Inference on Haplotype Effects in Case-Control Studies Using Unphased Genotype Data (original) (raw)
Abstract
A variety of statistical methods exist for detecting haplotype-disease association through use of genetic data from a case-control study. Since such data often consist of unphased genotypes (resulting in haplotype ambiguity), such statistical methods typically apply the expectation-maximization (EM) algorithm for inference. However, the majority of these methods fail to perform inference on the effect of particular haplotypes or haplotype features on disease risk. Since such inference is valuable, we develop a retrospective likelihood for estimating and testing the effects of specific features of single-nucleotide polymorphism (SNP)-based haplotypes on disease risk using unphased genotype data from a case-control study. Our proposed method has a flexible structure that allows, among other choices, modeling of multiplicative, dominant, and recessive effects of specific haplotype features on disease risk. In addition, our method relaxes the requirement of Hardy-Weinberg equilibrium of haplotype frequencies in case subjects, which is typically required of EM-based haplotype methods. Also, our method easily accommodates missing SNP information. Finally, our method allows for asymptotic, permutation-based, or bootstrap inference. We apply our method to case-control SNP genotype data from the Finland–United States Investigation of Non-Insulin-Dependent Diabetes Mellitus (FUSION) Genetics study and identify two haplotypes that appear to be significantly associated with type 2 diabetes. Using the FUSION data, we assess the accuracy of asymptotic P values by comparing them with P values obtained from a permutation procedure. We also assess the accuracy of asymptotic confidence intervals for relative-risk parameters for haplotype effects, by a simulation study based on the FUSION data.
Introduction
Association-based statistical methods are likely to be required for the successful mapping of a genetic variant that influences a complex disease. Such methods generally are more powerful than linkage-based methods for identifying such a genetic variant (Risch 2000; Botstein and Risch 2003), particularly when the variant has only a moderate effect on disease risk (Risch and Merikangas 1996). In general, association-based methods attempt to identify a genetic variant that either directly predisposes to disease or is in linkage disequilibrium with such a causal variant. Since linkage disequilibrium among variants exists only over short genetic distances, association methods require a high-density map of markers for successful identification of a disease-predisposing variant. Therefore, many association analyses utilize a high-density map of biallelic SNPs, such as that published by the International SNP Map Working Group (2001).
A popular SNP-based association approach for disease mapping consists of collecting SNP and disease data from samples of unrelated individuals through use of a case-control study design. For such a design, one can apply traditional statistical methods to assess association between SNP allelic variants and disease. Power to detect such association will decrease as linkage disequilibrium between the tested variant and the disease-predisposing variant decreases. Since linkage disequilibrium exists over short genetic distances, these traditional association tests likely have limited power to identify disease-predisposing variants. Therefore, many studies utilize modified case-control association tests based on SNP-based haplotypes, which are specific combinations of allelic variants at a series of tightly linked SNPs on the same chromosome. Haplotype-based association methods should be inherently more powerful for gene mapping than methods based on single SNPs, since haplotype-based methods incorporate linkage disequilibrium information from multiple markers. Simulation studies (Akey et al. 2001; Zaykin et al. 2002) support this theory. In addition, unlike single SNPs, haplotypes can identify unique chromosomal segments that contain disease-influencing variants.
Haplotypes have an additional advantage over single SNPs when multiple disease-susceptibility variants occur within the same gene. Morris and Kaplan (2002) showed that haplotype-based association methods are more powerful than analogous allele-based methods when each susceptibility variant originates and predisposes to disease independently of the other susceptibility variants. Haplotypes are also useful when disease arises from the interaction of multiple _cis_-acting susceptibility variants found within the gene. Evidence suggests that a variety of diseases originate from multiple variant interaction, including neural tube defects (Joosten et al. 2001) and prostate cancer (Tavtigian et al. 2001). For such diseases, haplotype-based association methods will be preferable over single SNP-based association methods, since the former methods allow for the joint effect of multiple genetic variants, whereas the latter do not.
One difficulty in applying haplotype-based association methods to disease data is that the SNP data from the cases and controls often consist of unphased genotype data, which results in haplotype ambiguity. To resolve the ambiguity, one can apply molecular haplotyping techniques (Michalatos-Beloin et al. 1996; Eitan and Kashi 2002), but these procedures require substantial amounts of laboratory work. Alternatively, one can apply the expectation-maximization (EM) algorithm (Dempster et al. 1977) to infer haplotype frequencies from genotype data (Excoffier and Slatkin 1995; Hawley and Kidd 1995; Long et al. 1995), under the assumption that such frequencies are in Hardy-Weinberg equilibrium (HWE) within the sample. The EM algorithm can accommodate several SNP loci and does not require knowledge of recombination between SNPs. Fallin and Schork (2000) demonstrated the EM algorithm’s accuracy for estimating SNP-based haplotype frequencies using a wide variety of simulation designs.
For a case-control study design, several haplotype association methods exist that utilize EM-inferred haplotype frequencies. Early approaches (Zhao et al. 2000; Fallin et al. 2001) applied omnibus tests that compared estimated haplotype frequencies between cases and controls. Although such tests assess overall association between haplotypes and disease, they do not provide inference on the effects of specific haplotypes or haplotype features. Such inference is valuable for facilitating the identification of specific chromosomal segments that contain disease-predisposing variant(s). Therefore, we may wish to estimate and test the disease-predisposing effect of either a specific haplotype or a specific region shared by a subset of haplotypes. In addition, we might also wish to determine whether such chromosomal segments of interest act on disease in a multiplicative, dominant, or recessive fashion.
To address these issues, Schaid et al. (2002) and Zaykin et al. (2002) developed tests of specific haplotype effects based on the prospective likelihood of disease, conditional on the possible haplotypes. Both methods treat haplotypes as covariates in a regression model. To accommodate subjects with ambiguous haplotype covariates, these methods compute the expected value of the covariates conditional on the subject’s genotype data, using EM-inferred haplotype frequencies estimated in the pooled sample of cases and controls (under the assumption of HWE). Although appropriate under the null hypothesis of no haplotype-disease association, haplotype estimation in the pooled sample is problematic under the alternative hypothesis, since the frequencies are stratified with respect to disease status. Even if the control and case haplotype frequencies are separately in HWE, this stratification violates the EM algorithm’s assumption of HWE in the pooled sample, which may bias estimates of haplotype effect.
Stram et al. (2003_b_) investigated the bias in estimates of haplotype effect when naively using the prospective likelihood with case-control data while assuming HWE in the pooled sample. These authors determined that bias in haplotype-effect estimates were often pronounced when the genotype data failed to accurately predict the underlying haplotype-pair data. To quantify haplotype predictability from genotype data, the haplotype uncertainty measure of Stram et al. (2003_a_),
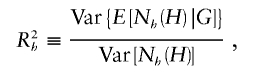
was used, where 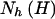 denotes the number of copies of haplotype h in haplotype pair H, and G denotes genotype data. Stram et al. (2003_b_) determined that the effect of a particular haplotype on disease is often biased when _R_2_h_⩽0.9.
denotes the number of copies of haplotype h in haplotype pair H, and G denotes genotype data. Stram et al. (2003_b_) determined that the effect of a particular haplotype on disease is often biased when _R_2_h_⩽0.9.
Since estimation of odds ratios for specific haplotypes or haplotype features is desirable, Stram et al. (2003_b_) and Zhao et al. (2003) developed separate approaches for both estimating and testing effects of haplotype features through use of case-control genotype data. Stram et al. (2003_b_) conditioned a prospective likelihood on known sampling probabilities of case and control subjects in the population. Although we might know such sampling probabilities for either a population-based or nested case-control–based study, we are unlikely to know these quantities in general. Zhao et al. (2003) applied a prospective estimating-equation approach that required only the HWE assumption of haplotype frequencies in the control sample. However, this approach estimated control haplotype frequencies using control genotype data only. As we will show, case genotype data can contribute information for improving the efficiency of haplotype frequency estimates in the control sample.
We propose a retrospective likelihood method for haplotype inference in a case-control study using unphased SNP genotype data that allows for both testing and estimation of haplotype effects. Our method relaxes the assumption of HWE in the case sample and easily accommodates missing SNP genotype information. We believe our method has advantages over the methods of Stram et al. (2003_b_) and Zhao et al. (2003). Unlike Stram et al.'s (2003_b_) method, our method does not require prior knowledge of the sampling probabilities of case and control subjects in the population. Compared with the estimating-equation approach of Zhao et al. (2003), our method’s likelihood-based framework may yield more efficient parameter estimates for a properly specified model and allows one to apply criteria such as the Akaike information criterion (AIC) (Akaike 1985) for model selection. Our method also has an additional benefit over the approach of Zhao et al. (2003), in that we estimate control haplotype frequencies through use of both control and case genotype data, which should increase statistical efficiency.
In subsequent sections, we develop the retrospective likelihood and describe estimation procedures and statistical tests for inference. For estimation, we maximize the retrospective likelihood using an expectation-conditional-maximization (ECM) algorithm, as described by Meng and Rubin (1993). We illustrate the use of our method by applying it to unphased SNP genotype data from the Finland–United States Investigation of Non-Insulin-Dependent Diabetes Mellitus (FUSION) Genetics study (Valle et al. 1998). Using the FUSION data, we assess the accuracy of asymptotic P values by comparing them to P values obtained from a permutation procedure. We also assess the accuracy of asymptotic CIs for relative-risk parameters for haplotype effects, by a simulation study based on the FUSION data.
Methods
Assumptions and Notation
Assuming a retrospective study design, we collect a sample of n unrelated subjects, consisting of c controls and d cases. We let D denote a subject’s disease outcome indicator (where 1 indicates disease and 0 indicates no disease). We assume that the n subjects are each genotyped at a series of L SNPs. Given complete genotype information at each locus, the number of possible multi-SNP genotypes in the sample is 3_L_. If we allow for missing SNP genotype data (under the assumption that subjects with missing genotype data at a SNP locus must lack both alleles), then this number increases to 4_L_ possible multi-SNP genotypes. For either situation, the total number of possible haplotypes is 2_L_. We let _G_=g denote a subject’s multi-SNP genotype and _H_=(h,_h_′) denote the subject’s pair of haplotypes h and _h_′. By allowing some genotypes g to include missing SNP information, we may assume that _G_=g is known for each subject. However, _H_=(h,_h_′) is unknown if the subject is heterozygous at >1 SNP or if any SNP genotype is missing. We let S(g) denote the set of haplotype pairs 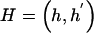 consistent with _G_=g. We adopt the convention that
consistent with _G_=g. We adopt the convention that 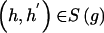 directly implies that
directly implies that 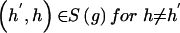 .
.
Observed-Data Likelihood
Our approach constructs the retrospective likelihood of the observed genotype data (which we define as “the observed-data likelihood,” or _L_OBS) as a function of the underlying haplotype data, conditional on disease status. We write _L_OBS as a product of multinomials of the genotype data:
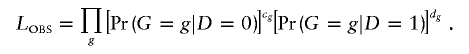
Here, 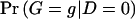 and
and 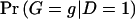 are the probabilities of genotype g in the control and case samples, respectively. c g and d g denote the numbers of control subjects and case subjects with genotype g in the sample.
are the probabilities of genotype g in the control and case samples, respectively. c g and d g denote the numbers of control subjects and case subjects with genotype g in the sample.
We can also express the likelihood _L_OBS as a function of haplotype pairs by writing 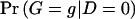 and
and 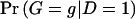 as the sum of the haplotype-pair frequencies that are consistent with genotype g. Let
as the sum of the haplotype-pair frequencies that are consistent with genotype g. Let 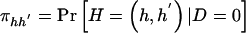 and
and 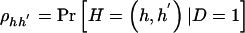 denote the frequency of haplotype pair _H_=(h,_h_′) in the control and case populations, respectively. We can write the frequency of genotype g as
denote the frequency of haplotype pair _H_=(h,_h_′) in the control and case populations, respectively. We can write the frequency of genotype g as 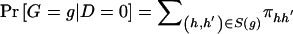 among control subjects and
among control subjects and 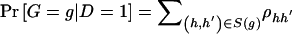 among case subjects. With this parameterization, _L_OBS becomes
among case subjects. With this parameterization, _L_OBS becomes
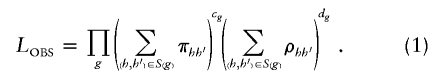
To facilitate inference of particular haplotype features, define
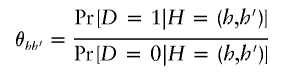
as the odds of disease for haplotype pair _H_=(h,_h_′). Following Satten and Kupper (1993) and Satten and Carroll (2000), we note that
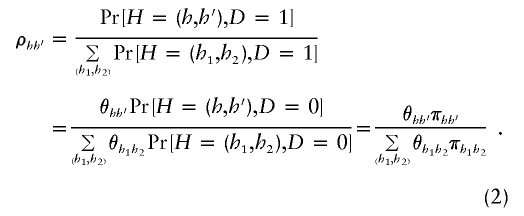
As a result, specification of π_hh_′ and θ_hh_′ fully determines ρ_hh_′. Using equation (2), we rewrite _L_OBS in equation (1) as
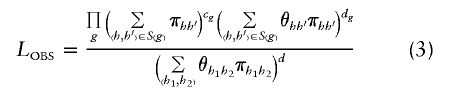
We wish to perform haplotype inference using the reparameterized _L_OBS in equation (3). Unfortunately, such inference is problematic when data consist of unphased genotypes. If external information is available that allows unambiguous determination of haplotype given genotype (e.g., if it is known that only a small number of haplotypes occur in a population and that no two haplotype pairs result in the same genotype), then S(g) can be restricted to the appropriate haplotype combinations and equation (3) can be used directly. However, without such external information and given genotype data only, no information exists to distinguish different haplotype pairs (h,_h_′) found in the same S(g). As a result, we cannot estimate all the π_hh_′ and θ_hh_′ as separate parameters.
To resolve this estimation problem, we must impose conditions to ensure identifiability of all the π_hh_′ and θ_hh_′. For π_hh_′_,_ we assume the haplotype pairs in the control population are in HWE, such that
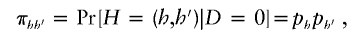
where p h denotes the frequency of haplotype h in the control population. We expect this HWE assumption in control subjects to hold when the disease is rare and when the susceptibility haplotypes have relatively low penetrance. If a rare highly penetrant haplotype exists, it should result in only a minor departure from the HWE assumption in the control population. If the disease is common or a common highly penetrant haplotype exists (again resulting in a common disease), then one would likely not employ a case-control study.
Although we assume HWE of the haplotypes in the control population, note that our method does not assume that haplotypes in the case sample are in HWE. We explicitly show this by rewriting ρ_hh_′ in equation (2) as
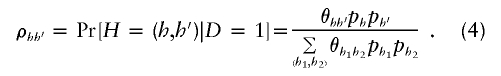
Equation (4) clearly shows that the haplotype frequencies in the cases do not follow HWE unless the effect of individual haplotypes on disease acts in multiplicative fashion (i.e., θ_hh_′=θ_h_θ_h_′, where θ h is the odds of disease, given haplotype h). We describe the benefits of relaxing this HWE assumption in the “Discussion” section.
We characterize identifiable models for θ_hh_′ in appendix A. To facilitate modeling, we write θ_hh_′=e X T _hh_′β, where β is an _R_-dimensional vector of disease relative-risk parameters, and X _hh_′ is an _R_-dimensional design vector that relates haplotype combinations to β. We provide examples of X _hh_′ for dominant, recessive, multiplicative, and general models for the effect of a single haplotype in later sections.
If we assume an identifiable model for all θ_hh_′ and impose HWE conditions in the control population, we can rewrite _L_OBS in equation (3) as
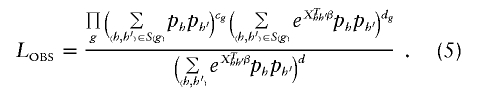
In this article, we will use _L_OBS in equation (5) for haplotype inference. Given haplotype ambiguity, inference of haplotype effect on disease may proceed by applying a missing-data maximization algorithm to _L_OBS. Instead of employing the popular EM algorithm, we apply an ECM algorithm to this likelihood (Meng and Rubin 1993) The ECM algorithm is a variant of the EM algorithm that replaces a (potentially unstable) joint maximization step of p and β with several computationally simpler conditional maximization steps. In appendix B, we provide details of the ECM algorithm for maximizing _L_OBS in equation (5).
Asymptotic Inference Methods
Using _L_OBS, we can test hypotheses about or construct estimators of relative-risk parameters β (e.g., _H_0:β=0 vs. H A:β≠0). For testing hypotheses, we first consider two statistics that appeal to asymptotic theory: a likelihood-ratio (LR) statistic and a robust score statistic. The LR statistic has the form 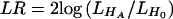 , where L H A and L _H_0 denote the value of _L_OBS under the alternative and null hypotheses, respectively. If we assume that no haplotype has estimated frequency 0 in the sample, the LR statistic asymptotically follows a _χ_2 distribution under H 0 , with degrees of freedom equal to the number of tested regression coefficients. We note here that, for a model in which each possible haplotype has a multiplicative effect on disease risk, the LR statistic is equivalent to the method of Fallin et al. (2001) and to an approach proposed by Zhao et al. (2000).
, where L H A and L _H_0 denote the value of _L_OBS under the alternative and null hypotheses, respectively. If we assume that no haplotype has estimated frequency 0 in the sample, the LR statistic asymptotically follows a _χ_2 distribution under H 0 , with degrees of freedom equal to the number of tested regression coefficients. We note here that, for a model in which each possible haplotype has a multiplicative effect on disease risk, the LR statistic is equivalent to the method of Fallin et al. (2001) and to an approach proposed by Zhao et al. (2000).
We can also use a score statistic to test hypotheses about relative-risk parameters. We use the robust score statistic of Boos (1992). Score statistics require the derivatives of log(L OBS) with respect to β and p h. For β, we obtain
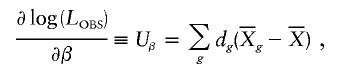
where
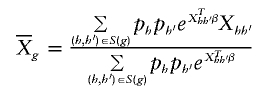
and
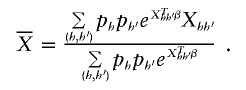
Calculation of the score statistic requires that each p h have estimated frequency >0 (otherwise, the information matrix is not invertible). If this requirement does not hold, we condition on the true haplotype frequency equaling 0 for each haplotype with estimated frequency 0. With this choice, if there are J haplotypes with nonzero frequency (assumed, without loss of generality, to be labeled 1–J), we rewrite all but one of the nonzero values of p h as
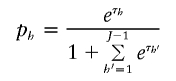
and set the final nonzero value of p h to be
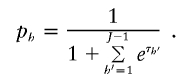
We then calculate the score function for τ_r_ as
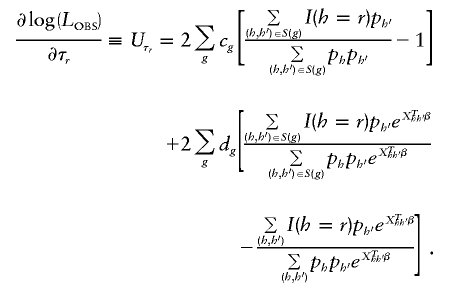
The robust score statistic also requires calculation of the observed information matrix H, which we evaluate by taking numerical derivatives of _U_β and 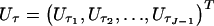 . For convenience, we write H in block-factored form
. For convenience, we write H in block-factored form
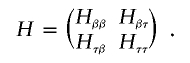
The robust score statistic also requires evaluation of the empirical variance-covariance matrix Σ of the score function 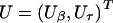 . Using H and Σ, we calculate the robust variance of _U_β as _V_=(I R,-_H_βτ_H_ττ)Σ(I R,-_H_βτ_H_ττ)T, where I R is an identity matrix with dimension equal to the dimension of β. We then use _U_β and V to construct robust score statistics to test β. For example, we construct a global score statistic for testing _H_0:β=0 as _S_=U _T_β=0_V_-1_U_β=0, which asymptotically follows a _χ_2 distribution under H 0 with degrees of freedom equal to R (the number components in β).
. Using H and Σ, we calculate the robust variance of _U_β as _V_=(I R,-_H_βτ_H_ττ)Σ(I R,-_H_βτ_H_ττ)T, where I R is an identity matrix with dimension equal to the dimension of β. We then use _U_β and V to construct robust score statistics to test β. For example, we construct a global score statistic for testing _H_0:β=0 as _S_=U _T_β=0_V_-1_U_β=0, which asymptotically follows a _χ_2 distribution under H 0 with degrees of freedom equal to R (the number components in β).
Choosing whether to apply an LR or score statistic for inference depends on many factors. One issue that affects this selection concerns the observed number of estimated haplotype frequency parameters (p h) in the sample. The calculation of score tests and asymptotic CIs requires inverting the information matrix, which has dimension equal to one less than the number of observed haplotypes plus the number of parameters in β. When this number is large, the matrix inversion may be numerically difficult to perform. The ECM algorithm used to maximize _L_OBS does not require inversion of large matrices (see appendix B), so we can easily calculate an LR statistic for inference. However, the validity of the LR statistic relies on correct specification of the model for the disease odds θ_hh_′. Robust score statistics are valid asymptotically even when one misspecifies this model. Further, robust score statistics require only null haplotype frequencies and are useful for situations in which maximization of _L_OBS is difficult.
Permutation and Bootstrap Inference Methods
In a typical haplotype analysis, one or more sample haplotype frequencies are estimated to be 0. In this situation, asymptotic inference using either the LR or robust score statistic proceeds assuming those haplotypes with estimated frequencies of 0 have a true (population) haplotype frequency equaling 0. If this assumption is questionable, we can apply permutation approaches for proper inference. We can apply a permutation test by shuffling assignments to case and control samples and calculating a test statistic for each permutation. For the LR statistic, we assess significance by comparing the test statistic for the observed data with the appropriate percentile of the distribution of test statistics calculated using the permuted data. For a score test, we assess significance by first obtaining the average score statistic (denoted by  ) over the permutations. We then center the score statistic for both the observed data and for each permutation by subtracting
) over the permutations. We then center the score statistic for both the observed data and for each permutation by subtracting 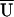 from _U_β=0 and calculating
from _U_β=0 and calculating
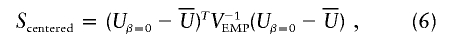
where _V_EMP is the empirical variance-covariance matrix of _U_β=0 from the permutation samples. The permutation-based P value is the proportion of times _S_centered, calculated using a permutation sample, exceeds the value of _S_centered calculated for the original data. The LR statistic is not amenable to centering in this way. Note that calculation of _S_centered requires only inversion of a matrix with dimension equal to the number of parameters in _U_β. Further, _S_centered may be valid in situations in which 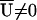 ; for example, when the HWE model for the control haplotype frequencies π_hh_′ does not hold.
; for example, when the HWE model for the control haplotype frequencies π_hh_′ does not hold.
Estimates of relative risk parameters β can be obtained using maximum likelihood. CIs can be constructed by inverting the observed information matrix. As with score tests, this approach is conditional on all haplotypes that have estimated frequency 0 having true frequency 0. We can also construct bootstrap CIs for parameters β by resampling with replacement from the original data (again, preserving the number of cases and controls), estimating β for each replicate data set, and using the percentiles of the estimated βs as confidence limits (Effron and Tibshirani 1998). The permutation approach is numerically less intensive than bootstrapping, because the estimated null hypothesis haplotype frequencies are identical for each permutation.
Application to FUSION Data
We applied our haplotype method to a subset of data from the FUSION study. A sample of 796 case subjects with type 2 diabetes and 415 control subjects were genotyped at five SNPs (distance between adjacent SNPs <300 kb) found along a putative susceptibility region on chromosome 22. We let 0 and 1 denote the two alleles of each SNP. Previous work from the FUSION study identified a putative susceptibility haplotype, 01100, that may yield increased odds of type 2 diabetes (L. Scott, personal communication).
The FUSION data set contained subjects with missing genotype data at one or more of the five SNPs. Within the sample, 131 (16.5%) of the case subjects and 82 (19.8%) of the control subjects were missing genotype information for at least one SNP. Missing SNP genotype rates in the total sample for SNPs 1–5 were 2.9%, 5.6%, 5.4%, 4.5%, and 2.3%, respectively.
We began our haplotype analysis of the FUSION data by applying the EM algorithm to the combined sample as well as separately to the case and control samples to determine haplotypes present in the data set. Using these frequencies, we determined the uncertainty of each haplotype in the genotype data by employing the R_2_h measure of Stram et al. (2003_a_). To account for missing SNP genotype data, we calculated R_2_h using
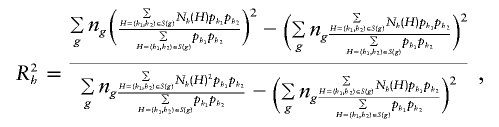
where n _g_=c g+d g, and N h(H) denotes the number of copies of haplotype h in H. Here, the multilocus genotypes in the sum can include the value “missing” at any of the individual loci.
We tested for association between each observed haplotype and type 2 diabetes status using 1-df asymptotic LR and robust score statistics based on _L_OBS in equation (5), under the assumption of a multiplicative model. To assess the accuracy of asymptotic results, we also calculated permutation-based P values for the LR statistic and _S_centered from equation (6). Each permutation-based P value was calculated using 10,000 random permutations of case and control status.
Using the LR and robust score statistics, we identified those haplotypes with significant associations and included them in more extensive analyses that fit recessive, dominant, multiplicative, and general (two-parameter) models for the odds of disease 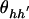 . If we let δ_hh_′ be an indicator function that equals 1 when h_=h_′ and 0 otherwise, a model for the effect of a specific haplotype h* takes the form θ_hh_′=e_β0+β1δ_hh*δ_h_′_h* for a recessive model, θ_hh_′=e_β0+β1(δ_hh*+δ_h_′_h*-δ_hh_*δ_h_′h*) for a dominant odds model, θ_hh_′=e_β0+β1(δ_hh*+δ_h_′h*) for a multiplicative odds model, and θ_hh_′=e_β0+β1(δ_hh*+δ_h_′h*-δ_hh_*δ_h_′h*)+β2δ_hh_*δ_h_′h* for a general odds model. Here, _β_1 (and _β_2 in the general odds model) is the effect of h* on disease, and _β_0 is the intercept. We calculated the AIC for each model and inferred the mechanism of genetic action by choosing the model with the lowest AIC value (Akaike 1985). Finally, we used the observed pattern of risk and protective haplotypes to suggest an overall model for the effect of haplotypes on the risk of disease in these data.
. If we let δ_hh_′ be an indicator function that equals 1 when h_=h_′ and 0 otherwise, a model for the effect of a specific haplotype h* takes the form θ_hh_′=e_β0+β1δ_hh*δ_h_′_h* for a recessive model, θ_hh_′=e_β0+β1(δ_hh*+δ_h_′_h*-δ_hh_*δ_h_′h*) for a dominant odds model, θ_hh_′=e_β0+β1(δ_hh*+δ_h_′h*) for a multiplicative odds model, and θ_hh_′=e_β0+β1(δ_hh*+δ_h_′h*-δ_hh_*δ_h_′h*)+β2δ_hh_*δ_h_′h* for a general odds model. Here, _β_1 (and _β_2 in the general odds model) is the effect of h* on disease, and _β_0 is the intercept. We calculated the AIC for each model and inferred the mechanism of genetic action by choosing the model with the lowest AIC value (Akaike 1985). Finally, we used the observed pattern of risk and protective haplotypes to suggest an overall model for the effect of haplotypes on the risk of disease in these data.
We computed asymptotic CIs for relative risk parameters by inverting the observed information matrix. To determine whether these CIs had appropriate coverage, we simulated data sets using parameters that match estimates from the FUSION data and plotted the empirical coverage of CIs (the proportion of intervals containing the true parameter) as a function of the nominal coverage of the CI. A straight line for this plot indicates appropriate coverage.
Results
Application of the EM algorithm to the case and control samples uncovered 17 haplotypes in the sample from the FUSION data set. Table 1 gives the frequency of each haplotype in the case and control samples. Table 1 also provides the R_2_h values for each of the observed 17 haplotypes in the data set. The R_2_h value for each observed FUSION haplotype was ⩽0.7322, which indicates considerable haplotype uncertainty, given the genotype data. On the basis of these R_2_h results, application of a prospective model that assumed HWE in the study population would likely yield biased estimates of haplotype effect (Stram et al. 2003_b_).
Table 1.
Haplotype Frequencies and Association Test Statistics for FUSION Data Set[Note]
| Frequency | LR Statistic | Robust Score Statistic | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Haplotype | Control | Case | R_2_h | LR StatisticValuea | Permutation-Based_P_ Value | Asymptotic_P_ Value | Robust ScoreStatistic Value | Permutation-Based_P_ Value | Asymptotic_P_ Value |
| 00011 | .0042 | .0066 | .0155 | .2910 | .6145 | .5896 | .3154 | .6175 | .5744 |
| 00100 | .0035 | .0034 | .0131 | .0092 | .8866 | .9238 | .0093 | .8801 | .9213 |
| 00110 | .0018 | .0007 | .0024 | .1927 | .8010 | .6607 | .1615 | .8085 | .6878 |
| 01011 | .1292 | .1344 | .4503 | .1953 | .6548 | .6585 | .2108 | .6708 | .6461 |
| 01100 | .2514 | .3183 | .7322 | 12.7004 | .0008 | .0004 | 13.1913 | .0009 | .0003 |
| 01101 | .0012 | <10−6 | .0010 | 2.1258 | .2305 | .1448 | 1.0020 | .2306 | .3168 |
| 01110 | <10−6 | .0046 | .0088 | 3.5869* | .1000 | .0582 | 3.0910 | .1299 | .0787 |
| 01111 | .0019 | <10−6 | .0019 | 3.4588 | .0944 | .0629 | 1.1913 | .0944 | .2751 |
| 10000 | .0136 | .0139 | .0342 | .1510 | .7009 | .6976 | .1497 | .6988 | .7005 |
| 10010 | <10−6 | .0012 | .0025 | 1.1656* | .5503 | .2803 | 1.5557 | .5509 | .2123 |
| 10011 | .3574 | .2884 | .7036 | 12.1775 | .0007 | .0005 | 11.8723 | .0007 | .0006 |
| 10100 | .0520 | .0596 | .3154 | .2555 | .6049 | .6133 | .2770 | .6237 | .5987 |
| 10110 | .0317 | .0319 | .2680 | .0644 | .8004 | .7997 | .0655 | .8081 | .7981 |
| 11011 | .1391 | .1290 | .3875 | .8258 | .3585 | .3635 | .8394 | .3622 | .3596 |
| 11100 | .0110 | .0092 | .0289 | .0160 | .9036 | .8993 | .0166 | .9187 | .8976 |
| 11110 | <10−6 | .0013 | .0019 | 1.4940* | .2907 | .2216 | .8190 | .2977 | .3655 |
| 11111 | .0020 | <10−6 | .0015 | 3.7632 | .0041 | .0524 | 1.1953 | .0041 | .2743 |
In addition to haplotype frequencies and R_2_h values, the table provides the 1-df LR statistic and robust score statistic values for each haplotype, under the assumption of a multiplicative model, calculated using both asymptotic theory and a Monte Carlo approximation to the permutation distribution with 10,000 random reassignments of case and control status. In the absence of multiple testing issues, we can compare each test with a prespecified cutoff P value (.05 or .01). Given that we test 17 hypotheses in table 1, the Bonferroni procedure corresponds to comparing each P value with .05/17≈.003 or .01/17≈.0006.
Our results in table 1 show that we observed some haplotypes only in case subjects (e.g., 01110), whereas we observed other haplotypes only in control subjects (e.g., 01101). Although we estimate the relative risk parameters β to be infinite in such situations, we found that none of these haplotypes were significantly associated with disease after adjusting for multiple comparisons. We also found that our ECM algorithm had difficulty converging when modeling the effect of a haplotype found only in case subjects. This occurs because ρ_hh_′∝θ_hh_′π_hh_′, so that, as estimates of π_hh_′ decrease, estimates of θ_hh_′ must increase, but in such a way that their product gives a finite value for ρ_hh_′. This difficulty does not arise for haplotypes found only in controls, because estimates of π_hh_′ are finite and both θ_hh_′ and ρ_hh_′ can tend towards 0. As a result, for haplotypes found only in cases, we reverse the roles of case and control and estimate θ-1_hh_′, the odds of being disease-free given the haplotype, instead. Because this trick requires the case population to be in HWE, we can apply it only for the multiplicative model. We indicate LR statistics calculated using this approach with an asterisk in table 1. We note that our score statistics are invariant when we switch the roles of case and control subjects.
Asymptotic and permutation-based P values generally agreed and were meaningfully different only when a haplotype was absent in either case subjects or control subjects. The only exception to this finding was for haplotype 00110, which has such a low frequency that replicate data sets generated in the permutation procedure were likely to have that haplotype appear only in cases or only in controls.
Examination of the LR and score statistic values in table 1 revealed that only haplotypes 01100 and 10011 were significantly associated with disease at a Bonferroni-corrected P value of .003. We incorporated these two haplotypes in more extensive analyses to determine which model (recessive, dominant, multiplicative, or general) best describes each haplotype’s effect on type 2 diabetes. We present the results of these analyses for haplotypes 01100 and 10011 in tables 2 and 3, respectively. For both haplotypes, we determined that a multiplicative model had the lowest AIC value. Note that, for the general model, the effect of the first copy of the haplotype (0.33 for 01100 and −0.35 for 10011) was nearly equal to the effect of the second copy of the haplotype (0.40 for 01100 and −0.28 for 10011), which also suggests a multiplicative model for each haplotype.
Table 2.
Model Selection for Risk Haplotype 01100 from FUSION Data Set
| Model | β (95% CI) | AIC |
|---|---|---|
| Recessive | .32 (.03 to .60) | 6641.3 |
| Dominant | .27 (.05 to .48) | 6640.0 |
| Multiplicative | .35 (.15 to .54) | 6633.2 |
| General | .33 (.10 to .55); .40 (.11 to .69) | 6635.1 |
Table 3.
Model Selection for Protective Haplotype 10011 from FUSION Data Set
| Model | β (95% CI) | AIC |
|---|---|---|
| Recessive | −.22 (−.52 to .08) | 6643.9 |
| Dominant | −.33 (−.55 to −.11) | 6637.0 |
| Multiplicative | −.33 (−.52 to −.15) | 6633.8 |
| General | −.35 (−.57 to −.14); −.28 (−.59 to .02) | 6635.6 |
Figure 1 shows the empirical coverage of CIs for the relative-risk parameter of haplotype 01100. We assumed that haplotype 01100 had a multiplicative effect on disease, with relative risk parameter β=0.35 corresponding to the value in table 2. We simulated 10,000 data sets with the same numbers of case and control subjects and the same haplotype frequencies as the FUSION study. The straight line in figure 1 suggests that the CIs in tables 2 and 3 are reliable.
Figure 1.

Empirical coverage of CIs for the relative-risk parameter β of haplotype 01100. Results are based on 10,000 simulated data sets with the same haplotype frequencies as the FUSION data. Haplotype 01100 has a multiplicative effect on disease risk, with β=0.35.
Results in table 2 indicate that haplotype 01100 is a susceptibility haplotype that increases the odds of diabetes (since the values of β in table 2 are positive). This result supports the previous finding from the FUSION study. However, table 3 shows that haplotype 10011 is protective against diabetes (since the values of β in table 3 are negative). It is interesting to note that these two haplotypes have no SNP allelic variants in common, which suggests that we consider a model with an overall risk score corresponding to the number of SNP variants in common with the disease-susceptibility haplotype 01100. Results from the model show that each additional SNP variant that agrees with that of haplotype 01100 increases the risk (on the log scale) by 0.087 (95% CI .045 to .129), so that the odds ratio of diabetes for an individual with two copies of haplotype 01100 relative to an individual with two copies of haplotype 10011 is _e_10*(0.087)≈2.39. This model yielded an AIC of 6629.3, which is lower than any individual model shown in tables 2 and 3. This finding suggests this model fits the FUSION data better than any previous model in tables 2 and 3. We also fit a two-parameter model that allowed for independent multiplicative action of haplotypes 01100 and 10011. This two-parameter model yielded an AIC of 6631.4, so we prefer the model that counts agreements with the risk haplotype.
Discussion
We have developed a unified likelihood-based framework for estimating and testing the effects of specific haplotypes or haplotype features on disease under the assumption of a case-control study design. We believe that our proposed method will facilitate the identification of genetic variants that influence complex disease. Our approach can accommodate, test, and estimate multiple haplotype effects under a variety of different genetic mechanisms. In addition to a simple and natural likelihood formulation, our approach also allows us to characterize which models for the effect of haplotype on disease risk are identifiable. Although derived within the context of our approach, these results should be applicable to other haplotype inference approaches as well, since they are based solely on the effect of changes in the disease risk model and the probability of the observed genotype.
One attractive feature of our approach is that our parameterization of the likelihood is retrospective and properly accounts for the case-control sampling design. There are a number of advantages of a retrospective approach over a prospective approach. First, many prospective-likelihood methods (such as those developed by Schaid et al. [2002] and Zaykin et al. [2002]) are limited to hypothesis testing, because haplotype frequencies are stratified by disease status under the alternative hypothesis. Even if a multiplicative model holds, so that both case and control populations are in HWE, the study (pooled) population is not in HWE except under the null hypothesis. Stram et al. (2003_b_) demonstrated that application of a prospective likelihood to case-control data yields biased estimates of haplotype frequencies and odds ratio parameters under the alternative hypothesis when substantial haplotype ambiguity exists in the sample. In our approach (as in that of Zhao et al. 2003), we assume HWE only in the control population.
A second potential advantage to a retrospective approach involves efficiency. Carroll et al. (1995) showed that variance estimates obtained from fitting a prospective model to retrospective data may be larger than those obtained from fitting a proper retrospective model when one restricts the distribution of (H,G) in some way. In haplotype analyses, the assumption of Hardy-Weinberg equilibrium in the sample is such a restriction. Note that, if we knew H unambiguously, we could efficiently test the null hypothesis of no haplotype-disease association in the case-control samples using the prospective likelihood _Pr_[D|_H_] (Prentice and Pyke 1979). Because our approach is based on the retrospective likelihood that describes the way the study data were collected, it is (asymptotically) optimally efficient.
Finally, because our approach is likelihood-based, we can apply model selection criteria such as the AIC (Akaike 1985) to determine the best model for haplotype effects on disease risk. We have illustrated this approach in our analysis of data from the FUSION study. The question of the best way to select a haplotype model when one uses a large number of SNPs is of great importance. Although the approach we have presented here is a starting point, we believe additional work is needed in this area.
Although our retrospective method has some appealing features for haplotype analysis, it also has limitations. A major assumption in our approach is that haplotypes from control subjects are in HWE. To determine the effect of HWE departure on our method, we performed additional simulations in the context of the FUSION data set. We simulated haplotype data through use of the same haplotype frequencies and numbers of case and control subjects as in the FUSION study, but we used a common fixation index, _F_=0.05, for each haplotype pair in the control population (resulting in a departure from HWE in the control sample). We then simulated models in which haplotype 01100 acted according to a multiplicative, dominant, or recessive mechanism with the disease relative-risk parameter β=0, −0.35, and 0.35. We generated 500 data sets for each disease model and calculated asymptotic and permutation-based P values of the robust score statistic for testing _H_0:β=0. We calculated permutation-based P values using 1,000 random permutations generated under the null hypothesis for each data set.
Table 4 presents the simulation results when the HWE assumption is violated. We see that departure from HWE has negligible effect for the multiplicative model. Further, parameter estimates from the multiplicative model remained unbiased (results not shown). However, for dominant and recessive models, the asymptotic P values of the robust score statistic were markedly inflated under the null hypothesis, and estimates of β were noticeably biased downward for dominant models and upward for recessive models (results not shown). In contrast, the centered permutation score statistic described in equation (6) had appropriate size and still had good power to detect alternatives. On the basis of these results, we recommend that all P values for dominant or recessive models be validated using the centered permutation score test. Further, we caution that parameter estimates for nonmultiplicative models may be suspect when asymptotic and permutation-based P values disagree. The approach of Zhao et al. (2003) may be more robust to departure from HWE in the control population; further study of this issue is warranted.
Table 4.
Effect of HWE Departure in Control Subjects for Testing _H_0: β=0[Note]
| P Values of Robust Score Statistic at Nominal α=.05 | ||
|---|---|---|
| Model and β | Asymptotic | Permutation |
| Multiplicative: | ||
| .0 | .050 | .061 |
| .35 | .962 | .966 |
| −.35 | .938 | .936 |
| Recessive: | ||
| .0 | .200 | .054 |
| .35 | .918a | .334 |
| −.35 | .194a | .374 |
| Dominant: | ||
| .0 | .108 | .050 |
| .35 | .636a | .788 |
| −.35 | .962a | .764 |
A second limitation of our method relative to other haplotype methods is that it does not allow currently for environmental covariates. Although we believe we can extend our approach to incorporate covariates, this extension is nontrivial, whereas the approach of Zhao et al. (2003) easily accounts for covariates. We will consider this extension in a future manuscript.
Our analysis of the FUSION data suggests some guidelines as to when asymptotic results are reliable and when a resampling approach is necessary. In general, we found that asymptotic P values were accurate when the proposed model included only those haplotypes that are frequent enough such that permutation- or bootstrap-based replicate data sets are unlikely to assign such haplotypes exclusively to either case subjects or control subjects. However, for modeling the effect of low frequency haplotypes, we recommend a resampling-based approach.
Although we use an iterative algorithm to maximize the likelihood, it is sufficiently fast to allow for large-scale simulation studies. Analyses of 10,000 replicates for determining the permutation-based significance level of haplotype 01100 in the FUSION data set under a multiplicative model took ∼1 h on a Dell Latitude C840 with an Intel Pentium 4 processor. We note that estimation of relative-risk parameters for haplotype analyses can take substantially longer when there is a great imbalance in the haplotype frequencies between cases and controls. Our software is available upon request.
In this article and in our software implementation, we have considered haplotypes comprised of SNPs. In fact, the approach presented here is not limited to SNPs and is applicable to any marker loci. Genotypes corresponding to microsatellite loci, however, result in much less phase uncertainty. As a result, the strategy of reconstructing the individual haplotypes and analyzing the reconstructed data as if phase information were known incurs a smaller error when using microsatellite loci relative to SNPs.
Acknowledgments
We thank the members of the FUSION study for allowing us to present results from the analysis of FUSION data. We thank Dr. Laura Scott for her useful conversations regarding the FUSION data. We also thank Dr. Michael Boehnke and Dr. Paul Rathouz for their helpful comments on a previous version of the manuscript. Finally, we thank the reviewers for their constructive comments.
Appendix A: Identifiability Conditions of θ
For a given set of parameters β and p, nonidentifiability in the model for θ_hh_′=e X T _hh_′β occurs when a change in the parameter vector β does not produce a concomitant change in 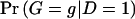 for at least one genotype g that is observed among the cases. Using equation (4), we can write
for at least one genotype g that is observed among the cases. Using equation (4), we can write 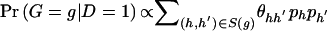 . For
. For 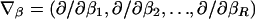 and some vector γ,
and some vector γ, 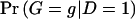 remains unchanged if
remains unchanged if 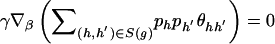 or if
or if 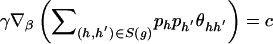 for every genotype g. Define the gradient vector
for every genotype g. Define the gradient vector
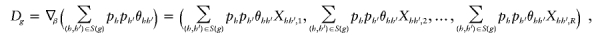
where X _hh_′,r is the _r_th element of X _hh_′. Let D be the matrix whose _g_th row is D g. Then the conditions we wish to impose are (1) _D_γ≠0 and (2) _D_γ⊥1 for any γ≠0, where 1 is the vector with all components equal to 1. We can verify the first condition by ensuring that D T D has full rank—that is, that the eigenvalues of D T D are all nonzero. We ensure the second condition by confirming that D _T_1=0.
Appendix B: The ECM Algorithm for Updating β and p
In this appendix, we describe the ECM (Meng and Rubin 1993) algorithm used to maximize the observed likelihood _L_OBS. The ECM algorithm is a variant of the EM algorithm that proceeds iteratively, with each iteration consisting of an E step and two CM (conditional maximization) steps. The E step imputes missing haplotype data, given current parameter estimates and observed genotype data. The first CM step updates β (conditional on fixed p), and the second CM step updates p (conditional on fixed β). Our ECM algorithm consists of cycling between these three steps. In the standard EM algorithm, the parameters β and p would be updated simultaneously.
Full-Data Likelihood (_L_FULL)
If phase information were known, we could write the likelihood as
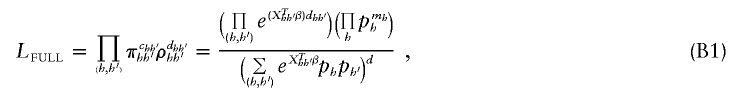
where c _hh_′ and d _hh_′ denote the number of controls and cases with haplotype pair (h,_h_′), respectively, and m h denotes the number of copies of haplotype h among cases and controls combined.
E Step
At the start of the (k+1)th step, we have available estimates of parameters β and p from the previous iteration, which we denote by β(k) and p(k). The E step estimates the number of control subjects with haplotype combination (h,_h_′) to be
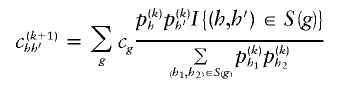
and the number of case subjects with haplotype combination (h,_h_′) to be
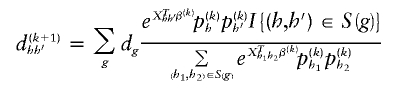
Here, 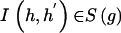 equals 1 when the haplotype pair is consistent with genotype g and equals 0 otherwise.
equals 1 when the haplotype pair is consistent with genotype g and equals 0 otherwise.
CM Step to Update β
Using 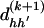 and
and 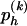 , we update β(k+1) by maximizing the log-likelihood of equation (B1) with respect to β. The log-likelihood of the part of likelihood (B1) that is proportional to β is
, we update β(k+1) by maximizing the log-likelihood of equation (B1) with respect to β. The log-likelihood of the part of likelihood (B1) that is proportional to β is
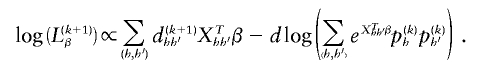
We maximize this log-likelihood through use of a quasi-Newton algorithm that incorporates the relevant score equations of β. The score vector is given by
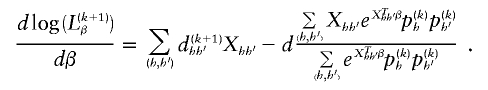
Because of the similarity between this maximization and logistic regression with an offset term, we find this optimization to be numerically stable. Unlike Newton-Raphson or Fisher-Scoring algorithms, this quasi-Newton algorithm does not require inversion of Hessian or information matrices when updating β.
CM Step to Update p
Unlike the CM step to update β, some care must be exercised in updating p. The log of the likelihood (B1) that depends on p is
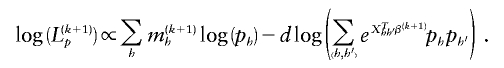
The score equations corresponding to maximizing this log-likelihood subject to the constraint 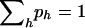 are
are
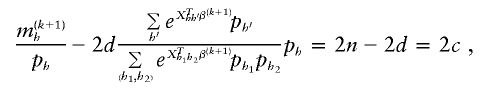
where 2_c,_ 2_d,_ and 2_n_ are the number of control haplotypes, case haplotypes, and total sample haplotypes, respectively. We can rewrite these equations as
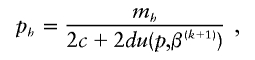
where
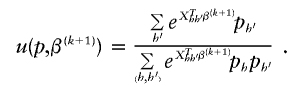
We solve for p iteratively, using p(k)≡p(k,0) as a starting value, then calculating p(k,s+1)h using
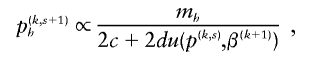
and then normalizing p(k,s+1)h. It is not necessary to carry this iteration to completion for each CM step to increase speed of computation. In our simulations, we used two iterations at each CM step. Note that, because m h and u are always nonnegative, estimates of p from our algorithm always form a proper probability density function.
Convergence of the ECM Algorithm
To start the ECM algorithm, we first estimate parameters p, under the assumption that β=0, using our implementation of the standard EM algorithm proposed by Excoffier and Slatkin (1995) and by others. Following Fallin and Schork (2000), we restarted this null-model EM algorithm 10 times at randomly chosen starting values. Then, starting at the null values of p, we iterated the ECM steps until the parameters β and p converge. Early simulation results suggest that a convergence criterion of
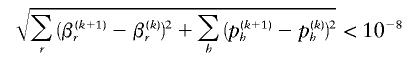
is adequate for termination of the ECM algorithm. This value is smaller than the values (10−5 or 10−6) chosen by Fallin and Schork (2000). However, when we applied our approach using these typical criteria, our empirical type I error rates for five-SNP haplotype data sets were anticonservative and were often double or triple the nominal type I error rate (data not shown). Moreover, our estimates of haplotype frequencies and effect size were less accurate when ɛ<10-6 was used, compared with when ɛ<10-8 was used (data not shown). An alternative strategy is to base convergence on the derivative of _L_OBS, which we can easily compute.
References
- Akaike H (1985) Prediction and entropy. In: Atkinson AC, Fienberg SE (eds) A celebration of statistics. Springer, New York, pp 1–24 [Google Scholar]
- Akey J, Jin L, Xiong M (2001) Haplotypes vs. single marker linkage disequilibrium tests: what do we gain? Eur J Hum Genet 9:291–300 10.1038/sj.ejhg.5200619 [DOI] [PubMed] [Google Scholar]
- Boos DD (1992) On generalized score tests. Am Stat 46:327–333 [Google Scholar]
- Botstein D, Risch N (2003) Discovering genotypes underlying human phenotypes: past successes for Mendelian disease, future approaches for complex disease. Nat Genet Suppl 33:228–237 10.1038/ng1090 [DOI] [PubMed] [Google Scholar]
- Carroll RJ, Wang S, Wang CY (1995) Prospective analysis of logistic case-control studies. J Am Stat Assoc 90:157–169 [Google Scholar]
- Dempster AP, Laird NM, Rubin DB (1977) Maximum likelihood estimation from incomplete data via the EM algorithm. J R Stat Soc 39:1–38 [Google Scholar]
- Effron B, Tibshirani RJ (1998) An introduction to the bootstrap. Chapman & Hall/CRC, Boca Raton [Google Scholar]
- Eitan Y, Kashi Y (2002) Direct micro-haplotyping by multiple double PCR amplifications of specific alleles (MD-PASA). Nucleic Acids Res 30:e62 10.1093/nar/gnf062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Slatkin M (1995) Maximum-likelihood estimation of molecular haplotyping frequencies in a diploid population. Mol Biol Evol 12:921–927 [DOI] [PubMed] [Google Scholar]
- Fallin D, Cohen A, Essioux L, Chumakov I, Blumenfield M, Cohen D, Schork N (2001) Genetic analysis of case/control data using estimated haplotype frequencies: application to APOE locus variation and Alzheimer’s disease. Genome Res 11:143–151 10.1101/gr.148401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallin D, Schork NJ (2000) Accuracy of haplotype frequency estimation for biallelic loci, via the expectation-maximization algorithm for unphased diploid genotype data. Am J Hum Genet 67:947–959 10.1086/303069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley M, Kidd K (1995) HAPLO: a program using the EM algorithm to estimate the frequencies of multi-site haplotypes. J Hered 86:409–411 [DOI] [PubMed] [Google Scholar]
- International SNP Map Working Group, The (2001) A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409:928–933 10.1038/35057149 [DOI] [PubMed] [Google Scholar]
- Joosten PH, Toepoel M, Mariman EC, Van Zoelen EJ (2001) Promoter haplotype combinations of the platelet-derived growth factor alpha-receptor gene predispose to human neural tube defects. Nat Genet 27:215–217 10.1038/84867 [DOI] [PubMed] [Google Scholar]
- Long JC, Williams RC, Urbanek M (1995) An E-M algorithm and testing strategy for multiple-locus haplotypes. Am J Hum Genet 56:799–810 [PMC free article] [PubMed] [Google Scholar]
- Meng X-L, Rubin DB (1993) Maximum likelihood estimation via the ECM algorithm: a general framework. Biometrika 80:267–278 [Google Scholar]
- Michalatos-Beloin S, Tishkoff S, Bentley K, Kidd K, Ruano G (1996) Molecular haplotyping of genetic markers 10 kb apart by allele-specific long-range PCR. Nucleic Acids Res 24:4841–4843 10.1093/nar/24.23.4841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RW, Kaplan NL (2002) On the advantage of haplotype analysis in the presence of multiple disease susceptibility alleles. Genet Epidemiol 23:221–233 10.1002/gepi.10200 [DOI] [PubMed] [Google Scholar]
- Prentice RL, Pyke R (1979) Logistic disease incidence models and case-control studies. Biometrika 66:403–411 [Google Scholar]
- Risch N (2000) Searching for genetic determinants in the new millennium. Nature 405:847–856 10.1038/35015718 [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517 [DOI] [PubMed] [Google Scholar]
- Satten GA, Carroll RJ (2000) Conditional and unconditional categorical regression models with missing covariates. Biometrics 56:384–388 [DOI] [PubMed] [Google Scholar]
- Satten GA, Kupper LL (1993) Inferences about exposure-disease associations using probability-of-exposure information. J Am Stat Assoc 88:200–208 [Google Scholar]
- Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA (2002) Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet 70:425–434 10.1086/338688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stram DO, Haiman CA, Hirschhorn JN, Altshuler D, Kolonel LN, Henderson BE, Pike ML (2003_a_) Choosing haplotype-tagging SNPs based on unphased genotype data from a preliminary sample of unrelated subjects with an example from the Multiethnic Cohort Study. Hum Hered 55:27–36 10.1159/000071807 [DOI] [PubMed] [Google Scholar]
- Stram DO, Leigh Pearce C, Bretsky P, Freedman M, Hirschhorn JN, Altshuler D, Kolonel LN, Henderson BE, Thomas DC (2003_b_) Modeling and E-M estimation of haplotype-specific relative risks from genotype data for a case-control study of unrelated individuals. Hum Hered 55:179–190 10.1159/000073202 [DOI] [PubMed] [Google Scholar]
- Tavtigian S, Simard J, Teng D, Abtin V, Baumgard M, Beck A, Camp N, et al (2001) A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet 27:172–180 10.1038/84808 [DOI] [PubMed] [Google Scholar]
- Valle T, Tuomilehto J, Bergman RN, Ghosh S, Hauser ER, Eriksson J, Nylund SJ, Kohtamaki K, Toivanen L, Vidgren G, Tuomilehto-Wolf E, Ehnholm C, Blaschak J, Langefeld CD, Watanabe RM, Magnuson V, Ally DS, Hagopian WA, Ross E, Buchanan TA, Collins F, Boehnke M (1998) Mapping genes for NIDDM: design of the Finland-United States Investigation of NIDDM Genetics (FUSION) study. Diabetes Care 21:949–958 [DOI] [PubMed] [Google Scholar]
- Zaykin DV, Westfall PH, Young SS, Karnoub MA, Wagner MJ, Ehm MG (2002) Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum Hered 53:79–91 10.1159/000057986 [DOI] [PubMed] [Google Scholar]
- Zhao JH, Curtis D, Sham PC (2000) Model-free analysis and permutation tests for allelic associations. Hum Hered 50:133–139 10.1159/000022901 [DOI] [PubMed] [Google Scholar]
- Zhao LP, Li SS, Khalid N (2003) A method for the assessment of disease associations with single-nucleotide polymorphism haplotypes and environmental variables in case-control studies. Am J Hum Genet 72:1231–1250 10.1086/375140 [DOI] [PMC free article] [PubMed] [Google Scholar]