Association of MHC and rheumatoid arthritis: HLA polymorphisms in phenotypic variants of rheumatoid arthritis (original) (raw)
Abstract
Genes in the human leukocyte antigen (HLA) region remain the most powerful disease risk genes in rheumatoid arthritis (RA). Several allelic variants of HLA-DRB1 genes have been associated with RA, supporting a role for T-cell receptor-HLA-antigen interactions in the pathologic process. Disease-associated HLA-DRB1 alleles are similar but not identical and certain allelic variants are preferentially enriched in patient populations with defined clinical characteristics. Also, a gene dosing effect of HLA-DRB1 alleles has been suggested by the accumulation of patients with two RA-associated alleles, especially in patient subsets with a severe disease course. Therefore, polymorphisms in HLA genes are being explored as tools to dissect the clinical heterogeneity of the rheumatoid syndrome. Besides HLA polymorphisms, other risk genes will be helpful in defining genotypic profiles correlating with disease phenotypes. One such phenotype is the type of synovial lesion generated by the patient. HLA genes in conjunction with other genetic determinants may predispose patients to a certain pathway of synovial inflammation. Also, patients may or may not develop extraarticular manifestations, which are critical in determining morbidity and mortality. HLA genes, complemented by other RA risk genes, are likely involved in shaping the T-cell repertoire, including the emergence of an unusual T-cell population characterized by the potential of vascular injury, such as seen in extraarticular RA.
Similar content being viewed by others

Genetics of rheumatoid arthritis
Article Open access 27 January 2022

Introduction
A patient is diagnosed with RA if four of seven American College of Rheumatology criteria are satisfied [1]. Some of these criteria are vague, particularly the key criterion of arthritis, which is established by inspecting and palpating joints. Interestingly, histomorphologic analysis of the arthritic lesion is not required, which stands quite in contrast to other fields of medicine where tissue diagnosis is mandatory. For outside observers, it comes as a surprise that classification criteria, mostly relying on subjective evaluation, have proven to be quite useful. From a pathophysiological point of view, it is inevitable that the diagnostic category of RA includes more than a single disease entity. Molecular genetics have now created the opportunity to readdress this critical issue. Rheumatoid arthritis is a classical example of a complex genetic disease characterized by incomplete penetrance, genetic heterogeneity and, almost certainly, a role for multiple disease genes. In this paper, it is proposed that differences in genetic determinants and diversity of disease-risk gene combinations generate discernible and clinically relevant variants of the rheumatoid syndrome [2]. The current review will attempt to utilize the knowledge of the HLA association with RA to differentiate disease subtypes and to generate a hypothesis of how different dimensions of RA could be affected by HLA-dependent immune pathways [3].
RA - a heterogeneous syndrome
The concept that RA is a heterogeneous syndrome encompassing multiple disease phenotypes is supported by clinical and pathophysiologic evidence. Clinicians have long known that the disease course, pattern of involvement, and responsiveness to therapeutic interventions vary considerably [4]. Rheumatoid arthritis can take a mild expression with irreversible damage only appearing after decades, or bony erosions can be present in early disease. Another useful approach to dissecting disease subtypes relies on the absence or presence of extraarticular manifestations. In population based studies, 56% of male patients and 42% of female patients had clinical overt rheumatoid organ disease [5]. Extraarticular RA shortens the life span [6]; however, extrasynovial manifestations may only surface after a substantial lag period, emphasizing the need for predictive molecular markers. The rheumatoid process can target almost any organ. The most frequent forms of extraarticular RA, rheumatoid nodules, are typical granulomas initiated by vasculopathy. Clinically, Felty's syndrome, pulmonary disease, ischemic heart disease, scleritis, and rheumatoid vasculitis are more threatening, particularly when affecting mediumsized blood vessels. Manifestations in different organ systems can occur independently, again emphasizing the multidimensional nature of the disease. Constitutional symptoms are more difficult to define; however, patients with active disease may develop a wasting syndrome. Also, patients with RA have an increased risk for infections [7], and it is not known whether this reflects a genuine complication of autoimmunity or an iatrogenic component of disease.
From a pathophysiological view, the most prominent autoimmune manifestation, the production of rheumatoid factor (RF) autoantibodies, is only found in a subset of patients. Such patients have more aggressive disease, and almost all patients with extraarticular complications are RF-positive. Whether other autoantibodies have prognostic value has remained an issue of debate [8,9]. Finally, an additional level of complexity is introduced by variability of the synovial lesion. In a series of synovial biopsies, different patterns of inflammation, closely associated with different combinations of tissue cytokines, were identified [10]. One third of the patients had formed follicular structures, resembling germinal centers. In 19% of the synovial membranes, classical granulomas were found; 48% of the biopsies revealed a type of diffuse synovitis lacking additional lymphoid microstructures.
HLA genes - one of multiple genetic factors modulating disease risk and expression
As documented by numerous associations and recently by genome-wide linkage studies, genes in the HLA region play an important role in disease susceptibility [11,12,13]. Association studies have provided evidence that a sequence of amino acids, termed the shared epitope, are involved in the disease process [14]. This sequence stretch is encountered in different alleles, bordered by distinct hypervariable regions of the HLA-DRB1 gene.
A second important feature that distinguishes the HLA component of RA from some other HLA-associated diseases is a gene dose effect [15]. Inheritance of two copies of RA-associated HLA alleles increased disease risk significantly. Obvious questions are whether the context of the HLA-DRβ1 chain in which the sequence cassette is inherited, whether gene dosing influences disease expression, and whether more than one HLA function is important in the different dimensions of the disease.
HLA molecules control a variety of functions in the immune system. They are critically involved in restricting the recognition of antigenic peptides by T cells. Considering the dominance of T cells in the synovial lesion, it has been proposed that disease-associated HLA molecules selectively bind and present arthritogenic antigens. This model does not satisfactorily explain the gene dose effect. Gene dosing is compatible with a role of HLA polymorphisms in mechanisms shaping the T cell repertoire [16]. Repertoire abnormalities have been described in RA patients. Based upon the utilization of T cell receptor BV-BJ combinations, RA patients can be clearly distinguished from controls [17]. Also, in RA patients, the diversity of the T cell repertoire is contracted [18]; and large clonal expansion of CD4 [19], as well as CD8 T cells [20], is frequently encountered.
HLA polymorphisms and the synovial lesion
Generation of synovial inflammation is almost certainly influenced by HLA polymorphisms; synovitis is ultimately dependent on T lymphocytes that regulate the production of proinflammatory monokines and tissue-injurious metalloproteinases [21]. Tissue-infiltrating T cells undergo clonal expansion in situ, with selection of identical T cells in different joints, strongly suggesting antigen-driven T cell activation [22,23]. Finally, in rheumatoid synovitis, follicular structures with germinal center reactions are formed. These sophisticated lymphoid microstructures specialize in the generation of high-affinity antibodies and are absolutely T cell dependent [24,25]. Thus, there is overwhelming evidence that immune processes in the synovium are directed by antigen recognition. The precise nature and number of the antigens remain to be elucidated [26].
Considering that the joint-destructive lesion reflects adaptive immune responses, it is a natural question whether polymorphisms of HLA genes influence the inflammatory reaction; specifically, whether HLA polymorphisms modulate the aggressiveness of rheumatoid synovitis. A series of studies suggests that this may be the case. Retrospective studies have indicated that HLA genotyping provides information about the aggressiveness of rheumatoid synovitis [27]. HLA-DR B1*01 alleles are preferentially found in patients with RF-negative RA, a variant of disease known to progress more slowly. HLA-DRB1*0404 was enriched among RF-negative and RF-positive patients, whereas HLA-DRB1*0401 was the dominant allele in RF-producing patients [28]. Thus, HLA-DRB1*0401 appears to confer risk for the formation of a more rapidly progressive synovial lesion. Patients combining HLA-DRB1*04 and HLA-DRB1*01 required joint arthroplasty earlier than patients inheriting only a single copy of RA-associated HLA alleles [15]. In prospective studies, assessing the use of HLA typing for predicting progression of joint disease, HLA-DRB1*04+ patients were more likely to develop early erosive disease [29,30].To what extent HLA polymorphisms can be used as biomarkers to predict disease course, however, is still unanswered [31,32,33]. So far, no prospective study with sufficient patient numbers to control for disease and treatment variables, and to assess the impact of polymorphisms of disease-associated alleles and gene dosing is available.
How could polymorphic HLA residues influence the character of the synovial lesion? Mechanisms of T cell repertoire formation, as well as local peptide selection and presentation, could be relevant [16]. T cells utilizing BV-BJ combinations overrepresented in the rheumatoid repertoire are also preferentially encountered in lesional T cells undergoing in situ proliferation [34]. Remarkably, antigen recognition in the synovium is not uniform, but is associated with the formation of complex lymphoid microstructures, such as germinal centers and granulomas, in subsets of patients [10]. Available data indicate that granulomas and germinal centers do not coexist in the same synovial sample. It is likely that HLA polymorphisms dictate which lymphoid microstructure is formed. Possible involvement of HLA class I molecules is suggested by the accumulation of CD8+CD40L+ cells at the periphery of germinal centers [35]. These CD8 cells lack expression of perforin but carry intracellular IFNγ, identifying them as possible helper cells in the germinal center reaction.
HLA polymorphisms and repertoire abnormalities in extraarticular RA
The highest frequencies of RA-associated HLA-DR molecules are seen in patients with extraarticular RA. However, not all shared epitope-containing alleles are equally enriched. HLA-DRB1*01, while a risk factor for RA joint disease, does not contribute to the risk of extraarticular RA, whereas a combination of two HLA-DR B1*04 alleles seems to optimize the risk [15]. The combination of HLA-DRB1*0401 and HLA-DRB1*0404 has been detected in patients with Felty's syndrome [36] and patients with rheumatoid nodules. Among patients with rheumatoid vasculitis, individuals homozygous for HLA-DRB1*0401 are clearly overrepresented. Studies with sufficient numbers of patients stratified for the clinical phenotype of extraarticular disease are required to define the effect of different HLA polymorphisms and allelic combinations on the targeting of RA to different organ systems. Preliminary evidence suggests that extraarticular manifestations in different tissues do not have a tendency to co-occur; for example, patients with arthritis do not necessarily develop rheumatoid lung disease, and leukocytoclastic vasculitis manifests in a different patient subset than rheumatoid arteritis of medium-sized arteries (unpublished observations).
A second marker of abnormality in rheumatoid vasculitis is the expansion of CD4+CD28null T cells [37]. These unusual T cells are infrequent in healthy individuals [38]; they are expanded in RA patients, with a clear correlation between the size of the CD4+CD28null compartment and the clinical pattern of extraarticular disease. The inheritance of two RA-associated HLA-DR alleles may facilitate the generation of CD4+CD28null T cells.
What are the possible mechanisms through which CD4+CD28null cells influence RA and increase the likelihood of vascular complications? These cells are cytotoxic via a granzyme/perforin mechanism [39] and produce large quantities of IFNγ [40]. A direct role of these T cells in vascular injury is suggested by the finding that patients with acute coronary syndrome, caused by surface rupture of atherosclerotic plaque, also carry expanded frequencies of CD4+CD28null cells [41]. In patients with fatal coronary ischemia, CD4+CD28null cells preferentially infiltrated into the ruptured but not the stable plaque, suggesting direct involvement in vascular wall damage.
Summary
RA is a disease with a multitude of clinical phenotypes. Disease heterogeneity is introduced by variations in synovitis (Fig. 1) as well as diversity of extraarticular manifestations (Fig. 2). Variability of the synovial lesion, as demonstrated by the creation of lymphoid organizations and correlated cytokine profiles, is likely to be regulated by differences in T cell function, and thus is predicted to be HLA dependent. HLA polymorphisms also have been associated with production of RF, autoantibodies correlated with the aggressiveness of articular and extraarticular disease. Shared epitope-containing DRB1*04 alleles confer risk for more destructive synovitis with accelerated joint damage when DRB1*04 and DRB1*01 are combined.
Figure 1
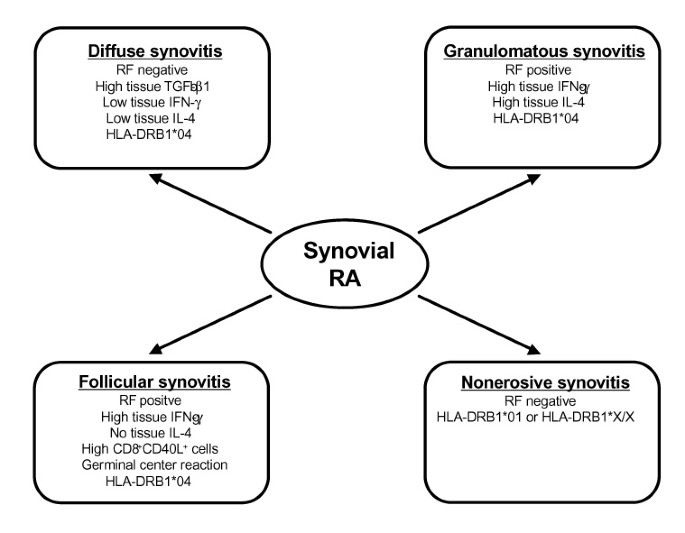
Heterogeneity of rheumatoid synovitis. Risk factors known to predispose for particular phenotypes are listed.
Figure 2
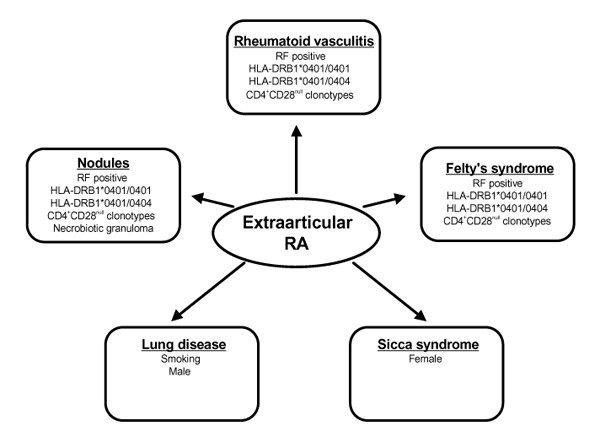
Phenotypic variants of extraarticular rheumatic arthritis and associated risk factors.
Combinations of two RA-associated HLA-DRB1*04 alleles mainly affect the risk for extraarticular RA. Polymorphic HLA genes and their combinations may have a role in modulating the targeting of rheumatoid inflammation outside the joint. Precise mechanisms of combined action of HLA alleles in influencing the expression of extraarticular RA are incompletely understood, but abnormalities in T cell repertoire formation, such as contraction of diversity and T cell clonopathy, have been associated with vascular injury. HLA polymorphisms may prove extremely helpful in dissecting clinically defined phenotypes of RA, and may thus not only have clinical impact as biomarkers, but could also provide clues toward the different pathogenic pathways in the disease process.
References
- Arnett FC, Edworthy SM, Bloch DA: The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988, 31: 315-324.
Article PubMed Google Scholar - Weyand CM, Klimiuk PA, Goronzy JJ: Heterogeneity of rheumatoid arthritis: from phenotypes to genotypes. Springer Semin Immunopathol. 1998, 20: 5-22.
Article PubMed Google Scholar - Weyand CM, Goronzy JJ: Pathogenesis of rheumatoid arthritis. Med Clin North Am. 1997, 81: 29-55.
Article PubMed Google Scholar - Harris ED: . Rheumatoid Arthritis. Philadelphia: W.B. Saunders Company. 1997
Google Scholar - Weyand CM, Schmidt D, Wagner U, Goronzy JJ: The influence of sex on the phenotype of rheumatoid arthritis. Arthritis Rheum. 1998, 41: 817-822.
Article PubMed Google Scholar - Turesson C, Jacobsson L, Bergstrom U: Extra-articular rheumatoid arthritis: prevalence and mortality. Rheumatol (Oxford). 1999, 38: 668-674.
Article Google Scholar - Symmons DP: Mortality in rheumatoid arthritis. Br J Rheumatol. 1988, 27 (suppl 1): 44-54.
Google Scholar - Aho K, Palusuo T, Kurki P: Marker antibodies of rheumatoid arthritis: diagnostic and pathogenetic implications. Semin Arthritis Rheum. 1994, 23: 379-387.
Article PubMed Google Scholar - Kirwan JR, Quilty B: Prognostic criteria in rheumatoid arthritis: can we predict which patients will require specific anti-rheumatoid treatment?. Clin Exp Rheumatol. 1997, 15 (suppl 17): S15-S25.
Google Scholar - Klimiuk PA, Goronzy JJ, Bjornsson J, Beckenbaugh RD, Weyand CM: Tissue cytokine patterns distinguish variants of rheumatoid synovitis. Am J Pathol. 1997, 151: 1311-1319.
PubMed PubMed Central Google Scholar - Reveille JD: The genetic contribution to the pathogenesis of rheumatoid arthritis. Curr Opin Rheumatol. 1998, 10: 187-200.
Article PubMed Google Scholar - Auger I, Roudier J: HLA-DR and the development of rheumatoid arthritis. Autoimmunity. 1997, 26: 123-128.
Article PubMed Google Scholar - Nepom GT: Major histocompatibility complex-directed susceptibility to rheumatoid arthritis. Adv Immunol. 1998, 68: 315-332.
Article PubMed Google Scholar - Gregersen PK, Silver J, Winchester RJ: The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30: 1205-1213.
Article PubMed Google Scholar - Weyand CM, Hicok KC, Conn DL, Goronzy JJ: The influence of HLA-DRB1 genes on disease severity in rheumatoid arthritis. Ann Intern Med. 1992, 117: 801-806.
Article PubMed Google Scholar - Goronzy JJ, Zettl A, Weyand CM: T cell receptor repertoire in rheumatoid arthritis. Int Rev Immunol. 1998, 17: 339-363.
Article PubMed Google Scholar - Walser-Kuntz DR, Weyand CM, Weaver AJ, O'Fallon WM, Goronzy JJ: Mechanisms underlying the formation of the T cell receptor repertoire in rheumatoid arthritis. Immunity. 1995, 2: 597-605.
Article PubMed Google Scholar - Wagner UG, Koetz K, Weyand CM, Goronzy JJ: Perturbation of the T cell repertoire in rheumatoid arthritis. Proc Natl Acad Sci USA. 1998, 95: 14447-14452.
Article PubMed PubMed Central Google Scholar - Schmidt D, Goronzy JJ, Weyand CM: CD4+ CD7- CD28- T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J Clin Invest. 1996, 97: 2027-2037.
Article PubMed PubMed Central Google Scholar - Fitzgerald JE, Ricalton NS, Meyer AC: Analysis of clonal CD8+ T cell expansions in normal individuals and patients with rheumatoid arthritis. J Immunol. 1995, 154: 3538-3547.
PubMed Google Scholar - Klimiuk PA, Yang H, Goronzy JJ, Weyand CM: Production of cytokines and metalloproteinases in rheumatoid synovitis is T cell dependent. Clin Immunol. 1999, 90: 65-78. 10.1006/clim.1998.4618.
Article PubMed Google Scholar - Rittner HL, Zettl A, Jendro MC, Bartz-Bazzanella P, Goronzy JJ, Weyand CM: Multiple mechanisms support oligoclonal T cell expansion in rheumatoid synovitis. Mol Med. 1997, 3: 452-465.
PubMed PubMed Central Google Scholar - Ikeda Y, Masuko K, Nakai Y: High frequencies of identical T cell clonotypes in synovial tissues of rheumatoid arthritis patients suggest the occurrence of common antigen-driven immune responses. Arthritis Rheum. 1996, 39: 446-453.
Article PubMed Google Scholar - Tarlinton D: Germinal centers: form and function. Curr Opin Immunol. 1998, 10: 245-251.
Article PubMed Google Scholar - Przylepa J, Himes C, Kelsoe G: Lymphocyte development and selection in germinal centers. Curr Top Microbiol Immunol. 1998, 229: 85-104.
PubMed Google Scholar - Cope AP, Sonderstrup G: Evaluating candidate autoantigens in rheumatoid arthritis. Springer Semin Immunopathol. 1998, 20: 23-39.
Article PubMed Google Scholar - Weyand CM, Goronzy JJ: HLA-DRB1 alleles as severity markers in RA. Bull Rheum Dis. 1994, 43: 5-8.
PubMed Google Scholar - Weyand CM, McCarthy TG, Goronzy JJ: Correlation between disease phenotype and genetic heterogeneity in rheumatoid arthritis. J Clin Invest. 1995, 95: 2120-2126.
Article PubMed PubMed Central Google Scholar - Wagner U, Kaltenhauser S, Sauer H: HLA markers and prediction of clinical course and outcome in rheumatoid arthritis. Arthritis Rheum. 1997, 40: 341-351.
Article PubMed Google Scholar - Seidl C, Koch U, Buhleier T: HLA-DRB1*04 subtypes are associated with increased inflammatory activity in early rheumatoid arthritis. Br J Rheumatol. 1997, 36: 941-944.
Article PubMed Google Scholar - Gough A, Faint J, Salmon M: Genetic typing of patients with inflammatory arthritis at presentation can be used to predict outcome. Arthritis Rheum. 1994, 37: 1166-1170.
Article PubMed Google Scholar - Suarez-Almazor ME, Tao S, Moustarah F, Russell AS, Maksymowych W: HLA-DR1, DR4, and DRB1 disease related subtypes in rheumatoid arthritis. Association with susceptibility but not severity in a city wide community based study. J Rheumatol. 1995, 22: 2027-2033.
PubMed Google Scholar - Mottonen T, Paimela L, Leirisalo-Repo M, Kautiainen H, Ilonen J, Hannonen P: Only high disease activity and positive rheumatoid factor indicate poor prognosis in patients with early rheumatoid arthritis treated with 'sawtooth' strategy. Ann Rheum Dis. 1998, 57: 533-539.
Article PubMed PubMed Central Google Scholar - Yang H, Rittner H, Weyand CM, Goronzy JJ: Aberrations in the primary T-cell receptor repertoire as a predisposition for synovial inflammation in rheumatoid arthritis. J Investig Med. 1999, 47: 236-245.
PubMed Google Scholar - Wagner UG, Kurtin PJ, Wahner A: The role of CD8+CD40L+ T cells in the formation of germinal centers in rheumatoid synovitis. J Immunol. 1998, 161: 6390-6397.
PubMed Google Scholar - Wordsworth P, Pile KD, Buckely JD: HLA heterozygosity contributes to susceptibility to rheumatoid arthritis. Am J Hum Genet. 1992, 51: 585-591.
PubMed PubMed Central Google Scholar - Martens PB, Goronzy JJ, Schaid D, Weyand CM: Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum. 1997, 40: 1106-1114.
Article PubMed Google Scholar - Vallejo AN, Nestel AR, Schirmer M, Weyand CM, Goronzy JJ: Agingrelated deficiency of CD28 expression in CD4+ T cells is associated with the loss of gene-specific nuclear factor binding activity. J Biol Chem. 1998, 273: 8119-8129.
Article PubMed Google Scholar - Namekawa T, Wagner UG, Goronzy JJ, Weyand CM: Functional subsets of CD4 T cells in rheumatoid synovitis. Arthritis Rheum. 1998, 41: 2108-2116.
Article PubMed Google Scholar - Park W, Weyand CM, Schmidt D, Goronzy JJ: Co-stimulatory pathways controlling activation and peripheral tolerance of human CD4+CD28- T cells. Eur J Immunol. 1997, 27: 1082-1090.
Article PubMed Google Scholar - Liuzzo G, Kopecky SL, Frye RL: Perturbation of the T-cell repertoire in patients with unstable angina. Circulation. 1999, 100: 2135-2139.
Article PubMed Google Scholar
Acknowledgments
The authors are indebted to Tammy J Dahl and James W Fulbright for their assistance.
Author information
Authors and Affiliations
- Mayo Clinic Foundation, Rochester, Minnesota, USA
Cornelia M Weyand & Jörg J Goronzy
Authors
- Cornelia M Weyand
You can also search for this author inPubMed Google Scholar - Jörg J Goronzy
You can also search for this author inPubMed Google Scholar
Rights and permissions
About this article
Cite this article
Weyand, C.M., Goronzy, J.J. Association of MHC and rheumatoid arthritis: HLA polymorphisms in phenotypic variants of rheumatoid arthritis.Arthritis Res Ther 2, 212 (2000). https://doi.org/10.1186/ar90
- Received: 19 January 2000
- Accepted: 29 February 2000
- Published: 19 April 2000
- DOI: https://doi.org/10.1186/ar90