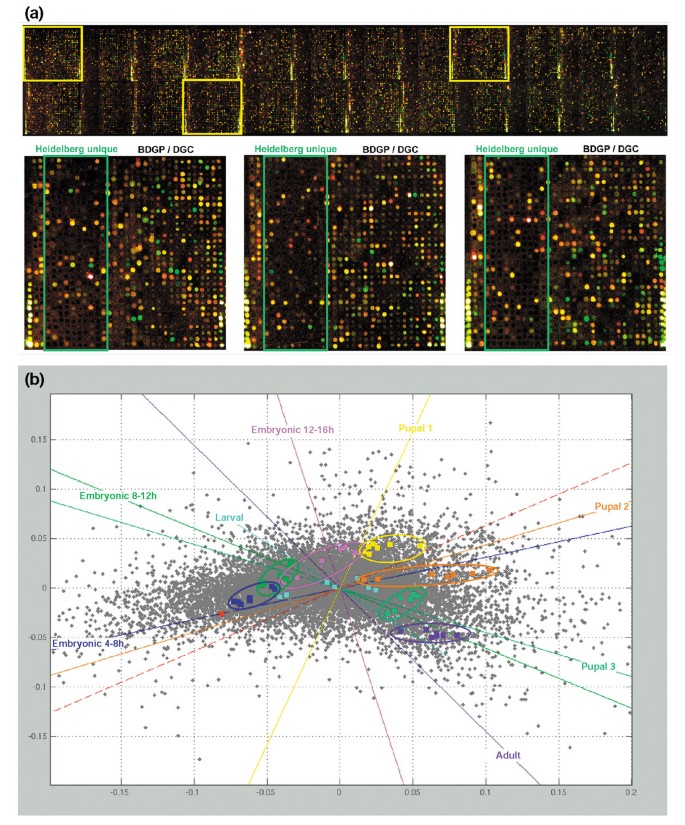
Developmental profiling. (a) Two-color hybridization (green: adult stage; red: 4-8 h old embryo) on the Heidelberg FlyArray directly showing the expression of genes unique to the Heidelberg Prediction (see lower part, spots within the green rectangle). (b) Correspondence cluster analysis of the developmental profiling. Samples from nine different stages of the Drosophila life-cycle were hybridized to the Heidelberg FlyArray. Each experiment was performed at least in triplicate, including a dye reversal to avoid bias. In the resulting plot, each hybridization of an individual developmental stage is depicted as a colored square for each replicate present on the slide. They all form distinct clusters (except for the larval stage), indicating the degree of reproducibility and specificity between them. As a consequence of the normalization process, only the median of all control hybridizations (0-4 h) is shown in the diagram as a single red square. Genes are shown as grey dots if they exhibited significant differential transcription levels. The distance between dots is low when their expression profiles show similar shape, independent of their absolute values. Colored guiding lines are displayed that correspond to the transcription profiles of virtual genes that would exhibit a signal in one condition only.
Expression profiling II - expression status of common and predictions unique to the Heidelberg Collection
After data processing and stringent filtering, we found 13,927 genes to be expressed during the Drosophila life-cycle, of which 10,378 genes belong to the BDGP predicted set and 3,497 are newly predicted in the Heidelberg Collection R1 (Table 2, Additional data file 3). Thus, we see experimental evidence for 76.5% of the conservative BDGP annotation and 47.8% of the novel ab initio predictions. These numbers show an intriguing similarity to the detection rate seen in studies performed on subsets of predicted human genes [14]. In this analysis, about 80-85% of the known genes but only 58% of the predicted genes could be validated by microarray analysis. The 76.5% validation rate of the BDGP genes may thus represent the detection limits of our microarray analysis, arguing that most of the BDGP annotations will be 'real' genes.
Table 2 Summary for the Heidelberg Collection R1
Re-evaluation based on FlyBase Release 3.1
The latest FlyBase Release 3.1 not only resulted in structural changes to 85% of the transcripts and 45% of the predicted proteins [9] but also takes care of most of the genes that have been reported missing in BDGP Release 1 and 2. For example, most of the testes-specific ESTs absent in Release 1 [6] now match their corresponding UTRs in Release 3.1. Likewise, only approximately 280 of the 1,042 novel predicted genes (compared to Release 2) of Gopal et al. [5] are still missing. For these reasons, we re-evaluated the combination of the published annotation data using FlyBase Release 3.1 and the original Heidelberg Prediction. The total number of genes in the new Heidelberg Collection R2 was 19,879 (Table 3, Additional data file 3), of which 6,224 are unique to the Heidelberg Prediction and 13,050 are common to both. The HD FlyArray already contains amplicons for most (92.7%) of the FlyBase Release 3.1 genes as, in the meantime, many former HDC unique predictions have been included. Our microarray analysis provides evidence for the expression of 9,908 (78.3%) of the FlyBase Release 3.1 genes that are represented on the array and 2,636 (42.9%) of the novel genes. In addition, the array contains amplicons made from 286 predictions unique to BDGP Release 2, of which 160 (55.9%) are expressed.
Table 3 Summary for the Heidelberg Collection R2
Validation by RT-PCR and _in situ_hybridization
Despite the fact that the developmental expression profiling was thoroughly filtered and statistically analyzed, we decided to define the lower limit for the number of novel genes by an additional level of validation. We therefore performed RT-PCR analyses for a semi-random selection (with respect to chromosomal order, see Methods) of newly predicted genes as well as of some previously known ones. For the latter, we confirmed the microarray results at 93.8% (136/145), and confirmed 74.4% (218/293) of the uniquely predicted novel genes. While we cannot exclude the fact that the RT-PCR erroneously missed some weakly expressed genes in the RNA pool made from all stages analyzed in the microarray experiments, this result clearly points to the existence of at least 2,000 additional genes (Table 3, Additional data file 3).
The additional analysis of gene expression by in situ hybridization adds spatial information; thus the detection of a variety of distinct patterns for the newly predicted genes may not only point to possible functions but will also further substantiate them. Therefore we performed in situ hybridization experiments for a subset of the HDC unique predictions that showed expression during embryonic stages. Of 213 genes analyzed, 82 (38.5%) were confirmed by this analysis (Figure 3, see also Additional data file 3). The low success rate of the in situ analysis compared to that of the BDGP Gene Expression project (approximately 80%) [23] is due to the use of small PCR fragments (median size: 385 bp) for probe generation and a similarly low success rate was observed for BDGP/FlyBase predicted genes included in our study. We further analyzed a subset of these in situ negative genes by RT-PCR and confirmed their expression (13/17) at the respective developmental stages. We found that many developmentally expressed genes being present in a variety of different tissues (Table 4) had escaped the annotation process so far. For example, HDC09253 is located on chromosome arm 3L in a region where no other gene is predicted (Figure 3a, left). Whilst no expression is observed during early stages (Figure 3a, top), the gene is expressed in the posterior spiracles and the ectoderm from stage 12 onwards (Figure 3a, middle and bottom). HDC04256 is located on the second chromosome, in a locus where no other genes are predicted (Figure 3b, left). The HDC04256 transcript is detected from stage 11 on in a subset of the trunk mesoderm (Figure 3b, middle), while it is restricted to the gonads during later stages (Figure 3b, bottom). The gene HDC02494 is located on chromosome 2L, in the second intron of the wb gene, but is transcribed in opposite direction. Expression starts in the mesoderm anlage as well as in the head furrow at stage 7 (Figure 3c, top), before showing ubiquitous transcription during later stages (Figure 3c, middle and bottom). Photographs of the in situ hybridization patterns observed at different developmental stages for all novel genes analyzed so far can be accessed on our website [[24](/articles/10.1186/gb-2003-5-1-r3#ref-CR24 "The Heidelberg Prediction in situ patterns. [ http://HDFlyArray.zmbh.uni-heidelberg.de/cgi-bin/insitu.pl
]")\]. Finally, we found good agreement between the microarray based expression profiling data (Figure [4g](/articles/10.1186/gb-2003-5-1-r3#Fig4)), northern blotting (Figure [4h](/articles/10.1186/gb-2003-5-1-r3#Fig4)) and the _in situ_ hybridization results (Figure [4a-f](/articles/10.1186/gb-2003-5-1-r3#Fig4)) as exemplified for the novel annotation HDC13470.Figure 3
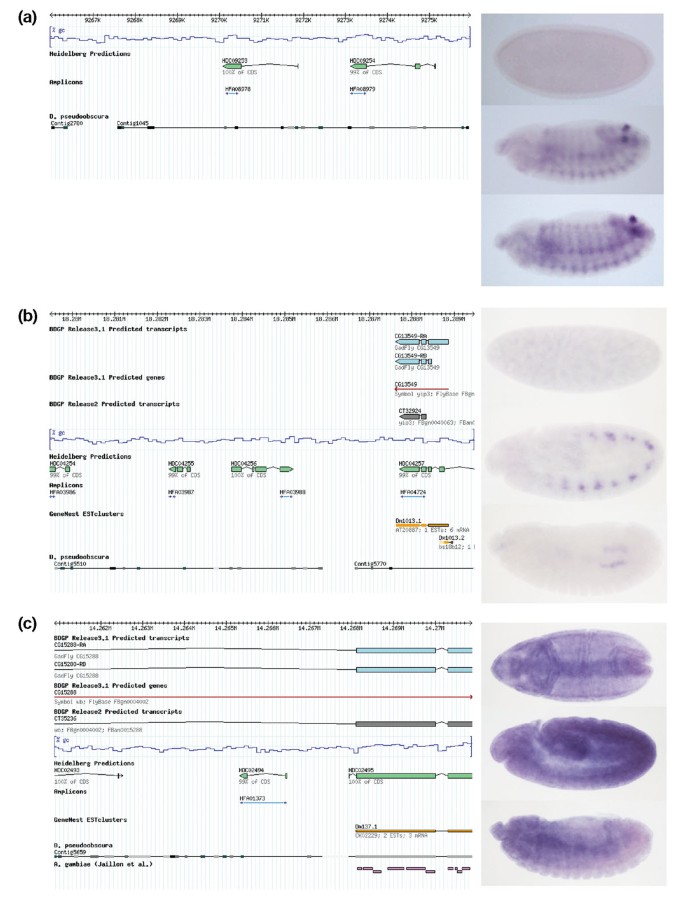
Genomic location and expression patterns of Heidelberg unique predictions. The left part of the figure visualizes the genomic region (10 kb of sequence) for some examples of the novel Heidelberg Predictions. In addition, here is the corresponding amplicon present on the microarray as well as information on conserved regions (D. pseudoobscura in gray, A. gambiae in pink) and ESTs (orange). (a) HDC09253 and (b) HDC04256 lie within regions missing any BDGP/FlyBase prediction. HDC02494 is predicted within known FlyBase predictions but is located on the opposite strand (c). On the right, the in situ pictures show the expression patterns at three different time points of development, 0-4 h (top), 4-8 h (middle) and 8-12 h (bottom), respectively. Embryos are shown in (a, b) lateral view, (c) top: ventral view, middle and bottom: lateral view; anterior is always to the left.
Table 4 Expression patterns obtained by in situ hybridization
Figure 4
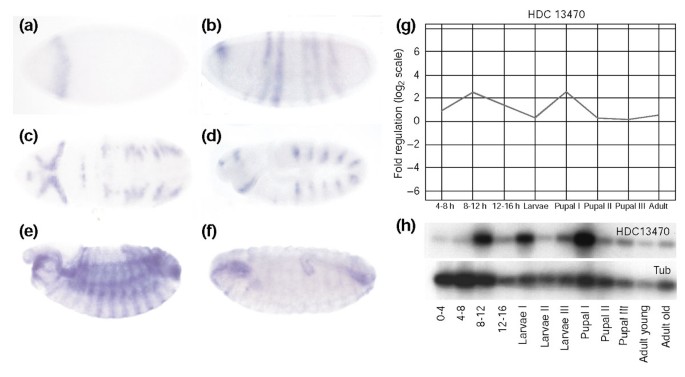
In situ hybridization for HDC13470. (a-f) In situ hybridization of various stages of embryonic development using HDC13470 as probe. (g) The microarray-based expression profile (all stages compared to 0-4 h) is nicely reproduced by (h) the result of the northern analysis. Tub, tubulin. Embryos (a, b, d-f) are shown in lateral view, (c) is a ventral view, with the anterior always to the left.
Are the novel genes pseudogenes?
One important step during the design of the amplicon set was to exclude regions that showed significant homologies to other regions in the genome. Not only should this step exclude amplicons that would represent conserved protein motifs but it should also prevent most of the transposable elements and repeat structures from being represented in our set. Nevertheless, regions of low stringency identity might have been included in our amplicon set and thus part of the observed expression might result from cross-hybridization of a real gene to, for example, an amplicon representing a non-expressed, degenerated pseudogene. We therefore re-analyzed all amplicons for HDC unique predictions that scored positive in our expression profiling for the existence of additional low stringency blast hits and found no match for 86.5% of them. Careful comparison of the expression profiles for the remaining 13.5% with their second site hits showed that only 15.4% of them were co-regulated, demonstrating that no more than 2% of all novel genes might represent false-positives due to cross-hybridization.
Confirmation by genome-wide RNAi
As previously mentioned, the design of HD FlyArray enables further uses of our amplicon set, such as the expression of peptide representatives for each gene for use in antibody production or, as exemplified by the work performed by Boutros et al. (M.B., A. Kiger, S. Armknecht, K. Kerr, M.H, S.A.H., B.K, HDFlyArray Consortium, R.P., and R.N. Perrimon, unpublished results), for the generation of dsRNA templates for genome-wide RNAi studies. This integrated approach allows for validation of screening results by other techniques based on the same set of DNA fragments. Accordingly, we obtained further evidence for the validity of the Heidelberg Collection from genome-wide RNAi studies on cell viability/lethality. This analysis showed that, after stringent filtering, 369 (2.9%) out of the 12,655 FlyBase R3.1 genes and 68 (1.1%) out of 6,143 HDC unique predictions showed lethality. Assuming that the ratio of lethal genes remains the same for both subsets, this functional screen argues that approximately 2,330 (38%) of the HDC unique predictions are expressed genes, a number in good accord of the results obtained by our developmental profiling experiments.
We further analyzed this screen to obtain an estimate of how many of the novel predictions may constitute additional exons for genes already predicted by the FlyBase annotation. To this end, we tested whether FlyBase R3.1 predicted genes neighboring a HDC unique prediction also showed lethality in the RNAi screen. Including 25 kb upstream or downstream, we found such a FlyBase gene for 8/68 (11.8%) of the HDC unique predictions. For 1/68, we saw another HDC unique prediction. Expanding the search space to 50 kb, about 14.7% of the novel genes might be additional exons to a FlyBase gene and 3/68 novel genes might consist of two HDC unique predictions. These numbers are essentially identical to the results obtained for the 369 FlyBase predicted genes influencing cell viability in the RNAi screen. For these, we found 13% (48/369) within 25 kb and 16% (59/369) within 50 kb that could be interpreted as additional exons. We conclude that the vast majority of the HDC unique predictions are not additional exons of genes predicted by FlyBase.
Gene models
In contrast to the extensive corrections and additions that significantly improved the genome annotation Release 3.1, we did not rework the original set of gene models predicted by Fgenesh as in some cases the arbitrary separation of exons into different genes may have been advantageous in predicting genes that lie within the intron of another gene. Nevertheless, as our expression data not only validates the existence of transcriptional units but also excludes cross-hybridization and the assignment of exons into separate genes as the primary source for most of our expressed novel predictions, we believe it is appropriate to call such transcriptional active regions genes. Ultimately, the correct gene structure for all Drosophila genes, including the BDGP/FlyBase predictions, will await the completion of the Drosophila gene collection (DGC) project [25] and possibly also an ORFeome project similar to that of Reboul et al. [26]. This group has shown for the very well studied C. elegans genome that more than 50% of the computational predicted genes needed corrections in their intron-exon structures. In the latter type of project, our set of verified predictions will be a good basis for the design of an almost complete primer set to clone and sequence the whole Drosophila ORFeome.
Until such experimental confirmation for all gene models exists, the comparison of different predictions may offer a good starting point for judging the reliability of the computed gene structures. We therefore used the Generic Genome Browser (GBrowse) [27] platform to establish a comparative view of the different genome annotations based on the BDGP annotations Release 2, FlyBase Release 3.1 and the Heidelberg Prediction (Figure 5). Moreover, our website [[28](/articles/10.1186/gb-2003-5-1-r3#ref-CR28 "The Heidelberg FlyArray GBrowser. [ http://HDFlyArray.zmbh.uni-heidelberg.de/cgi-bin/gbrowse
]")\] also offers a view of the amplicons present on the HD FlyArray as well as the GeneNest EST clustering \[[17](/articles/10.1186/gb-2003-5-1-r3#ref-CR17 "Haas SA, Beissbarth T, Rivals E, Krause A, Vingron M: GeneNest: automated generation and visualization of gene indices. Trends Genet. 2000, 16: 521-523. 10.1016/S0168-9525(00)02116-8.")\] and a genome-wide sequence comparison to _D. pseudoobscura_ \[[13](/articles/10.1186/gb-2003-5-1-r3#ref-CR13 "Drosophila pseudoobscura Genome Project. [
http://www.hgsc.bcm.tmc.edu/projects/drosophila/
]")\] and _A. gambiae_ \[[29](/articles/10.1186/gb-2003-5-1-r3#ref-CR29 "Jaillon O, Dossat C, Eckenberg R, Eiglmeier K, Segurens B, Aury JM, Roth CW, Scarpelli C, Brey PT, Weissenbach J, Wincker P: Assessing the Drosophila melanogaster and Anopheles gambiae genome annotations using genome-wide sequence comparisons. Genome Res. 2003, 13: 1595-1599. 10.1101/gr.922503."), [30](/articles/10.1186/gb-2003-5-1-r3#ref-CR30 "Exofish between the genome of Anopheles and the genome of Drosophila. [
http://www.genoscope.cns.fr/externe/Fly/
]")\]. Additional information is available for BDGP/FlyBase annotated genes by linkage to their respective FlyBase entries \[[31](/articles/10.1186/gb-2003-5-1-r3#ref-CR31 "The FlyBase Consortium: The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res. 2003, 31: 172-175. 10.1093/nar/gkg094.")\]. Additionally, all EST clusters are connected via GeneNest \[[17](/articles/10.1186/gb-2003-5-1-r3#ref-CR17 "Haas SA, Beissbarth T, Rivals E, Krause A, Vingron M: GeneNest: automated generation and visualization of gene indices. Trends Genet. 2000, 16: 521-523. 10.1016/S0168-9525(00)02116-8."), [32](/articles/10.1186/gb-2003-5-1-r3#ref-CR32 "Krause A, Haas SA, Coward E, Vingron M: SYSTERS, GeneNest, SpliceNest: exploring sequence space from genome to protein. Nucleic Acids Res. 2002, 30: 299-300. 10.1093/nar/30.1.299.")\] to SpliceNest \[[32](/articles/10.1186/gb-2003-5-1-r3#ref-CR32 "Krause A, Haas SA, Coward E, Vingron M: SYSTERS, GeneNest, SpliceNest: exploring sequence space from genome to protein. Nucleic Acids Res. 2002, 30: 299-300. 10.1093/nar/30.1.299."), [33](/articles/10.1186/gb-2003-5-1-r3#ref-CR33 "Coward E, Haas SA, Vingron M: SpliceNest: visualizing gene structure and alternative splicing based on EST clusters. Trends Genet. 2002, 18: 53-55. 10.1016/S0168-9525(01)02525-2.")\], a web-based graphical tool for exploring gene structure, including alternative splicing, based on a genomic mapping of EST consensus sequences, and to SYSTERS \[[32](/articles/10.1186/gb-2003-5-1-r3#ref-CR32 "Krause A, Haas SA, Coward E, Vingron M: SYSTERS, GeneNest, SpliceNest: exploring sequence space from genome to protein. Nucleic Acids Res. 2002, 30: 299-300. 10.1093/nar/30.1.299."), [34](/articles/10.1186/gb-2003-5-1-r3#ref-CR34 "Krause A, Stoye J, Vingron M: The SYSTERS protein sequence cluster set. Nucleic Acids Res. 2000, 28: 270-272. 10.1093/nar/28.1.270.")\], a protein family database. All amplicons are linked to information about their sequence, transcription status (expression profiling, RT-PCR) as well as to the observed _in situ_ hybridization pattern if available.Figure 5
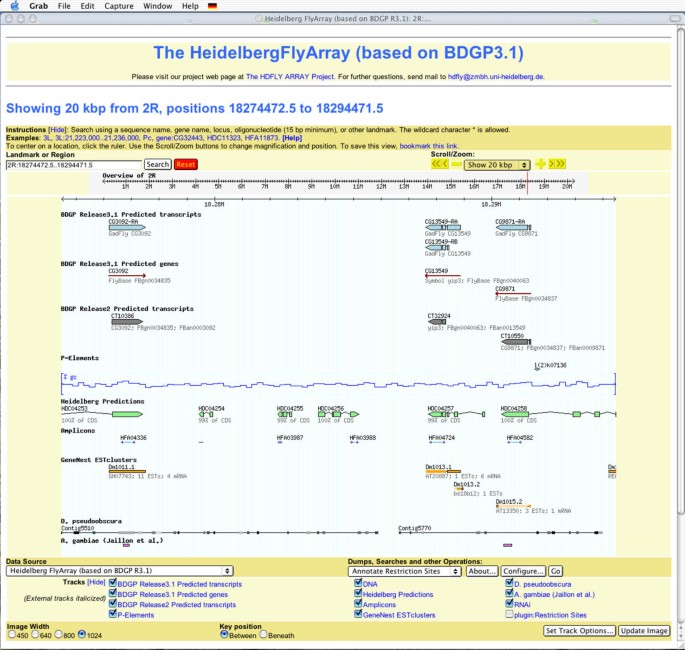
The Heidelberg FlyArray website. Screen shot of the Heidelberg FlyArray website based on the GBrowse platform. After selecting the genomic region of interest, for example by gene name, amplicon name or position, the user is offered a comparative view of the different gene models from the BDGP genome annotations Release 2, FlyBase Release 3.1 and the Heidelberg Prediction, as well as the placement of the amplicons chosen for the Heidelberg FlyArray. In addition, researchers find a comparison to D. pseudoobscura and A. gambiae along with a novel EST clustering and information on known P-element insertions.
Conclusions
Our integrated in silico and 'wet biology' approach offers the advantage of being less restrictive than previous predictions in including ab initio gene predictions and thereby allows the detection of a more complete gene set. Confirming this, our data not only provide in silico but also experimental evidence for over 2,000 additional Drosophila genes. Thus, assuming that all FlyBase annotations are real, the gene count in Drosophila must be raised to at least 16,000 genes or even up to 17,000, applying the observed detection rate of the microarray to the HDC unique predictions. The successful application of this integrated approach should not be limited to Drosophila - it may also be a good starting point for other organisms, such as mouse, rat or human, for which the huge size of the genome prohibits - at least for the near future - pure tiling path approaches.
In addition, the fact that most of the newly identified genes show no significant homology to known proteins (comparison to SwissProt) or domains/motifs (InterPro search) and also lack considerable conservation between species (D. pseudoobscura, A. gambiae) demonstrates the importance of our experimental scheme. The future detailed study of these novel genes will not only result in the identification of novel protein motifs and thus functions in Drosophila but may also improve future homology based in silico genome annotation approaches in other organisms by offering a more complete dataset as basis.
In addition to its value in microarray production, the Heidelberg amplicon set also proved to be a valuable tool for genome-wide RNAi studies. The possibility of using the same set of fragments for both expression profiling and genome-wide RNAi experiments will be of great benefit for further studies on genetic networks.
Methods
Annotation
The complete Heidelberg Prediction is available via download as a FASTA formatted file. Each entry consists of the CDS position information, the strand orientation as well as the sequence of genomic region spanning the prediction. For details on the Fgenesh software and the parameters used refer to [16]. Only limited filtering of the resulting ab initio annotation was performed: firstly we removed predictions coding for less than 30 amino acids and then genes with a total exon score <15 were excluded. Please note that the 20,622 genes predicted may include some pseudogenes as well as mobile elements.
Combining gene predictions
We extended the BDGP set by genes of the Drosophila Gene Collection (DGC), which were not represented in BDGP, based on their gene names. Sets of predicted genes (for example, DGC/BDGP and Fgenesh) were combined by comparing the overlap in exon sequences of the appropriate orientation. Two gene predictions were defined as reflecting the same gene if the number of common exonic base pairs exceeded 30% of the length of the shorter prediction. If two gene predictions out of one set were covered by a single prediction of the second set we took the shorter predictions as representatives. Finally, we manually added 71 genes that were not included in the genomic sequence.
Primer design strategy
Since the amplicons should be unique to the gene they represent, the GenomePRIDE software [18, [35](/articles/10.1186/gb-2003-5-1-r3#ref-CR35 "GenomePride. [ http://pride.molgen.mpg.de
]")\] used for the design of the PCR primers also performed an all-against-all comparison of the exonic sequences of all genes. This BLAST search (default settings) allowed the detection of similarity regions showing >70% identity. All similarity regions longer than 40 bp were flagged not to be included in an amplicon. This way, domains or conserved parts within gene families and also repetitive elements are likely to be excluded from the amplicon set when these elements appear in at least two different genes. The overall strategy of designing gene-specific PCR primers is divided into two phases. Given the annotation of the exon structure of the gene of interest, GenomePRIDE first screens for the optimal target region within those exons. In a pre-processing step, GenomePRIDE splices the genomic sequence covering the target gene in order to generate an artificial sequence consisting of a concatenation of all related exons followed by all-spliced intron sequences. GenomePRIDE then computes a quality for every potential fragment of a user-defined length (in the current project, 500 bp) within the exonic sequence by evaluating the fraction of homology to other genes, the fraction of intronic sequence, and by measuring the location of the putative target region with respect to the preferred location defined by the user. If no target region reaches a quality above a certain threshold, the optimal fragment length is automatically reduced to 50% in order to increase the likelihood of finding a target region of good quality. For example, in cases where all exons are shorter than the user-defined fragment length, the fraction of intron sequence would be high, leading to the automatic reduction of optimal fragment length to 50% of the original length. This procedure is repeated as long as no region above the quality cut-off is found, and the reduced length is still longer than the minimal fragment length. However, if none of the target regions reach the quality threshold, a region of the original optimal length will be selected.After defining the optimal region within a gene, GenomePRIDE computes both PCR primers independently. The optimal position of a primer is hereby defined by the boundaries of the previously selected target region, aiming to amplify a fragment of optimal length. Similar to the PRIDE software used for sequencing, the design of a single primer using GenomePRIDE is based on the evaluation of the thermodynamic stability, strength of the most stable secondary binding site, formation of primer dimers, and the position of the primer (now with respect to the preselected target). The evaluation of potential secondary binding sites of each primer not only includes all exons and introns of the respective gene, but also includes the sequences flanking the gene. We used the default value of 10 kb for the length of these flanking regions, which is generally sufficient to avoid the design of primers that may give rise to a secondary PCR product. Primers were synthesized by Eurogentec (Seraing, Belgium).
PCR amplification
PCR amplifications were performed in 96-well microtiter plates. The fragments for all genes of the Heidelberg Collection R1 were initially PCR-amplified from genomic DNA of D. melanogaster (OregonR) using gene-specific primers, all of which contained one of 10 different, unique tag sequences of 15 nucleotide length at their 5' ends. Subsequent re-amplification was carried out using the matching tag-primer pairs. The first PCR round was performed in 50 μl reactions containing 100 ng of genomic DNA, 1x QIAGEN PCR buffer (1.5 mM MgCl2, Qiagen, Hilden, Germany), 40 μM each dNTP, 1U QIAGEN Taq polymerase (Qiagen, Hilden, Germany) and 20 pmol of each primer. The plates were incubated for 5 min at 94°C, followed by 35 cycles of denaturation at 94°C for 30 s, annealing for 30 s and elongation at 72°C for 90 s. The annealing temperature was lowered during the first 10 cycles from 65°C to 55°C to increase specificity of the amplification. In the last 20 cycles the elongation time was prolonged by 5 s in each cycle to compensate for decreasing enzymatic activity. Re-amplification was initiated by inoculating a 100 μl PCR reaction with 1 μl of first round reaction containing 1x QIAGEN PCR buffer (1.5 mM MgCl2, Qiagen, Hilden, Germany), 0.1 mM each dNTP, 2U QIAGEN Taq polymerase (Qiagen, Hilden, Germany) and 50 pmol of each primer. The plates were incubated for 5 min at 94°C, followed by 35 cycles of denaturation at 94°C for 30 s, annealing for 30 s at 60°C and elongation at 72°C for 90 s. In the last 20 cycles the elongation time was prolonged by 5 s in each cycle to compensate for decreasing enzymatic activity. Amplification success for first and second PCRs was checked on 1% agarose gels and negative PCRs (incorrect size, no product, several bands) were repeated.
Microarray construction
The second round PCR-product was spotted in 3x SSC, 150 mM NaPO4, 1.5 M betaine onto QMT Amino slides (Quantifoil, Jena, Germany) using a MicroGrid II arrayer (BioRobotics, Cambridge, UK) and SMP3 pins (TeleChem International Inc., Sunnyvale, USA). Each PCR-product was spotted twice at different positions on the microarray. As controls, PCR-products of Arabidopsis cDNAs, genomic Drosophila DNA and C. elegans cDNAs were spotted. The DNA was UV-crosslinked (250 mJ/cm2) and baked for 4 h at 80°C. In total, the Heidelberg FlyArray contains 47,616 features, representing the 21,306 ORF-amplicons and 2,502 controls.
Sample collection
Embryo samples were collected as four-hour egg lays, which were allowed to develop for the desired interval and then snap frozen in liquid nitrogen. A small aliquot was DAPI-stained and inspected for correct staging. The different larval stages as well as the different pupal stages were handpicked and separately snap frozen. Adults were collected as male and female flies and also snap frozen. Total RNA was isolated from all samples using the Trizol reagent (Invitrogen, Karlsruhe, Germany). The concentration and quality was analyzed by separating the samples in an RNA 6000 Nano Assay on the Agilent Bioanalyzer 2100 (Agilent Technologies, Palo Alto, USA). At least three different RNA preparations were pooled for each developmental stage. For larval stage sample, equal amounts of the original pools made from the three separate larval stages were mixed. Likewise, for adult stages, equal amounts of male and female pools were mixed.
Microarray hybridization
At least three independent experiments were performed for each of the eight different stages (embryonic stage 4-8 h, embryonic stage 8-12 h, embryonic stage 12-16 h, pooled larval stage, pupal stage I, pupal stage II, pupal stage III and adult stage) in competitive hybridizations against the common control, which was a sample made from RNA isolated from embryonic stage 0-4 h. At least one dye swap was included in each of the repetitions. We used the indirect labeling method, with 9 μg random hexamer primer (Invitrogen, Karlsruhe, Germany) added to 20 μg total RNA. First-strand cDNA synthesis was performed using 1x dNTP-Mix (25 mM dATP, dCTP, dGTP, 15 mM dTTP, 10 mM amino-allyl-dUTP (Sigma, Heidelberg, Germany), 400 U Superscript II RT (Invitrogen, Karlsruhe, Germany), 1x first-strand buffer (Invitrogen, Karlsruhe, Germany), 0.1 M DTT and incubated at 42°C for 2 h. After purification with QIAquick columns (Qiagen, Hilden, Germany), the cDNA was eluted in 1 M KPO4, pH 8.5, and dried. Then, Cy3 or Cy5 dye-esters (Amersham Pharmacia Biotech, Freiburg, Germany) in 4.5 μl DMSO (Fluka, Buchs, Switzerland) were added to the cDNA in 0.1 M Na2CO3 and incubated in the dark at room temperature for 2 h. After purification on QIAquick columns, the concentration of the labeled cDNA and the dye incorporation rate were determined. Pre-hybridization of the QMT Amino slides was done in 1% BSA, 5x SSC, 0.1% SDS at 55°C for 45 min in order to block the amino-coated glass surface. For hybridization, the labeled cDNAs were taken up in SlideHyb buffer #1 (Ambion, Woodward, USA), denatured at 95°C for 5 min, and applied to the array. Hybridization was performed in appropriate chambers (TeleChem) in a waterbath at 55°C for 16 h. Washing of the slides after hybridization was performed according to the manufacturer's instructions.
Filtering and data acquisition
The hybridized glass slides were scanned on a ScanArray 5000 (Perkin Elmer, Wellesley, USA) using the ScanArray software (Version 3.1). Directly fluorescence-labeled external controls spotted on the array were used as a reference for the alignment of laser and photomultiplier (PMT) settings. The resulting images (TIFF format, 16-bit grayscale) for each channel-Cy5 (632 nm) and Cy3 (532 nm)-were analyzed further with the GenePix software (Version 4.0; Axon Instruments, Union City, USA). The images were combined and quantified giving rise to the results files (gpr-format). These files contain information about the gene names and/or clone IDs, linked this information to the respective microarray features and the relevant signal intensities.
A first quality filter was applied directly after scanning the array. As each microarray contains two replicates for each gene, we used the standard deviation (SD) of their dye-ratio (Cy5/Cy3) to filter for reproducible hybridizations. Only if at least 30% of all genes on the array had a SD of log(632/532) of less than one third between the replicates, was the microarray included for further analysis. The raw data of these experiments were analyzed further using the M-CHiPS software package [[36](/articles/10.1186/gb-2003-5-1-r3#ref-CR36 "Microarray data warehouse and analysis tools. [ http://www.mchips.org
]"), [37](/articles/10.1186/gb-2003-5-1-r3#ref-CR37 "Fellenberg K, Hauser NC, Brors B, Hoheisel JD, Vingron M: Microarray data warehouse allowing for inclusion of experiment annotations in statistical analysis. Bioinformatics. 2002, 18: 423-433. 10.1093/bioinformatics/18.3.423.")\]. Linear regression normalization was used, which calculates the 5% quantile of each hybridization as additive offset. Since we used intensities corrected by background subtraction, the mean intensity of each channel subtracted by the respective background value (background mean × number of feature pixels) was extracted for normalization. Subsequently, the data had to be filtered. In M-CHiPS, the filter criteria intensity threshold, reproducibility and ratio of the fitted intensities were applied to select for differentially regulated genes.Correspondence analysis
Correspondence analysis (CA) is an exploratory projection method which displays the associations between genes and hybridizations in a plot diagram. It is well suited for analyzing large data sets - representing transcription intensities together with the corresponding hybridizations in one high dimensional space [19]. As a consequence, the quality of hybridizations in multiconditional experiments can be validated when repeated hybridizations from one condition (for example, 'developmental stage') form clusters. Genes with similar expression profiles have small distances in the plot and are associated with these clusters. Eventually, 24 hybridizations that meet the conditions mentioned above were chosen for the analysis, also performed with the M-CHiPS package. The log2 ratio for each gene was calculated dividing each stage median intensity by the median-fitted universal control intensity for further analysis.
RT-PCR
For an independent validation of the expression profiling data we re-used our original primer set and selected several complete 96-well microtiter plates enriched in primer pairs specific for Heidelberg unique predictions. Although this selection is only semi-random, as the resulting amplicons are ordered along the chromosome, no specific bias may be expected. RNA was isolated as described above for Microarray Hybridization. For RT-PCR, RNA from all stages was pooled. Contaminating genomic DNA was removed by digestion of 20 μg pooled RNA in a 50 μl reaction containing 1x NEB restriction buffer 2, 50U RNasin (Promega, Mannheim, Germany) and 25U RNase-free DNase I (Roche Diagnostics, Mannheim, Germany) for 1 h at 37°C. After phenol-chloroform and chloroform extraction and precipitation with 2.5vol of ethanol, RNA was re-dissolved at 1 μg/μl and used for reverse transcription. One microgram of RNA was incubated with 30 pmol random primers (Roche Diagnostics, Mannheim, Germany) at 65°C for 10 min, chilled on ice, and supplemented with 2 μl of 100 mM DTT, 2 μl of dNTPs (10 mM each, Roche Diagnostics, Mannheim, Germany), 10U of RNasin (Promega, Mannheim, Germany), 4 μl of 5x Expand RT-buffer (Roche Diagnostics, Mannheim, Germany), and 50U of Expand reverse transcriptase (Roche Diagnostics, Mannheim, Germany) to a final reaction volume of 20 μl. After 10 min at 30°C, the reaction was incubated for 1 h at 42°C followed by 10 min at 55°C. PCR was performed in a 50 μl reaction inoculated with 1 μl of the RT reaction containing 1x QIAGEN PCR buffer (1.5 mM MgCl2, Qiagen, Hilden, Germany), 0.4 mM each dNTP, 2U QIAGEN Taq polymerase (Qiagen, Hilden, Germany) and 100 pmol of each primer. The plates were incubated for 5 min at 94°C, followed by 35 cycles of denaturation at 94°C for 30 s, annealing for 30 s and elongation at 72°C for 90 s. The annealing temperature was lowered during the first ten cycles from 65°C to 55°C to increase specificity of the amplification. In the last 20 cycles the elongation time was prolonged by 5 s in each cycle to compensate for decreasing enzymatic activity. For all RT-PCRs, a control lacking reverse transcriptase addition (RT-) to detect contamination with genomic DNA was included. PCR amplification success was checked on 1% agarose gels and only scored if the RT-control was negative.
_In situ_hybridization
A subset of genes was verified by in situ hybridization provided that our amplicon was in a size range from 150 bp to 1.5 kb and the gene showed expression during embryonic stages as measured by the microarray. RNA probes were generated based on the amplicons of the Heidelberg Collection. The necessary T7 promoter site was added by re-amplification of the first PCR products from the microarray set using tag-primers containing the T7 recognition site. In vitro transcription was performed in 96-well plates by adding 1 μg of the purified PCR product to a mixture of 10 mM NTPs, 3.5 mM digoxigenin-11-UTP, 1x transcription buffer (Roche Diagnostics, Mannheim, Germany), 20U RNasin (Promega, Mannheim, Germany), and 40U of T7 Polymerase (Roche Diagnostics, Mannheim, Germany). After overnight incubation, 40U DNAse (Roche Diagnostics, Mannheim, Germany) were added and probes were incubated another 20 min, followed by a LiCl precipitation. Washed pellets were resuspended in 100 μl of 50% formamide in H2O. Success of the in vitro transcription was tested by gel electrophoresis. OregonR embryos from overnight lays were collected and supplemented by 0-4 h collections to give an even distribution of all stages. Embryos were then dechorionated, devitellinized, fixed and stored in methanol at -20°C. Re-hydrated and post-fixed embryos were incubated for 1 h in hybridization buffer (50% formamide, 5x SSC, 100 μg/ml herring-sperm DNA, 50 μg/ml heparin, 0.1% Tween20). 50 μl of embryos were placed in a 1.5 ml tube. 10 μl of the Digoxigenin-labelled RNA probe was added and the embryos were incubated overnight at 60°C. Embryos were then washed four times with pre-hybridization buffer (50% formamide, 5x SSC, pH 5) at 60°C, rinsed three times with PBT (PBS, 0.1% Tween20) at room temperature, and then washed four times with PBT for 10 min each. 100 ml of preabsorbed antibody (anti-DIG Fab Fragment, AP coupled, Roche Diagnostics, Mannheim, Germany, 1:200 diluted in PBT/5% goat serum) was added and embryos were incubated for 90 minutes. Following three rinses and three 10 min washes in PBT, followed by two rinses in AP buffer (50 mM MgCl2, 100 mM NaCl, 20 mM Tris pH 9.5), the NBT/BCIP color substrates were used to detect the hybridized probes. After staining was complete, embryos were washed in PBT three times followed by three washes in 100% ethanol. For inspection, embryos were mounted in 70% glycerol in PBS.
Mapping and comparison of sequence features
All genome sequence related data are presented in the Generic Genome Browser web interface (GBrowse) [27, [28](/articles/10.1186/gb-2003-5-1-r3#ref-CR28 "The Heidelberg FlyArray GBrowser. [ http://HDFlyArray.zmbh.uni-heidelberg.de/cgi-bin/gbrowse
]")\]. The positional information of the underlying data (BDGP Release 2 and FlyBase Release 3.1 \[[38](/articles/10.1186/gb-2003-5-1-r3#ref-CR38 "Berkeley Drosophila Genome Project. [
http://www.fruitfly.org/
]")\], _D. pseudoobscura_ \[[13](/articles/10.1186/gb-2003-5-1-r3#ref-CR13 "Drosophila pseudoobscura Genome Project. [
http://www.hgsc.bcm.tmc.edu/projects/drosophila/
]")\], P insertions \[[39](/articles/10.1186/gb-2003-5-1-r3#ref-CR39 "BDGP Download Miscellaneous Drosophila Sequences. [
http://www.fruitfly.org/sequence/dlMisc.shtml
]")\], _A. gambiae_ \[[30](/articles/10.1186/gb-2003-5-1-r3#ref-CR30 "Exofish between the genome of Anopheles and the genome of Drosophila. [
http://www.genoscope.cns.fr/externe/Fly/
]")\]) was either extracted directly from the respective sources or was generated by comparing the appropriate DNA sequences against the genomic sequence using NCBI-BLAST (default settings, minimal score: 80). In case of multiple hits the best match defined the position of the respective feature within the genome. For _D. pseudoobscura_ we modified the BLAST options (q = 1) in order to detect also matches down to 50% identity. All mapping information was translated into gff-format. The overlap between different sequence features was computed based on these gff-files by counting the common base pairs. The search for protein domains was performed via an Interpro (Release 6.2) scan.Accession numbers
The microarray data described in this article have been submitted to the GEO data library under the accession numbers GPL517, GSM10917-GSM10940. The validated predictions described in this paper have been submitted to the TPA data library at GenBank under the accession numbers BK001800-BK003945.
Additional data files
All primary data are available, including the original Heidelberg Prediction (Additional data file 1, in fasta-format); the Heidelberg Primer set (Additional data file 2) and all microarray result files (gpr-format) as well as resulting data sets such as the Heidelberg Collection R1 and R2; correspondence analysis results; RT-PCR and in situ hybridization results (all included in Additional data file 3 and Additional data file 4); and the gff-files used for setting up the GBrowse website.
References
- Hogenesch JB, Ching KA, Batalov S, Su AI, Walker JR, Zhou Y, Kay SA, Schultz PG, Cooke MP: A comparison of the Celera and Ensembl predicted gene sets reveals little overlap in novel genes. Cell. 2001, 106: 413-415.
Article PubMed CAS Google Scholar - Daly MJ: Estimating the human gene count. Cell. 2002, 109: 283-284.
Article PubMed CAS Google Scholar - Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al: The genome sequence of Drosophila melanogaster. Science. 2000, 287: 2185-2195. 10.1126/science.287.5461.2185.
Article PubMed Google Scholar - Karlin S, Bergman A, Gentles AJ: Genomics. Annotation of the Drosophila genome. Nature. 2001, 411: 259-260. 10.1038/35077152.
Article PubMed CAS Google Scholar - Gopal S, Schroeder M, Pieper U, Sczyrba A, Aytekin-Kurban G, Bekiranov S, Fajardo JE, Eswar N, Sanchez R, Sali A, et al: Homology-based annotation yields 1,042 new candidate genes in the Drosophila melanogaster genome. Nat Genet. 2001, 27: 337-340. 10.1038/85922.
Article PubMed CAS Google Scholar - Andrews J, Bouffard GG, Cheadle C, Lu J, Becker KG, Oliver B: Gene discovery using computational and microarray analysis of transcription in the Drosophila melanogaster testis. Genome Res. 2000, 10: 2030-2043. 10.1101/gr.10.12.2030.
Article PubMed CAS PubMed Central Google Scholar - Posey KL, Jones LB, Cerda R, Bajaj M, Huynh T, Hardin PE, Hardin SH: Survey of transcripts in the adult Drosophila brain. Genome Biol. 2001, 2: research0008.1-0008.16. 10.1186/gb-2001-2-3-research0008.
Article Google Scholar - Morin X, Daneman R, Zavortink M, Chia W: A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc Natl Acad Sci USA. 2001, 98: 15050-15055. 10.1073/pnas.261408198.
Article PubMed CAS PubMed Central Google Scholar - Misra S, Crosby MA, Mungall CJ, Matthews BB, Campbell KS, Hradecky P, Huang Y, Kaminker JS, Millburn GH, Prochnik SE, et al: Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome Biol. 2002, 3: research0083.1-0083.22. 10.1186/gb-2002-3-12-research0083.
Article Google Scholar - Reese MG, Kulp D, Tammana H, Haussler D: Genie - gene finding in Drosophila melanogaster. Genome Res. 2000, 10: 529-538. 10.1101/gr.10.4.529.
Article PubMed CAS PubMed Central Google Scholar - Burge C, Karlin S: Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997, 268: 78-94. 10.1006/jmbi.1997.0951.
Article PubMed CAS Google Scholar - Holt RA, Subramanian GM, Halpern A, Sutton GG, Charlab R, Nusskern DR, Wincker P, Clark AG, Ribeiro JM, Wides R, et al: The genome sequence of the malaria mosquito Anopheles gambiae. Science. 2002, 298: 129-149. 10.1126/science.1076181.
Article PubMed CAS Google Scholar - Drosophila pseudoobscura Genome Project. [http://www.hgsc.bcm.tmc.edu/projects/drosophila/]
- Shoemaker DD, Schadt EE, Armour CD, He YD, Garrett-Engele P, McDonagh PD, Loerch PM, Leonardson A, Lum PY, Cavet G, et al: Experimental annotation of the human genome using microarray technology. Nature. 2001, 409: 922-927. 10.1038/35057141.
Article PubMed CAS Google Scholar - Rubin GM, Hong L, Brokstein P, Evans-Holm M, Frise E, Stapleton M, Harvey DA: A Drosophila complementary DNA resource. Science. 2000, 287: 2222-2224. 10.1126/science.287.5461.2222.
Article PubMed CAS Google Scholar - Salamov AA, Solovyev VV: Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000, 10: 516-522. 10.1101/gr.10.4.516.
Article PubMed CAS PubMed Central Google Scholar - Haas SA, Beissbarth T, Rivals E, Krause A, Vingron M: GeneNest: automated generation and visualization of gene indices. Trends Genet. 2000, 16: 521-523. 10.1016/S0168-9525(00)02116-8.
Article PubMed CAS Google Scholar - Haas SA, Hild M, Wright APH, Hain T, Talibi D, Vingron M: Genome-scale design of PCR primers and long oligomers for DNA microarrays. Nucleic Acids Res. 2003, 31: 5576-5581. 10.1093/nar/gkg752.
Article PubMed CAS PubMed Central Google Scholar - Fellenberg K, Hauser NC, Brors B, Neutzner A, Hoheisel JD, Vingron M: Correspondence analysis applied to microarray data. Proc Natl Acad Sci USA. 2001, 98: 10781-10786. 10.1073/pnas.181597298.
Article PubMed CAS PubMed Central Google Scholar - Arbeitman MN, Furlong EE, Imam F, Johnson E, Null BH, Baker BS, Krasnow MA, Scott MP, Davis RW, White KP: Gene expression during the life-cycle of Drosophila melanogaster. Science. 2002, 297: 2270-2275. 10.1126/science.1072152.
Article PubMed CAS Google Scholar - White KP, Rifkin SA, Hurban P, Hogness DS: Microarray analysis of Drosophila development during metamorphosis. Science. 1999, 286: 2179-2184. 10.1126/science.286.5447.2179.
Article PubMed CAS Google Scholar - Furlong EE, Andersen EC, Null B, White KP, Scott MP: Patterns of gene expression during Drosophila mesoderm development. Science. 2001, 293: 1629-1633. 10.1126/science.1062660.
Article PubMed CAS Google Scholar - Tomancak P, Beaton A, Weiszmann R, Kwan E, Shu S, Lewis SE, Richards S, Ashburner M, Hartenstein V, Celniker SE, Rubin GB: Systematic determination of patterns of gene expression during Drosophila embryogenesis. Genome Biol. 2002, 3: research0088.1-0088.14. 10.1186/gb-2002-3-12-research0088.
Article Google Scholar - The Heidelberg Prediction in situ patterns. [http://HDFlyArray.zmbh.uni-heidelberg.de/cgi-bin/insitu.pl]
- Stapleton M, Liao G, Brokstein P, Hong L, Carninci P, Shiraki T, Hayashizaki Y, Champe M, Pacleb J, Wan K, et al: The Drosophila gene collection: identification of putative full-length cDNAs for 70% of D. melanogaster genes. Genome Res. 2002, 12: 1294-1300. 10.1101/gr.269102.
Article PubMed PubMed Central Google Scholar - Reboul J, Vaglio P, Rual JF, Lamesch P, Martinez M, Armstrong CM, Li S, Jacotot L, Bertin N, Janky R, et al: C. elegans ORFeome version 1.1: experimental verification of the genome annotation and resource for proteome-scale protein expression. Nat Genet. 2003, 34: 35-41. 10.1038/ng1140.
Article PubMed Google Scholar - Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A, et al: The generic genome browser: a building block for a model organism system database. Genome Res. 2002, 12: 1599-1610. 10.1101/gr.403602.
Article PubMed CAS PubMed Central Google Scholar - The Heidelberg FlyArray GBrowser. [http://HDFlyArray.zmbh.uni-heidelberg.de/cgi-bin/gbrowse]
- Jaillon O, Dossat C, Eckenberg R, Eiglmeier K, Segurens B, Aury JM, Roth CW, Scarpelli C, Brey PT, Weissenbach J, Wincker P: Assessing the Drosophila melanogaster and Anopheles gambiae genome annotations using genome-wide sequence comparisons. Genome Res. 2003, 13: 1595-1599. 10.1101/gr.922503.
Article PubMed CAS PubMed Central Google Scholar - Exofish between the genome of Anopheles and the genome of Drosophila. [http://www.genoscope.cns.fr/externe/Fly/]
- The FlyBase Consortium: The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res. 2003, 31: 172-175. 10.1093/nar/gkg094.
Article Google Scholar - Krause A, Haas SA, Coward E, Vingron M: SYSTERS, GeneNest, SpliceNest: exploring sequence space from genome to protein. Nucleic Acids Res. 2002, 30: 299-300. 10.1093/nar/30.1.299.
Article PubMed CAS PubMed Central Google Scholar - Coward E, Haas SA, Vingron M: SpliceNest: visualizing gene structure and alternative splicing based on EST clusters. Trends Genet. 2002, 18: 53-55. 10.1016/S0168-9525(01)02525-2.
Article CAS Google Scholar - Krause A, Stoye J, Vingron M: The SYSTERS protein sequence cluster set. Nucleic Acids Res. 2000, 28: 270-272. 10.1093/nar/28.1.270.
Article PubMed CAS PubMed Central Google Scholar - GenomePride. [http://pride.molgen.mpg.de]
- Microarray data warehouse and analysis tools. [http://www.mchips.org]
- Fellenberg K, Hauser NC, Brors B, Hoheisel JD, Vingron M: Microarray data warehouse allowing for inclusion of experiment annotations in statistical analysis. Bioinformatics. 2002, 18: 423-433. 10.1093/bioinformatics/18.3.423.
Article PubMed CAS Google Scholar - Berkeley Drosophila Genome Project. [http://www.fruitfly.org/]
- BDGP Download Miscellaneous Drosophila Sequences. [http://www.fruitfly.org/sequence/dlMisc.shtml]
Acknowledgements
We would like to thank A. Kiger, S. Armknecht, K. Kerr and N. Perrimon for sharing information before publication and for a stimulating and open collaboration over the course of this project. We are grateful to M. Leptin for help in analyzing the in situ hybridization patterns. Further we are indebted to S Hagar and J Werdermann for excellent technical support and J. Jerecic and L. Ringrose for comments on the manuscript. This work was supported by a grant from the BMBF-funded German Human Genome Project to F.S., J.D.H. and R.P.
Author information
Author notes
Authors and Affiliations
- Zentrum für Molekulare Biologie Heidelberg (ZMBH), University of Heidelberg, Im Neuenheimer Feld 282, 69120, Heidelberg, Germany
M Hild, B Koch, F Sauer & R Paro - Division of Functional Genome Analysis, Deutsches Krebsforschungszentrum (DKFZ), Im Neuenheimer Feld 580, 69120, Heidelberg, Germany
B Beckmann, C Busold, K Fellenberg & JD Hoheisel - Max Planck Institute for Molecular Genetics, Ihnestraße 73, 14195, Berlin, Germany
SA Haas & M Vingron - Softberry, Inc., 116 Radio Circle, Suite 400, Mount Kisko, NY, 10549, USA
V Solovyev - Deutsches Krebsforschungszentrum (DKFZ), Im Neuenheimer Feld 580, 69120, Heidelberg, Germany
M Boutros - Department of Biochemistry, University of California, Riverside, CA, 92521, USA
F Sauer
Authors
- M Hild
You can also search for this author inPubMed Google Scholar - B Beckmann
You can also search for this author inPubMed Google Scholar - SA Haas
You can also search for this author inPubMed Google Scholar - B Koch
You can also search for this author inPubMed Google Scholar - V Solovyev
You can also search for this author inPubMed Google Scholar - C Busold
You can also search for this author inPubMed Google Scholar - K Fellenberg
You can also search for this author inPubMed Google Scholar - M Boutros
You can also search for this author inPubMed Google Scholar - M Vingron
You can also search for this author inPubMed Google Scholar - F Sauer
You can also search for this author inPubMed Google Scholar - JD Hoheisel
You can also search for this author inPubMed Google Scholar - R Paro
You can also search for this author inPubMed Google Scholar
Corresponding authors
Correspondence toM Vingron, F Sauer, JD Hoheisel or R Paro.
Additional information
M Hild, B Beckmann, SA Haas contributed equally to this work.
Electronic supplementary material
Authors’ original submitted files for images
Rights and permissions
About this article
Cite this article
Hild, M., Beckmann, B., Haas, S. et al. An integrated gene annotation and transcriptional profiling approach towards the full gene content of the Drosophila genome.Genome Biol 5, R3 (2003). https://doi.org/10.1186/gb-2003-5-1-r3
- Received: 22 August 2003
- Revised: 13 October 2003
- Accepted: 19 November 2003
- Published: 22 December 2003
- DOI: https://doi.org/10.1186/gb-2003-5-1-r3