Moving toward a system genetics view of disease (original) (raw)
Abstract
Testing hundreds of thousands of DNA markers in human, mouse, and other species for association to complex traits like disease is now a reality. However, information on how variations in DNA impact complex physiologic processes flows through transcriptional and other molecular networks. In other words, DNA variations impact complex diseases through the perturbations they cause to transcriptional and other biological networks, and these molecular phenotypes are intermediate to clinically defined disease. Because it is also now possible to monitor transcript levels in a comprehensive fashion, integrating DNA variation, transcription, and phenotypic data has the potential to enhance identification of the associations between DNA variation and diseases like obesity and diabetes, as well as characterize those parts of the molecular networks that drive these diseases. Toward that end, we review methods for integrating expression quantitative trait loci (eQTLs), gene expression, and clinical data to infer causal relationships among gene expression traits and between expression and clinical traits. We further describe methods to integrate these data in a more comprehensive manner by constructing coexpression gene networks that leverage pairwise gene interaction data to represent more general relationships. To infer gene networks that capture causal information, we describe a Bayesian algorithm that further integrates eQTLs, expression, and clinical phenotype data to reconstruct whole-gene networks capable of representing causal relationships among genes and traits in the network. These emerging network approaches, aimed at processing high-dimensional biological data by integrating data from multiple sources, represent some of the first steps in statistical genetics to identify multiple genetic perturbations that alter the states of molecular networks and that in turn push systems into disease states. Evolving statistical procedures that operate on networks will be critical to extracting information related to complex phenotypes like disease, as research goes beyond a single-gene focus. The early successes achieved with the methods described herein suggest that these more integrative genomics approaches to dissecting disease traits will significantly enhance the identification of key drivers of disease beyond what could be achieved by genetic association studies alone.
Similar content being viewed by others
Introduction
Genetics is at the dawn of a new era with maturing technologies that enable low-cost, high-throughput genotyping of hundreds of thousands of DNA markers that in turn can be tested for association to complex traits of interest like disease and drug response. A number of studies have already leveraged the availability of such technologies to identify polymorphisms in genes that associate with diseases like age-related macular degeneration (Edwards et al. 2005; Haines 2005; Klein 2005), diabetes (Grant 2006; Sladek 2007), and obesity (Herbert 2006), to name just a few. In addition, there are scores of similar genome-wide association studies that are ongoing and that promise to deliver scores of genes that harbor variations that associate with diseases like obesity and diabetes. While these types of genetic discoveries provide a peek into pathways that underlie disease, they are usually devoid of context, so that elucidating the functional role such genes play in disease can take years, or even decades, as has been the case for ApoE, an Alzheimer’s susceptibility gene identified nearly 15 years ago (Peacock et al. 1993).
Information that defines how variations in DNA that associate with disease actually impact the complex physiologic processes underlying disease flows through transcriptional and other molecular, cellular, tissue, and organism networks (Fig. 1). In the past the ability to comprehensively assess intermediate phenotypes that comprise the hierarchy of networks that drive disease was not possible. However, today DNA microarrays have radically changed the way we study genes, enabling a more comprehensive look at the role they play in everything from the regulation of normal cellular processes to complex diseases like obesity and diabetes. In their typical use, microarrays allow researchers to screen thousands of genes for differences in expression or differences in how genes are connected in molecular networks (Schadt and Lum 2006) between experimental conditions of interest. These data are often used to discover genes that differ between normal and disease-associated tissue, to model and predict continuous or binary measures, to predict patient survival, and to classify disease or tumor subtypes. Because gene expression levels in a given sample are measured simultaneously, researchers are able to identify genes whose expression levels are correlated, implying an association under specific conditions or more generally.
Fig. 1
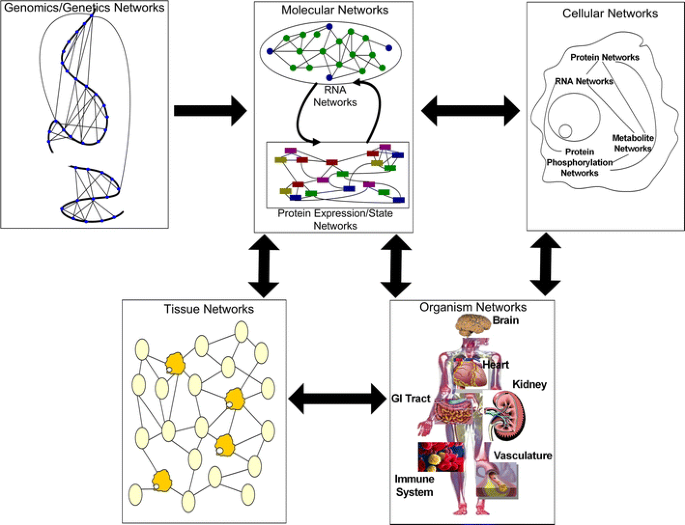
High-level view of the flow of information in biological systems through a hierarchy of networks. Each panel highlights a different set of networks at play in a biological system. Genomics networks represent interactions among DNA sequences that may give rise to longer-range as well as more local chromosome structures that modulate gene activity, in addition to inducing synergistic effects on higher-order phenotypes. Genomics networks drive molecular networks composed of RNA, protein, metabolites, and other molecules in the system. Molecular networks are components of cellular networks in which the complex web of interactions among these networks gives rise to the complex phenotypes that define living systems. Tissue networks comprise cellular networks that are clearly influenced by the molecular and genomics networks, and organism networks comprise tissue networks that are clearly defined by the component cellular and molecular networks. Complex phenotypes like disease emerge from this complex web of interacting networks, given genetic and environmental perturbations to the system
Integrating genetic and functional genomic data can provide a path to inferring causal associations between genes and disease. In the past, causal associations between genes and traits have been investigated using time series experiments, gene knockouts or transgenics that overexpress a gene of interest, RNAi-based knockdown or viral-mediated overexpression of genes of interest, and chemical activation or inhibition of genes of interest. A more systematic and arguably relevant source of perturbation to make such inferences regarding genes and disease are DNA polymorphisms, where gene expression and other molecular phenotypes in a number of species have been shown to be significantly heritable and at least partially under the control of specific genetic loci (Brem et al. 2002; DeCook et al. 2006; Hubner et al. 2005; Jin et al. 2001; Klose et al. 2002; Monks et al. 2004; Morley et al. 2004; Oleksiak et al. 2002; Schadt et al. 2003; Stranger et al. 2005). By examining the effects that naturally occurring variations in DNA have on variations in gene expression traits in human or experimental populations, other phenotypes (including disease) can be examined with respect to these same DNA variations and ultimately ordered with respect to genes to infer causal control (Fig. 2) (Kulp and Jagalur 2006; Lum et al. 2006; Mehrabian et al. 2005; Schadt et al. 2005). The power of this integrative genomics strategy rests in the molecular processes that transcribe DNA into RNA and then RNA into protein, so that information on how variations in DNA impact complex physiologic processes often flows directly through transcriptional networks. As a result, integrating DNA variation, transcription, and phenotypic data has the potential to enhance identification of the associations between DNA variation and disease and characterize those parts of the molecular networks that drive disease.
Fig. 2
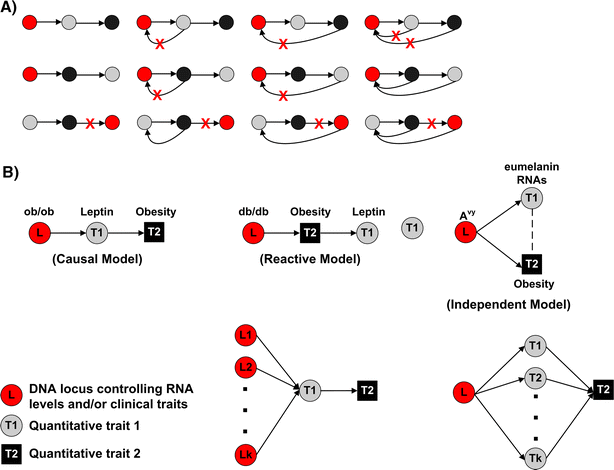
Possible relationships between phenotypes with and without genetic information. Edges between nodes in each of the graphs represent an association between the nodes. A directed edge indicates a causal association between the nodes. A A subset of the number of possible relationships between three variables. In the case where one of the three nodes in the network is a DNA locus (red nodes), many of the graphs are no longer possible, given that directed edges from expression trait to DNA locus are not possible. The red Xs highlight edges that would not be allowed if the red node were a DNA locus. B The first three graphs represent the set of possible relationships between two traits and a controlling genetic locus when feedback mechanisms are ignored. The final two graphs represent more complicated scenarios in which multiple genetic loci control a given trait that in turn drives a second trait or a single genetic locus drives multiple traits that collectively drive another trait
Here we review different approaches for integrating expression quantitative trait loci (eQTLs), expression, and clinical data to infer causal relationships among gene expression traits and between expression and disease traits. We further review methods to integrate these data in a more comprehensive manner by constructing coexpression gene networks that leverage pairwise gene interaction data to represent more general relationships. This type of network provides a useful construct for characterizing the topologic properties of biological networks and for partitioning such networks into functional units (modules) that underlie complex phenotypes like disease. However, these networks are, by design, undirected and so do not explicitly capture causal relationships among genes. To infer gene networks that capture causal information, we review Bayesian network reconstruction algorithms that, like the methods operating on only two or three expression traits and/or clinical traits mentioned above, integrate eQTLs, expression, and clinical phenotype data to reconstruct whole-gene networks capable of representing direction along the edges of the network. Here, directionality among the edges corresponds to causal relationships among genes and between genes and clinical phenotypes related to disease. These emerging high-dimensional data analysis approaches that integrate large-scale data from multiple sources represent the first steps in statistical genetics, moving away from considering one trait at a time and toward operating in a network context. Evolving statistical procedures that operate on networks will be critical to extracting information related to complex phenotypes like disease as research goes beyond the single-gene focus. The early successes achieved with some of the methods described herein suggest that these more integrative genomics approaches to dissecting disease traits will significantly enhance the identification of key drivers of disease beyond what could be achieved by genetics alone.
Leveraging the heritability of expression as a path to reconstructing networks
Gene transcripts have been identified that are associated with complex disease phenotypes (Karp et al. 2000; Schadt et al. 2003), are alternatively spliced (Johnson et al. 2003), elucidate novel gene structures (Mural et al. 2002; Schadt et al. 2004; Shoemaker et al. 2001), can serve as biomarkers of disease or drug response (DePrimo et al. 2003), lead to the identification of disease subtypes (Mootha et al. 2003; Schadt et al. 2003; van’t Veer et al. 2002), and elucidate mechanisms of drug toxicity (Waring et al. 2001). Changes in gene expression often reflect changes in a gene’s activity and the impact a gene has on different phenotypes. Because gene expression is a quantitative trait, linkage and association methods can be directly applied to such traits to identify genetic loci that control them. In turn, genetic loci that control for expression traits may also associate with higher-order phenotypes affected by expression changes in the gene of interest, providing a path to directly identify genes controlling for phenotypes of interest. Therefore, identifying the heritable traits and the extent of their genetic variability provides insight about the evolutionary forces contributing to the changes in expression that associate with biological processes that underlie diseases like obesity and diabetes, beyond what can be gained by looking at the transcript abundance data alone.
It is now well established that gene expression is a significantly heritable trait (Alberts et al. 2005; Brem et al. 2002; Chesler et al. 2005; Cheung et al. 2005; Jansen and Nap 2001; Monks et al. 2004; Morley et al. 2004; Petretto et al. 2006a, b; Schadt et al. 2003, 2005). If a gene expression trait is highly correlated with a disease trait of interest, and if the corresponding gene physically resides in a region of the genome that is associated with the disease trait, then knowing that the expression trait is also genetically linked to a region coincident with its physical location provides an objective and direct path to identify candidate causal genes for the disease trait (Alberts et al. 2005; Brem et al. 2002; Chesler et al. 2005; Cheung et al. 2005; Jansen and Nap 2001; Monks et al. 2004; Morley et al. 2004; Petretto et al. 2006a, b; Schadt et al. 2003, 2005). The genetic information therefore enables the dissection of the covariance structure for two traits of interest into genetic and nongenetic components, and the genetic component can then be leveraged to support whether an expression and disease trait are related in a causal, reactive, or independent manner (with respect to the expression trait). Elucidating causal relationships is possible in this setting given the unambiguous flow of information from changes in DNA to changes in RNA and protein function (Fig. 1). That is, given that two traits are linked to the same DNA locus and a few important simplifying assumptions, there are a limited number of ways in which these two traits can be related with respect to a given locus (GuhaThakurta et al. 2006; Schadt 2005; Schadt et al. 2005), whereas in the absence of such genetic information, many indistinguishable relationships would be possible, so that additional data would be required to establish the correct relationships.
Leveraging DNA variation information to reconstruct gene networks supposes that we are able to systematically identify genetic loci that at least partially control transcript abundances for genes of interest. This of course is straightforward given that transcript abundance or gene expression traits are quantitative measures that can be analyzed like any other quantitative trait in a genetics context. However, the difficulty in analysis and interpretation comes with the large number of traits examined. Microarrays are capable of monitoring tens of thousands (or hundreds of thousands) of transcripts simultaneously. Therefore, methods to compute eQTLs must consider computational tractability given the need to run the analyses potentially millions of times. In addition, significance thresholds must take into account multiple testing. Multiple testing issues relate not only to the number of transcripts tested but also to the number of markers or proportion of the genome tested. However, the strong correlation structure that exists among many of the expression traits monitored in a segregating population can be leveraged to enhance the power to detect relationships among genes.
A number of methods have been developed and applied to gene expression traits in segregating populations to identify eQTLs and to establish relationships among genes and between genes and disease traits, where multiple traits at a time can be considered. Typical approaches to the joint analysis of genetic traits involve mapping each gene expression trait individually and inferring the genetic correlation between pairs or sets of expression traits based on pairwise Pearson correlation, eQTL overlaps, and/or tests for pleiotropy. Using a family-based sample, Monks et al. (2004) estimated the genetic correlation between pairs of traits using a bivariate variance-component-based segregation analysis and showed that the genetic correlation was better able to distinguish clusters of genes in pathways than correlations based on the observed expression traits. This type of method can be extended to perform bivariate and multivariate QTL analyses, which can be more highly powered to detect QTLs when traits are correlated. Clusters of correlated gene expression traits can often contain hundreds or thousands of genes, which would be computationally prohibitive in a joint analysis. Kendziorski et al. (2006) approached this problem in a different way by employing a Bayesian mixture model to exploit the increased information from the joint mapping of correlated gene expression traits, which is computationally tractable for large sets of genes. Instead of doing a linkage scan by computing LOD scores at positions along the genome, Kendziorski et al. (2006) computed the posterior probability that a particular gene expression trait maps to marker m for each marker, as well as the posterior probability that the trait maps nowhere in the genome. Nonlinkage in this setting is declared for a transcript if the posterior probability of nonlinkage exceeds a threshold that bounds the posterior expected false discovery rate (FDR). One benefit to this approach is that it controls false discovery for the number of expression traits being tested, whereas assessing the appropriate significance cutoffs in single-transcript linkage analysis often requires data permutation analyses. The drawback of this method is that it assumes that linkage occurs at either one or none of the markers tested and it lacks a well-defined method for the case when multiple eQTLs control an expression trait.
In a study of inbred strain crosses, the only valid way of estimating the extent of genetic control of a given trait is to explicitly model each eQTL, including any epistatic interactions if they exist. Brem and Kruglyak (2005) showed that epistatic interactions were prevalent in the gene expression levels in yeast, and similar suggestions have been made in other species as well (Schadt et al. 2005), but more definitive studies are needed to characterize the extent of epistasis among eQTLs in these other species. In the absence of epistasis, the genetic contribution for each transcript has been estimated by summing contributions for each individual eQTL, assuming that little or no allelic association exists between the eQTLs. In the presence of epistasis, however, this practice cannot yield a valid estimate, and multilocus models are instead required to obtain valid estimates. In addition, multilocus modeling can identify loci contributing to expression traits that would have been missed in single-locus eQTL scans (Brem and Kruglyak 2005; Storey et al. 2005).
Integrating eQTLs and clinical trait linkage mapping to infer causality
While understanding the mechanisms of RNA expression is in itself important for understanding biological processes, the ultimate use of this information is identifying the relationship between variation in expression levels and disease phenotypes in an organism of interest. Microarray experiments are commonly used to explore differential expression between disease and normal tissue samples or between samples from different disease subtypes. These studies are designed to detect association between gene expression and disease-associated traits, which in turn can lead to the identification of biomarkers of disease or disease subtypes. However, in the absence of supporting experimental data, these data alone are not able to distinguish genes that drive disease from those that respond. As discussed above, eQTL mapping can aid traditional clinical trait QTL (cQTL) mapping by narrowing the set of candidate genes underlying a given cQTL peak and by identifying expression traits that are causally associated with the clinical traits.
Expression traits detected as significantly correlated with a clinical phenotype may reflect a causal relationship between the traits, either because the expression trait contributes to, or is causal for, the clinical phenotype, or because the expression trait is reactive to, or a marker of, the clinical phenotype. However, correlation may also exist in cases when the two traits are not causally associated. Two traits may appear correlated due to confounding factors such as tight linkage of causal mutations (Schadt et al. 2005) or may arise independently from a common genetic source. The A y mouse provides an example of correlations between eumelanin RNA levels and obesity phenotypes induced by an allele that acts independently on these different traits, causing both decreased levels of eumelanin RNA and an obesity phenotype. More generally, a clinical and expression traits for a particular gene may depend on the activity of a second gene in such a manner that conditional on the second gene, the clinical and expression traits are independent.
Correlation data alone cannot indicate which of the possible relationships between gene expression traits and a clinical trait are true. For example, given two expression traits and a clinical trait detected as correlated in a population of interest, there are 112 ways to order the traits with respect to one another. If we consider the traits as nodes in a network, then there are five possible ways the traits (or nodes) can be connected: (1) connected by an undirected edge, (2) connected by a directed edge moving left to right, (3) connected by a directed edge moving right to left, (4) connected by a directed edge moving right to left and a directed edge moving left to right, and (5) not connected by an edge. Since there are three pairs of nodes, there are 5 × 5 × 5 = 125 possible graphs. However, because we start with the assumption that the traits are all correlated with one another, we exclude 12 of the 125 possible graphs in which one node is not connected to either of the other two nodes, in addition to excluding the graph in which none of the nodes are connected, leaving us with 112 possible graphs (Fig. 2A). The joint trait distribution induced by these different graphs are often statistically indistinguishable from one another (i.e., they are Markov equivalent, so that their distributions are identical), making it nearly impossible in most cases to infer the true relationship. On the other hand, when the two traits are at least partially controlled by the same genetic locus and when more complicated methods of control (e.g., feedback loops) are ignored, the number of relationships between the QTLs and the two traits of interest can be reduced to three basic models illustrated graphically in Fig. 2B. The dramatic reduction in the number of possible graphs to consider is mainly driven by the fact that changes in DNA drive changes in phenotypes and not vice versa. That is, while it may be possible that changes in RNA or protein lead to changes in DNA at a high enough frequency to detect associations between germ-line transmitted DNA changes and phenotype in segregating populations, it seems extremely unlikely.
It is important to note here that when we use the term causality, it is perhaps meant in a more nonstandard sense than most researchers in the life sciences may be accustomed to. In the molecular biology or biochemistry setting, claiming a causal relationship between, say, two proteins usually means that one protein has been determined experimentally to physically interact with or to induce processes that directly affect another protein and that in turn leads to a phenotypic change of interest. In such instances, an understanding of the causal factors relevant to this activity are known, and careful experimental manipulation of these factors subsequently allows for the identification of genuine causal relationships. However, in the present setting, the term “causal” is used from the standpoint of statistical inference, where statistical associations between changes in DNA, changes in expression (or other molecular phenotypes), and changes in complex phenotypes like disease are examined for patterns of statistical dependency among these variables that allows directionality to be inferred among them, where the directionality then provides the source of causal information (highlighting putative regulatory control as opposed to physical interaction). The graphical models (networks) described here, therefore, are necessarily probabilistic structures that use the available data to infer the correct structure of relationships among genes and between genes and clinical phenotypes (Schadt and Lum 2006). In a single experiment with one time point measurement, these methods cannot easily model more complex regulatory structures that are known to exist, like negative feedback control. However, the methods can be useful in providing a broad picture of correlation and causative relationships, and while the more complex structures may not be explicitly represented in this setting, they are captured nevertheless given that they represent observed states that are reached as a result of more complicated processes like feedback control.
Distinguishing proximal (“_cis_”) eQTL effects from distal (“_trans_”)
All genes expressed in living systems are _cis_-regulated at some level and so are under the control of various _cis_-acting elements such as promoters and TATA boxes (Fig. 3). In this context, expression as a quantitative trait for eQTL mapping presents a unique situation in quantitative trait genetics because the expression trait corresponds to a physical location in the genome (the structural gene that is transcribed, giving rise to the expression trait). The transcription process operates on the structural gene, and so DNA variations in the structural gene that affect transcription will be identified as eQTLs in the mapping process. In such cases eQTLs would be identified as _cis_-acting, given that the most reasonable explanation for seeing an eQTL coincident with the physical location of the gene will be that variations within the gene region itself give rise to variations in its expression (Doss et al. 2005). However, because we cannot guarantee that the eQTL is truly _cis_-acting (i.e., it could arise from variation in a gene that is closely linked to the gene expression trait in question), it is more accurate to refer to such eQTLs as proximal, given that they are close to the gene corresponding to the expression trait. Because the _cis_-regulated components of expression traits are among the most proximal traits in a biological system with respect to the DNA (given that RNA is transcribed from DNA), we might expect that true _cis_-acting genetic variance components of expression traits are among the easiest components to detect via QTL analysis, if they exist. This indeed has been observed in a number of studies in which proximal (presumably _cis_-acting) eQTLs have been identified that explain unprecedented proportions of a trait’s overall variance (several published studies highlight examples where greater than 90% of the overall variation was explained by a single _cis_-acting eQTL) (Brem et al. 2002; Cervino et al. 2005; Cheung et al. 2005; Lum et al. 2006; Monks et al. 2004; Schadt et al. 2003).
Fig. 3
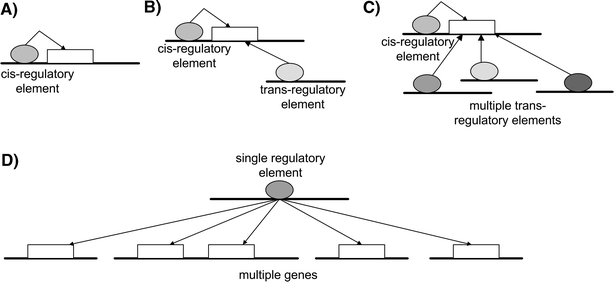
Mapping proximal and distal eQTLs for gene expression traits. The white rectangles represent genes that are controlled by transcriptional units. The ellipses represent the transcriptional control units, which could be transcription regulatory sites, other genes that control the expression of the indicated gene, and so on. A _Cis_-acting control unit acting on a gene. DNA variations in this control unit that affected the gene’s expression would lead to a _cis_-acting (proximal) eQTL. B Cis and trans control units regulating the indicated gene. DNA variations in these control units that affected the gene’s expression would lead to proximal and distal eQTLs. C Cis control unit and multiple trans control units regulating the indicated genes. DNA variations in these control units would lead to a complex eQTL signature for the gene. D A single control unit regulating multiple genes. DNA variations in this single control unit could lead to a cluster of distal eQTLs (an eQTL hot spot)
Variations in expression levels induced by DNA variations in or near the gene itself may in turn induce changes in the expression levels of other genes (Fig. 3). Each of these genes in a population of interest may not harbor any DNA variation in their structural gene so that they do not give rise to true _cis_-acting eQTL, but they nevertheless would give rise to eQTLs that link to the gene region inducing changes in their expression. Therefore, we see that the individual variation in gene expression can be of two fundamental types. The first, termed proximal, often results from DNA variations of a gene that directly influence transcript levels of that gene. The second, termed _trans_-acting or distal, does not involve DNA variations of the gene in question but rather is secondary to alterations of other true _cis_-acting genetic variations (Fig. 3). In reality, variation in expression traits may be due to variation in _cis_-acting elements and/or one or multiple _trans_-acting elements. In addition, master regulators of transcription, which affect the expression of many traits in trans (Fig. 3), may exist, though the evidence on this is not conclusive at this point in all species, given the limited number of studies and small sample sizes for all studies published to date.
In most cases it is not possible to infer the true regulatory effects (i.e., cis vs. trans) of an eQTL without complex bioinformatics study (GuhaThakurta et al. 2006) and experimental validation. As a result, eQTLs have been categorized into proximal and distal types based on the distance between the eQTL and the location of the structural genes. Obviously, if these are on different chromosomes the eQTLs are distal, but if they fall on the same chromosome then they are considered proximal only if the distance between the structural gene and the eQTLs do not exceed some threshold. The exact threshold is a function of the number of meioses and extent of recombination in a given population data set. In a completely outbred population where LD mapping has been used to fine-map the eQTLs, it has been reasonable to require the distance between the proximal eQTL and structural gene to be less than 1 Mb (Cheung et al. 2005). However, in an F2 intercross population constructed from two inbred lines of mice, the extent of LD will be extreme given that all animals are descended from a single F1 founder, with only two meiotic events separating any two mice in the population. In such cases the resolution of linkage peaks is quite low, requiring the threshold of peak-to-physical gene distance to be more relaxed, so that eQTLs that are within 20 or 30 Mb could be potentially _cis_-acting (Doss et al. 2005; Schadt et al. 2003). While the proximal eQTLs provide an easy path to making causal inference, given that the larger effect sizes commonly associated with proximal eQTLs make them easier to detect (Brem et al. 2002; Cervino et al. 2005; Cheung et al. 2005; Lum et al. 2006; Monks et al. 2004; Schadt et al. 2003), the methods discussed above work for distal as well as proximal eQTLs. In fact, if a given gene sits more centrally in a given gene network that drives disease, it may capture a larger percentage of the genetic variation associated with the disease (Fig. 2B), making the gene easier to identify and associate with disease. This was the case in one of the first studies to explicitly leverage DNA and RNA changes to map genes for obesity (Schadt et al. 2005). In that study three genes (C3ar1, Tgfbr2, and Zfp90) were identified and validated as causal for obesity, and in all three cases the QTLs that facilitated identification of the causal association were all distally acting with respect to the expression traits.
More generally leveraging eQTL data to reconstruct gene networks
The classic reductionist view applied to genetics has motivated the identification of single genes associated with disease as one means of getting a foot into disease pathways. However, even in cases where genes are involved in pathways that are well known, it is unclear whether the gene causes disease via the known pathway or whether the gene is involved in other pathways or more complex networks that lead to disease. This was the case with TGFBR2, a recently identified and validated obesity susceptibility gene (Schadt et al. 2005). The classic view of the signaling pathways involving the superfamily of transforming growth factor β (TGF-β) proteins is that TGF-β acts through receptor serine/threonine kinases to phosporylate regulatory proteins of the Smad family, which then move into the nucleus where they bind DNA to activate specific sets of target genes (Alberts 2002) (Fig. 4A). Although the number of biological functions this cascade ultimately impacts is large, the classic pathway is simplistic, involving only a limited number of genes, with little insight provided into the vast network of gene interactions that potentially modulate key players in this pathway.
Fig. 4
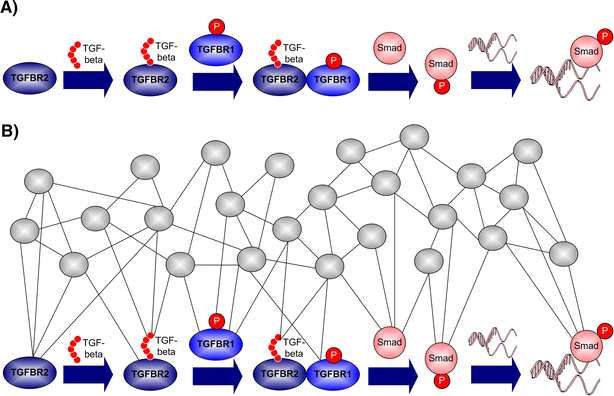
Genes comprising simple linearly ordered pathways operate in a network context. A The classic view of TGF-β signaling (Alberts 2002) involves Tgfbr2 as a key component. Tgfbr2 was recently identified and validated as an obesity susceptibility gene. B The genes comprising the TGF-β signaling pathway are correlated with hundreds of other genes in the liver network (Schadt et al. 2005) so that components of this pathway affect and are affected by many different parts of the network
RNA levels of the type II TGF-β receptor (TGFBR2) were recently shown to be very significantly correlated with thousands of other gene expression traits in the liver transcriptional network of a cross between two inbred lines of mice (referred to here as the BXD cross) (Schadt et al. 2003, 2005) This set of genes associated with TGFBR2 was enriched for a broad range of biological functions known to be associated with the classic TGF-β signaling pathway and with metabolic disease traits such as obesity. Furthermore, TGFBR2 RNA levels in the BXD cross were also found to be significantly correlated with many obesity-related traits like fat mass, percent body fat, and weight. Taking a view that a complex network of gene interactions underlies obesity phenotypes in the BXD cross, genotypic and gene expression data were systematically integrated to assess whether changes in DNA sequence at a given location in the genome (reflected as genotypes in the cross animals) leading to changes in transcript abundances for a given gene supported an independent, causative, or reactive function of that gene relative to various obesity phenotypes like fat mass (Schadt et al. 2005). In partitioning the thousands of genes associated with obesity in this way, TGFBR2 was one of 40 genes predicted as causal for obesity in the BXD cross. TGFBR2 and two other genes selected for validation were all validated as causal for obesity in this study (Schadt et al. 2005). These data directly demonstrated that TGFBR2 and other genes in this signaling pathway are involved in a more general gene network (Schadt et al. 2005a, b), so that it is possible that perturbations in these other genes or in TGFBR2 itself may drive diseases like obesity by influencing other parts of the network beyond the TGF-β signaling pathway (Fig. 4B). Therefore, considering single genes in the context of a whole-gene network may provide the necessary context within which to interpret the disease role a given gene may play.
Networks provide a convenient framework for exploring the context within which single genes operate. Networks are simply graphical models comprising nodes and edges. For gene networks associated with biological systems, the nodes in the network typically represent genes, and edges (links) between any two nodes indicate a relationship between the two corresponding genes. For example, an edge between two genes may indicate that the corresponding expression traits are correlated in a given population of interest (Zhu et al. 2004), that the corresponding proteins interact (Kim et al. 2005), or that changes in the activity of one gene lead to changes in the activity of the other gene (Schadt et al. 2005). Interaction or association networks have recently gained more widespread use in the biological community, where networks are formed by considering only pairwise relationships between genes, including protein interaction relationships [49], coexpression relationships (Gargalovic et al. 2006; Ghazalpour et al. 2006), and other straightforward measures that may indicate association between two genes.
Genetic data can aid in the construction of association networks by helping to reduce artifactual correlations between expression traits. Significant artifactual correlations can arise because of correlated noise structures between array-based experiments networks. One way to leverage the eQTL data in this setting is to simply filter out gene-gene correlations in which the expression traits are not at least partially explained by common genetic effects (Lum et al. 2006). For example, we can connect two genes with an edge in a coexpression network if (1) the p value for the Pearson correlation coefficient between the two genes is less than some prespecified threshold, and (2) the two genes had at least one eQTL in common. This can be taken a step further by formally assessing whether two expression traits driven by a common QTL are related in a causal or reactive fashion, filtering out correlations driven by expression traits that are independently driven by common or closely linked QTLs (Doss et al. 2005; Schadt et al. 2005).
As has been discussed, multiple traits driven by common QTLs is a central idea that can be leveraged to construct networks. One intuitive way to establish whether two genes share at least one eQTL is to perform single-trait eQTL mapping for each expression trait and then consider eQTLs for each trait overlapping if the corresponding LOD for the eQTLs are above some threshold and if the eQTLs are in close proximity to one another. The significance of the statistic corresponding to the strength of association between two genes in the coexpression networks is then chosen such that the resulting network exhibits the scale-free property (Gargalovic et al. 2006; Ghazalpour et al. 2006; Lum et al. 2006) and the false discovery rate for the gene-gene pairs represented in the network is constrained. Beyond the simple, albeit intuitively appealing, eQTL overlap method, we can formally test whether two overlapping eQTLs represent a single eQTL or closely linked eQTLs by employing a pleiotropy effects test (PET), such as that originally described by Jiang and Zeng (1995) and Zeng et al. (2000). The formation of gene clusters by simultaneously considering gene-gene and marker-gene correlations also promises to provide a more comprehensive characterization of shared genetic effects (Lee et al. 2006).
Identifying modules of highly interconnected genes in coexpression networks
Given the scale-free and hierarchical nature of coexpression networks (Barabasi and Oltvai 2004; Ghazalpour et al. 2006; Lum et al. 2006), one of the key problems is to identify the network modules, or functional units, in the network that represent those hub nodes (nodes that are significantly correlated with many other nodes) that are highly interconnected with one another but that are not as highly connected with other hub nodes. Figure 5 illustrates a topological connectivity map for the most highly connected genes in the adipose tissue of the BXH cross (E. E. Schadt et al., unpublished). After hierarchically clustering both dimensions of this plot, the network is seen to break out into clearly identifiable modules. Gene-gene coexpression networks are highly connected, and the clustering results shown in Fig. 5 illustrate that there are gene modules arranged hierarchically within these networks.
Fig. 5
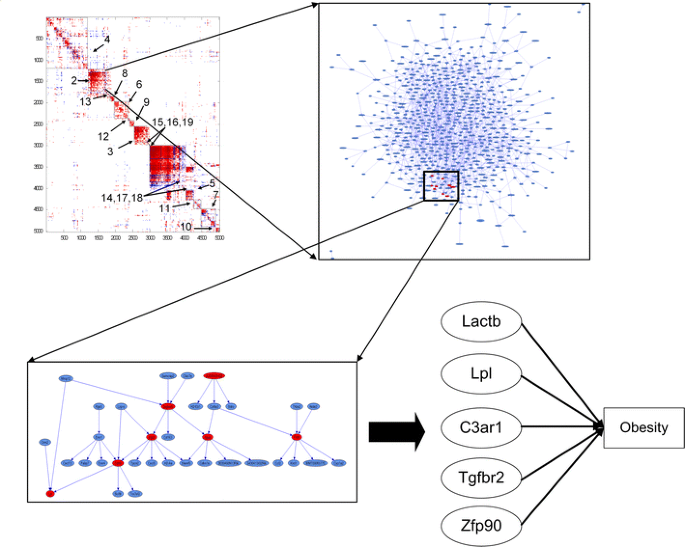
Coexpression and Bayesian networks from adipose expression data collected in a murine F2 intercross population. The upper-left panel is a topological overlap map view of the adipose coexpression network. All pairs of correlations among the 5000 most highly connected genes in the adipose data are plotted in the color matrix display (red indicates positive correlation, blue indicates negative correlation, and white indicates correlation not significant at the p < 10-20 level). The genes are ordered along the x and y axes using an agglomerative hierarchical clustering algorithm. Tightly correlated groups of genes (modules) clearly emerge from this plot. Modules are identified as described in the text. The upper-right panel is the Bayesian network corresponding to genes in module 2 highlighted in the topological overlap map. The lower-left panel represents a subnetwork consisting of 36 genes that contain the genes Lpl and Lactb recently validated as causal for obesity (E. E. Schadt et al., unpublished). More generally, module 2 highlighted in the topological overlap map contains a number of genes validated as causal for obesity (lower-right panel), indicating that disease-causing genes may cluster into functionally coherent sets in the network
Ravasz et al. (2002) used manually selected height cutoff to separate tree branches after hierarchical clustering, in contrast to Lee et al. (2004) who formed maximally coherent gene modules with respect to gene ontology (GO) functional categories. Another strategy is to employ a measure similar to that used by Lee et al. (2004) but without the dependence on the GO functional annotations, given it is of interest to determine independently whether coexpression modules are enriched for GO functional annotations (Lum et al. 2006). The modules identified in this way are informative for identifying the functional components of the network that are associated with disease (Lum et al. 2006). It has been demonstrated that the types of modules depicted in Fig. 5 are enriched for known biological pathways, for genes that associate with disease traits, and for genes that are linked to common genetic loci (Ghazalpour et al. 2006; Lum et al. 2006). In this way, one can identify those key groups of genes that are perturbed by genetic loci that lead to disease, and that therefore define the intermediate steps that actually define disease states.
Using eQTL data to reconstruct probabilistic networks
Coexpression networks are useful constructs for characterizing gross topological properties of biological networks, highlighting nodes that are highly connected, and identifying functional modules that aid in the characterization of subnetworks associated with disease. The edges in these networks, however, are undirected so that they do not provide explicit details on the connectivity structure among genes in the network. As suggested above, one way to incorporate causal information into the coexpression networks would be to define direction using the causality procedures described in Fig. 2B. However, such a method would be limited by considering only pairs of genes at a time. The naturally occurring variations in DNA can be leveraged more generally as a systematic source of perturbations to infer causal associations among gene expression traits and between gene expression and clinical traits, moving us toward the ultimate goal of reconstructing whole-gene networks that drive disease, so that for any given gene a more complete context is defined. Zhu et al. (2004) were among the first to formally incorporate genetic data into the reconstruction of whole-gene networks using Bayesian network reconstruction methods. Bayesian networks are directed acyclic graphs that, while limited with respect to representing temporal information or feedback loops, allow for the explicit representation of causal associations among nodes in the network. With Bayesian network reconstruction methods taking gene expression data as the only source of input, many relationships between genes in such a setting will be Markov equivalent (symmetric), similar to what was discussed for three-node graphs in Fig. 2. This means one cannot statistically distinguish whether a given gene causes another gene to change or vice versa. To break this symmetry, Zhu et al. (2004) incorporated eQTL data as prior information to establish more reliably the correct direction among expression traits.
Bayesian network methods have been applied previously to reconstruct networks comprising only expression traits, and to networks comprising both expression and disease traits, where the aim has been to identify those portions of the network that are driving a given disease trait. Forming candidate relationships among genes was performed using an extension of standard Bayesian network reconstruction methods (Chiellini et al. 2002). In the first approach to extend this method using genetic data, QTL information for the transcript abundances of each gene considered in the network was incorporated into the reconstruction process. It is well known that searching for the best possible network linking a moderately sized set of genes is an NP-hard problem. Exhaustively searching for the optimal network with hundreds of genes is presently a computationally intractable problem. Therefore, various simplifications are typically applied to reduce the size of the search space and to reduce the number of parameters that need to be estimated from the data. Two simplifying assumptions to achieve such reductions are commonly employed. First, while any gene in a biological system can control many other genes, a given gene can be restricted so that it is allowed to be controlled by a reduced set of genes. Second, the set of genes that can be considered possible causal drivers (parent nodes) for a given gene can be restricted using the type of causality arguments discussed in previous sections, as opposed to allowing for the possibility of any gene in the complete gene set to serve as a parent node. The eQTL data in this case can be leveraged as prior information to restrict the types of relationships that can be established among genes and the QTL information can be more intimately integrated into the network reconstruction process. As indicated in previous sections, correlation measures are symmetric and so can indicate association but not causality. However, QTL mapping information for the gene expression traits can be used to help sort out causal relationships. The different tests described in the section on making causal inferences between pairs of traits provides one way to explicitly sort out such relationships. Zhu et al. (2004) leveraged the eQTL data in a different way to make similar types of inferences in their network reconstruction algorithm.
With the various constraints and measures defined above, the goal in reconstructing whole-gene networks is to find a graphical model M (a gene network) that best represents the relationships between genes, given a gene expression data set D of interest. That is, given data D, we seek to find the model M with the highest posterior probability P(M|D). The prior probability P(M) of model M is
P(M)=prodlimitsXrightarrowYP(XtoY){P(M) = {\prod\limits_{{X} \rightarrow {Y}} {P(X \to Y)} }}P(M)=prodlimitsXrightarrowYP(XtoY)
where the product is taken over all paths in the network (M) under consideration. The algorithm Zhu et al. (2004) employed to search through all possible models to find the network that best fits the data is similar to the local maximum search algorithm implemented by Friedman et al. (2000). Zhu et al. (2007) recently demonstrated via simulation of biologically realistic networks that the integration of genetic and expression data in this fashion to reconstruct gene networks leads to networks that are more predictive than networks reconstructed from expression data alone.
The Bayesian network reconstruction algorithm can be used to elucidate the module connectivity structure depicted in Fig. 5. Because reconstruction of Bayesian networks is an NP-hard problem (Garey and Johnson 1979), the number of nodes that can be considered in the network and the extent of connections (edges) among these nodes must be reduced (over what can be considered in reconstructing coexpression networks) to make the problem tractable, thereby making such networks more sparse compared with coexpression networks. Toward this end, Fig. 5 shows the result of the Bayesian network reconstruction algorithm discussed above applied to module 2 of the coexpression network depicted in Fig. 5. Further highlighted in Fig. 5 is a subnetwork containing the gene Lpl, a gene recently identified as causal for obesity in the BXH cross (E. E. Schadt et al., unpublished). In fact, the module 2 subnetwork contains a number of genes recently identified and validated as causal for obesity (Fig. 5). The more detailed structure provided by the different networks depicted in Fig. 5 allows for the examination of the context in which specific genes like Lpl operate, providing insights into which parts of the network may impact a given gene’s function and what other parts of the network may be impacted by the gene’s function.
Conclusions
The identification of DNA polymorphisms that associate with diseases like obesity and diabetes can be considered only as the beginning in a long series of steps needed to elucidate disease pathways and to establish the specific role individual genes may play in the process. Diseases like obesity are diseases of the system, potentially involving many different pathways operating in many different tissues and ultimately giving rise to not only different disease subtypes but to different comorbidities of the disease as well. The integration of gene expression (and other molecular profiling data more generally) and genotypic data will be critical if we are ever to understand how genetic and environmental perturbations to a given system lead to complex traits like disease. If common forms of these diseases represent states of a network, then focusing on single-gene perturbations will likely never reveal the most effective ways to treat or prevent disease.
The integration of the diverse sets of molecular data now being generated in population settings is only in its infancy. Many of the methods employed to date toward this end are more heuristic in nature and so will benefit from a more formal treatment. In addition, little to date has been done to integrate expression data from multiple tissues to dissect how modules in one tissue may communicate with modules in another tissue. The types of interactions considered along with eQTL data so far have been restricted to RNA-RNA association data, despite the availability of large-scale DNA-protein and protein-protein interaction data. The predictive power of the types of networks discussed in this review could be enhanced by more systematically integrating protein-protein interactions, protein-DNA interactions, protein-RNA interactions, RNA-RNA interactions, protein state information, methylation state, and interactions with metabolites as these types of data become available. These developments promise to take us beyond the single-gene view of disease and move us closer to the type of systems level view, depicted in Fig. 1, that may be needed to fully understand the complexity of common human diseases like obesity and diabetes. Of course, further study and experimentation are needed to demonstrate more convincingly that understanding the state of a given molecular network, interactions among molecular networks, and how the states of such networks change in response to different genetic and environmental contexts is tractable enough to take us beyond the reductionist approach, which to date has achieved great success in elucidating the complexity of living systems more generally.
References
- Alberts B (2002) Molecular biology of the cell (New York: Garland Science), p xxxiv
- Alberts R, Terpstra P, Bystrykh LV, de Haan G, Jansen RC (2005) A statistical multiprobe model for analyzing cis and trans genes in genetical genomics experiments with short-oligonucleotide arrays. Genetics 171:1437–1439
CAS PubMed PubMed Central Google Scholar - Barabasi AL, Albert R (1999) Emergence of scaling in random networks. Science 286:509–512
CAS PubMed Google Scholar - Barabasi AL, Oltvai ZN (2004) Network biology: understanding the cell’s functional organization. Nat Rev Genet 5:101–113
CAS PubMed Google Scholar - Brem RB, Kruglyak L (2005) The landscape of genetic complexity across 5,700 gene expression traits in yeast. Proc Natl Acad Sci U S A 102:1572–1577
CAS PubMed PubMed Central Google Scholar - Brem RB, Yvert G, Clinton R, Kruglyak L (2002) Genetic dissection of transcriptional regulation in budding yeast. Science 296:752–755
CAS PubMed Google Scholar - Cervino AC, Li G, Edwards S, Zhu J, Caurie C, et al. (2005) Integrating QTL and high-density SNP analyses in mice to identify Insig2 as a susceptibility gene for plasma cholesterol levels. Genomics 86:505–517
CAS PubMed Google Scholar - Chesler EJ, Lu L, Shou S, Qu Y, Gu J, et al. (2005) Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat Genet 37:233–242
CAS PubMed Google Scholar - Cheung VG, Spielman RS, Ewens KG, Weber TM, Morley M, et al. (2005) Mapping determinants of human gene expression by regional and genome-wide association. Nature 437:1365–1369
CAS PubMed PubMed Central Google Scholar - Chiellini C, Brtacca A, Novelli SE, Gorgun CZ, Ciccarone A, et al. (2002) Obesity modulates the expression of haptoglobin in the white adipose tissue via TNFalpha. J Cell Physiol 190:251–258
CAS PubMed Google Scholar - DeCook R, Lall S, Nettleton D, Howell SH (2006) Genetic regulation of gene expression during shoot development in Arabidopsis. Genetics 172:1155–1164
CAS PubMed PubMed Central Google Scholar - DePrimo SE, Wong LM, Khatry DB, Nicholas SL, Manning WC, et al. (2003) Expression profiling of blood samples from an SU5416 Phase III metastatic colorectal cancer clinical trial: a novel strategy for biomarker identification. BMC Cancer 3:3
PubMed PubMed Central Google Scholar - Doss S, Schadt EE, Drake TA, Lusis AJ (2005) Cis-acting expression quantitative trait loci in mice. Genome Res 15:681–691
CAS PubMed PubMed Central Google Scholar - Edwards AO, Ritter R 3rd, Abel KJ, Manning A, Panhuysen C, et al. (2005) Complement factor H polymorphism and age-related macular degeneration. Science 308:421–424
CAS PubMed Google Scholar - Friedman N, Linial M, Nachman I, Pe'er D (2000) Using Bayesian networks to analyze expression data. J Comput Biol 7:601–620
CAS PubMed Google Scholar - Garey MR, Johnson DS (1979) Computers and intractability: a guide to the theory of NP-completeness (San Francisco: W. H. Freeman), p x, 338 pp
- Gargalovic PS, Imura M, Zhang B, Gharavi NM, Clark MJ, et al. (2006) Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci U S A 103:12741–12746
CAS PubMed PubMed Central Google Scholar - Ghazalpour A, Doss S, Zhang B, Wang S, Plaisier C, et al. (2006) Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet 2(8):e130
PubMed PubMed Central Google Scholar - Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, et al. (2006) Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet 38:320–323
CAS PubMed Google Scholar - GuhaThakurta D, Xie T, Anand M, Edwards SW, Li G, et al. (2006) Cis-regulatory variations: a study of SNPs around genes showing cis-linkage in segregating mouse populations. BMC Genomics 7:235
PubMed PubMed Central Google Scholar - Haines JL, Hauser MA, Schnidt S, Scott WK, Olson LM, et al. (2005) Complement factor H variant increases the risk of age-related macular degeneration. Science 308:419–421
CAS PubMed Google Scholar - Han JD, Bertin N, Hao T, Goldberg DS, Berriz GF, et al. (2004) Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 430:88–93
CAS PubMed Google Scholar - Herbert A, Gerry NP, McQueen MB, Heid IM, Pfeufer A, et al. (2006) A common genetic variant is associated with adult and childhood obesity. Science 312:279–283
CAS PubMed Google Scholar - Hubner N, Wallace CA, Zimdahl H, Petretto E, Schulz H, et al. (2005) Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat Genet 37:243–253
CAS PubMed Google Scholar - Jansen RC, Nap JP (2001) Genetical genomics: the added value from segregation. Trends Genet 17:388–391
CAS PubMed Google Scholar - Jiang C, Zeng ZB (1995) Multiple trait analysis of genetic mapping for quantitative trait loci. Genetics 140:1111–1127
CAS PubMed PubMed Central Google Scholar - Jin W, Riley RM, Wolfinger RD, White KP, Passador-Gurgel G, et al. (2001) The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat Genet 29:389–395
CAS PubMed Google Scholar - Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, et al. (2003) Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302:2141–2144
CAS PubMed Google Scholar - Karp CL, Grupe A, Schadt E, Ewert SL, Keane-Moore M, et al. (2000) Identification of complement factor 5 as a susceptibility locus for experimental allergic asthma. Nat Immunol 1:221–226
CAS PubMed Google Scholar - Kendziorski CM, Chen M, Yuan M, Lan H, Attie AD (2006) Statistical methods for expression quantitative trait loci (eQTL) mapping. Biometrics 62:19–27
CAS PubMed Google Scholar - Kim JK, Gabel HW, Kamath RS, Tewari M, Pasquinelli A, et al. (2005) Functional genomic analysis of RNA interference in C. elegans. Science 308:1164–1167
CAS PubMed Google Scholar - Klein RJ, Zeis C, Chew EY, Tsai JY, Sackler RS, et al. (2005) Complement factor H polymorphism in age-related macular degeneration. Science 308:385–389
CAS PubMed PubMed Central Google Scholar - Klose J, Nock C, Herrmann M, Stuhler K, Marcus K, et al. (2002) Genetic analysis of the mouse brain proteome. Nat Genet 30:385–393
CAS PubMed Google Scholar - Kulp DC, Jagalur M (2006) Causal inference of regulator-target pairs by gene mapping of expression phenotypes. BMC Genomics 7:125
PubMed PubMed Central Google Scholar - Lee I, Date SV, Adai AT, Marcotte EM (2004) A probabilistic functional network of yeast genes. Science 306:1555–1558
CAS PubMed Google Scholar - Lee SI, Pe’er D, Dudley AM, Church GM, Koller D (2006) Identifying regulatory mechanisms using individual variation reveals key role for chromatin modification. Proc Natl Acad Sci U S A 103:14062–14067
CAS PubMed PubMed Central Google Scholar - Lum PY, Chen Y, Zhu J, Lamb J, Melmed S, et al. (2006) Elucidating the murine brain transcriptional network in a segregating mouse population to identify core functional modules for obesity and diabetes. J Neurochem 97(Suppl 1):50–62
CAS PubMed Google Scholar - Mehrabian M, Allayee H, Stockton J, Lum PY, Drake TA, et al. (2005) Integrating genotypic and expression data in a segregating mouse population to identify 5-lipoxygenase as a susceptibility gene for obesity and bone traits. Nat Genet 37:1224–1233
CAS PubMed Google Scholar - Monks SA, Leonardson A, Zhu H, Cundiff P, Pietrusiak P, et al. (2004) Genetic inheritance of gene expression in human cell lines. Am J Hum Genet 75:1094–1105
CAS PubMed PubMed Central Google Scholar - Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, et al. (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34:267–273
CAS PubMed Google Scholar - Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, et al. (2004) Genetic analysis of genome-wide variation in human gene expression. Nature 430:743–747
CAS PubMed PubMed Central Google Scholar - Mural RJ, Adams MD, Myers EW, Smith HO, Miklos GL, et al. (2002) A comparison of whole-genome shotgun-derived mouse chromosome 16 and the human genome. Science 296:1661–1671
CAS PubMed Google Scholar - Oleksiak MF, Churchill GA, Crawford DL (2002) Variation in gene expression within and among natural populations. Nat Genet 32:261–266
CAS PubMed Google Scholar - Peacock ML, Warren JT Jr, Roses AD, Fink JK (1993) Novel polymorphism in the A4 region of the amyloid precursor protein gene in a patient without Alzheimer’s disease. Neurology 43:1254–1256
CAS PubMed Google Scholar - Petretto E, Mangion J, Dickens NJ, Cook SA, Kumaran MK, et al. (2006a) Heritability and tissue specificity of expression quantitative trait loci. PLoS Genet 2:e172
PubMed PubMed Central Google Scholar - Petretto E, Mangion J, Pravanec M, Hubner N, Aitman TJ (2006b) Integrated gene expression profiling and linkage analysis in the rat. Mamm Genome 17:480–489
CAS PubMed Google Scholar - Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabasi AL (2002) Hierarchical organization of modularity in metabolic networks. Science 297:1551–1555
CAS PubMed Google Scholar - Schadt EE (2005) Exploiting naturally occurring DNA variation and molecular profiling data to dissect disease and drug response traits. Curr Opin Biotechnol 16:647–654
CAS PubMed Google Scholar - Schadt EE, Lum PY (2006) Reverse engineering gene networks to identify key drivers of complex disease phenotypes. J Lipid Res 47(12):2601-2613
CAS PubMed Google Scholar - Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, et al. (2003) Genetics of gene expression surveyed in maize, mouse and man. Nature 422:297–302
CAS PubMed Google Scholar - Schadt EE, Edwards SW, GuhaThakurta D, Holder D, Ying L, et al. (2004) A comprehensive transcript index of the human genome generated using microarrays and computational approaches. Genome Biol 5:R73
PubMed PubMed Central Google Scholar - Schadt EE, Lamb J, Yang X, Zhu J, Edwards S, et al. (2005a) An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet 37:710–717
CAS PubMed PubMed Central Google Scholar - Schadt EE, Sachs A, Friend S (2005b) Embracing complexity, inching closer to reality. Sci STKE 2005:pe40
- Shoemaker DD, Schadt EE, Armour CD, He YD, Garrett-Engele P, et al. (2001) Experimental annotation of the human genome using microarray technology. Nature 409:922–927
CAS PubMed Google Scholar - Sladek R, Rocheleau G, Rung J, Dina C, Shen L, et al. (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445:881–885
CAS PubMed Google Scholar - Storey JD, Akey JM, Kruglyak L (2005) Multiple locus linkage analysis of genomewide expression in yeast. PLoS Biol 3:e267
PubMed PubMed Central Google Scholar - Stranger BE, Forrest MS, Clark AG, Minichiello MJ, Deutsch S, et al. (2005) Genome-wide associations of gene expression variation in humans. PLoS Genet 1:e78
PubMed PubMed Central Google Scholar - van’t Veer LJ, Dai H, van de Vijver MJ, He YD, et al. (2002) Gene expression profiling predicts clinical outcome of breast cancer. Nature 415:530–536
Google Scholar - Waring JF, Jolly RA, Ciurlionis R, Lum PY, Praestgaard JT, et al. (2001) Clustering of hepatotoxins based on mechanism of toxicity using gene expression profiles. Toxicol Appl Pharmacol 175:28–42
CAS PubMed Google Scholar - Zeng ZB, Liu J, Stam LF, Kao CH, Mercer JM, et al. (2000) Genetic architecture of a morphological shape difference between two Drosophila species. Genetics 154:299–310
CAS PubMed PubMed Central Google Scholar - Zhu J, Lum PY, Lamb J, GuhaThakurta D, Edwards SW, et al. (2004) An integrative genomics approach to the reconstruction of gene networks in segregating populations. Cytogenet Genome Res 105:363–374
CAS PubMed Google Scholar - Zhu J, Wiener MC, Zhang C, Fridman A, Minch E, et al. (2007) Increasing the power to detect causal associations among genes and between genes and complex traits by combining genotypic and gene expression data in segregating populations. PLOS Comput Biol 3(4):e69
PubMed PubMed Central Google Scholar
Author information
Authors and Affiliations
- Rosetta Inpharmatics, LLC, 401 Terry Avenue N., Seattle, Washington, 98109, USA
Solveig K. Sieberts & Eric E. Schadt
Authors
- Solveig K. Sieberts
You can also search for this author inPubMed Google Scholar - Eric E. Schadt
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toEric E. Schadt.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sieberts, S.K., Schadt, E.E. Moving toward a system genetics view of disease.Mamm Genome 18, 389–401 (2007). https://doi.org/10.1007/s00335-007-9040-6
- Received: 23 March 2007
- Accepted: 21 May 2007
- Published: 01 July 2007
- Issue Date: July 2007
- DOI: https://doi.org/10.1007/s00335-007-9040-6