Stress-Induced Biological Aging: A Review and Guide for Research Priorities (original) (raw)
. Author manuscript; available in PMC: 2023 Oct 23.
Published in final edited form as: Brain Behav Immun. 2022 May 31;104:97–109. doi: 10.1016/j.bbi.2022.05.016
Abstract
Exposure to chronic adverse conditions, and the resultant activation of the neurobiological response cascade, has been associated with an increased risk of early onset of age-related disease and, recently, with an older biological age. This body of research has led to the hypothesis that exposure to stressful life experiences, when occurring repeatedly or over a prolonged period, may accelerate the rate at which the body ages. The mechanisms through which chronic psychosocial stress influences distinct biological aging pathways to alter rates of aging likely involve multiple layers in the physiological-molecular network. In this review, we integrate research using animal, human, and in vitro models to begin to delineate the distinct pathways through which chronic psychosocial stress may impact biological aging, as well as the neuroendocrine mediators (i.e., norepinephrine, epinephrine, and glucocorticoids) that may drive these effects. Findings highlight key connections between stress and aging, namely cellular metabolic activity, DNA damage, telomere length, cellular senescence, and inflammatory response patterns. We conclude with a guiding framework and conceptual model that outlines the most promising biological pathways by which chronic adverse conditions could accelerate aging and point to key missing gaps in knowledge where future research could best answer these pressing questions.
Keywords: psychosocial stress, biological aging, cellular senescence, telomere length, DNA damage, inflammation
1. Introduction
Exposure to chronic adverse conditions that activate a neurobiological stress response has been associated with increased risk for morbidity and mortality1,2, including earlier onset of the most common diseases of aging (e.g., cardiovascular disease, diabetes, hypertension, and cognitive decline). This body of research has led to the hypothesis that the key mechanism driving associations between stress and these health outcomes is an acceleration of the biological aging process, including increases in systemic inflammation and telomere shortening.3–5 However, emerging research on the biology of aging offers additional insights into the specific mechanisms that might drive accelerated aging under chronic adverse conditions.
Psychosocial stress is a broad term that encompasses many different experiences and exposures across the lifespan, including early life adversity, low socioeconomic status, stressful life events (e.g., loss of a loved one), caregiving, work-related stress, financial strain, discrimination, low social support, interpersonal conflict, and loneliness. These experiences of chronic stress and adversity are biologically impactful when an individual perceives that the environmental demands of a situation tax or exceed their ability to adapt or cope.6 Thus, stressful experiences may not exact a lasting physiological toll if an individual has adequate reserves that enable successful response to and/or recovery from a stressor. It is when the demands of a situation are in excess of an individual’s coping capacity that the pathophysiological processes that drive aging occur. This excess stress activates key neuroendocrine responses, including the sympathetic nervous system (SNS) and the hypothalamus-adrenal-pituitary (HPA) axis. Activation of the SNS results in the release of catecholamines (e.g., norepinephrine) from neuronal axon terminals that innervate tissue in immune and other organ systems (e.g., lymph nodes, thymus, spleen), and the release of both norepinephrine (NE) and epinephrine (E) from the adrenal medulla into circulation via blood vessels.7,8 The HPA axis regulates the release of glucocorticoids (GC), commonly referred to as cortisol in humans and corticosterone in rodents, from the adrenal cortex into blood vessels to circulate throughout the body. These neuroendocrine mediators alter numerous physiological processes that may play an important role in biological aging. This review and guide for research priorities aims to delineate the distinct pathways through which chronic psychosocial stress and the repeated activation of the SNS and HPA may directly influence biological aging pathways, and in turn, impact the rate of biological aging. By understanding how stress can impact the pathophysiological process of biological aging, we can also identify places to intervene and promote healthy aging. We begin by describing current conceptual models of biological aging, and then apply these models to the extant literature linking stress and SNS and HPA activation to biological aging processes. We conclude by highlighting key areas with limited existing evidence, opportunities to intervene and promote healthy aging, and discuss future research priorities.
2. Conceptual Models of Biological Aging
Biological aging can be described as an iterative and gradual process of decline in optimal physiology that occurs over decades of life. This process consists of biological changes at the molecular, genomic, and cellular levels, that result in the accumulation of damage and lead to pathophysiology that disrupts higher level functions that are necessary to sustain life.9 As damage accumulation reaches a level that limits function, the organism begins to exhibit aging-related phenotypes, including onset of age-related diseases and functional limitations.10 In their seminal review paper, López-Otín and colleagues identified several hallmarks of the biological aging process.9 They proposed that genomic instability (e.g., DNA damage), telomere attrition, epigenetic alterations, and loss of proteostasis are the primary causes of cellular damage. Secondary cellular changes that occur after prolonged damage accumulation include deregulated nutrient sensing, poor energy production from mitochondrial dysfunction, and cellular senescence. These hallmarks may initially mitigate damage but, if they become chronic, they can further promote damage through the release of reactive oxygen species (ROS) and inflammatory signals from senescent cells, termed the senescence-associated secretory phenotype (SASP), and damaged or necrotic tissue (e.g., damage-associated molecular patterns [DAMPs]).11–15 López-Otín and colleagues identified that stem cell exhuastion and altered intercellular communication (e.g., inflammtion) were ultimately responsible for the observed aging-related phenotypes. We describe each of these hallmarks in more detail in Table 1.
Table 1.
Hallmarks of Aging (Lopez-Otín et al., 2013)
| Hallmark | Definition | Measurement |
|---|---|---|
| Genomic instability | Accumulation of DNA damage, including point mutations, translocations, chromosomal gains and losses, telomere shortening, and gene disruption | γ-H2AX, 8-OHdG, Comet assay18,20,158 |
| Telomere attrition | Accumulation of DNA damage to telomeres, the protective sequences of DNA at the end of chromosomes | Telomere length, telomerase enzyme activity159 |
| Epigenetic alterations | Age-related DNA modifications, including histone acetylation, methylation, and chromatin remodeling | DNA methylation and histone modification via next generation sequencing149 |
| Loss of proteostasis | Disruption to cellular systems that refold or degrade unfolded or misfolded proteins and stabilize correctly folded proteins | Heat shock proteins, accumulation of unfolded or misfolded proteins (e.g., Alzheimer’s disease), failure in autophagy160,161 |
| Deregulated nutrient sensing | Age-related changes in the nutrient-sensing pathways that regulate metabolism | IGF-1, mTOR, AMPK, sirtuins, FOXO |
| Mitochondrial dysfunction | Decreased ability of mitochondria to produce ATP and increased production of reactive oxygen species | ROS production, mitochondrial respiratory capacity, mtDNA damage105,109 |
| Cellular senescence | Cell cycle arrest accompanied by the release of inflammatory cytokines, chemokines, and damage-associated molecular patterns (DAMPs) referred to as the senescence-associated secretory phenotype (SASP) | p16INK4a, p21, p53, senescence-associated β-galactosidase, SASP11,136 |
| Stem cell exhaustion | Decreased stem cell function, including renewal, immune function, and blood production | FGF2, reduced proliferation in culture, cell lineage skewing (e.g., fewer adaptive immune cells and naive T cells)162 |
| Altered intercellular communication | Increased inflammatory signaling, release of pro-inflammatory cytokines and DAMPs; Endocrine communication failure | NF-κB, IL-6, TNF-α,HMGB1, NLRP3 Inflammasomes , SASP13,65,163 |
Although aging is a natural process that occurs gradually over time as a direct result of living, evidence also suggests that it may be modifiable. Geroscience is an emerging field of scientific inquiry that posits that biological aging is a common driver of age-related diseases and declines and that interventions that directly impact or modify biological aging pathways can slow, prevent, or even reverse accelerated aging and delay the onset of age-related disease.16 Indeed, research on pharmacological and behavioral interventions that target these pathways has provided evidence that biological aging can be delayed, with the additional benefit of extending the healthspan (number of years free from disease) and lifespan.16,17 Importantly, in this review we use the hallmarks of biological aging as a framework to summarize the growing literature on psychosocial stress as a biobehavioral modifier of accelerated aging. Specifically, we review evidence from pre-clinical in vitro and animal models and a growing body of observational and experimental human studies to begin to delineate the distinct biological aging mechanisms through which chronic psychosocial stress and activation of the stress response system (i.e., release of neuroendocrine mediators) can directly impact pathways to accelerated aging.
3. Effects of Stress on Biological Aging Pathways
3.1. Cellular Stress and DNA Damage
One of the first processes in the biological aging pathway is the generation of damaging agents. The most frequent form of damage within a cell is a result of the production of oxidants—a process common to all respiratory life.18 Oxidative stress refers to a state of cellular imbalance in which the production of oxidants exceeds the antioxidant capacity of a cell, and it is often a consequence of the over-production of reactive oxygen species (ROS) during cell replication, mitochondrial respiration, and the breakdown of biogenic amines.18 Specifically, during oxidative phosphorylation—the process by which healthy mitochondria produce energy—stored carbohydrates and fats are broken down into adenosine triphosphate (ATP) and carbon dioxide (CO2), and electrons partially reduce oxygen into superoxide, which is an ROS. This form of ROS production can be further amplified under conditions of psychological stress when catecholamines bind to _β_2-adrenoreceptors on the cell surface, leading to an up-regulation of enzyme protein kinase A (PKA) activity, which increases oxidative phosphorylation and creates more ROS.19,20 Increased activity of the mitochondrial enzyme monoamine oxidase-a (MAO-A) during stress also produces ROS by breaking down catecholamines and other biogenic amines, leading to MAO-A dependent production of H2O2. This activity has been shown to damage cardiomyocytes and cardiac tissue in mice and to play a role in age-related disease.21–23
Given that ROS are unstable singlet oxygen molecules, they stabilize themselves by seeking out and binding to nearby molecules. When ROS bind to DNA molecules, that region of the DNA becomes damaged.18 ROS-induced damage occurs in vulnerable regions of the chromosome such as the telomeres but can also occur across the whole genome and within mitochondrial DNA.11,24 Importantly, excess levels of DNA damage in a cell can activate a DNA damage response (DDR) pathway that initiates cellular arrest to allow the damage to be repaired before cellular replication occurs.15 Sustained and/or elevated levels of DNA damage that cannot be repaired can initiate apoptosis, necrosis, or cellular senescence, the latter being a permanent state of cellular replication arrest that occurs through activation of the p53 pathway.11,25 Through this process, damaged cells prevent mutations from being carried through to daughter cells, and function as a tumor suppressor by preventing oncogenic mutations from being replicated. As evidence of this, a recent study demonstrated that deletion of a gene that encodes a key DNA repair enzyme in mice caused increased DNA damage and immunosenescence that led to widespread senescence and damage to tissues and organ systems.26 Importantly, when damage to the telomeric region of the DNA is not repaired prior to cell replication, it can accelerate the shortening of telomeres and activate cellular senescence when telomeres reach a critically short length,27,28 highlighting an important second outcome of cellular stress.
Research with humans and animals has linked chronic psychosocial stress to increased levels of DNA damage. For instance, observational human studies suggest that academic stress, bereavement, and informal caregiving are associated with elevated DNA damage and reduced DNA repair capacity in peripheral blood.18,29–34 In experimental models, individuals exposed to a stressful experience showed elevated markers of DNA damage, RNA damage, and lipid peroxidation, suggesting a causal effect of acute stress on these pathways.18,34 More striking findings have been reported in rodent models, where repeated exposure to stressors resulted in increased DNA damage across a number of stressor types (e.g., social isolation, restraint stress, forced swimming) and tissues (e.g., peripheral blood, bone marrow cells, liver, gut mucosa).35–40 In accordance with this, mice subjected to restraint stress showed an up-regulation of genes related to DNA damage response signaling pathways in T-lymphocytes; however, the absolute levels of DNA damage in the cells did not change, suggesting that the damage was repaired.41 Other research has demonstrated that rodents exposed to psychosocial stress show increased DNA damage and reduced DNA repair in the brain, including the prefrontal cortex, amygdala, and hippocampal regions.38,42–44
Several studies have demonstrated that stress hormones such as NE and isoproterenol (a synthetic catecholamine) can directly increase DNA damage in different types of cells, including 3T3, U2OS, and ovarian cancer and epithelial cell lines,20,45–48 however, one study found that NE exposure was associated with decreased DNA damage. As an in vivo model of chronic stress, mice that received isoproterenol for 4 weeks also showed increased DNA damage in the thymus.20 Other research has linked elevated cortisol to increased levels of oxidative stress and decreased DNA damage repair activity in human peripheral blood and 3T3 cells.18,45,49 The combined effect of multiple stress hormones has also been examined. For instance, 3T3 cells (an immortalized mouse embryonic fibroblast cell line) treated with NE, E, or cortisol showed an increase in DNA damage and a decrease in DNA repair capacity, as well as altered expression of genes associated with DNA damage or repair.45 This effect was also observed in multiple breast cancer cell lines, along with increased ROS.19
Taken together, the evidence that psychosocial stress and associated stress hormone mediators are drivers of aging and that this process occurs through the accumulation of DNA damage is striking. Most research to date indicates that these pathways are causal and likely involve a direct increase in ROS through cellular respiration and break down of catecholamines and increased DNA damage and reduced DNA repair activity.19,20,45–49
3.2. Telomere Maintenance
Telomere maintenance is an important mechanism for preventing cellular aging.9 Telomeres are DNA complexes that cap the ends of chromosomes and naturally shorten over time with cell replication. The enzyme telomerase plays an important role in telomere maintenance, as it functions to repair and elongate telomeres to protect them from shortening.50,51 As mentioned above, telomeres are particularly vulnerable to damage accumulation, and critically short telomeres that lack sufficient telomerase capping can trigger cellular senescence.28 Telomerase activity can also decrease ROS and protect mitochondrial DNA under conditions of cellular stress.52,53 Epel and colleagues were the first to link psychosocial stress to telomere maintenance, finding that maternal caregivers of children with and without a disability who reported higher perceived stress had shorter telomeres and lower telomerase activity in peripheral blood mononuclear cells (PBMCs), and higher levels of a circulating marker of oxidative stress than those who reported lower stress.54 Since this seminal study, numerous reports have linked many different experiences of chronic stress and adversity across the lifespan, including low socioeconomic status, early life adversity, and low social support, with shortened telomere length in peripheral blood leukocytes, saliva (composed predominantly of leukocytes), and buccal cells (for review, see Rentscher, Carroll, & Mitchell, 2020).55 In addition, studies have also found evidence of potential intergenerational effects of stress on telomere maintenance, such that higher maternal stress during pregnancy was associated with shorter newborn and child telomere length.56–58 In an animal model of chronic stress, prairie voles that were socially isolated had higher corticosterone, shorter telomere length, and increased oxidative damage than those that were not.59
Observational and experimental studies have also linked stress hormones with shortened telomere length. For instance, in a study of maternal caregivers and controls, higher levels of nocturnal NE, E, and cortisol were associated with shorter telomere length in PBMCs.60 Similarly, in a study of women caregiving for a partner with dementia, higher levels of nocturnal E following an acute stressor and flatter diurnal cortisol slopes were associated with shorter PBMC telomere length.61 Other research has found that shorter telomere length was associated with higher E in women,62,63 and a higher cortisol awakening response in women, with the opposite pattern observed in men64 Maternal cortisol levels may also impact their offspring’s telomere length, with higher cortisol output relating to shorter newborn telomere length.65 There is some evidence that stress hormones may also modify telomerase activity. In a study of maternal caregivers and controls, higher levels of nocturnal E were associated with lower telomerase levels in PBMCs. In an in vitro model, human T cells that were treated with hydrocortisone at levels equivalent to those reached during stress exposure showed decreased telomerase levels, suggesting that cortisol may inhibit telomerase enzyme activity and impair telomere maintenance.66
Together, these findings suggest that many forms of psychosocial stress are associated with shorter telomere length in humans, with some evidence linking stress hormones to lower telomerase levels and shortened telomere length. However, most of these studies are observational and additional research is needed that investigates the causal pathways linking stress and stress hormones to these telomere maintenance mechanisms.
3.3. Mitochondrial Dysfunction
Mitochondrial dysfunction is a secondary cellular change that results from the accumulation of damage and is characterized by enhanced oxidative damage, increased mitochondrial biogenesis, and decreased mitophagy. As mentioned previously, normal respiratory processes such as oxidative phosphorylation create ROS that can also lead to mitochondrial damage and a decline in function. Mitochondrial DNA (mtDNA) is a preferential target of ROS due to its physical proximity, lack of efficient repair mechanisms, and limited protection, as mtDNA lacks histones.24 Research has suggested a vicious cycle whereby damaged mitochondria in cardiomyocytes produced up to 10 times more hydrogen peroxide (H2O2) than healthy mitochondria, which then lead to exaggerated increases in oxidative damage, acting to further accelerate aging.67 In accordance with this, another study found that mitochondria in senescent fibroblasts produced more ROS than mitochondria in younger fibroblasts.68 Mitochondrial dysfunction is an important process in the biological aging pathway because oxidative damage caused by mitochondria is thought to drive telomere shortening and contribute to increased cellular senescence through cellular stress pathways.24,68,69 One study demonstrated that damaged mtDNA can lead to accelerated aging in mice, finding that mice without the ability to repair mtDNA had 3 to 8 times more mtDNA mutations, aged faster phenotypically, and had a shorter lifespan than wild-type mice; however, it is interesting to note that mice with more mutations did not show an increase in H2O2 or markers of RNA and DNA oxidative damage.70
Research also suggests that cells that have entered a senescent state have increased mitochondrial biogenesis, which has been termed senescence-associated mitochondrial dysfunction (SAMD), that can lead to further ROS production.24 Mitochondrial biogenesis is mediated by peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC-1α), a gene that regulates cellular energy metabolism.24 As evidence of this, studies have found that senescent cells show increased expression of PGC-1α and other genes associated with mitochondrial biogenesis.24 In addition, senescent mouse lung fibroblasts and cultured epithelial cells have shown up-regulation of genes associated with mitochondrial biogenesis, such as mammalian target of rapamycin (mTOR) and PGC-1α, and increased ROS production, indicating an increase in mitochondrial biogenesis.71
In aging cells, the accumulation of dysfunctional mitochondria can also be attributed to decreased mitophagy (mitochondrial autophagy), the process by which cells clear dead or dysfunctional mitochondria.72 Researchers have proposed several mechanisms through which mitophagy can be disrupted. For instance, studies have found that elevated cytoplasmic p53 (which increases with DNA damage) decreased mitochondrial fission (in which new mitochondria are broken down while dysfunctional mitochondria remain fused and avoid degradation),72 and the persistent activation of mitochondrial enzyme MAO-A (which inhibits Parkin, a mitochondrial degradation signaling protein) lead to an increase in the number of dysfunctional mitochondria.73 Another study found that MAO-A inhibited the later stages of the autophagy-lysosome pathway (responsible for the breakdown of mitochondria), causing the accumulation of protein degradation signaling molecules.74 In this study, MAO-A also impaired lysosomal acidification and function and impaired lysosomal biogenesis by inhibiting the master regulator of the autophagy-lysosome pathway, transcription factor EB. This accumulation of dysfunctional mitochondria increases the amount of ROS in the cell, leading to enhanced transcription of pro-inflammatory genes in the NF-кB pathway and production of pro-inflammatory cytokines.72,75,76 As a caveat, much of this research has been conducted with cardiomyocytes as a model of cardiovascular disease; research is needed to determine if these findings are applicable to many other tissues, including lymphocytes, neuronal cells, vascular endothelium, liver, intestinal walls, osteocytes, and muscle.
The contribution of psychosocial stress and associated stress hormones to mitochondrial dysfunction in cells is beginning to be defined, and a well-developed conceptual model has been published to guide this area of research.75,77,78 In this paper, we focus on mitochondrial dysfunction as one pathway through which stress may alter rates of aging; however, interested readers are referred to these reviews for a more in-depth discussion of this topic.75,77,78 As mentioned previously, increased activity of the mitochondrial enzyme MAO-A during stress can increase ROS production through the breakdown of catecholamines. In addition, research suggests that the breakdown of catecholamines can lead to mtDNA deletions in the adrenal medulla and adrenal cortex, and that this process increases with age.79 The effect of cortisol on cellular metabolism is thought to have an inverted-U shape, in which short-term exposure increases mitochondrial biogenesis but long-term exposure increases ROS production and respiratory chain dysfunction.80
Experimental studies with rodents have demonstrated that several forms of chronic stress (e.g., restraint stress, noise, unpredictable stress) are associated with decreased mitochondrial function, energy production, and activity on respiratory chain complexes.78 For instance, in mouse and rat models, chronic stress increased mitochondrial damage in the brain by inhibiting respiratory chain complexes I-III and decreasing mitochondrial membrane potential81–83. Other studies showed that respiratory chain complex IV (COX) had a 0–80% reduced activity.78 Tellingly, COX is encoded by mtDNA, indicating that functional changes could occur from mtDNA defects. Whereas much of the current research with animals has measured facets of mitochondrial function in specific tissues after exposure to acute and chronic stressors, research with humans has been observational and has not measured mitochondrial function directly.78 To date, a few studies have linked experiences of chronic stress and adversity, including caregiving and early life adversity, to higher mtDNA copy number and higher free circulating mtDNA,84,85 and one study reported increased free floating mtDNA in circulation after exposure to an acute laboratory stressor.86
Much of the research on stress hormones and mitochondria has examined the effects of dysfunctional mitochondria on the SNS and HPA axes; however, a few studies have focused on the effects of stress hormones on mitochondrial function.77 In a study with humans, the binding of glucocorticoid receptors to mtDNA enhanced the transcription of mitochondrial genes, increasing the energy production capacity of the mitochondria.87 In an in vitro model, rat liver tissue that was exposed to E increased mitochondrial biogenesis and oxidative damage to lipids and proteins.88 Importantly, once mitochondria become dysfunctional, they become even more sensitive to mediators such as ROS and stress hormones, acting as an accelerator of aging in response to further stress exposures.89 Considerable research remains to be conducted in order to link psychosocial stress and associate neuroendocrine mediators to the mitochondrial pathway of aging.
3.4. Cellular Senescence
Cellular senescence is a state of permanent cell growth arrest in which cells are unable to proliferate but remain metabolically active.11,90 In younger organisms, cellular senescence works as a tumor-suppression mechanism by stopping malignant cells from proliferating;91 however, in older organisms, the accumulation of senescent cells is thought to drive a majority of age-related diseases, including diabetes, arthritis, cancer, dementia, and cardiovascular disease.92 Senescence can be caused by oncogene mutations, severe DNA damage, telomere shortening, and mitochondrial dysfunction, but is dependent on activation of the p53/p21 and pRB/p16 tumor suppressor pathways to sustain a senescent state.11,13,15 When senescence is initiated by telomere shortening or repeated replication, it is referred to as replicative senescence.13,93 Cellular senescence that is initiated by DNA damage is commonly mediated by the p53/p21 pathway with secondary engagement of the pRB/p16 pathway; however, certain stimuli such as oncogenic RAS can induce solely the pRB/p16 pathway.11 Thus, both pathways are important for initiating cellular senescence in vivo.
Similar to aging, there are several hallmarks of cellular senescence that can be categorized as altered signaling pathways and morphological changes.93 The most commonly studied altered signaling pathways include activation of the DNA damage response (DDR), cyclin-dependent kinase (CDK) cell-cycle inhibitors, and senescence-associated secretory phenotype (SASP).93 Although classified independently, many of these signaling pathways interact. The DDR is a network of cellular pathways that detects and signals the presence of DNA damage and promotes DNA repair.94 CDKs regulate proteins involved in cell cycle progression, with p16INK4a and p21 the most commonly associated with senescence.93 Whereas p21 is more generally associated with the DDR and is regulated by p53, p16INK4a is consistently activated in senescent cells.93 The SASP is a set of inflammatory cytokines, chemokines, and DAMPs that varies by cell type; yet, the majority of cells exhibit a discrete shift in the inflammatory agents they release upon entering a senescent state, and these factors are believed to be critical in driving age-related diseases.11–15,95 The SASP is mediated by transcription factor NF-кB, which is activated by the DDR.93 Many SASP factors are associated with low grade, chronic inflammation and can cause neighboring cells to mimic the behavior of aggressive cancer cells, including basement layer invasion, proliferation, and migration.13,15 In terms of morphological alterations, senescent cells display an enlarged, flattened, and irregular shape, an increased lysosomal content, and a greater number of mitochondria. It is important to note that the morphological alterations are not exclusive to the senescent state, making senescent cells difficult to identify.11,93,96
Research that examines whether psychosocial stress and associated neuroendocrine mediators can directly impact the accumulation of senescent cells and alter their secretory phenotype is not well developed. To date, one study in humans has investigated the association between psychosocial stress and cellular senescence, finding that chronic stress exposure, perceived stress, and accumulated daily stress appraisals were each associated with expression of the p16INK4a-encoding gene CDKN2A, as a marker of senescence in leukocytes.97 In addition, two experimental studies with mice demonstrated that exposure to chronic stress was associated with increased p53 and a trend toward increased p16INK4a expression in the liver and spleen98 and increased p16INK4a and p21 expression in bone marrow leukocytes.40
A handful of in vivo and in vitro studies have investigated the effects of stress hormones on cellular senescence. For instance, mice that were administered isoproterenol (a synthetic catecholamine) showed up-regulated p53 and p21 in cardiac tissue and bone marrow cells, suggesting an increase in senescent cells.99 As a model of chronic stress, mice that received isoproterenol for 4 weeks showed decreased p53 in the thymus.20 As mentioned previously, research has demonstrated that when NE is broken down by MAO-A, it can increase the production of ROS, activate p53 and the DDR, and initiate cellular senescence, confirmed by the presence of senescence-associated β-galactosidase-positive cells.73 As additional evidence of this, cells incubated with isoproterenol showed increased levels of ROS, p53, and p21; however, this study did not test whether this also resulted in senescence.99 In addition, U2OS cells and mouse embryonic fibroblasts cultured with isoproterenol every 12 hours for 3 days showed decreased p53.20 With regards to regulation of the SASP, one study also found that cortisol suppressed expression of the SASP in senescent cells, consistent with the anti-inflammatory role of cortisol.100
Together, although extant findings suggest that adrenergic signaling by NE and, to a lesser extent, E, up-regulate metabolic activity that can activate cellular stress pathways,20,73,99,100 very few studies have extended these findings to the full biological aging pathway to examine whether elevated or prolonged exposure to neuroendocrine mediators induces cellular senescence. Likewise, cortisol may play an important role in this pathway; however, the effects of chronic exposure have not yet been tested. To date, very few studies have examined whether experiences of chronic stress and adversity and stress hormone mediators impact cellular senescence, pointing to a gap in current knowledge.
3.5. Inflammation
Inflammation has a myriad of effects on cellular processes. Of paramount relevance to biological aging, inflammation is a key source of damage and inflammatory cells are destructive not only to pathogens (intended target), but also to nearby cells and tissues (unintended target). Importantly, this collateral damage from immune activity is thought to be a driver of the aging process.101 Inflammation is known to increase with chronological age—a process that has been termed ‘inflammaging’102—and is a hallmark of aging. Psychosocial stress and associated neuroendocrine mediators can directly activate these cellular processes and the pathways through which stress promotes inflammation have been well characterized in both human and animal research.103 The SNS and HPA axes can modulate inflammation, and although they sometimes have opposing effects, they have also been shown to work in tandem to regulate the inflammatory response. These processes have been described in several excellent reviews and will be highlighted here as they relate to biological aging; however, readers are directed to additional reading for a more in-depth discussion.5,8,103–105 Specifically, the SNS regulates the inflammatory response via the production and transportation of NE.106 In response to stress, NE interacts with beta-adrenergic receptors on the surface of immune cells, which results in a signaling cascade characterized by enhanced activity of transcription factors NF-κB, AP-1/JUN, and CREB.103 NF-кB is a nuclear transcription factor that mediates the pro-inflammatory response by regulating genes involved in the production of pro-inflammatory cytokines such as Interleukin (IL)-1B, IL-6 and Tumor Necrosis Factor (TNF)-α, among many others.103 The HPA axis acts in a reciprocal fashion through the production of GC and activation of glucocorticoid receptors, which inhibit NF-кB activity and the expression of pro-inflammatory cytokines.103 Although acute increases in GC inhibit the transcription of pro-inflammatory cytokines, chronic increases in GC are thought to lead to glucocorticoid receptor insensitivity or down-regulation of glucocorticoid receptors and an enhanced pro-inflammatory response.107–110
A sizeable literature has drawn direct links between chronic psychosocial stress and adversity and inflammation.103 In observational and experimental studies, stress exposure has been associated with an increase in NF-кB transcription factor activity and the production of key inflammatory cytokines (e.g., IL-6, TNF-α, C-reactive protein [CRP]), chemokines (e.g., MCP-1, CXCL1), and DAMPs (e.g., HMGB1, Hsp72) in humans and animals.111–116 Research also suggests that chronic stress is associated with glucocorticoid insensitivity (i.e., resistance) in immune cells. For instance, individuals with greater stress related to loneliness and caregiving had fewer glucocorticoid receptors, lower glucocorticoid receptor and higher NF-кB transcription factor activity, and enhanced expression of CRP, IL-1RA, and IL-6.110,117,118 Among parents of children with cancer, greater stress was associated with reduced ability of cortisol and dexamethasone (a synthetic glucocorticoid) to suppress IL-6 production following stimulation with lipopolysaccharide (LPS) in vitro.107,119 In addition, early life adversity has been associated with lower cortisol output, down-regulation of glucocorticoid receptor activity, and up-regulation of adrenergic responsive CREB and NF-кB.120 Experimental studies with animals have also demonstrated that exposure to social stressors induces glucocorticoid insensitivity in splenocytes and peripheral immune cells.121–128
Several studies have also demonstrated that stress hormones NE and E can increase inflammation in both in vivo and in vitro models. In response to acute stress challenges such as LPS stimulation or recalling an event that caused anger, an enhanced catecholamine response was associated with a significant increase in circulating IL-6, IL-10, and CRP in humans and rodents.129,130 Other studies have found that increasing doses of NE and E in vivo, particularly over longer periods of time, led to increased NF-кB and IL-6 expression in mice and rats.131,132 These results are further supported by in vitro findings. Across different cell types, from lymphocytes to hepatoma cells, studies have demonstrated that treatment with NE or E results in increased expression of IL-6, CRP, and TNF-α.133–137 In addition, NE has been found to alter the activation and release of inflammatory cytokines from CD8 T cells.137,138
Research has also shown that the production of GC has a classical anti-inflammatory effect and decrease the production of pro-inflammatory cytokines in in vivo and in vitro models. For instance, adult humans with a higher cortisol response to an acute social stressor showed a less pronounced NF-кB response.139 In a population-based study, individuals with a lower cortisol awakening response and flatter cortisol slopes over the day had higher levels of IL-6 and TNF-α, although the association between cortisol slopes and TNF-α was attenuated when controlling for other inflammatory risk factors.140 In addition, adults reporting higher perceived stress had flatter diurnal cortisol slopes and elevated circulating inflammatory markers.141 Research has also demonstrated that cortisol and various synthetic GC decrease intra-cellular production of pro-inflammatory cytokines (most notably IL-6 and TNF-α) in response to LPS stimulation in vitro and in ex vivo cells.142–144 Conversely, research has also shown that the binding of GC to glucocorticoid receptors can activate the NF-кB signaling pathway and pro-inflammatory cytokine production,109 and dexamethasone (a corticosteroid) enhances the induction of toll-like receptor 2 (TLR-2) by TNF-α,145 causing an inflammatory immune response. These findings are supported by a review of genome-wide microarray studies that identified more than 800 genes that are co-regulated by GC and TNF-α.146
Taken together, the research linking psychosocial stress and associated neuroendocrine mediators to inflammatory activity is well defined. This may be one key lynch pin in how experiences of chronic stress and adversity drive biological aging. Further work that builds on this significant foundation of evidence to connect these stress-induced inflammatory pathways to specific hallmarks of biological aging and aging phenotypes, along with manipulation or blockade of these pathways with anti-inflammatory agents, are critical next steps in research on the effects of stress on accelerated aging and disease risk.
4. An Integrated Conceptual Model of Stress and Biological Aging
The body of research summarized in this review suggests that psychosocial stress and the associated cascade of stress hormone mediators contribute to a systemic physiological stress response that could accelerate biological aging through multiple pathways simultaneously. This systemic response is characterized by promotion of oxidative stress, DNA damage, inflammation, and telomere shortening, mitochondrial dysfunction, and ultimately, cellular senescence, which then in turns feeds back on this pathway to further promote inflammation and oxidative stress.
Based on this growing body of research, we propose an integrated conceptual model to guide future research on stress-induced biological aging (Figure 1). This model proposes that experiences of psychosocial stress and adversity contribute to accelerated aging through two primary and parallel pathways. First, psychological stress activates the SNS and HPA axis, which release stress hormones from sympathetic nerve fibers (NE), and adrenal glands (NE, E, and GC). Binding of NE and E to adrenergic receptors on the surface of cells increases energy production and mitochondrial respiration. This increased cellular metabolism results in production of metabolic byproducts, including ROS. Excess production of oxidants that exceed the antioxidant capacity of the cell results in cellular stress and damage accumulation. If unresolved, damage to the nuclear (i.e., telomeric and non-telomeric regions) and mitochondrial DNA can lead to activation of the DDR and p53/p21 pathway.27,75 Concomitant suppression of telomerase activity by GC prevents repair and lengthening of telomere ends. In the mitochondria, mtDNA damage contributes to an increase in the p53/p21 pathway and the breakdown of mitophagy (mitochondrial autophagy). Importantly, p53 activation suppresses expression of PGC-1α, the master regulator of mitochondrial biogenesis, which decreases mitochondrial biogenesis.147,148 This lack of biogenesis, accompanied by impaired mitophagy, increases the number of dysfunctional mitochondria in the cell and further increases ROS production.72–74,149 Activation of the p53 tumor suppressor pathway also feeds back to increase activation of pro-inflammatory transcription factor NF-κB. Finally, excess DNA damage, telomere shortening, and mitochondrial dysfunction can all lead to cellular apoptosis, necrosis, or cellular senescence. These senescent cells exhibit a shift in their secretory pattern involving a heightened release of pro-inflammatory factors, termed the senescence-associated secretory phenotype (SASP). The SASP is comprised of dozens of factors, including an extensive list of common inflammatory cytokines (e.g., IL-6, IL-1, TNF-α), chemokines (e.g., IL-8, MCP-2, MCP-4, MIP-1α, MIP-3α), and other inflammatory and growth factors (e.g., GM-CSF, VEGF, IGFBPs, MMPs, ICAMs).13 In parallel, dead necrotic cells can further promote inflammatory activity through release of intracellular debris that act as DAMPs and trigger receptors (e.g., TLR-4) on the surface of immune cells, acting as a further promotor of inflammation and tissue damage.113
Figure 1. Integrated Conceptual Model of Stress and Biological Aging.
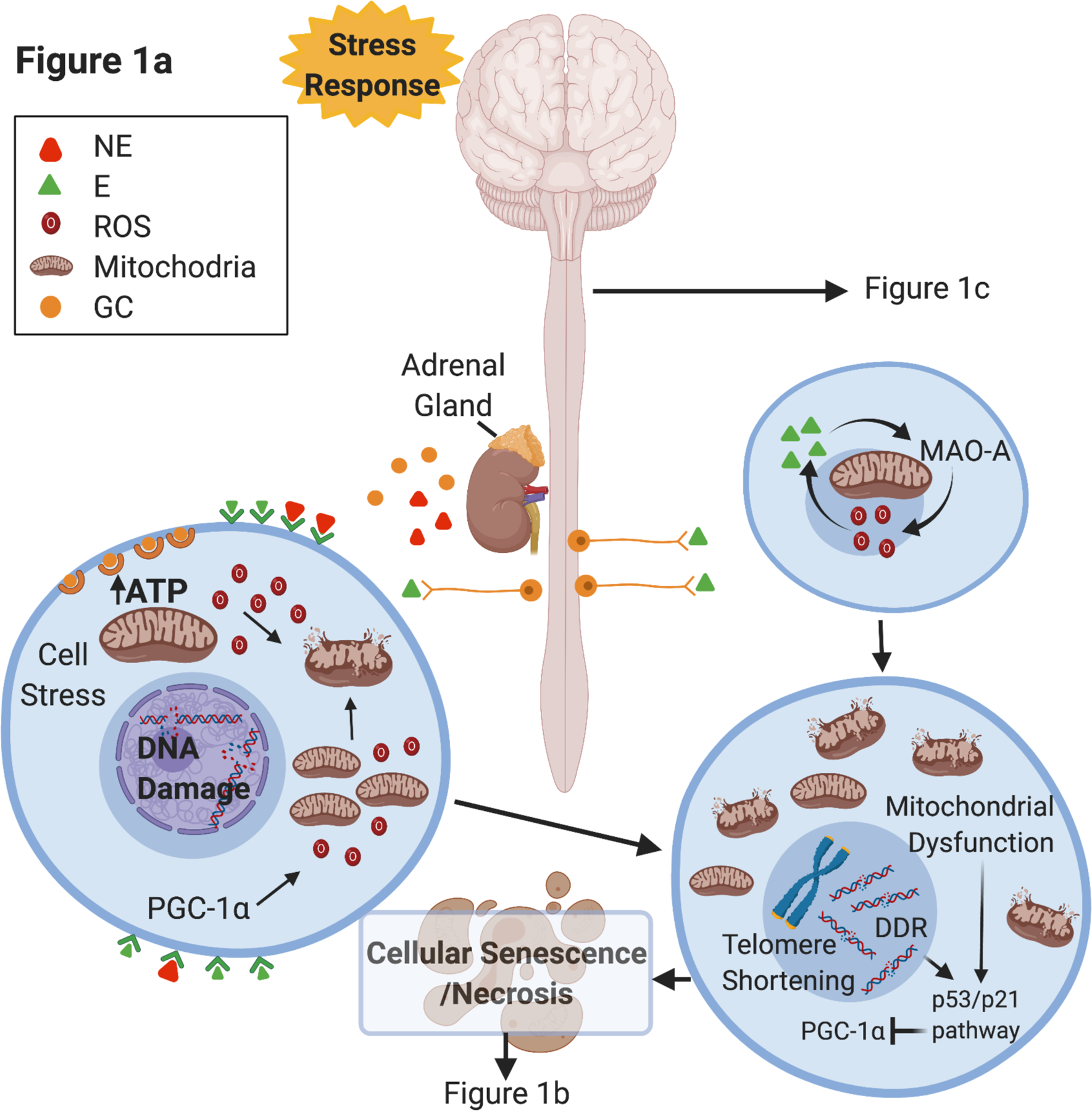
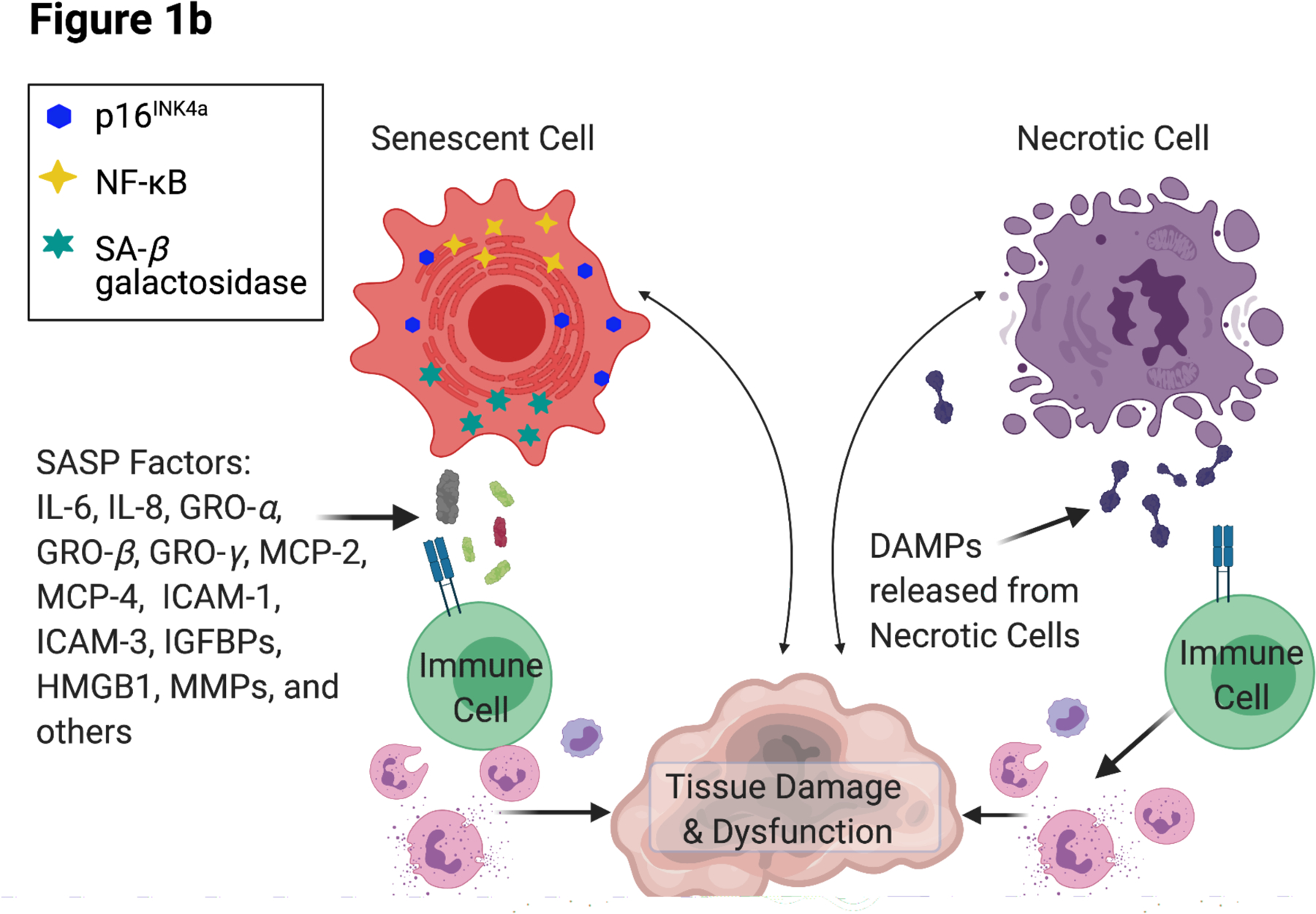
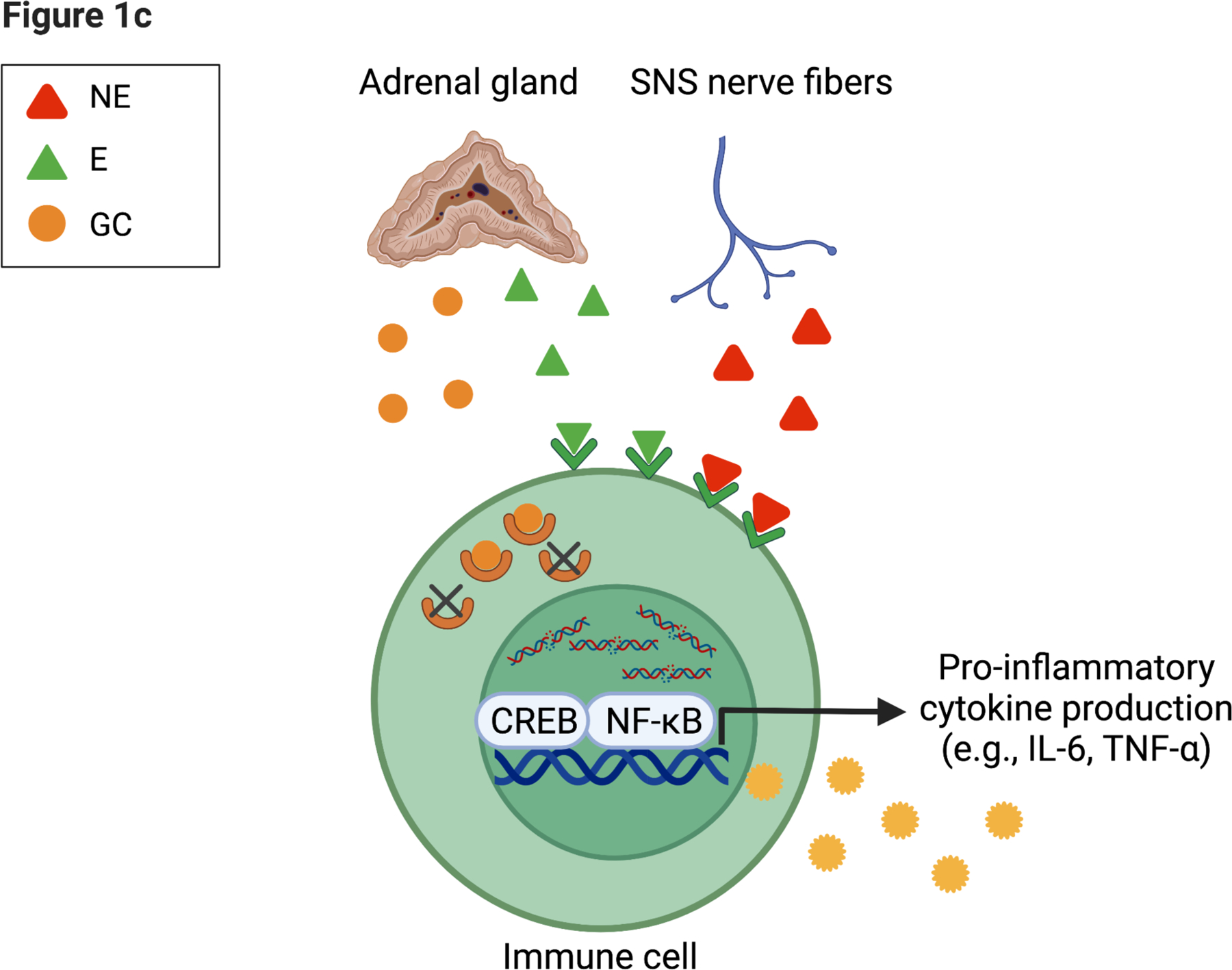
Integrated conceptual model depicting two primary and parallel pathways through which experiences of psychosocial stress and adversity contribute to accelerated aging.
(a) Following activation of the stress response, norepinephrine (NE) is released at sympathetic nerve terminals and epinephrine (E) is released via the adrenal gland into circulation. NE and E stimulate cells to increase metabolic activity of the mitochondria, elevating adenosine triphosphate (ATP) and reactive oxygen species (ROS). Excess ROS can result in DNA damage within the nucleus and the mitochondria. Telomere shortening and DNA damage response pathways are activated and can result in cellular senescence or necrosis. NE and E also lead to activation of MAO-A, which can further increase ROS and inhibit mitophagy. Mitochondrial damage leads to dysfunction that can also exacerbate cellular damage and activate cell stress pathways.
(b) Progressive accumulation of senescent and necrotic cells can further promote systemic inflammation via the release of damage-associated molecular patterns (DAMPs) and senescence-associated secretory phenotype (SASP) factors, acting to further drive tissue damage.
(c) Direct innervation of sympathetic fibers on immune tissue can activate a pro-inflammatory response (e.g., CREB transcription factor) within immune cells. Subsequent release of glucocorticoids (GC) via the HPA axis can have an anti-inflammatory effect; however, prolonged exposure to is thought to result in insensitivity (e.g., glucocorticoid resistance)107, where the receptors for GC no longer signal to the nucleus an effective anti-inflammatory response. Figure created with BioRender.com.
In a parallel pathway, NE increases the activation of pro-inflammatory transcription factor NF-кB and production of circulating inflammatory markers (e.g., TNF-α, IL-6). In addition, prolonged release of GC can decrease glucocorticoid receptor elements, reducing their anti-inflammatory function, which further heightens the inflammatory response. A detailed description of this pathway has been discussed previously.103
5. Future Directions
This review summarizes a considerable body of research that points to a biologically plausible pathway through which psychosocial stress may drive biological aging. However, no research to date has fully demonstrated the complete pathway beginning with induction of psychological stress that results in damage accumulation and leads to cellular senescence, including whether stress-induced senescence then results in earlier aging phenotypes and mortality. Future preclinical and clinical research paradigms in psychoneuroimmunology could harness existing expertise in chronic stress exposure and stress hormone manipulation of cell biology to begin to understand this highly relevant pathway to disease. In this section, we outline three key research domains that are necessary to further our understanding of this pathway and include potential applications for other psychoneuroimmunology research domains that have yet to examine the role of aging biology.
The first key research domain involves the use of in vitro preclinical models to test the effects of stress hormones on biological aging. There is a need for basic research that focuses on the molecular and cellular mechanisms involved in these aging pathways across different tissue types (e.g., immune cells, fibroblasts, neuronal, endothelial, cardiac, etc.) to build our understanding of the molecular machinery that is impacted by stress hormones and drives aging processes. For example, it is not clear whether NE exposure results in damage accumulation specifically in the nucleus, mitochondria, or both, and whether the damage is sufficient to force cells into senescence. It will also be important for future research to test whether specific factors (e.g., physical activity, diet, pharmaceuticals) that are known to promote resistance to cellular stress and damage, such as those that increase antioxidant availability, prevent senescence initiation pathways, alter glucose or MAO metabolism, or control mitochondrial metabolism, could block this effect, and modify the biological aging pathway. Another potential application of this work is to examine whether, under conditions that are known to induce DNA damage such as exposure to cytotoxic agents (e.g., radiation, chemotherapeutics), the additional exposure to NE or GC might impair repair pathways and promote further damage accumulation and cellular senescence. This basic research could shed light on key pathways to target in clinical settings.
The second key research domain involves testing these concepts in animal models where stress physiology and its effects on biological aging can be examined within the interactive levels of the organism. Research gaps are substantial in this domain. For example, one particularly compelling transgenic mouse model (p16–3MR) could be leveraged to better characterize how chronic stress exposure alters biological aging pathways and aging outcomes. With this model, cells that express p16INK4a luminesce and can be selectively removed with a pharmacological agent to examine the reversal of aging.150 This model could therefore be used to induce chronic stress, observe cellular senescence and related inflammation, and then remove senescent cells to examine whether inflammation or observed aging phenotypes are altered. A multitude of research questions could be addressed using this paradigm, including whether additional biological modifications through inhibitors or pharmacological agents (e.g., mTOR regulators, carcinogens, mitochondrial regulators, inflammatory blockade) accelerate or decelerate this aging process, whether these processes enable cancer initiation, progression, and metastasis, and whether senescence and the SASP play a role in stress-induced exacerbation of conditions like chronic pain, chronic fatigue syndrome, arthritis, dementia/Alzheimer’s disease, anxiety, insomnia, and depression.8,104,151–154 We would like to note that this review predominantly included research examining the role of stress on peripheral tissue. Stress-induced biological aging likely also acts within the central nervous system via direct activity, as well as indirect activity through the blood-brain barrier. How these aging processes occur in the periphery and the central nervous system are important areas for future research on these topics. Other directions for future research include using knockout mice to modify key inflammatory pathways to exacerbate the impact of chronic stress exposure and test the role that inflammation alone might play in driving accelerated aging phenotypes observed under chronic stress. It will also be important to consider how stress hormones might impact hallmarks of aging differentially in younger compared to older animals, as the observed effects are likely to be nuanced with increased aging of the system. Finally, animal models that examine whether and how stress hormones might influence the efficacy of novel pharmaceutical interventions that target aging pathways, such as senolytics, for the treatment of specific aging-related diseases would contribute important insights to this emerging area of Geroscience research.
The last key domain to advance research in this area involves developing robust clinical models of stress and biological aging. As outlined earlier, correlational studies suggest that psychosocial stress is associated with elevated DNA damage and disrupted repair pathways and shorter telomere length in humans; however, to our knowledge, only one study has linked chronic stress exposure and perceived stress to elevated expression of cellular senescence marker p16INK4a in humans.97 In addition, causal models have not been tested, outside of a few studies examining associations between stress and longitudinal changes in telomere length. It will be important for future research to adopt experimental (e.g., intervention) designs and/or repeated assessments of chronic stress and markers of biological aging to better understand how experiences of chronic stress and adversity impact the biological aging process over time and how this relates to age-related declines and diseases, as well as how intervention could potentially reverse aging. Further refinement of biological markers of aging in human models will also be key to accelerating progress in this area, such as recent advances in epigenetic markers of aging155–158 and multisystem indicators of age acceleration.159 Other hallmarks of aging that are relevant to this model but few studies have investigated in humans include mitochondrial function and characterization of the SASP in circulating blood and diseased tissues. Application of this work is important for understanding early aging that may result from childhood adversity, as well as accelerated aging during middle and late adulthood and within the context of specific diseases (e.g., cancer, diabetes, cardiovascular disease, dementia).160–162 Finally, empirically supported behavioral (e.g., mind-body) interventions that target the stress response may also be effective in modifying biological aging pathways to slow, prevent, or reverse accelerated aging and delay the onset of age-related disease.163 These interventions may also be beneficial in the context of ongoing clinical trials of senolytic agents (e.g., NCT04063124, NCT04685590, NCT02848131, NCT04733534) for the treatment of age-related diseases. Although the senolytic drugs target and eliminate senescent cells to reduce disease burden, aging cells that are not yet senescent may still be vulnerable to progression in patients experiencing acute and chronic stress and adversity; therefore, a dual intervention that improves the management of stress responses while also targeting senescent cells may have the greatest benefit.
6. Conclusion
In summary, this review has integrated research using animal, human, and in vitro models to demonstrate how experiences of chronic stress and adversity that elicit a stress response might drive biological aging. The findings highlight several key hallmarks of aging that are modified by exposure to chronic stressors and stress hormones, including cellular stress, DNA damage, telomere shortening, inflammation, mitochondrial function, and cellular senescence. This growing literature has led to the development of an integrated conceptual model that begins to delineate the distinct pathways through which psychosocial stress may drive accelerated biological aging. We have concluded by discussing critical gaps in the literature that must be filled to validate the proposed pathways and extend this work to various relevant disease models. Research on aging biology and broader the field of Geroscience is well-positioned to exponentially grow in breadth and depth by integrating behavioral scientists and basic researchers in psychoneuroimmunology into this fascinating field.16,17
Highlights.
- Psychosocial stress influences multiple biological aging pathways.
- Stress hormone mediators may drive the effects of stress on biological aging.
- Biological aging pathways can further promote damage in response to additional stressors.
Funding
This work was supported by the National Institutes of Health [grant numbers K01AG065485, R01CA237535, R01AG052655, and R35CA197289] and the UCLA Cousins Center for Psychoneuroimmunology.
Footnotes
Declarations of Interest
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shields GS, Slavich GM. Lifetime stress exposure and health: A review of contemporary assessment methods and biological mechanisms. Soc Personal Psychol Compass. 2017;11(8):e12335. doi: 10.1111/spc3.12335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schneiderman N, Ironson G, Siegel SD. Stress and health: Psychological, behavioral, and biological determinants. Annu Rev Clin Psychol. 2005;1(1):607–628. doi: 10.1146/annurev.clinpsy.1.102803.144141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cole SW. The Conserved Transcriptional Response to Adversity. Curr Opin Behav Sci. 2019;28:31–37. doi: 10.1016/j.cobeha.2019.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Epel ES. Psychological and Metabolic Stress: A Recipe for Accelerated Cellular Aging? Vol 8.; 2009. doi: 10.14310/horm.2002.1217 [DOI] [PubMed] [Google Scholar]
- 5.Miller AH. Inflammation Versus Glucocorticoids as Purveyors of Pathology During Stress: Have We Reached the Tipping Point? Biol Psychiatry. 2008;64(4):263–265. doi: 10.1016/j.biopsych.2008.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen S, Janicki-Deverts D, Miller GE. Psychological stress and disease. J Am Med Assoc. 2007;298(14):1685–1687. doi: 10.1001/jama.298.14.1685 [DOI] [PubMed] [Google Scholar]
- 7.Kaye JM, Lightman SL. Psychological stress and endocrine axes. In: Vedhara K, Irwin MR, eds. Human Psychoneuroimmunology. Oxford University Press; 2005:25–52. [Google Scholar]
- 8.Sloan EK, Capitanio JP, Cole SW. Stress-induced remodeling of lymphoid innervation. Brain Behav Immun. 2008;22(1):15–21. doi: 10.1016/j.bbi.2007.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194. doi: 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferrucci L, Levine ME, Kuo P-L, Simonsick EM. Time and the Metrics of Aging. Circ Res. 2018;123(7):740–744. doi: 10.1161/CIRCRESAHA.118.312816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campisi J, D’Adda Di Fagagna F. Cellular senescence: When bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–740. doi: 10.1038/nrm2233 [DOI] [PubMed] [Google Scholar]
- 12.Collado M, Blasco MA, Serrano M. Cellular Senescence in Cancer and Aging. Cell. 2007;130(2):223–233. doi: 10.1016/j.cell.2007.07.003 [DOI] [PubMed] [Google Scholar]
- 13.Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu Rev Pathol Mech Dis. 2010;5(1):99–118. doi: 10.1146/annurev-pathol-121808-102144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Effros RB, Dagarag M, Spaulding C, Man J. The role of CD8+ T-cell replicative senescence in human aging. Immunol Rev. 2005;205(27):147–157. doi: 10.1111/j.0105-2896.2005.00259.x [DOI] [PubMed] [Google Scholar]
- 15.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192(4):547–556. doi: 10.1083/jcb.201009094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kennedy BK, Berger SL, Brunet A, et al. Geroscience: Linking aging to chronic disease. Cell. 2014;159(4):709–713. doi: 10.1016/j.cell.2014.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sierra F The emergence of geroscience as an interdisciplinary approach to the enhancement of health span and life span. Cold Spring Harb Perspect Med. 2016;6(4). doi: 10.1101/cshperspect.a025163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aschbacher K, O’Donovan A, Wolkowitz OM, Dhabhar FS, Su Y, Epel E. Good stress, bad stress and oxidative stress: Insights from anticipatory cortisol reactivity. Psychoneuroendocrinology. 2013;38(9):1698–1708. doi: 10.1016/j.psyneuen.2013.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flaherty RL, Owen M, Fagan-Murphy A, et al. Glucocorticoids induce production of reactive oxygen species/reactive nitrogen species and DNA damage through an iNOS mediated pathway in breast cancer. Breast Cancer Res. 2017;19(1). doi: 10.1186/s13058-017-0823-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hara MR, Kovacs JJ, Whalen EJ, et al. A stress response pathway regulates DNA damage through β2- adrenoreceptors and β-arrestin-1. Nature. 2011;477(7364):349–353. doi: 10.1038/nature10368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaludercic N, Takimoto E, Nagayama T, et al. Monoamine oxidase a-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res. 2010;106(1):193–202. doi: 10.1161/CIRCRESAHA.109.198366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maurel A, Hernandez C, Kunduzova O, et al. Age-dependent increase in hydrogen peroxide production by cardiac monoamine oxidase A in rats. Am J Physiol - Hear Circ Physiol. 2003;284(4 53–4):1460–1467. doi: 10.1152/ajpheart.00700.2002 [DOI] [PubMed] [Google Scholar]
- 23.Santin Y, Resta J, Parini A, Mialet-Perez J. Monoamine oxidases in age-associated diseases: New perspectives for old enzymes. Ageing Res Rev. 2021;66:101256. doi: 10.1016/j.arr.2021.101256 [DOI] [PubMed] [Google Scholar]
- 24.Passos JF, Von Zglinicki T. Mitochondria, telomeres and cell senescence. Exp Gerontol. 2005;40(6):466–472. doi: 10.1016/j.exger.2005.04.006 [DOI] [PubMed] [Google Scholar]
- 25.Maggiorani D, Manzella N, Edmondson DE, et al. Monoamine Oxidases, Oxidative Stress, and Altered Mitochondrial Dynamics in Cardiac Ageing. Oxid Med Cell Longev. 2017;2017. doi: 10.1155/2017/3017947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yousefzadeh MJ, Flores RR, Zhu Y, et al. An aged immune system drives senescence and ageing of solid organs. Nature. 2021;594(7861):100–105. doi: 10.1038/s41586-021-03547-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Von Zglinicki T Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27(7):339–344. doi: 10.1016/S0968-0004(02)02110-2 [DOI] [PubMed] [Google Scholar]
- 28.Blackburn EH. Telomere states and cell fates. Nature. 2000;408(6808):53–56. doi: 10.1038/35040500 [DOI] [PubMed] [Google Scholar]
- 29.Cohen L, Marshall GD, Cheng L, Agarwal SK, Wei Q. DNA repair capacity in healthy medical students during and after exam stress. J Behav Med. 2000;23(6):531–544. doi: 10.1023/A:1005503502992 [DOI] [PubMed] [Google Scholar]
- 30.Forlenza M, Latimer JJ, Baum A. The effects of stress on DNA repair capacity. Psychol Heal. 2000;15:881–891. Accessed November 30, 2017. http://www.tandfonline.com/doi/pdf/10.1080/08870440008405589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Irie M, Asami S, Nagata S, Miyata M, Kasai H. Relationships between Perceived Workload, Stress and Oxidative DNA Damage. Vol 74.; 2001. doi: 10.1007/s004200000209 [DOI] [PubMed] [Google Scholar]
- 32.Irie M, Asami S, Nagata S, Ikeda M, Miyata M, Kasai H. Psychosocial factors as a potential trigger of oxidative DNA damage in human leukocytes. Japanese J Cancer Res. 2001;92(3):367–376. doi: 10.1111/j.1349-7006.2001.tb01104.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sivoňová M, Žitňanová I, Hlinčíková L, Škodáček I, Trebatická J, Ďuračková Z. Oxidative stress in university students during examinations. Stress. 2004;7(3):183–188. doi: 10.1080/10253890400012685 [DOI] [PubMed] [Google Scholar]
- 34.Knickelbein KZ, Flint M, Jenkins F, Baum A. Psychological stress and oxidative damage in lymphocytes of aerobically fit and unfit individuals. J Appl Biobehav Res. 2008;13(1):1–19. doi: 10.1111/j.1751-9861.2008.00025.x [DOI] [Google Scholar]
- 35.Nishio Y, Nakano Y, Deguchi Y, et al. Social Stress Induces Oxidative DNA Damage in Mouse Peripheral Blood Cells. Vol 29.; 2007. doi: 10.3123/jemsge.29.17 [DOI] [Google Scholar]
- 36.Fischman HK, Pero RW, Kelly DD. Psychogenic stress induces chromosomal and DNA damage. Int J Neurosci. 1996;84(1–4):219–227. doi: 10.3109/00207459608987267 [DOI] [PubMed] [Google Scholar]
- 37.Fischman HK, Kelly DD. Chromosomes and stress. Int J Neurosci. 1999;99(1–4):201–219. doi: 10.3109/00207459908994325 [DOI] [PubMed] [Google Scholar]
- 38.Van Campen LE, Murphy WJ, Franks JR, Mathias PI, Toraason MA. Oxidative DNA damage is associated with intense noise exposure in the rat. Hear Res. 2002;164(1–2):29–38. doi: 10.1016/S0378-5955(01)00391-4 [DOI] [PubMed] [Google Scholar]
- 39.Bagchi D, Carryl OR, Tran MX, et al. Acute and chronic stress-induced oxidative gastrointestinal mucosal injury in rats and protection by bismuth subsalicylate. Mol Cell Biochem. 1999;196(1–2):109–116. doi: 10.1023/A:1006978431521 [DOI] [PubMed] [Google Scholar]
- 40.Rentscher KE, Carroll JE, Polsky LR, Lamkin DM. Chronic stress increases transcriptomic indicators of biological aging in mouse bone marrow leukocytes. Brain, Behav Immun - Heal. Published online April 2022:100461. doi: 10.1016/j.bbih.2022.100461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flint MS, Carroll JE, Jenkins FJ, Chambers WH, Han ML, Baum A. Genomic profiling of restraint stress-induced alterations in mouse T lymphocytes. J Neuroimmunol. 2005;167(1–2):34–44. doi: 10.1016/j.jneuroim.2005.06.012 [DOI] [PubMed] [Google Scholar]
- 42.Consiglio AR, Ramos ALLP, Henriques JAP, Picada JN. DNA brain damage after stress in rats. Prog Neuro-Psychopharmacology Biol Psychiatry. 2010;34(4):652–656. doi: 10.1016/j.pnpbp.2010.03.004 [DOI] [PubMed] [Google Scholar]
- 43.Hara MR, Sachs BD, Caron MG, Lefkowitz RJ. Pharmacological blockade of a β2AR-β-arrestin-1 signaling cascade prevents the accumulation of DNA damage in a behavioral stress model. Cell Cycle. 2013;12(2):219–224. doi: 10.4161/cc.23368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forsberg K, Aalling N, Wörtwein G, et al. Dynamic regulation of cerebral DNA repair genes by psychological stress. Mutat Res - Genet Toxicol Environ Mutagen. 2015;778:37–43. doi: 10.1016/j.mrgentox.2014.12.003 [DOI] [PubMed] [Google Scholar]
- 45.Flint MS, Baum A, Chambers WH, Jenkins FJ. Induction of DNA damage, alteration of DNA repair and transcriptional activation by stress hormones. Psychoneuroendocrinology. 2007;32(5):470–479. doi: 10.1016/j.psyneuen.2007.02.013 [DOI] [PubMed] [Google Scholar]
- 46.Flint MS, Baum A, Episcopo B, et al. Chronic exposure to stress hormones promotes transformation and tumorigenicity of 3T3 mouse fibroblasts. Stress. 2013;16(1):114–121. doi: 10.3109/10253890.2012.686075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patel PR. Norepinephrine Reduces Reactive Oxygen Species (ROS) and DNA Damage in Ovarian Surface Epithelial Cells. J Bioanal Biomed. 2015;07(03). doi: 10.4172/1948-593x.1000127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lamboy-Caraballo R, Ortiz-Sanchez C, Acevedo-Santiago A, Matta J, Monteiro ANA, Armaiz-Pena GN. Norepinephrine-induced DNA damage in ovarian cancer cells. Int J Mol Sci. 2020;21(6):2250. doi: 10.3390/ijms21062250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Brien SN, Larcom LL, Baxley EG. Correlates of Plasma Cortisol and Dna Repair in Human Peripheral Lymphocytes:Suppression of Repair in Women Taking Estrogen. Vol 39.; 1993. doi: 10.1159/000182743 [DOI] [PubMed] [Google Scholar]
- 50.Greider CW, Blackburn EH. Identification of a Specific Telomere Terminal Transferase Activity in Tetrahymena Extracts. Vol 43.; 1985. doi: 10.1016/0092-8674(85)90170-9 [DOI] [PubMed] [Google Scholar]
- 51.Greider CW, Blackburn EH. The Telomere Terminal Transferase of Tetrahymena Is a Ribonucleoprotein Enzyme with Two Kinds of Primer Specificity. Vol 51.; 1987. doi: 10.1016/0092-8674(87)90576-9 [DOI] [PubMed] [Google Scholar]
- 52.Ahmed S, Passos JF, Birket MJ, et al. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J Cell Sci. 2008;121(7):1046–1053. doi: 10.1242/jcs.019372 [DOI] [PubMed] [Google Scholar]
- 53.Haendeler J, Dröse S, Büchner N, et al. Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arterioscler Thromb Vasc Biol. 2009;29(6):929–935. doi: 10.1161/ATVBAHA.109.185546 [DOI] [PubMed] [Google Scholar]
- 54.Epel ES, Blackburn EH, Lin J, et al. Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci U S A. 2004;101(49):17312–17315. doi: 10.1073/pnas.0407162101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rentscher KE, Carroll JE, Mitchell C. Psychosocial stressors and telomere length: A current review of the science. Annu Rev Public Heal. 2020;41. doi: 10.1146/annurev-publhealth [DOI] [PubMed] [Google Scholar]
- 56.Carroll JE, Mahrer NE, Shalowitz M, Ramey S, Dunkel Schetter C. Prenatal maternal stress prospectively relates to shorter child buccal cell telomere length. Psychoneuroendocrinology. 2020;121:104841. doi: 10.1016/j.psyneuen.2020.104841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marchetto NM, Glynn RA, Ferry ML, et al. Prenatal stress and newborn telomere length. Am J Obstet Gynecol. 2016;215(1):94.e1–94.e8. doi: 10.1016/j.ajog.2016.01.177 [DOI] [PubMed] [Google Scholar]
- 58.Entringer S, Epel ES, Lin J, et al. Maternal psychosocial stress during pregnancy is associated with newborn leukocyte telomere length. Am J Obstet Gynecol. 2013;208(2):134.e1–134.e7. doi: 10.1016/j.ajog.2012.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stevenson JR, McMahon EK, Boner W, Haussmann MF. Oxytocin administration prevents cellular aging caused by social isolation. Psychoneuroendocrinology. 2019;103:52–60. doi: 10.1016/j.psyneuen.2019.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Epel ES, Lin J, Wilhelm FH, et al. Cell aging in relation to stress arousal and cardiovascular disease risk factors. Psychoneuroendocrinology. 2006;31(3):277–287. doi: 10.1016/j.psyneuen.2005.08.011 [DOI] [PubMed] [Google Scholar]
- 61.Tomiyama AJ, O’Donovan A, Lin J, et al. Does cellular aging relate to patterns of allostasis?. An examination of basal and stress reactive HPA axis activity and telomere length. Physiol Behav. 2012;106(1):40–45. doi: 10.1016/j.physbeh.2011.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parks CG, DeRoo LA, Miller DB, McCanlies EC, Cawthon RM, Sandler DP. Employment and work schedule are related to telomere length in women. Occup Environ Med. 2011;68(8):582–589. doi: 10.1136/oem.2010.063214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parks CG, Miller DB, McCanlies EC, et al. Telomere length, current perceived stress, and urinary stress hormones in women. Cancer Epidemiol Biomarkers Prev. 2009;18(2):551–560. doi: 10.1158/1055-9965.EPI-08-0614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thomas N, Hudaib AR, Romano-Silva M, et al. Influence of cortisol awakening response on telomere length: Trends for males and females. Eur J Neurosci. 2021;00:1–10. doi: 10.1111/ejn.14996 [DOI] [PubMed] [Google Scholar]
- 65.Bosquet Enlow M, Sideridis G, Bollati V, Hoxha M, Hacker MR, Wright RJ. Maternal cortisol output in pregnancy and newborn telomere length: Evidence for sex-specific effects. Psychoneuroendocrinology. 2019;102:225–235. doi: 10.1016/j.psyneuen.2018.12.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Choi J, Fauce SR, Effros RB. Reduced telomerase activity in human T lymphocytes exposed to cortisol. Brain Behav Immun. 2008;22(4):600–605. doi: 10.1016/j.bbi.2007.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marzetti E, Csiszar A, Dutta D, Balagopal G, Calvani R, Leeuwenburgh C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: From mechanisms to therapeutics. Am J Physiol - Hear Circ Physiol. 2013;305(4). doi: 10.1152/ajpheart.00936.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Passos JF, Saretzki G, Ahmed S, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5(5):1138–1151. doi: 10.1371/journal.pbio.0050110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Anderson R, Lagnado A, Maggiorani D, et al. Length‐independent telomere damage drives post‐mitotic cardiomyocyte senescence. EMBO J. 2019;38(5):e100492. doi: 10.15252/embj.2018100492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kujoth CC, Hiona A, Pugh TD, et al. Medicine: Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science (80- ). 2005;309(5733):481–484. doi: 10.1126/science.1112125 [DOI] [PubMed] [Google Scholar]
- 71.Summer R, Shaghaghi H, Schriner DL, et al. Activation of the mTORC1/PGC-1 axis promotes mitochondrial biogenesis and induces cellular senescence in the lung epithelium. Am J Physiol - Lung Cell Mol Physiol. 2019;316(6):L1049–L1060. doi: 10.1152/ajplung.00244.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Korolchuk VI, Miwa S, Carroll B, von Zglinicki T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine. 2017;21:7–13. doi: 10.1016/j.ebiom.2017.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Manzella N, Santin Y, Maggiorani D, et al. Monoamine oxidase-A is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. Aging Cell. 2018;17(5). doi: 10.1111/acel.12811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Santin Y, Sicard P, Vigneron F, et al. Oxidative Stress by Monoamine Oxidase-A Impairs Transcription Factor EB Activation and Autophagosome Clearance, Leading to Cardiomyocyte Necrosis and Heart Failure. Antioxidants Redox Signal. 2016;25(1):10–27. doi: 10.1089/ars.2015.6522 [DOI] [PubMed] [Google Scholar]
- 75.Picard M, Juster RP, McEwen BS. Mitochondrial allostatic load puts the “gluc” back in glucocorticoids. Nat Rev Endocrinol. 2014;10(5):303–310. doi: 10.1038/nrendo.2014.22 [DOI] [PubMed] [Google Scholar]
- 76.Nelson G, Kucheryavenko O, Wordsworth J, von Zglinicki T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech Ageing Dev. 2018;170:30–36. doi: 10.1016/j.mad.2017.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Picard M, McEwen BS. Psychological Stress and Mitochondria: A Conceptual Framework. Psychosom Med. 2018;80(2):126–140. doi: 10.1097/PSY.0000000000000544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Picard M, McEwen BS. Psychological Stress and Mitochondria: A Systematic Review. Psychosom Med. 2018;80(2):141–153. doi: 10.1097/PSY.0000000000000545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Neuhaus JFG, Baris OR, Kittelmann A, Becker K, Rothschild MA, Wiesner RJ. Catecholamine Metabolism Induces Mitochondrial DNA Deletions and Leads to Severe Adrenal Degeneration during Aging. Neuroendocrinology. 2017;104(1):72–84. doi: 10.1159/000444680 [DOI] [PubMed] [Google Scholar]
- 80.Manoli I, Alesci S, Blackman MR, Su YA, Rennert OM, Chrousos GP. Mitochondria as key components of the stress response. Trends Endocrinol Metab. 2007;18(5):190–198. doi: 10.1016/j.tem.2007.04.004 [DOI] [PubMed] [Google Scholar]
- 81.Madrigal JLM, Olivenza R, Moro MA, et al. Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology. 2001;24(4):420–429. doi: 10.1016/S0893-133X(00)00208-6 [DOI] [PubMed] [Google Scholar]
- 82.Gong Y, Chai Y, Ding JH, Sun XL, Hu G. Chronic mild stress damages mitochondrial ultrastructure and function in mouse brain. Neurosci Lett. 2011;488(1):76–80. doi: 10.1016/j.neulet.2010.11.006 [DOI] [PubMed] [Google Scholar]
- 83.Rezin GT, Cardoso MR, Gonçalves CL, et al. Inhibition of mitochondrial respiratory chain in brain of rats subjected to an experimental model of depression. Neurochem Int. 2008;53(6–8):395–400. doi: 10.1016/j.neuint.2008.09.012 [DOI] [PubMed] [Google Scholar]
- 84.Tyrka AR, Parade SH, Price LH, et al. Alterations of Mitochondrial DNA Copy Number and Telomere Length with Early Adversity and Psychopathology. Biol Psychiatry. 2016;79(2):78–86. doi: 10.1016/j.biopsych.2014.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Picard M, Prather AA, Puterman E, et al. A Mitochondrial Health Index Sensitive to Mood and Caregiving Stress. Biol Psychiatry. 2018;84(1):9–17. doi: 10.1016/j.biopsych.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Trumpff C, Marsland AL, Basualto-Alarcón C, et al. Acute psychological stress increases serum circulating cell-free mitochondrial DNA. Psychoneuroendocrinology. 2019;106:268–276. doi: 10.1016/j.psyneuen.2019.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Psarra AMG, Sekeris CE. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells. Role of the mitochondrial glucocorticoid receptor. Biochim Biophys Acta - Mol Cell Res. 2011;1813(10):1814–1821. doi: 10.1016/j.bbamcr.2011.05.014 [DOI] [PubMed] [Google Scholar]
- 88.Napolitano G, Barone D, Di Meo S, Venditti P. Adrenaline induces mitochondrial biogenesis in rat liver. J Bioenerg Biomembr. 2018;50(1):11–19. doi: 10.1007/s10863-017-9736-6 [DOI] [PubMed] [Google Scholar]
- 89.Picard M, Trumpff C, Burelle Y. Mitochondrial psychobiology: foundations and applications. Published online 2019. doi: 10.1016/j.cobeha.2019.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25(3):585–621. doi: 10.1016/0014-4827(61)90192-6 [DOI] [PubMed] [Google Scholar]
- 91.Campisi J Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell. 2005;120(4):513–522. doi: 10.1016/j.cell.2005.02.003 [DOI] [PubMed] [Google Scholar]
- 92.Kirkland JL, Tchkonia T. Cellular Senescence: A Translational Perspective. EBioMedicine. 2017;21:21–28. doi: 10.1016/j.ebiom.2017.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018;28(6):436–453. doi: 10.1016/j.tcb.2018.02.001 [DOI] [PubMed] [Google Scholar]
- 94.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–1078. doi: 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Özcan S, Alessio N, Acar MB, et al. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging (Albany NY). 2016;8(7):1316–1329. doi: 10.18632/aging.100971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Matjusaitis M, Chin G, Sarnoski EA, Stolzing A. Biomarkers to identify and isolate senescent cells. Ageing Res Rev. 2016;29:1–12. doi: 10.1016/j.arr.2016.05.003 [DOI] [PubMed] [Google Scholar]
- 97.Rentscher KE, Carroll JE, Repetti RL, Cole SW, Reynolds BM, Robles TF. Chronic stress exposure and daily stress appraisals relate to biological aging marker p16 INK4a. Psychoneuroendocrinology. 2019;102:139–148. doi: 10.1016/j.psyneuen.2018.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Razzoli M, Nyuyki-Dufe K, Gurney A, et al. Social stress shortens lifespan in mice. Aging Cell. 2018;17(4):e12778. doi: 10.1111/acel.12778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Katsuumi G, Shimizu I, Yoshida Y, et al. Catecholamine-induced senescence of endothelial cells and bone marrow cells promotes cardiac dysfunction in mice. Int Heart J. 2018;59(4):837–844. doi: 10.1536/ihj.17-313 [DOI] [PubMed] [Google Scholar]
- 100.Laberge RM, Zhou L, Sarantos MR, et al. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell. 2012;11(4):569–578. doi: 10.1111/j.1474-9726.2012.00818.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Franceschi C, Campisi J. Chronic inflammation (Inflammaging) and its potential contribution to age-associated diseases. Journals Gerontol - Ser A Biol Sci Med Sci. 2014;69(S1):S4–S9. doi: 10.1093/gerona/glu057 [DOI] [PubMed] [Google Scholar]
- 102.Franceschi C, Campisi J. Chronic inflammation (Inflammaging) and its potential contribution to age-associated diseases. Journals Gerontol - Ser A Biol Sci Med Sci. 2014;69(S1):S4–S9. doi: 10.1093/gerona/glu057 [DOI] [PubMed] [Google Scholar]
- 103.Irwin MR, Cole SW. Reciprocal regulation of the neural and innate immune systems. Nat Rev Immunol. 2011;11(9):625–632. doi: 10.1038/nri3042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46–56. doi: 10.1038/nrn2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Slavich GM. Social Safety Theory: A Biologically Based Evolutionary Perspective on Life Stress, Health, and Behavior. Annu Rev Clin Psychol. 2020;16(1):265–295. doi: 10.1146/annurev-clinpsy-032816-045159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McEwen BS. Physiology and neurobiology of stress and adaptation: Central role of the brain. Physiol Rev. 2007;87(3):873–904. doi: 10.1152/physrev.00041.2006 [DOI] [PubMed] [Google Scholar]
- 107.Miller GE, Cohen S, Ritchey AK. Chronic psychological stress and the regulation of pro-inflammatory cytokines: A glucocorticoid-resistance model. Heal Psychol. 2002;21(6):531–541. doi: 10.1037/0278-6133.21.6.531 [DOI] [PubMed] [Google Scholar]
- 108.Cohen S, Janicki-Deverts D, Doyle WJ, et al. Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc Natl Acad Sci U S A. 2012;109(16):5995–5999. doi: 10.1073/pnas.1118355109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pace TWW, Hu F, Miller AH. Cytokine-effects on glucocorticoid receptor function: Relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun. 2007;21(1):9–19. doi: 10.1016/j.bbi.2006.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Miller GE, Murphy MLM, Cashman R, et al. Greater inflammatory activity and blunted glucocorticoid signaling in monocytes of chronically stressed caregivers. Brain Behav Immun. 2014;41(1):191–199. doi: 10.1016/j.bbi.2014.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: Implications for health. Nat Rev Immunol. 2005;5(3):243–251. doi: 10.1038/nri1571 [DOI] [PubMed] [Google Scholar]
- 112.Hänsel A, Hong S, Cámara RJA, von Känel R. Inflammation as a psychophysiological biomarker in chronic psychosocial stress. Neurosci Biobehav Rev. 2010;35(1):115–121. doi: 10.1016/j.neubiorev.2009.12.012 [DOI] [PubMed] [Google Scholar]
- 113.Fleshner M Stress-evoked sterile inflammation, danger associated molecular patterns (DAMPs), microbial associated molecular patterns (MAMPs) and the inflammasome. Brain Behav Immun. 2013;27(1):1–7. doi: 10.1016/j.bbi.2012.08.012 [DOI] [PubMed] [Google Scholar]
- 114.Frank MG, Weber MD, Watkins LR, Maier SF. Stress sounds the alarmin: The role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming. Brain Behav Immun. 2015;48:1–7. doi: 10.1016/j.bbi.2015.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Maslanik T, Mahaffey L, Tannura K, Beninson L, Greenwood BN, Fleshner M. The inflammasome and danger associated molecular patterns (DAMPs) are implicated in cytokine and chemokine responses following stressor exposure. Brain Behav Immun. 2013;28:54–62. doi: 10.1016/j.bbi.2012.10.014 [DOI] [PubMed] [Google Scholar]
- 116.Cheng Y, Pardo M, Armini R de S, et al. Stress-induced neuroinflammation is mediated by GSK3-dependent TLR4 signaling that promotes susceptibility to depression-like behavior. Brain Behav Immun. 2016;53:207–222. doi: 10.1016/j.bbi.2015.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cole SW, Hawkley LC, Arevalo JM, Sung CY, Rose RM, Cacioppo JT. Social regulation of gene expression in human leukocytes. Genome Biol. 2007;8(9):1–13. doi: 10.1186/gb-2007-8-9-r189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Miller GE, Chen E, Sze J, et al. A Functional Genomic Fingerprint of Chronic Stress in Humans: Blunted Glucocorticoid and Increased NF-κB Signaling. Biol Psychiatry. 2008;64(4):266–272. doi: 10.1016/j.biopsych.2008.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Walsh CP, Ewing LJ, Cleary JL, et al. Development of glucocorticoid resistance over one year among mothers of children newly diagnosed with cancer. Brain Behav Immun. 2018;69:364–373. doi: 10.1016/j.bbi.2017.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Miller GE, Chen E, Fok AK, et al. Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. Proc Natl Acad Sci U S A. 2009;106(34):14716–14721. doi: 10.1073/pnas.0902971106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Avitsur R, Stark JL, Sheridan JF. Social stress induces glucocorticoid resistance in subordinate animals. Horm Behav. 2001;39(4):247–257. doi: 10.1006/hbeh.2001.1653 [DOI] [PubMed] [Google Scholar]
- 122.Cole SW, Mendoza SP, Capitanio JP. Social stress desensitizes lymphocytes to regulation by endogenous glucocorticoids: Insights from in vivo cell trafficking dynamics in rhesus macaques. Psychosom Med. 2009;71(6):591–597. doi: 10.1097/PSY.0b013e3181aa95a9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cole SW, Capitanio JP, Chun K, Arevalo JMG, Ma J, Cacioppo JT. Myeloid differentiation architecture of leukocyte transcriptome dynamics in perceived social isolation. Proc Natl Acad Sci U S A. 2015;112(49):15142–15147. doi: 10.1073/pnas.1514249112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Niraula A, Wang Y, Godbout JP, Sheridan JF. Corticosterone production during repeated social defeat causes monocyte mobilization from the bone marrow, glucocorticoid resistance, and neurovascular adhesion molecule expression. J Neurosci. 2018;38(9):2328–2340. doi: 10.1523/JNEUROSCI.2568-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Powell ND, Sloan EK, Bailey MT, et al. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via β-adrenergic induction of myelopoiesis. Proc Natl Acad Sci U S A. 2013;110(41):16574–16579. doi: 10.1073/pnas.1310655110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Quan N, Avitsur R, Stark JL, et al. Molecular mechanisms of glucocorticoid resistance in splenocytes of socially stressed male mice. J Neuroimmunol. 2003;137(1–2):51–58. doi: 10.1016/S0165-5728(03)00042-0 [DOI] [PubMed] [Google Scholar]
- 127.Reber SO, Birkeneder L, Veenema AH, et al. Adrenal insufficiency and colonic inflammation after a novel chronic psycho-social stress paradigm in mice: Implications and mechanisms. Endocrinology. 2007;148(2):670–682. doi: 10.1210/en.2006-0983 [DOI] [PubMed] [Google Scholar]
- 128.Stark JL, Avitsur R, Padgett DA, Campbell KA, Beck FM, Sheridan JF. Social stress induces glucocorticoid resistance in macrophages. Am J Physiol - Regul Integr Comp Physiol. 2001;280(6 49–6). doi: 10.1152/ajpregu.2001.280.6.r1799 [DOI] [PubMed] [Google Scholar]
- 129.Kop WJ, Weissman NJ, Zhu J, et al. Effects of Acute Mental Stress and Exercise on Inflammatory Markers in Patients With Coronary Artery Disease and Healthy Controls. Am J Cardiol. 2008;101(6):767–773. doi: 10.1016/j.amjcard.2007.11.006 [DOI] [PubMed] [Google Scholar]
- 130.Szabó C, Haskó G, Zingarelli B, et al. Isoproterenol regulates tumour necrosis factor, interleukin-10, interleukin-6 and nitric oxide production and protects against the development of vascular hyporeactivity in endotoxaemia. Immunology. 1997;90(1):95–100. doi: 10.1046/j.1365-2567.1997.00137.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bierhaus A, Wolf J, Andrassy M, et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci U S A. 2003;100(4):1920–1925. doi: 10.1073/pnas.0438019100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.DeRijk RH, Boelen A, Tilders FJH, Berkenbosch F. Induction of plasma interleukin-6 by circulating adrenaline in the rat. Psychoneuroendocrinology. 1994;19(2):155–163. doi: 10.1016/0306-4530(94)90005-1 [DOI] [PubMed] [Google Scholar]
- 133.Aninat C, Seguin P, Descheemaeker PN, Morel F, Malledant Y, Guillouzo A. Catecholamines induce an inflammatory response in human hepatocytes. Crit Care Med. 2008;36(3):848–854. doi: 10.1097/CCM.0B013E31816532BE [DOI] [PubMed] [Google Scholar]
- 134.Black PH. Stress and the inflammatory response: A review of neurogenic inflammation. Brain Behav Immun. 2002;16(6):622–653. doi: 10.1016/S0889-1591(02)00021-1 [DOI] [PubMed] [Google Scholar]
- 135.Cole SW, Arevalo JMG, Takahashi R, et al. Computational identification of gene-social environment interaction at the human IL6 locus. Proc Natl Acad Sci U S A. 2010;107(12):5681–5686. doi: 10.1073/pnas.0911515107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fu YC, Chi CS, Yin SC, Hwang B, Chiu YT, Hsu SL. Norepinephrine induces apoptosis in neonatal rat cardiomyocytes through a reactive oxygen species-TNFα-caspase signaling pathway. Cardiovasc Res. 2004;62(3):558–567. doi: 10.1016/j.cardiores.2004.01.039 [DOI] [PubMed] [Google Scholar]
- 137.Slota C, Shi A, Chen G, Bevans M, Weng N ping. Norepinephrine preferentially modulates memory CD8 T cell function inducing inflammatory cytokine production and reducing proliferation in response to activation. Brain Behav Immun. 2015;46:168–179. doi: 10.1016/j.bbi.2015.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Strell C, Sievers A, Bastian P, et al. Divergent effects of norepinephrine, dopamine and substance P on the activation, differentiation and effector functions of human cytotoxic T lymphocytes. BMC Immunol. 2009;10(1):62. doi: 10.1186/1471-2172-10-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wolf JM, Rohleder N, Bierhaus A, Nawroth PP, Kirschbaum C. Determinants of the NF-κB response to acute psychosocial stress in humans. Brain Behav Immun. 2009;23(6):742–749. doi: 10.1016/j.bbi.2008.09.009 [DOI] [PubMed] [Google Scholar]
- 140.DeSantis AS, DiezRoux AV, Hajat A, et al. Associations of salivary cortisol levels with inflammatory markers: The Multi-Ethnic Study of Atherosclerosis. Psychoneuroendocrinology. 2012;37(7):1009–1018. doi: 10.1016/j.psyneuen.2011.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Knight EL, Jiang Y, Rodriguez-Stanley J, Almeida DM, Engeland CG, Zilioli S. Perceived stress is linked to heightened biomarkers of inflammation via diurnal cortisol in a national sample of adults. Brain Behav Immun. 2021;93:206–213. doi: 10.1016/j.bbi.2021.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.DeRijk R, Michelson D, Karp B, et al. Exercise and Circadian Rhythm-Induced Variations in Plasma Cortisol Differentially Regulate Interleukin-1β (IL-1β), IL-6, and Tumor Necrosis Factor-α (TNFα) Production in Humans: High Sensitivity of TNFα and Resistance of IL-6. J Clin Endocrinol Metab. 1997;82(7):2182–2191. doi: 10.1210/jcem.82.7.4041 [DOI] [PubMed] [Google Scholar]
- 143.Petrovsky N, McNair P, Harrison LC. Diurnal rhythms of pro-inflammatory cytokines: Regulation by plasma cortisol and therapeutic implications. Cytokine. 1998;10(4):307–312. doi: 10.1006/cyto.1997.0289 [DOI] [PubMed] [Google Scholar]
- 144.Waage A, Slupphaug G, Shalaby R. Glucocorticoids inhibit the production of IL 6 from monocytes, endothelial cells and fibroblasts. Eur J Immunol. 1990;20(11):2439–2443. doi: 10.1002/eji.1830201112 [DOI] [PubMed] [Google Scholar]
- 145.Hermoso MA, Matsuguchi T, Smoak K, Cidlowski JA. Glucocorticoids and Tumor Necrosis Factor Alpha Cooperatively Regulate Toll-Like Receptor 2 Gene Expression. Mol Cell Biol. 2004;24(11):4743–4756. doi: 10.1128/mcb.24.11.4743-4756.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cruz-Topete D, Cidlowski JA. One hormone, two actions: Anti- And pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation. 2014;22(1–2):20–32. doi: 10.1159/000362724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Villeneuve C, Guilbeau-Frugier C, Sicard P, et al. P53-PGC-1α pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-a upregulation: Role in chronic left ventricular dysfunction in mice. Antioxidants Redox Signal. 2013;18(1):5–18. doi: 10.1089/ars.2011.4373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sahin E, Colla S, Liesa M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470(7334):359–365. doi: 10.1038/nature09787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hoshino A, Mita Y, Okawa Y, et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4. doi: 10.1038/ncomms3308 [DOI] [PubMed] [Google Scholar]
- 150.Chang J, Wang Y, Shao L, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22(1):78–83. doi: 10.1038/nm.4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Grace PM, Tawfik VL, Svensson CI, Burton MD, Loggia ML, Hutchinson MR. The neuroimmunology of chronic pain: From rodents to humans. J Neurosci. 2021;41(5):855–865. doi: 10.1523/JNEUROSCI.1650-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Miller AH, Raison CL. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16(1):22–34. doi: 10.1038/nri.2015.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Irwin MR. Sleep and inflammation: partners in sickness and in health. Nat Rev Immunol. 2019;19(11):702–715. doi: 10.1038/s41577-019-0190-z [DOI] [PubMed] [Google Scholar]
- 154.Davis MC, Zautra AJ, Younger J, Motivala SJ, Attrep J, Irwin MR. Chronic stress and regulation of cellular markers of inflammation in rheumatoid arthritis: Implications for fatigue. Brain Behav Immun. 2008;22(1):24–32. doi: 10.1016/j.bbi.2007.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Levine ME, Hosgood HD, Chen B, Absher D, Assimes T, Horvath S. DNA methylation age of blood predicts future onset of lung cancer in the women’s health initiative. Aging (Albany NY). 2015;7(9):690–700. doi: 10.18632/aging.100809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Horvath S DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. doi: 10.1186/gb-2013-14-10-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lu AT, Quach A, Wilson JG, et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY). 2019;11(2):303–327. doi: 10.18632/aging.101684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Belsky DW, Caspi A, Arseneault L, et al. Quantification of the pace of biological aging in humans through a blood test, the DunedinPoAm DNA methylation algorithm. Elife. 2020;9:1–56. doi: 10.7554/eLife.54870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Belsky DW, Caspi A, Houts R, et al. Quantification of biological aging in young adults. Proc Natl Acad Sci U S A. 2015;112(30):E4104–E4110. doi: 10.1073/pnas.1506264112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mandelblatt JS, Ahles TA, Lippman ME, et al. Applying a Life Course Biological Age Framework to Improving the Care of Individuals With Adult Cancers. JAMA Oncol. Published online August 5, 2021. doi: 10.1001/jamaoncol.2021.1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sedrak MS, Gilmore NJ, Carroll JE, Muss HB, Cohen HJ, Dale W. Measuring Biologic Resilience in Older Cancer Survivors. https://doi.org/101200/JCO2100245. 2021;39(19):2079–2089. doi: 10.1200/JCO.21.00245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Armenian SH, Gibson CJ, Rockne RC, Ness KK. Premature Aging in Young Cancer Survivors. JNCI J Natl Cancer Inst. 2019;111(3):226–232. doi: 10.1093/JNCI/DJY229 [DOI] [PubMed] [Google Scholar]
- 163.Black DS, Slavich GM. Mindfulness meditation and the immune system: a systematic review of randomized controlled trials. Ann N Y Acad Sci. 2016;1373(1):13–24. doi: 10.1111/nyas.12998 [DOI] [PMC free article] [PubMed] [Google Scholar]