Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads (original) (raw)
Background
Adapter sequences are short oligonucleotides used to be ligated to the ends of DNA fragments of interest, so that they can be combined with primers for amplification. When the sequencing read length is greater than that of the target DNA, the adapter sequence is read out, sometimes partially, next to the unknown target DNA sequence. To recover the target DNA sequence, it is essential to identify the adapter sequence and trim it.
Adapter trimming was first used in small RNA (sRNA) sequencing, where typical lengths of the target fragments range from 18 nucleotides (nt) to 30 nt, while the typical read length is 36 nt. Another important application for adapter trimming is DNase-Seq, which is a high-resolution technique used to profile hypersensitive sites that are frequently bound by transcription factors. Recent studies showed that the sequencing of short reads (50–100 base pairs (bp)) gives better results [1].
For all the next-generation sequencing (NGS) applications including chromosomal DNA sequencing or complementary DNA (cDNA) sequencing, double-stranded DNA is first fragmented using nebulization or ultrasonics to obtain lengths of several hundreds bp. The ends of the fragments are then repaired and ligated with adapters. After purification, the adapter-ligated fragments are either poured on slides/chips as water-in-oils or attached to flow-cells for cluster generation. After several cycles of emulsion-PCR or bridge-PCR, the amplified templates are ready for sequencing. Fragmentation is a stochastic process that is influenced by the varied force field and thermodynamic stability of different parts of the sequence; e.g. parts with different GC content. To improve the enrichment rate and to cover the target genome or transcriptome more evenly, the sample preparation protocol usually requires fragments to be enriched in a specified length range. Even when longer average fragment lengths are chosen, the inherent nature of fragment size selection cannot always prevent short fragments and primer-dimmers from going on to the next stage. Therefore, there are nearly always reads that need to be trimmed. For the newly emerged Nextera long mate-pair (LMP) protocol, junction adapters that connect paired target fragments exist in all properly constructed fragments; thus, adapter trimming is essential to gain correct paired reads.
Adapter trimming is different from contaminant removal and is usually associated with NGS protocols where adapters are synthesized and specified by the reagent vendors. Given a known adapter pattern and a read sequence, adapter trimming is usually modeled as a semi-global sequence alignment, also called end-space free alignment, where any space at the end of or beginning of the alignment does not incur penalties.
Semi-global sequence alignment can be performed using the Smith-Waterman algorithm [2] with minor revisions of the boundary condition as implemented in _Cutadapt_[3], which has a time complexity of O(m n), where m is the pattern length, and n is the sequence length. A trickier solution, i.e. Ukkonen’s algorithm for _k_-difference matching [4], has an expected time of O(k n), where k<m is the maximum number of differences allowed. Further improvements in Ukkonen’s algorithm by bitwise parallelism were proposed by Myer [5] and implemented in Btrim [6], which has a time complexity of O(m n/w), where w is the word length of the computer; e.g. w equals 64 for a 64-bit machine. In practice, Myer’s bit-vector dynamic programming algorithm is the fastest _k_-difference matching algorithm currently available. However, Myer’s algorithm is more appropriate for searching patterns in text and is difficult to adapt to deal with sequencing quality values in NGS data.
Other popular adapter trimmers are available; e.g. Fastx_clipper in the FastX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/), SeqTrim (https://github.com/dariogf/SeqtrimNext) [7], _TagCleaner_[8], and EA-Tools (http://code.google.com/p/ea-utils/). Recently published trimmers are: SeqPrep (https://github.com/jstjohn/SeqPrep) which focuses on paired-end (PE) reads; _Flexbar_[9], a flexible barcode demultiplexer that uses the Needleman-Wunsch algorithm [10] for pair-wise global sequence alignments, which has the same time complexity as that of Smith-Waterman algorithm; Trimmomatic (http://www.usadellab.org/cms/index.php?page=trimmomatic), which is a part of an integrated tool _RobiNA_[11]; Scythe (https://github.com/vsbuffalo/scythe), which uses a Naive Bayesian approach to classify contaminants in reads; TrimGalore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), which internally invokes Cutadapt with an extension to handle PE reads; _AdapterRemoval_[12], which is carefully tuned for trimming adapters from both single-end (SE) or PE reads; _AlienTrimmer_[13], which is based on _k_-mer decomposition for contaminant detection; and _NextClip_[14], which is dedicated to trimming adapters within Nextera LMP reads.
For sRNA sequencing data, a simple script can handle the adapter trimming task with acceptable accuracy and speed. However, trimming adapter sequences from genome sequencing data requires tools that are much more efficient, because the volume of data is much larger. Some very fast tools such as Btrim and Trimmomatic tend to over-simplify the underlying model. Btrim neglects the quality values associated with base calls for adapter trimming although it does use quality values for quality trimming [6]. Trimmomatic adopts a hash-based search followed by a simple score-based search, both of which neglect insertions and deletions for adapter matching (http://www.usadellab.org/cms/index.php?page=trimmomatic). To gain sufficient accuracy, most of the adaptor trimming algorithms use conventional pair-wise alignment algorithms such as the Smith-Waterman or Needleman-Wunsch algorithms; however, these alignment algorithms are inefficient for adapter trimming compared with more sophisticated algorithms [5].
In a typical application of adapter trimming, e.g. for sRNA sequencing, usually only a short prefix of the full-length adapter is scanned to reduce run time. However, this strategy may increase the possibility of random hits and cause biases in the trimmed sequences. Even when the specified pattern is of sufficient length, for reads where the 3’ end overlaps with a short prefix of the adaptor pattern, it is hard to judge whether the overlap is from finding the adapter sequence or from sequence homology. On the one hand, over-trimming of a bona fide part of the sequence causes loss of information; on the other hand, leaving an adapter untrimmed causes noise in the downstream analysis.
In this paper, we propose a bit-masked k-difference matching dynamic programming algorithm with O(k n) expected time and O(m) space in which the information within the adapter sequences is transferred into bits. We developed a carefully designed statistical scheme that incorporates quality values (and PE information when applicable), and implemented an industry-standard Linux program called Skewer to address the trimming problem accurately and efficiently.
Results and discussions
Features
The algorithm and statistical scoring schemes are implemented as a Linux program Skewer using C++. A comparison of the main features of Skewer with those of existing mainstream adapter trimmers are presented in Table 1.
Table 1 Main features of various adapter trimmers
Experiment environment
The server that was used for the experiments had 4 × 8-core Intel®; 2.67GHz CPUs, 1T memory, and RAID with bandwidths of 266MB/s and 262MB/s for reading and writing respectively. The operating system (OS) was the Red Hat Enterprise Linux Server release 6.3.
Experiments on simulated data
General information
We simulated 10 million 100 bp + 100 bp PE Solexa reads from the Arabidopsis thaliana genome using ART, a NGS read simulator [15], with some revision on the source codes for simulating adapter-contaminated reads (http://sourceforge.net/projects/skewer/files/Simulator/). The trained profile was from the real sequencing data of A. thaliana where about 36% of the reads were contaminated with adapters. We compared Skewer with mainstream adapter trimmers that can handle PE reads as well as four representative adapter trimmers that can handle only SE reads.
To assess trimming quality, we defined the following metrics: FP (false positive) as the number of reads that were over-trimmed, either for trimming non-contaminant reads (false trimming), noted as F P_ f t, or for over-trimming contaminant reads, noted as F P_ o t; FN (false negative) as the number of reads that were under-trimmed, either for not trimming contaminant reads (false retaining), noted as F N_ f r, or under-trimming contaminant reads, noted as F N_ u t; and TN (true negative) as the number of untrimmed non-contaminant reads.
From these numbers, we defined the positive predictive value (PPV) as the ratio of the number of correctly trimmed reads to the number of trimmed reads; sensitivity (Sen) as the ratio of the number of correctly trimmed reads to the number of contaminant reads; and specificity (Spec) as the ratio of the number of untrimmed non-contaminant reads to the number of non-contaminant reads as follows:
PPV = TP / ( TP + FP _ ft + FP _ ot + FN _ ut )
(1)
Sen = TP / ( TP + FN _ fr + FN _ ut + FP _ ot )
(2)
Spec = TN / ( TN + FP _ ft )
(3)
Finally, we defined the Matthew’s correlation coefficient (mCC), which is a quality measure for pattern recognition, as:
mCC= TP · TN − FP · FN ( TP + FP ) ( TP + FN ) ( TN + FP ) ( TN + FN )
(4)
Primary result
Each method was run with its default parameters, except that the minimum output fragment length and the thread number (if applied) were set to 1.
The results obtained from these runs are listed in Table 2 and details are available in Table S1 of Additional file 1. FastX, an earlier and widely adopted NGS adapter trimmer, had a relatively low mCC (0.6683) and a low processing speed (0.92_M_ b p/s); SeqTrim had a similar overall performance as FastX (0.6618), but it had the slowest processing speed (0.03_M_ b p/s) of all the trimmers tested despite its extensive logging utility; TagCleaner was clearly the most conservative of the trimmers (F P_=0), but it had the lowest sensitivity (45.50%) and was notably slower than FastX (58.7%_ of the speed); EA-Tools had the highest sensitivity (99.72_%) for processing SE reads and was orders of magnitude faster (13_X_∼400_X) than the slow trimmers; Cutadapt, the most widely accepted adapter trimmer, exhibited a good compromise between sensitivity and specificity (96.27_%_ vs. 96.93_%), and had the highest mCC (0.9286) among the existing tools for processing SE reads; TrimGalore, a wrapper for Cutadapt, had a performance that was equivalent to EA-tools with default settings, but it was considerably slower than EA-tools (28.2%∼31.6%_ of the speed); SeqPrep, a dedicated PE reads adapter trimmer and merger, had the highest mCC (0.9975) among the existing tools for processing PE reads, but it was slow (0.64_M_ b p/s); Btrim had the highest speed (23.63_M_ b p/s) for adapter trimming, but it had low sensitivity (53.44_%); Scythe had an mCC similar to that of Cutadapt for SE reads adapter trimming, but was more conservative; Flexbar had slightly lower metrics and about 20% lower processing speed than TrimGalore; Trimmomatic was among the most conservative ones, but it had an acceptable sensitivity (72.31%) and a relatively high speed (16.73_M b p/s); AlienTrimmer had similar metrics to Btrim, but was much slower (1.64_M_ b p/s); and AdapterRemoval had a similar overall performance as SeqPrep for PE reads processing, but unlike SeqPrep it can also handle SE reads.
Table 2 Performance of adapter trimmers on 2Gbp simulated data
The results of the runs listed in Table 2 show that Skewer outperformed all the mainstream tools in terms of mCC for both SE and PE trimming (0.9291 and 0.9989 respectively), although Skewer was only marginally better than Cutadapt and Scythe in SE trimming. Furthermore, Skewer was substantially faster (one times faster for SE and more than 12 times faster for PE trimming) than the tools that had comparative performances.
Trimmomatic and AlienTrimmer both used above 2_G_ bytes peak memory. Most of the other trimmers used less than 50_M_ bytes memory except SeqTrim, which used 115.7_M_ bytes. Although Skewer did not have the least memory usage, its memory consumption (less than 35_M_ bytes, see Table S2 of Additional file 1 for details) was far from a bottleneck on a 64-bit computer. In fact, Skewer uses additional memory to facilitate the processing of IUPAC (International Union of Pure and Applied Chemistry) characters and for parallel computing.
Scalability for parallel computing
We ran various adapter trimmers that support multi-threading to compare their scalability under a parallel computing environment. SeqTrim was excluded because it is too slow to gain comparative speed even with tens of threads. In addition, Cutadapt, AdapterRemoval, and other tools were not included because they currently lack a multi-threading function. In the eight threads case, for both the uncompressed and compressed inputs, Skewer achieved the highest speedup among the adapter trimmers tested (7.87 for uncompressed input, and 4.54 for compressed input) (see Table S2 of Additional file 1 for details).
Receiver operating characteristic (ROC) curves
ROC curves for various adapter trimmers under different stringencies were plotted and are shown in Figure 1 and Figure 2 (see Table S3 of Additional file 1 for details).
Figure 1
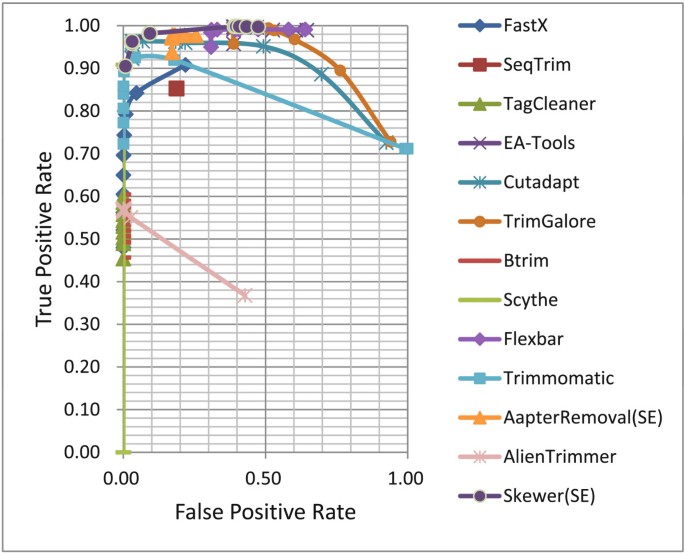
ROC curves of various adapter trimmers for processing single-end reads of simulated data. ROC: receiver operating characteristic.
Figure 2
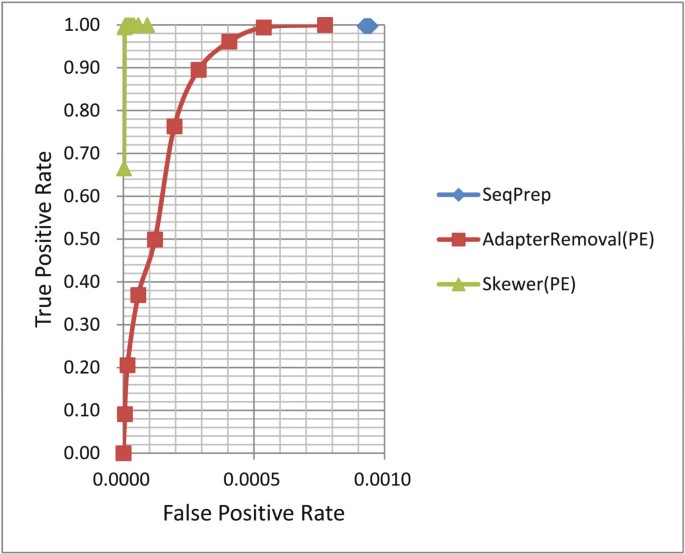
ROC curves of various adapter trimmers for processing paired-end reads of simulated data. ROC: receiver operating characteristic.
In high stringency (left) regions, both Trimmomatic and Cutadapt performed well in that they had low FPRs (false positive rates) and high TPRs (true positive rates); however, as the stringency decreases, both their performance degrade gradually (Figure 1). A similar trend was seen for TrimGalore where the ROC curve shifted to the upper-right region. This implies that these trimmers greedily picked up the first candidate that met the stringency rather than select the optimal one. The ROC curve for AlienTrimmer was similar to the above ones, but with a worse performance. FastX may adopt some optimization technique, however, its performance was worse than those of Trimmomatic and Cutadapt within all the stringency range. Other adapter trimmers showed advantages on a specific metric; e.g. AdapterRemoval, Flexbar, and EA-Tools were the most sensitive, while TagCleaner, Btrim, and Scythe were the most conservative. SeqTrim appeared only as a dot in the ROC curves plot, because it does not provide a stringency threshold. Skewer outperformed the other adapter trimmers in that it had the least FPR to gain a specific TPR, when TPR > 95%.
ROC curves for the adapter trimmers that are aware of PE information were plotted and are shown in Figure 2. Other trimmers that can process PE reads have worse ROC curves than corresponding ROC curves for processing SE reads since the second reads usually have lower sequencing qualities. From Figure 2, we can see that Skewer had a nearly perfect ROC curve close to the upper-left corner. For example, it achieved a TPR of 99.951% with a FPR of 0.001%.
Experiments on real data
sRNA sequencing data for Caenorhabditis elegans
A recently published real sRNA data set (short read archive [SRA:SRR014966]) [16], which includes 14,251,981 reads of small non-coding RNA (ncRNA) from C. elegans, was used to evaluate the adapter trimmers. Because it is hard to recover all the underlying sRNA fragments for sequencing, we aligned the trimmed reads to the reference genome and used delta of the number of uniquely aligned reads relative to the number of uniquely aligned raw reads, noted as TT (true trimming), as a substitute for true positive. We also used delta of number of non-uniquely aligned reads relative to the number of non-uniquely aligned raw reads, noted as FT (false trimming), as a substitute for false positive. The rationale was that correct-trimming tends to change unaligned fragments to uniquely aligned fragments (true positive), while over-trimming tends to change uniquely aligned fragments to non-uniquely aligned fragments (false positive). Note that these metrics tolerate tiny mistakes that can be rescued by the alignment software and are useful for practical evaluation.
To evaluate the performances under various trimming stringencies, all the tools were used to trim adapter sequences from the C. elegans data set using various trimming stringency. Next the processed reads were aligned to the C. elegans genome [17] (version 10) using Bowtie2 [18] (version 2.1.0). We then used the above metrics for final plotting, with higher FT representing lower stringency.
The results are presented in Table S4 and Table S5 of Additional file 2, and illustrated in Figure 3. AdapterRemoval and Flexbar exhibited similar performance curves, while AdapterRemoval was slightly better than Flexbar within all the tested stringency range; and TrimGalore and Cutadapt had similar curves, while TrimGalore was slightly better than Cutadapt at all the stringencies. Under high stringency, EA-Tools, Skewer, and TrimGalore shared the first rank in terms of low FT and high TT; Trimmomatic, AdapterRemoval, and Schythe were ranked second under middle stringency, middle low stringency, and low stringency respectively; and Skewer ranked first at all the stringencies.
Figure 3
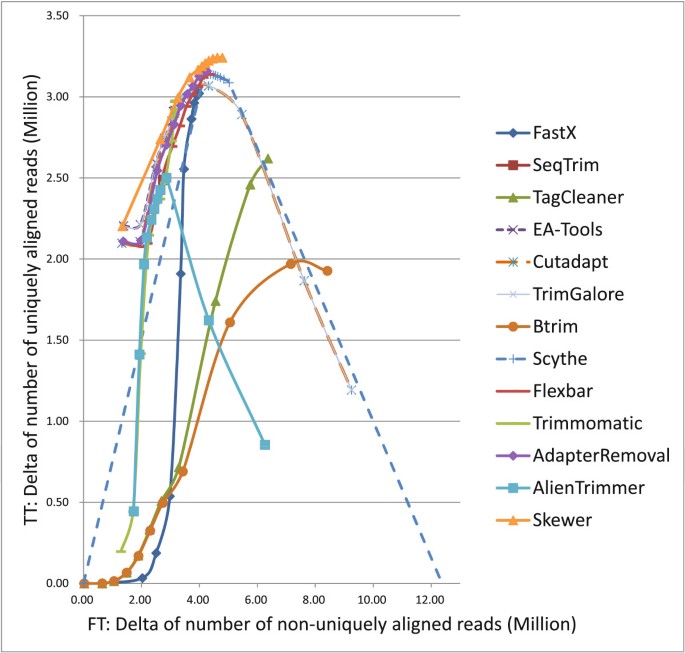
Performance of various adapter trimmers on real small RNA data [SRA:SRR014966].
Paired-end RNA sequencing data for Drosophila simulans
A real RNA-Seq data set with 27,005,344 pairs of 101 bp reads (short read archive [SRA:SRR330569]) from the gonads and carcasses of D. simulans was used to compare the performances of the adapter trimmers in trimming artificial contaminants from PE reads.
For the evaluation, we first used each of the tools that can deal with PE reads with default setting to trim adapters from the reads, with the exception that the minimum output fragment length was set to 20 and quality trimming was inhibited. We then used TopHat [19, 20] (version 2.0.10) to align the processed reads to the reference genome of _D. simulans_[21] (dsim revision 1.4). Finally the number of uniquely and concordantly aligned pairs was used as the performance metrics.
The results are presented in Table S6 of Additional file 3 and illustrated in Figure 4. Skewer outperformed the other adapter trimmers in terms of the number of uniquely and concordantly aligned pairs of the trimmed PE reads. Trimmomatic and AdapterRemoval, both of which performed well in processing the sRNA data, performed poorly in processing the long PE data. This finding implies that these tools may be tuned specifically for trimming adapters from sRNA data. Similarly, Btrim also performed less well with the PE data in this experiment. After investigating the processed data, we found that Btrim could recognize only the occurrence of the whole adapter sequence with a limited tolerance for insertions and deletions. It should be noted that all quality trimming was inhibited from these experiments to compare the adapter trimming performance alone. However, in real applications, quality trimming, which is outside of the scope of this paper, has been reported to improve the mapping rate and facilitate downstream data analysis [22].
Figure 4
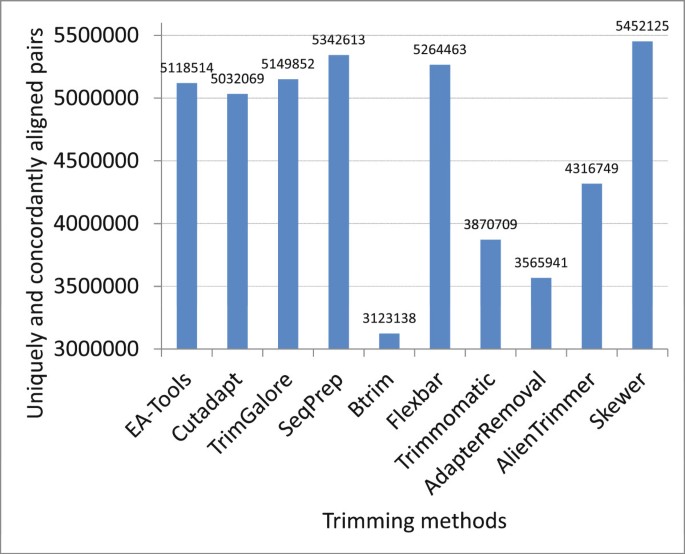
Performance of various adapter trimmers on real paired-end data [SRA:SRR330569].
Nextera long mate pair (LMP) data for Arabidopsis thaliana
A 5-kb insert size Nextera LMP library of A. thaliana Col-0 with 6,602,426 pairs of 251bp reads (European Nucleotide Archive [ENA:ERA264981]) and a 400-bp insert size Illumina HiSeq PE library of the same species with 17,341,797 pairs of 100 bp reads [ENA:SRR519624] were sequenced previously and used to demonstrate the utility of NextClip [14], a dedicated tool for trimming adapters from Nextera LMP libraries.
To compare Skewer with NextClip, we followed a validation procedure similar to the one described for NextClip [14]. Briefly, the LMP library was first trimmed using the adaptor trimmer. Then the trimmed LMP reads and the PE reads were de novo assembled using ABySS [23]. The time needed for the adapter trimming and the N50 lengths of the scaffolds were used as the metrics for the evaluation.
The result is listed in Table 3 (see Additional file 4 for relevant commands in detail), from which we can see that Skewer marginally outperforms NextClip in terms of assembly statistics (N50 length etc.) of the trimmed reads. In addition, Skewer is about 49% faster than NextClip in single thread mode.
Table 3 Comparison of NextClip and Skewer in processing Nextera long mate-pair (LMP) reads (ERA264981)
Conclusions
We presented a novel algorithm and applied it to adapter trimming. The inherent advantage of the proposed bit-masked k-difference matching dynamic programming algorithm makes it possible to search adapter sequence pattern in an exhaustive yet efficient manner. Moreover, by using carefully designed scoring schemes for adapter pattern matching in both SE and PE sequencing data, the resultant Skewer tool was shown to achieve accuracies that were not matched by other similar tools that are currently available. Importantly, Skewer was not optimized for specific applications (e.g. sRNA sequencing); however, compared with other adapter trimmers, it performed well over all NGS applications.
Read lengths and throughputs of NGS technologies are likely to keep increasing; therefore, efficient and accurate adapter trimming methods will continue to be important in the preprocessing steps in applications such as genome resequencing, de novo sequencing, transcriptome sequencing, as well as sRNA sequencing.
Methods
Problem definition
Notation
The Levenshtein distance between two strings _A_=_a_1_a_2… a|A|, B_=b_1_b_2…_b|B|, noted as ∥_A,_B_∥ l e v , is given by a recursive formula (assuming |A|>0,|B|>0):
A , B lev : = min a 1 a 2 … a | A | − 1 , b 1 b 2 … b | B | − 1 lev + δ a | A | , b | B | a 1 a 2 … a | A | − 1 , B lev + 1 A , b 1 b 2 … b | B | − 1 lev + 1
where δ a,_b_=1 if a_≠_b, otherwise δ a,b_=0. When m i n(|A|,|B|)=0, ∥_A,_B_∥ l e v :=m a x(|A|,|B|).
_k_-difference problem
Given a sequence _S_=s_1_s_2…_s n , a query pattern P_=p_1_p_2…_p m , and a threshold k(0≤_k<m), search all substrings of S, noted as {_P_′}, such that ∥_P_′,P_∥ l e v ≤_k.
Extended _k_-difference problem
Given a sequence _S_=s_1_s_2…_s n , a query pattern P_=p_1_p_2…_p m , and a threshold e(0≤_e<1,⌊_n_×_e_⌋=k), search all substrings of S, noted as {_P_′}, such that ∥_P_′,P_∥ l e v ≤_k; and all suffixes of S, noted as {_S_′}, such that ∃ a prefix of P, noted as _P_′, and ∥_S_′,_P_′∥ l e v ≤|S_′|×_e
Algorithms
For the k-difference problem, the classic approach [2] computes a (m+1)×(n+1) dynamic programming matrix C[0..m,0..n] using the following recurrence:
C [ i , j ] = min C [ i − 1 , j − 1 ] + δ ij C [ i − 1 , j ] + 1 C [ i , j − 1 ] + 1
where
δ ij = 0 , if p i = s j , 1 , otherwise .
with initialization at the upper boundary by _C_[0,_j_]=0, and at the left boundary by _C_[i,0]=i, for _i_=1,2,…,m and _j_=1,2,…,n. Finally, the algorithm tests the last row of the matrix, i.e. _C_[m,_j_], and reports those elements that are no greater than k. This algorithm has O(m n) time and O(m n) space complexity.
The space bound can be easily reduced to O(m) if matrix C is computed by columns, noted as C j for j_=1,2,…_n, and report a match each time C j [m_]≤_k, because computing column C j requires only the knowledge of previous column C _j_−1. With careful design, C j and C _j_−1 can share one column vector, as proposed by Ukkonen [4].
Ukkonen also observed that for columns that have the last element greater than k, there is a boundary index of C j , noted as l a c(C j ), such that C j [l a c(C j )]=k and C j [l_]>k for l_=l a c(C j )+1,…_m. It is easy to prove that l a c(C j )≤_l a c(C _j_−1)+1. Using this observation, Ukkonen reduced the time from O(m n) to expected O(k n) [4].
Our algorithm was developed from Ukkonen’s algorithm; however, we use a queue instead of an array to store all elements of current column above the boundary index. When there is a new element that corresponds to the topmost element of the new column, all elements in the queue shift automatically to the next (lower) position, just as elements transfer in the diagonals of matrix C. This process inherently keeps the basic properties of Ukkonen’s algorithm and facilitates subsequent improvements.
Lemma 1
In the dynamic programming matrix C for tackling the _k_-difference problem, the values of elements along each diagonal are monotonically non-decreasing.
The proof is provided in Additional file 5 Appendix A.
Theorem 1
All the matched elements of the query pattern and sequence are equal to their precursors in the diagonal and do not need to be updated in the dynamic programming process.
Proof.
This theorem is a direct consequence of Lemma 1 and the dynamic programming recurrence, when δ i j =0.
In other words, only mismatched elements need to be updated in the dynamic programming process. If bit-vectors that denote mismatched positions of comparison between the adapter sequence and each of the four nucleotide characters are pre-computed and a bit-vector that marks all positions of the queue elements that exceed the _k_-difference constraint is maintained, then unnecessary computations in updating the column vector can be inhibited. This is the key point that led to the main improvement of our algorithm over Ukkonen’s algorithm.
As listed in Algorithm 1, the _bit-masked k-difference matching algorithm_has the following characteristics:
- Use a queue instead of an array to store all elements of the current column above boundary index.
- In preprocessing, calculate for each of four nucleotide characters a bit-vector that denotes the mismatched positions compared with the adapter.
- Mark the internal cells that exceed the _k_-difference constraint by a bit-vector which shifts as the queue pushes it.
- When processing the column starting from each input nucleotide, update only the cells that mismatch and have not been marked.
This algorithm uses a queue of size m and several bit vectors of size ⌈m/_w_⌉, where w is the word length of the computer (for example w equals 64 for a 64-bit machine), and hence has a space of O(m). For each of the n characters in a target sequence, the character enters the queue once and exits from the queue at most once. For a random sequence, the expected size of the queue is O(k); hence, generally the algorithm has O(k n) expected time. However, because it is restricted by the bit-mask operations, each element in the queue usually updates at most k+1 times. Because bit operations are negligible compared with element update operations, this algorithm achieves O(k n) worst-case time in practice, which is better than the O(k n) expected time for Ukkonen’s algorithm.
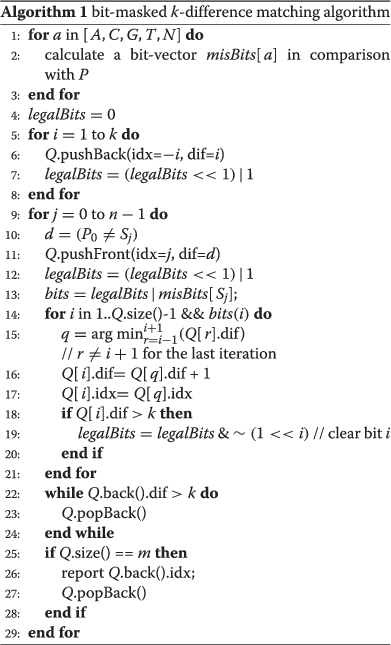
Algorithm 1 can be improved further by avoiding all unnecessary updates through constant time bit operations within each iteration cycle of the target nucleotide. The basic principle is that when an element in a diagonal of the original dynamic programming matrix has a value that is derived from an adjacent diagonal (i.e. an indel occurs in the corresponding path), the score and associated index of the element will remain unchanged if the precursor remains unchanged.
Although the above improvement can reduce the theoretical time complexity from expected O(k n) to worst-case O(k n), experiments on large volumes of real data showed that the reduced element update operations did not compensate for the additional bit operations.
For the extended k-difference problem, an additional step bounded by O(m) is performed to check all the elements remained in the queue if no hit was found in previous steps.
Deal with base-call qualities
The main advantage of the bit-masked k-difference matching algorithm over Myer’s bit-vector algorithm is that it can be extended to handle base-call quality values.
To handle base-call quality values, we introduce the following parameters: P m i n =−l o g_10(1/3), the minimum penalty for a mismatch; P m a x =−_l o _g_10(1040/(−10)/3), the maximum penalty for a mismatch; d e l t _a_=P m a x , the penalty for an insertion or deletion. The penalty of a mismatch with quality value q is calculated as:
P ( q ) = P min q ≤ 0 P min + q / 40 × ( P max − P min ) 0 < q < 40 P max q ≥ 40
It is trivial to prove that P(q)=−l o g_10(10_q/(−10)/3) when 0<q<40. This score is the negative logarithm of the probability that the corresponding base is actually a match to the adapter sequence with sequencing error.
Note that the scoring scheme herein only induces penalties for mismatches and insertions/deletions. For matches, because the possibility of false matching based on sequence homology reduces exponentially as the matching length increases, the false matching possibility can be reduced by setting a longer alignment length. When PE information is available, the false matching possibility can be reduced to a minimum.
The extended version of Algorithm 1 that can take quality values into consideration is outlined in Additional file 6 Appendix B.
Deal with paired-end information
Unlike the standard SE sequencing, PE sequencing reads out each DNA template twice, in opposite directions from different ends. The underlying fact is that all the PE reads that need to be trimmed must have the preserved paired sequences reverse-complement to each other, as illustrated in Figure 5 and Additional file 7 Appendix C.
Figure 5
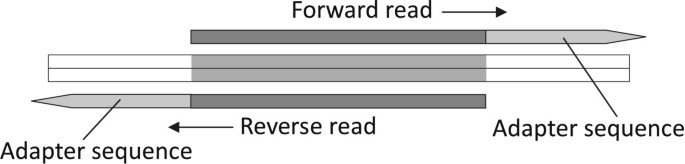
Layout of paired-end reads that have adapter contaminants.
Using this property, the program first finds all _k_-difference occurrences of adapters in both paired reads using the extended version of Algorithm 1 with quality values considered. Then the reverse-complementary property of each trimmed paired sequences is checked. Next, all candidates are evaluated with a scoring scheme that takes into account the fitness of adapter sequences in paired reads and the alignment of reverse-complementary counter-parts. Finally, the program outputs the optimal occurrence, if any.
The scoring scheme we used is as follows:
score ( idx ) = pscore ( read 1 [ idx … readLen − 1 ] , adapter 1 ) + pscore ( read 2 [ idx … readLen − 1 ] , adapter 2 ) + pscore ( read 1 [ 0 … idx − 1 ] , revComp ( read 2 [ 0 … idx − 1 ] ) )
where idx is the start position for trimming, p s c o r e(x,y)=|x|×P m a x −p e n a l t y(x,y), P m a x is the maximum penalty for a difference, p e n a l t y(x,y) is the penalty for matching x and y as calculated by the k-difference matching algorithms, and r e v C o m p(x) denotes the reverse complementary sequence of x. The goal is to find the idx that meets the _k_-difference requirement and maximizes the score function.
Deal with Nextera LMP information
Mate-pair library sequencing allows the generation of long-insert PE libraries that are useful in the scaffolding process of de novo genome assembly and in the detection of long-range genome structural variations. In the Nextera LMP library construction process, there are additional reactions called “tagmentation” and “circularization” before the normal PE library construction. The tagmentation reaction uses a specially engineered transposome to fragment the DNA sample and tag the DNA fragments by attaching a pair of biotinylated junction adapters simultaneously to the ends. Next, the tagmented DNA molecules are circularized and sheared by ultrasonics, and the sub-fragments containing the original junction parts are enriched via the biotin tag in the junction adapter.
Trimming adapters from Nextera LMP reads is like a reverse process of Nextera LMP library construction. To process Nextera mate-pair reads, the program first trims the adapters as if it is dealing with PE reads. Then, it trims junction adapters from the processed paired reads separately using the extended version of Algorithm 1.
Availability of supporting data
The data sets supporting the results of this article are available from high-throughput DNA and RNA sequence read archive: http://www.ncbi.nlm.nih.gov/sra/?term=SRR014966, http://www.ncbi.nlm.nih.gov/sra/?term=SRR330569, http:// www.ncbi.nlm.nih.gov/sra/?term=ERA264981, and http:// www.ncbi.nlm.nih.gov/sra/?term=SRR519624.
References
- He HH, Meyer CA, Hu SS, Chen MW, Zang C, Liu Y, Rao PK, Fei T, Xu H, Long H, Liu XS, Brown M: Refined dnase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nature Methods. 2014, 11 (1): 73-78.
Article PubMed Central PubMed CAS Google Scholar - Smith TF, Waterman MS: Identification of common molecular subsequences. J Mol Biol. 1981, 147 (1): 195-197.
Article PubMed CAS Google Scholar - Martin M: Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011, 17: 10-12.
Article Google Scholar - Ukkonen E: Finding approximate patterns in strings. J Algorithm. 1985, 6 (1): 132-137.
Article Google Scholar - Myers G: A fast bit-vector algorithm for approximate string matching based on dynamic programming. J ACM. 1999, 46 (3): 395-415.
Article Google Scholar - Kong Y: Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics. 2011, 98 (2): 152-153.
Article PubMed CAS Google Scholar - Falgueras J, Lara AJ, Fernandez-Pozo N, Canton FR, Perez-Trabado G, Claros MG: Seqtrim: a high-throughput pipeline for pre-processing any type of sequence read. BMC Bioinformatics. 2010, 11: 38-
Article PubMed Central PubMed Google Scholar - Schmieder R, Lim YW, Rohwer F, Edwards R: Tagcleaner: Identification and removal of tag sequences from genomic and metagenomic datasets. BMC Bioinformatics. 2010, 11: 341-
Article PubMed Central PubMed Google Scholar - Dodt M, Roehr JT, Ahmed R, Dieterich C: Flexbar–flexible barcode and adapter processing for next-generation sequencing platforms. Biology (Basel). 2012, 1 (3): 895-905.
Google Scholar - Needleman SB, Wunsch CD: A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970, 48 (3): 443-453.
Article PubMed CAS Google Scholar - Lohse M, Bolger AM, Nagel A, Fernie AR, Lunn JE, Stitt M, Usadel B: Robina: a user-friendly, integrated software solution for rna-seq-based transcriptomics. Nucleic Acids Res. 2012, 40 (Web Server issue): 622-627.
Article Google Scholar - Lindgreen S: Adapterremoval: easy cleaning of next-generation sequencing reads. BMC Res Notes. 2012, 5: 337-
Article PubMed Central PubMed Google Scholar - Criscuolo A, Brisse S: Alientrimmer: a tool to quickly and accurately trim off multiple short contaminant sequences from high-throughput sequencing reads. Genomics. 2013, 102 (5–6): 500-506.
Article PubMed CAS Google Scholar - Leggett RM, Clavijo BJ, Clissold L, Clark MD, Caccamo M: Nextclip: an analysis and read preparation tool for nextera long mate pair libraries. Bioinformatics. 2014, 30 (4): 566-568.
Article PubMed Central PubMed CAS Google Scholar - Huang W, Li L, Myers JR, Marth GT: Art: a next-generation sequencing read simulator. Bioinformatics. 2012, 28 (4): 593-594.
Article PubMed Central PubMed Google Scholar - Kato M, de Lencastre A, Pincus Z, Slack FJ: Dynamic expression of small non-coding rnas, including novel micrornas and pirnas/21u-rnas, during caenorhabditis elegans development. Genome Biol. 2009, 10 (5): 54-
Article Google Scholar - Hillier LW, Marth GT, Quinlan AR, Dooling D, Fewell G, Barnett D, Fox P, Glasscock JI, Hickenbotham M, Huang W, Magrini VJ, Richt RJ, Sander SN, Stewart DA, Stromberg M, Tsung EF, Wylie T, Schedl T, Wilson RK, Mardis ER: Whole-genome sequencing and variant discovery in c. elegans. Nat Methods. 2008, 5 (2): 183-188.
Article PubMed CAS Google Scholar - Langmead B, Salzberg SL: Fast gapped-read alignment with bowtie 2. Nat Methods. 2012, 9 (4): 357-359.
Article PubMed Central PubMed CAS Google Scholar - Trapnell C, Pachter L, Salzberg SL: Tophat: discovering splice junctions with rna-seq. Bioinformatics. 2009, 25 (9): 1105-1111.
Article PubMed Central PubMed CAS Google Scholar - Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL: Tophat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14 (4): 36-
Article Google Scholar - Consortium F: The flybase database of the drosophila genome projects and community literature. Nucleic Acids Res. 2003, 31 (1): 172-175.
Article Google Scholar - Del Fabbro C, Scalabrin S, Morgante M, Giorgi FM: An extensive evaluation of read trimming effects on illumina ngs data analysis. PLoS One. 2013, 8 (12): 85024-
Article Google Scholar - Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I: Abyss: a parallel assembler for short read sequence data. Genome Res. 2009, 19 (6): 1117-1123.
Article PubMed Central PubMed CAS Google Scholar