A Model Membrane Protein for Binding Volatile Anesthetics (original) (raw)
Abstract
Earlier work demonstrated that a water-soluble four-helix bundle protein designed with a cavity in its nonpolar core is capable of binding the volatile anesthetic halothane with near-physiological affinity (0.7 mM _K_d). To create a more relevant, model membrane protein receptor for studying the physicochemical specificity of anesthetic binding, we have synthesized a new protein that builds on the anesthetic-binding, hydrophilic four-helix bundle and incorporates a hydrophobic domain capable of ion-channel activity, resulting in an amphiphilic four-helix bundle that forms stable monolayers at the air/water interface. The affinity of the cavity within the core of the bundle for volatile anesthetic binding is decreased by a factor of 4–3.1 mM _K_d as compared to its water-soluble counterpart. Nevertheless, the absence of the cavity within the otherwise identical amphiphilic peptide significantly decreases its affinity for halothane similar to its water-soluble counterpart. Specular x-ray reflectivity shows that the amphiphilic protein orients vectorially in Langmuir monolayers at higher surface pressure with its long axis perpendicular to the interface, and that it possesses a length consistent with its design. This provides a successful starting template for probing the nature of the anesthetic-peptide interaction, as well as a potential model system in structure/function correlation for understanding the anesthetic binding mechanism.
INTRODUCTION
The use of anesthetics in modern medicine can be dated back approximately 150 years, but the mechanisms of action of inhalational anesthetic compounds remain elusive. A commonly accepted theory is that anesthetic compounds act on a small number of targets that function as ion channels in the central nervous system (Franks and Lieb, 1994, 1998; Krasowski and Harrison, 1999). Anesthetic compounds can exert direct effects by binding to the ion channels (Eckenhoff and Johansson, 1997; Franks and Lieb, 1994), and indirect effects by modulating the physiochemical properties of the host lipid bilayer (Cantor, 1997; Hauet et al., 2003). Also, there is direct evidence showing that anesthetic molecules can bind to hydrophobic pockets in ligand-gated ion channels (Chiara et al., 2003).
The investigation of the molecular basis of anesthetic binding to channel proteins remains a challenging task because 1), ion channels are transmembrane proteins that are difficult to isolate and purify; 2), experimental methods suitable for binding assays are limited, and are often complicated by the presence of detergents as the solubilizing agents; and 3), the presence of multiple intrinsic fluorophores, such as tryptophan, in the proteins render identification of binding sites difficult when using the fluorescent quenching technique (Ulmschneider and Sansom, 2001).
To circumvent the above difficulties, Johansson and co-workers employed a series of structurally defined, water-soluble four-helix bundle scaffolds with distinct hydrophobic cores (Johansson, 2001; Johansson et al., 2000, 1998, 1996) as a model system for studying anesthetic binding to proteins. Despite the obvious difference between water-soluble and membrane proteins, the use of a water-soluble, designed protein as the model system for the investigation of anesthetic binding is considered relevant, because anesthetic molecules have been shown to bind to the hydrophobic cavities in the membrane-spanning regions of many putative candidates, such as the acetylcholine receptor and the so-called background potassium channels (Johansson, 2003). More importantly, the hydrophobic cores of both membrane- and water-soluble proteins have been shown to be similar in terms of overall hydrophobicity (Spencer and Rees, 2002). Johansson and co-workers show that anesthetic binding sites can be engineered into the hydrophobic core of a water-soluble protein. Moreover, their results indicate that high anesthetic affinity can be achieved by optimizing the size of the cavity (Johansson et al., 1998) and the polarity of the side chains lining the binding site in the core (Johansson et al., 2000).
Although the work pioneered by Johansson and co-workers offers a powerful approach to the investigation of anesthetic binding, the application of a water-soluble model system is considered limited to some extent because it cannot precisely mimic all of the critical features of ion channels. In biology, ion channels are transmembrane proteins embedded in an impermeable signal-barrier provided by the lipid bilayer. They propagate the signals across the lipid bilayer via coordinated motions of various domains (Doyle et al., 1998; Jiang et al., 2003; Sixma and Smit, 2003; Xu et al., 2000).
As a first step toward engineering a transmembrane anesthetic-binding protein we have designed and synthesized a protein that is membrane-soluble, i.e., the halothane-binding amphiphilic protein (hbAP0), which possesses a hydrophilic domain based on a water-soluble halothane binding protein (A_α_2; Johansson et al., 1998) and a hydrophobic domain based on a synthetic proton channel protein (LS2; Lear et al., 1988), as used in the amphiphilic four-helix bundle peptide, AP0 (itself designed to selectively bind redox cofactors; Ye et al., 2004). Our results indicate that the affinity of hbAP0 for halothane is _K_d = 3.1 ± 0.6 mM versus _K_d = 0.71 ± 0.04 mM in the water-soluble analog A_α_2. We attribute the decrease in affinity to constraints imposed by the topology of the protein, which lead to a less optimal cavity volume. Absence of the cavity significantly increases the _K_d of hbAP0 for halothane analogous to that for A_α_2. X-ray reflectivity demonstrates that, at high surface pressures, the amphiphilic halothane binding protein orients at the air-water interface with the longitudinal bundle axes normal to the surface plane, the hydrophobic and hydrophilic domains pointing toward air and into the water, respectively. Efforts are currently underway to identify directly the localization and orientation of halothane with respect to the cavity binding site along the axis of the helical bundle (Strzalka et al., 2004a).
MATERIALS AND METHODS
Materials
Fluorenylmethoxycarbonyl (Fmoc)-protected L-_α_-amino acids, Fmoc-PEG-PAL-PS resin, hydroxydihydrobenzotriazine, and 1-hydroxybenzotriazole were purchased from Applied Biosystems (Foster City, CA). Halothane (2-bromo-2-chloro-1,1,1-trifluoroethane) was from Halocarbon Laboratories (Hackensack, NJ). N-octyl _β_-D-glucopyranoside (OG) was from Anatrace (Maumee, OH). All other solvents and reagents were either from Fisher Scientific (Springfield, NJ) or Sigma (St. Louis, MO).
Protein synthesis and preparation
The protein hbAP0 was assembled on an Applied Biosystems model 433A solid-phase protein synthesizer using the standard Fmoc/tBu protection strategy on an Fmoc-PEG-PAL-PS resin (Applied Biosystems) at 0.25-mmol scale. The proteins were acetylated at their N-termini in 1:1 (v/v) acetic anhydride-pyridine for 30 min and purified on a reversed phase C4 HPLC column (Vydac, Columbia, MD) using gradients of 6:3:1 isopropanol:acetonitrile:H2O and water containing 0.1% (v/v) 2,2,2-trifluoroacetic acid. Pure proteins (4.56 kDa molecular weight) were dimerized by oxidizing their C-terminal cysteines in 1:1 (v/v) 100 mM ammonium hydrogen carbonate buffer (pH 10.0) and methanol in air to form the 90-amino acid disulfide-linked protein dimer (9.12 kDa MW). The protein's identity and purity were confirmed by matrix-assisted laser desorption/ionization mass spectrometry.
Lyophilized hbAP0 proteins were first solubilized into 4.5% (w/v) OG, 50 mM potassium phosphate (KPi), pH 8.0 buffer, and then diluted fivefold with 50 mM KPi, pH 8.0 buffer to give a final 0.5% (w/v) OG solution. Analytical ultracentrifugation, circular dichroism, and intrinsic fluorescence experiments were performed with proteins solubilized in detergent buffer, whereas Langmuir monolayer deposition was done by dissolving proteins in methanol, to avoid the introduction of detergent molecules to the air-water interface.
Analytical ultracentrifugation
Sedimentation equilibrium experiments were performed at 25°C on hbAP0 proteins solubilized in OG micelles using a Beckman XLA/I analytical ultracentrifuge (Beckman, Fullerton, CA) as described previously (Noy et al., 2003). Samples were measured simultaneously in a series of buffered D2O/H2O solutions (v/v; 20%, 40%, 60%, 80%, 90%, and 100%, corresponding to solvent densities of 1.0205, 1.0420, 1.0635, 1.0849, 1.0957, and 1.1064 g/ml, respectively; calculated from buffer composition using the program SEDNTERP, available from the RASMB web site, http://www.bbri.org/RASMB/rasmb.html). The total protein concentration was 16 _μ_M. Radial profiles of absorbance at 280 nm were collected at 30,000, 35,000, and 45,000 rpm at 5°C for each sample. Data were collected for 14 and 16 h after setting the first speed, then 12 and 14 h after setting the next two speeds. Equilibrium conditions were assumed after verifying that the early and late data sets at each speed were the same.
Circular dichroism spectroscopy
CD experiments were carried out on an Aviv 62DS spectropolarimeter (Aviv, Lakewood, NJ). All measurements were made at 25°C in a quartz cuvette of 0.2-cm pathlength. Spectra were recorded over the far UV range of 180–260 nm with a time constant of 1 s, a spectral resolution of 1 nm, and a scan rate of 20 nm/min. The reference spectra of the respective media were subtracted. The fraction of residues in the _α_-helical conformation, _f_H, was estimated from the measured residue ellipticity at 222 nm, _θ_222, using the well-established method of Luo and Baldwin (1997) and Tatulian and Tamm (2000); _f_H = (_θ_222−_θ_c)/(_θ_H−_θ_c), where the temperature-dependent values for an infinite helix, _θ_H, and a random coil, _θ_c, are assumed to be −31,739 and −3400°/cm2 per dmol−1, respectively (Marvin et al., 1997).
Steady-state fluorescence measurements
Binding of halothane to the hbAP0 proteins was determined using steady-state intrinsic tryptophan fluorescence measurements on a K2 multifrequency cross-correlation phase and modulation spectrofluorometer (ISS, Champaign, IL). Tryptophan was excited at 280 nm (bandwidth 3 nm), and emission spectra (bandwidth 5 nm) were recorded with a maximum near 333 nm. A cutoff filter was used to minimize the effect of scattered excitation light below 305 nm in the measured emission spectrum. The quartz cell had a pathlength of 10 mm and a Teflon stopper. The cell holder was thermostatically controlled at 25.0 ± 0.1°C. Protein concentration was determined with a UV/Vis Spectrometer Lambda 2 (Perkin-Elmer, Norwalk, CT), taking _ɛ_280 for tryptophan = 5690 M−1 cm−1, calculated from the primary sequence with the ProtParam tool offered by the EXPASY server of the Swiss Institute of Bioinformatics (http://us.expasy.org/cgi-bin/protparam). Halothane-equilibrated hbAP0 protein in gas-tight Hamilton syringes (Reno, NV) was diluted with predetermined volumes of nonequilibrated protein (not exposed to anesthetic, but otherwise treated in the same manner) to achieve the final anesthetic concentrations indicated in the figures.
Quenching data is first normalized treating the highest fluorescence intensity as 1. As described previously (Johansson and Eckenhoff, 1996; Johansson et al., 1995, 1998), the quenched fluorescence (Q) is a function of the maximum possible quenching (_Q_max) at an infinite halothane concentration ([Halothane]) and the affinity of halothane for its binding site (_K_d) in the vicinity of the tryptophan residues. From mass law considerations, it then follows that
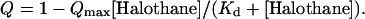 |
(1) |
|---|
Best-fit curves were generated using the Igor 4.09 program (WaveMetrics, Lake Oswego, OR), in which the _K_d and _Q_max are the unknown parameters. Data are expressed as mean ± SD. Data points are the averages of at least three experiments with separate samples.
Langmuir trough and isotherm measurements
The isotherm was recorded using a commercial Langmuir trough (Lauda, Lauda-Königshofen, Germany) equipped with a floating-barrier surface-pressure transducer. This trough gave reliable measurements at high surface pressure for these viscous monolayers. The paper Wilhelmy-plate surface-pressure transducer on the trough mounted on the liquid-surface spectrometer (below) would fail to hang vertically at high π, resulting in an artifactual plateau in the isotherm for π > 40 mN/m. The aqueous subphase contained 1 mM potassium phosphate and 10 mM KCl at pH 8.0, and was maintained at constant temperature of 20°C. The peptide was dissolved in methanol (typically 50 _μ_M) and spread onto the meniscus of a glass capillary passing through the air/water interface at an oblique angle. After spreading, we waited 10 min before compressing the monolayer at a constant rate.
Langmuir trough and reflectivity measurements
At the synchrotron, we mounted onto the sample stage of the liquid-surface spectrometer a Langmuir trough that has been described previously (Strzalka et al., 2000). The canister is equipped with an oxygen sensor that allowed us to measure when the air in the canister was completely replaced by moist helium. Purging the oxygen from the canister typically required ∼30 min after spreading the monolayer. After the purge, the monolayer was compressed at a constant rate until the desired surface pressure was achieved and the feedback constant-π control was engaged (for π ≤ 40 mN/m), or the barrier was simply stopped at the desired area/_α_-helix. Under constant pressure control, the area of the monolayer diminished by <2% during reflectivity measurements lasting ∼1 h. At high pressures, which could not be reliably measured at the synchrotron, we collected data at constant monolayer area. The observed pressure decayed <1 mN/m (∼2%) during the reflectivity measurements. The quality of the reflectivity data confirms that the monolayer remained stable during the course of the reflectivity scans.
Liquid-surface spectrometer
The x-ray reflectivity experiments were performed on beamline X-22B at the National Synchrotron Light Source at Brookhaven National Laboratory (Upton, NY). Details of the liquid-surface spectrometer have been reported elsewhere (Braslau et al., 1988; Helm et al., 1991). Here we give only a brief description. The synchrotron x-ray source was a bending-magnet in the electron storage ring operating at an energy of 2.8 GeV and currents of 150–250 mA. Monochromatic x rays were obtained via a horizontally reflecting Si (111) crystal monochromator to provide a wavelength λ = 1.546 Å. X rays were reflected downward onto the horizontal liquid-surface via a Ge (111) crystal to provide an angle of incidence α. Incident beam slits were set to collect the full horizontal width and vertically to limit the footprint on the liquid surface. A scintillation detector recorded the scattering from a thin Kapton film in the incident beam to provide an incident beam flux monitor. The specularly reflected beam from the liquid surface was measured at an angle β with respect to the liquid surface with another scintillation detector for α = β in the vertical scattering plane at 2_θ_xy = 0°. Scattered beam slits were set to accept the full specularly reflected beam. Off-specular background was measured at α = β with 2_θ_xy = ± 0.3°. The difference (specular minus off-specular background) provided the reflectivity R(_q_z) for photon momentum transfer q_z perpendicular to the liquid surface with q_z = (4_π/λ)sin_α.
Data analysis
The Fresnel-normalized specular x-ray reflectivity R(_q_z)/_R_F(_q_z) from a liquid surface arises from, in the first Born approximation, the modulus square of the Fourier transform of the gradient (or derivative) dρ(z)/_d_z of the electron density profile ρ(z) across the air-water interface averaged over the in-plane coherence length of the incident x rays (Als-Nielsen and Pershan, 1983; Helm et al., 1991), namely
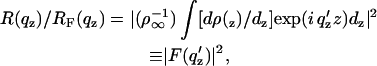 |
(2) |
|---|
where _R_F(_q_z) is the Fresnel reflectivity from a single infinitely sharp (ideal) interface, the electron density of the semi-infinite bulk subphase is _ρ_∞, and _q_c is _q_z at the critical angle for the subphase _α_c. This expression, Eq. 2, becomes progressively less valid as _q_z approaches _q_c, which is mitigated to some extent in the distorted-wave Born approximation by the use of _q_′z, where (_q_′z)2 = [(_q_z)2−(_q_c)2]. (Lösche et al., 1993) The normalized reflectivity data were analyzed by the box-refinement method, which requires no a priori assumptions and is therefore model-independent. This approach has been utilized previously by us and is presented in rigorous detail in a recent publication (Zheng et al., 2003).
RESULTS
Protein design
The hbAP0 is derived from the designed 62-residue helix-loop-helix protein A_α_2, with three heptads taken from the first three heptads of A_α_2. The sequence of A_α_2 is illustrated in Fig. 1. The two helices of A_α_2 only differ by seven residues. In aqueous solution, A_α_2 adopts an anti orientation (99%) (Johansson et al., 1998) to form a four-helix bundle, so that each layer of residues within the core along the bundle axis can be composed of four different residues. In contrast, the formation of a four-helix bundle architecture for hbAP0 is through four identical 40-residue helices, with each pair of helices being linked via N-terminal cysteine disulfide bridges to form a helix-loop-helix, presumably adopting a syn orientation in the membrane environment, i.e., at an interface between polar and nonpolar media. This means each layer of residues within the core along the bundle axis is composed of four identical residues. Both hbAP0 and A_α_2 share a layer of four Ala that form a cavity for binding halothane, when compared to mutants with four Leu residues in that layer, i.e., 4(_V_Leu−_V_Ala) = 228 Å3; the volume of halothane is 123 Å3.
FIGURE 1.
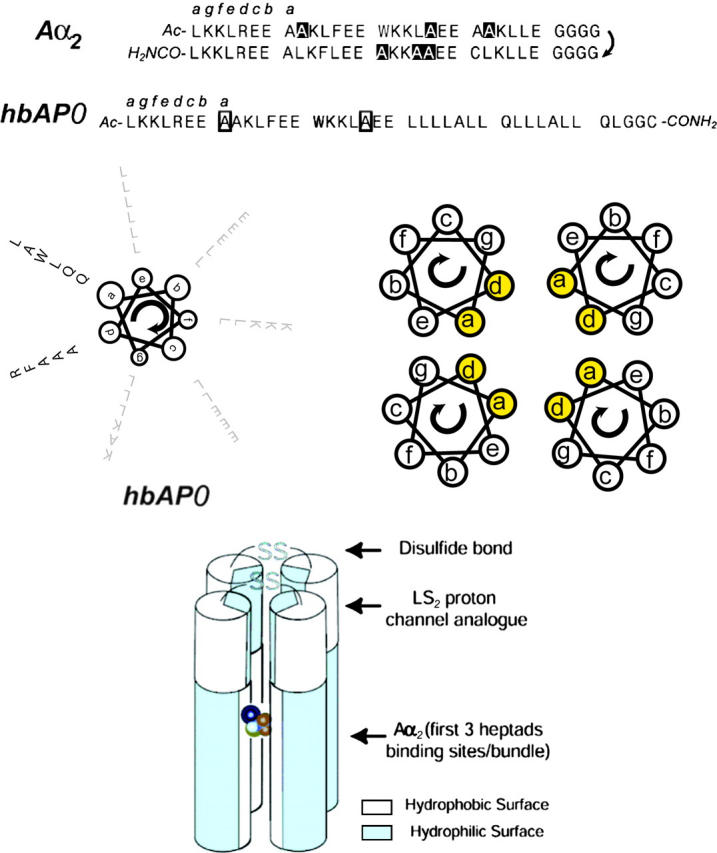
Schematic architecture of hbAP0. For comparison, we illustrate the sequence of the water-soluble halothane-binding peptide A_α_2. Mutation of the highlighted Ala residues to Leu results in the L_α_2 peptide, with a fourfold reduction in the binding affinity for halothane (L_α_2: _K_d = 3.1 ± 0.4 mM; A_α_2: _K_d = 0.71 ± 0.04 mM (Johansson et al., 1998). hbAP0 includes the first three heptads of A_α_2 in addition to the hydrophobic sequence derived from a synthetic proton channel LS2 (Lear et al., 1988). Two Gln in the hydrophobic sequence are aligned in _d_-positions of the hydrophobic core of the bundle. Exterior and interfacial side chains are gray-shaded to contrast with the side chains along the core region of the bundle. Halothane is displayed as a CPK model, with F in orange, Br in brown, Cl in green, and H in cyan. The location of halothane illustrates the binding pocket inside the bundle.
Secondary structure by CD
Before experiments, hbAP0 was dissolved in aqueous buffer in the presence of detergent, in which all subsequent physical characterizations in isotropic aqueous solution were conducted. We studied the secondary structure of hbAP0 in detergent micelles using CD spectroscopy. The far-UV circular dichroism spectrum in phosphate buffer with 0.9% OG shows the π → π* and n → π* transition at 208 and 222 nm, respectively; characteristics of typical _α_-helices (Fig. 2). The percentage of helical content is estimated to be 89%. Similarly, the spectrum from a sample of hbAP0 dissolved in methanol indicated approximately the same helical content, 93%.
FIGURE 2.

CD spectrum of hbAP0 in 0.9% OG, 50 mM KPi (pH 8.0) (solid line), and in methanol (dashed line). The characteristic maximum at 192 nm (not shown) and minima at 208 and 222 nm indicate that hbAP0 is _α_-helical in the presence of detergent micelles. The mean molar residue ellipticity at 222 nm suggests similar helix formation in detergent (89%) and in methanol (93%).
Halothane binding affinity by intrinsic tryptophan fluorescence
Before the binding assay, the environment surrounding the tryptophan was studied by fluorescence spectroscopy. The fluorescence spectra (Fig. 3 A) show a single peak located at 334 nm in the absence of halothane, and a slight blue-shift of 1–2 nm as halothane is introduced, except near complete quenching, when the maximum is slightly red-shifted by ∼3 nm. Our control experiment using N-acetyl-tryptophanamide in detergent buffer shows that water-exposed indole rings have a fluorescence maximum at 350 nm. This result indicates that the tryptophan in hbAP0 is located in a nonpolar environment (Johansson et al., 1995). The binding of halothane to the hydrophobic core of hbAP0 is monitored by the quenching of the intrinsic tryptophan fluorescence. The results are displayed in Fig. 3 B, which gives a _K_d of 3.1 ± 0.6 mM, and _Q_max of 1.2 ± 0.1. The binding isotherm indicates that halothane causes a concentration-dependent quenching of the tryptophan fluorescence without significantly changing the emission maximum, suggesting that the halothane binding is not accompanied by any substantial changes in the dielectric environment local to the indole rings (Johansson et al., 1995). Thus, the lack of a substantial red-shift in the tryptophan fluorescence emission maximum upon halothane binding suggests that the anesthetic does not promote unfolding of the bundle, which would lead to increased solvent-exposure of the indole rings. A mutant of hbAP0, in which the alanine residues forming the designed halothane binding cavity were mutated back to leucine, was also investigated analogous to the comparison of the water-soluble A_α_2 with L_α_2 studied previously (Johansson et al., 1998). The absence of the cavity similarly increased the _K_d for halothane binding to the hydrophobic core of the bundle by ∼2 mM.
FIGURE 3.
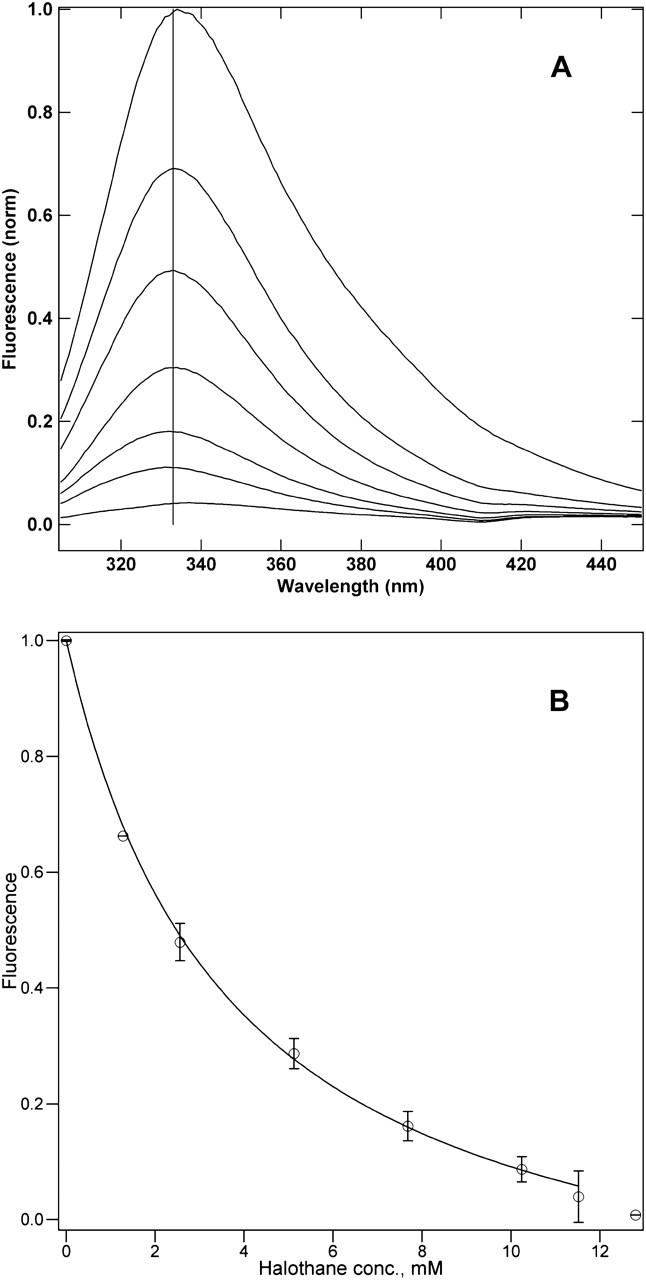
(A) Halothane concentration-dependent quenching of the hbAP0 bundle (1 _μ_M) fluorescence. Excitation was at 280 nm, the vertical line indicates 333 nm. The halothane concentration and wavelength of maximum emission for the spectra in order of decreasing fluorescence are 0 mM, 334 nm; 1.28 mM, 333 nm; 2.56 mM, 333 nm; 5.12 mM, 334 nm; 7.68 mM, 332 nm; 10.24 mM, 331 nm; and 11.52 mM, 337 nm. (B) The quenching profile for the hbAP0 bundle tryptophan fluorescence by halothane. The data points are the means of three experiments on separate samples, with the error bars representing the standard deviation. The line through the data points has the form of Eq. 2. The best fit shown yields a _K_d of 3.1 ± 0.4 mM, and _Q_max of 1.2 ± 0.1.
Aggregation state by analytical ultracentrifugation
The molecular mass of hbAP0 in aqueous solution in the presence of detergent was determined using analytical ultracentrifugation (Fig. 4). Simultaneous fits of different datasets gave a molecular weight for the sedimenting species of 19.5 ± 0.6 kDa (versus 18.25 kDa anticipated for a four-helix bundle) and 29 ± 7 detergent molecules associated with the sedimenting species, when the partial specific volume of the peptide was input as 0.70 ml/g, 10% lower than the theoretically calculated value (0.78 ml/g) based on the amino acid sequence (EXPASY server). The fitting similarly yields a partial specific volume of 0.68 ml/g, if we fix the molecular weight at 18.25 kDa for a four-helix bundle. This apparent discrepancy between theoretically calculated and experimental partial specific volume values is consistent with the small decrease in partial specific volume caused by the presence of OG (Noy et al., 2003). Overall, our results indicate that the oligomerization state of hbAP0 is consistent with the formation of a four-helix bundle.
FIGURE 4.
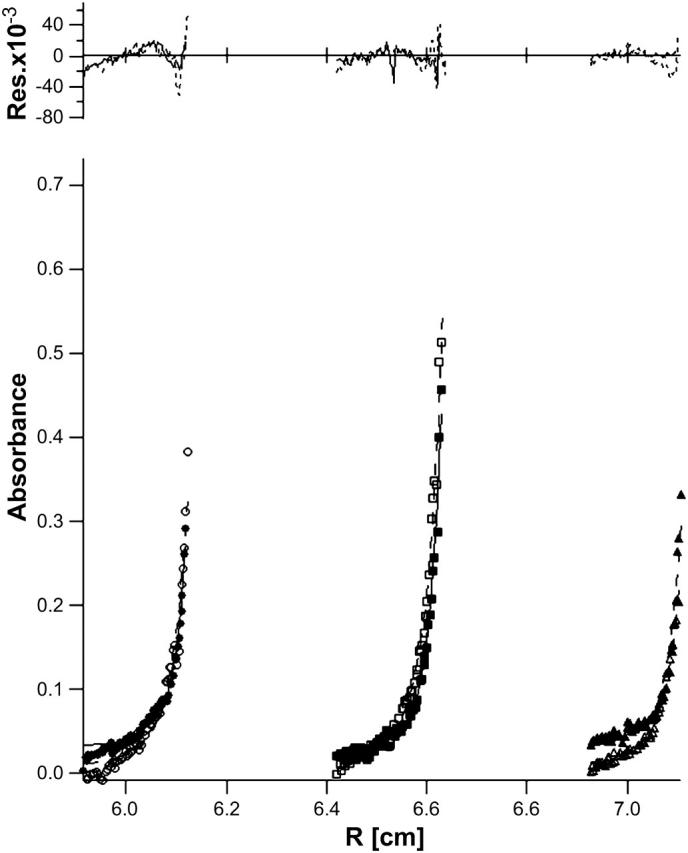
Simultaneous nonlinear fits of sedimentation equilibrium radial absorbance profiles of hbAP0 in 0.9% OG 10 mM KPi, 100 mM KCl pH = 8.0 buffer for raw data (see symbols) and their global fits (solid and dotted lines) at D2O/H2O 20% (•), 40% (▪), 60% (▴), 80% (○), 90% (□), and 100% (▵) at 45,000 RPM. The residuals for each fit appear above the radial absorbance profiles. The fitting of hbAP0 agrees with a single four-helix bundle species with a reasonable mole ratio of 29 ± 7 detergent/protein in the sedimenting species.
Pressure-area isotherm
The design of hbAP0 makes it a good amphiphile, as evidenced by the surface pressure-area isotherm (Fig. 5) and the stability of the surface pressure at constant area. Surface pressure first increases significantly at an area of ∼450 Å2/_α_-helix until it reaches a plateau-like region analogous to the feature in the isotherm of AP0 (Ye et al., 2004). At areas <∼200 Å2/_α_-helix, π increases more rapidly again. We did not observe an abrupt collapse of the monolayer, just a change in slope at the highest pressures recorded. We note that the minimum cross-sectional dimensions of a single helix derived from the analogous NMR structure of the peptide designated BB (Skalicky et al., 1999), the four-helix bundle peptide closely related to the hydrophilic domain of hbAP0, indicates a helical diameter of 12–13 Å, which provides a minimum cross-sectional area of 144–170 Å2. This value is still somewhat larger than that for close-packed, perfectly straight _α_-helices (namely ∼80 Å2 for a typical distance of closest approach of 10 Å) due to their bending to form a coiled-coil.
FIGURE 5.

The surface pressure-area (_π_-A) isotherm recorded while compressing a monolayer of pure hbAP0 spread from methanol solution on a subphase of 1 mM phosphate buffer with 10 mM KCl at pH 8 and 20°C. The letters indicate points at which x-ray reflectivity data was collected at constant pressure (π = 10, 20, 30, and 40 mN/m labeled a_–_d) or constant area (A = 190, 120, and 100 Å2/_α_-helix, equivalent to π = 44, 58, 62 mN/m, labeled e_–_g).
Orientation of bundles at the air-water interface by x-ray reflectivity
With reference to the isotherms described above, normalized x-ray reflectivity data R(_q_z)/_R_F(_q_z) for the pure hbAP0 monolayer appears in Fig. 6 A. At the lowest π of 10 mN/m, the data consist of a single broad maximum for momentum transfer _q_z < 0.7 Å−1. With increasing surface pressure, the maximum narrows and shifts to smaller _q_z, without developing subsidiary maxima/minima up to a pressure of 44 mN/m. With decreasing area/helix, the maximum narrows and shifts slightly to smaller _q_z, although now also developing more pronounced subsidiary maxima/minima. In Fig. 6 B, the inverse Fourier transforms of these data, which correspond to the autocorrelation of the gradient electron density profiles of the Langmuir monolayer, are shown. The results reveal that the thickness, or maximum extent, of the gradient profile of the monolayer increases dramatically between 30 mN/m and 40 mN/m. Below a surface pressure of 30 mN/m, the gradient electron density profile (and similarly, its integral, the electron density profile itself) contains no features separated by >20–30 Å (since the autocorrelation function is 0 for larger separations), whereas the gradient profile at 40 or 44 mN/m contains features separated by as much as 40–50 Å, although apparently without a well-defined peptide-subphase interface. At the highest π (smallest area/helix) investigated, the monolayer profile now extends further to ∼60 Å ∼70 Å, with a well-defined peptide-subphase interface as evidenced by the minimum in the autocorrelation function at that distance which is absent at lower pressures.
FIGURE 6.

(A) Fresnel-normalized x-ray reflectivity (circles) collected from monolayers of pure hbAP0 at different surface pressures, π, and curves drawn via box refinement. From top to bottom, π = 10, 20, 30, 40, 44, 58, and 62 mN/m. Datasets have been offset for clarity. (B) Patterson, or autocorrelation functions, computed from the inverse Fourier transform of the data in A. The data at lowest π produces a single, narrow minimum at low z that becomes broader as π reaches 40 mN/m and then develops a second minimum at large z at the highest π investigated, 58 (dotted) and 62 (bold) mN/m. (C) Profile structures for the hbAP0 monolayer at different π obtained by numerically integrating the profile gradients derived from box-refinement. At π = 10 mN/m, the profile structure contains a single maximum of 10 Å width at the air/water. At π = 20–30 mN/m, this maximum approximately doubles in thickness. At π ≈ 40 mN/m, the electron density distribution of the monolayer extends more deeply into the subphase to ∼z = −40 Å, but with a very broad peptide/subphase interface. At the highest π (bold), the profile has become a broad plateau of uniform density over −60 Å < z < −5 Å, consistent with all the helices of the ensemble oriented perpendicular to the interface. (D) Schematic showing pressure-induced orientation of hbAP0 protein indicated by both the autocorrelation functions and the absolute electron density profiles for the hbAP0 peptide monolayer at the air-water interface.
Fig. 6 C shows the monolayer electron density profiles derived from the normalized reflectivity data via the box-refinement method that requires no a priori assumptions to solve the well-known phase problem. At the surface pressure of 10 mN/m, the electron density profile contains a single maximum at the air-water interface consistent with the cross-section of a single _α_-helix oriented with the long axis lying in the plane of the air-water interface, i.e., the plane of the di-helix must also lie in the plane of the interface. At pressures of 20–30 mN/m, the plane of the di-helices rotates with the long axes of the helices remaining parallel to the plane of the interface, resulting in the maximum in the electron density profile of the monolayer approximately doubling in thickness. At a pressure of 40 or 44 mN/m, the electron density profile of the monolayer of hbAP0 extends more deeply into the subphase to ∼40 Å without a well-defined peptide-subphase interface (as consistent with the autocorrelation functions of the gradient profiles noted above), compared to the theoretical maximum of ∼55 Å expected for all of the helices oriented perpendicular to the surface. At the highest π (smallest area), the profile is now completely uniform over ∼55 Å between −60 Å < z < 0 Å, which shows clearly that all the helices of the ensemble are oriented perpendicular to the interface. The nature of this surface pressure-dependent orientational transition is shown schematically in Fig. 6 D.
DISCUSSION
Currently, little is known about the molecular interaction between anesthetic compounds and ion channels in the central nervous system. The design of water-soluble anesthetic-binding proteins pioneered by Johansson and co-workers have offered a powerful approach to the study of anesthetic-protein interactions. Our long-term goal is to engineer multidomain transmembrane proteins that mimic the functions of anesthetic-binding ion channels. In this work, we have designed and synthesized one amphiphilic anesthetic-binding protein. Hydrophobic amino acids were appended to the N-terminus of the water-soluble anesthetic-binding protein to facilitate insertion into lipid bilayers. This strategy had been successfully applied in the design of model integral membrane proteins capable of selectively binding redox cofactors (Discher et al., 2003) and their first realization in the peptide designated AP0 (Ye et al., 2004). It has been shown that choosing appropriate membrane sequences would not only facilitate the molecular assembly of the protein (Ye et al., 2004; B. Discher, D. Noy, S. Ye, C. Moser, J. Lear, J. Blasie, and P. Dutton, unpublished results), but also efficiently incorporate proteins into membrane media, such as lipid monolayers, detergent micelles and lipid vesicles (B. Discher, D. Noy, S. Ye, C. Moser, J. Lear, J. Blasie, and P. Dutton, unpublished results).
Like conventional membrane proteins, the driving force for the formation of a four-helix bundle is still not well understood; however, polar residues (i.e., glutamine) in the core region of the N-terminal hydrophobic sequence are considered to contribute to the assembly. This has been systematically investigated in de novo designed membrane proteins (Choma et al., 2000; Gratkowski et al., 2001; Lear et al., 1988), as well as observed in naturally occurring membrane proteins (Popot and Engelman, 2000).
The hydrophobic sequence in hbAP0 is derived from the LS2 synthetic ion channel (Lear et al., 1988), in which the three-heptad protein self-associates to form four-helix bundles in lipid membranes, resembling the ion channel of the acetylcholine receptor. The best structurally characterized example of a ligand-gated ion channel is the nAChR from Torpedo marmorata (Unwin, 1995), in which the transmembrane M2 sequence is the channel-lining segment. Although the pentameric construction of the pore in the AChR is changed to a tetrameric state in the LS2 synthetic ion channel, LS2 still exhibits ion permeability and a channel lifetime similar to the AChR when incorporated into lipid membrane (Lear et al., 1988). In our design, we replace the serine in the hydrophobic core with glutamine, as it is believed that Gln in the pore provides the narrow constriction associated with selectivity (Opella et al., 1999). This selectivity mechanism has also been observed in other ligand-gated ion channels such as the glycine receptors, which are also considered as a potential target for general anesthetics (Tang et al., 2002). In the future, we will examine the partitioning of the hbAP0 into lipid monolayers and bilayers, the ability of the protein to function as an ion channel, as well as the effect of anesthetic-binding on modulating the electrochemical properties.
According to the design, the Trp15 is at an _a_-position in the heptad repeat of a four-helix bundle, i.e., in the nonpolar core, and the fluorescence experiments indicate that the tryptophan is indeed located in a nonpolar environment. The calculated binding parameters are _K_d = 3.1 ± 0.6 mM, and _Q_max = 1.2 ± 0.1, implying that the fluorescence of all four tryptophan residues is quenched. Furthermore, the saturable manner of quenching can be interpreted to be a result of direct collisional interaction between halothane and hbAP0. However, the binding affinity decreases approximately fourfold compared to its water soluble counterpart A_α_2 (_K_d = 0.71 ± 0.04 mM, _Q_max = 1.06 ± 0.02).
Two competing effects could contribute to the change in affinity:
- Presumably, the cavity at the Ala19 position is the halothane binding site for both hbAP0 and A_α_2. The first three heptads of hbAP0 are copied from the water soluble region of A_α_2; however, the environment of the pockets in hbAP0 and A_α_2 are significantly different (Fig. 7). The interior residues adjacent to Ala19, layers V and VI, are all Trp or Leu in hbAP0, which are bulkier than the residues in the corresponding layers of A_α_2 by 167 Å3 and 47 Å3, respectively (Richards, 1974). This might decrease the cavity size, thereby making it somewhat less optimal.
- Trp is believed to introduce dipole-aromatic quadrupole interactions that would favor the halothane binding (Manderson and Johansson, 2002). Although the dominating structural features with regard to the change of the binding affinity need to be confirmed by, for example, a series of systematic mutations, the simple model membrane protein hbAP0 provides a promising system with which to probe the structural features of anesthetic binding sites in membrane proteins.
FIGURE 7.

Illustration of the hydrophobic core layers of hbAP0 (A) and A_α_2 (B). In hbAP0, all helices are parallel, whereas in A_α_2 helices I and IV are antiparallel to helices II and III. Only the side chains at heptad positions a and d are shown, and the amino acid position from the N-terminus is given. The potential halothane binding site is indicated with horizontal arrows.
At the air-water interface, the amphiphilic hbAP0 behaves as an integral membrane protein, the di-helices orienting perpendicular to the air-water interface at higher surface pressures and extending as essentially straight _α_-helices. We note here that analysis of grazing-incidence x-ray diffraction from Langmuir monolayers of the closely related amphiphilic peptide AP0 (Ye et al., 2004) mentioned in the Introduction indicates that it exists as a four-helix bundle at the air-water interface when similarly oriented at higher surface pressures with the helical axes perpendicular to the interface (J. Strzalka, S. Ye, I. Kuzmenko, T. Gog, and J. Blasie, unpublished results).
Note that GIXD data from Langmuir monolayers of the closely-related amphiphilic peptide AP0 (Ye et al., 2004) at higher surface pressures, where the helices are oriented perpendicular to the monolayer plane, show a broad maximum for momentum transfer parallel to the monolayer plane at q_xy ∼ 2_π/11 Å−1—which is absent in such data from the aqueous subphase itself and Langmuir monolayers of phospholipids on its surface. This diffraction arises from the interference between parallel helices, as is typical of GIXD from oriented multilayers of phospholipids containing integral membrane proteins whose transmembrane domains consist of a helical bundle. Modeling this GIXD data, and its inverse Fourier transform (namely the in-plane radial autocorrelation function, approximating the helices as straight rods of uniform electron density of ∼10 Å diameter) demonstrates that the di-helices aggregate to form four-helix bundles, which are rotationally disordered about the normal to the membrane plane with glass-like interbundle ordering in the monolayer plane. Other possible bundles arising from di-helices, e.g., two-helix, six-helix, etc., can be readily excluded on this basis because their respective GIXD and corresponding radial autocorrelation functions differ qualitatively well outside the signal/noise level from their experimental counterparts. The GIXD data from hbAP0 shows a similar maximum in position and shape at q_xy ∼ 2_π/10 Å−1.
Grazing-incidence x-ray diffraction data for hbAP0 (not shown) also at higher surface pressures is similar to that of AP0, suggesting that it too exists as a four-helix bundle under these conditions, namely in the absence of detergent which was employed to solubilize the peptide for the sedimentation equilibrium experiments. This makes it possible to further investigate protein partitioning into lipid monolayers and bilayers, as performed on other amphiphilic membrane proteins (B. Discher, D. Noy, S. Ye, C. Moser, J. Lear, J. Blasie, and P. Dutton, unpublished results). More importantly, this orientation at high surface pressure also provides a feasible way to investigate directly the position of the halothane binding site inside the amphiphilic four-helix bundle protein. Either nonresonance x-ray reflectivity, exploiting the five heavy halogen atoms of halothane, or resonance x-ray reflectivity (Strzalka et al., 2004a), exploiting the resonance scattering from halothane's bromine atom, can be utilized to determine the position of halothane within the profile structure of such a well-oriented protein monolayer (Ye et al., 2004). Polarized infrared spectroscopy can be used to probe the nature of the interface between the halothane ligand and the protein's individual amino acid residues via isotopic labeling. Furthermore, local conformational changes have been suggested by Gdn·HCl denaturation and terminal hydrogen exchange experiments (Johansson et al., 2000) upon halothane binding. By working with well-oriented protein monolayers composed of a series of peptides appropriately labeled with deuterated residues neighboring the halothane binding pocket of the protein, we can pursue neutron reflectivity to probe changes in the protein associated with halothane binding (Blasie and Timmins, 1999; Strzalka et al., 2004b).
Finally, the membrane protein design provides a successful template for future redesign, including positioning halothane binding cavities at different positions in the hydrophilic domain; for example, either proximal or distal to the ion-conducting hydrophobic domain or positioning the halothane binding cavity directly in the ion-conducting channel of the hydrophobic domain. The structural and dynamic consequences of anesthetic binding to such proteins in lipid monolayer or bilayer membranes are amenable to detailed structural analysis using surface spectroscopic and scattering approaches as well as functional consequences concerning the protein's ion channel activity, providing insights into how anesthetic complexation or membrane perturbation might alter protein function.
CONCLUSIONS
The design, assembly, and physical-chemical characterization of an amphiphilic four-helix bundle protein with specificity for volatile anesthetic binding has been described. The amphiphilicity allows for its unique vectorial orientation in a macroscopic ensemble at an interface between polar and nonpolar media, as provided by lipid monolayers and bilayers. This key advance provides a new laboratory for studying the nature of physicochemical interactions of the anesthetic ligands with the membrane protein via polarized surface spectroscopies and surface scattering techniques. It also serves as a model membrane protein for structural-functional studies of the effects of anesthetic ligands on its ion channel activity.
Acknowledgments
The authors thank Dror Noy and James Lear for assistance and valuable discussion of the ultracentrifugation experiment and data analysis; Ravi Pidikidi for help with peptide synthesis; Andrey Tronin for assistance with x-ray reflectivity data collection; Mike Sullivan for use of the support lab at beamline X9; Benjamin M. Ocko, Elaine Dimasi, and Scott Coburn for technical assistance at beamline X22-B at the National Synchrotron Light Source, Brookhaven National Laboratory; and Ivan Kuzmenko and Thomas Gog for technical assistance at Sector 9 at the Advanced Photon Source, Argonne National Laboratory.
This work was supported by the National Institutes of Health under GM55876. The National Synchrotron Light Source/Brookhaven National Laboratory and Advanced Photon Source/Argonne National Laboratory are supported by the U.S. Department of Energy.
References
- Als-Nielsen, J., and P. S. Pershan. 1983. Synchrotron x-ray-diffraction study of liquid surfaces. Nucl. Instrum. Methods. 208:545–548. [Google Scholar]
- Blasie, J. K., and P. Timmins. 1999. Neutron scattering in structural biology and biomolecular materials. MRS Bull. 24:40–47. [Google Scholar]
- Braslau, A., P. S. Pershan, G. Swislow, B. M. Ocko, and J. Als-Nielsen. 1988. Capillary waves on the surface of simple liquids measured by x-ray reflectivity. Phys. Rev. A. 38:2457–2470. [DOI] [PubMed] [Google Scholar]
- Cantor, R. S. 1997. The lateral pressure profile in membranes: a physical mechanism of general anesthesia. Biochemistry. 36:2339–2344. [DOI] [PubMed] [Google Scholar]
- Chiara, D. C., L. J. Dangott, R. G. Eckenhoff, and J. B. Cohen. 2003. Identification of nicotinic acetylcholine receptor amino acids photolabeled by the volatile anesthetic halothane. Biochemistry. 42:13457–13467. [DOI] [PubMed] [Google Scholar]
- Choma, C., H. Gratkowski, J. D. Lear, and W. F. DeGrado. 2000. Asparagine-mediated self-association of a model transmembrane helix. Nat. Struct. Biol. 7:161–166. [DOI] [PubMed] [Google Scholar]
- Discher, B. M., R. L. Koder, C. C. Moser, and P. L. Dutton. 2003. Hydrophilic to amphiphilic design in redox protein maquettes. Curr. Opin. Struct. Biol. 7:741–748. [DOI] [PubMed] [Google Scholar]
- Doyle, D. A., J. M. Cabral, R. A. Pfuetzner, A. L. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Eckenhoff, R. G., and J. S. Johansson. 1997. Molecular interactions between inhaled anesthetics and proteins. Pharmacol. Rev. 49:343–367. [PubMed] [Google Scholar]
- Franks, N. P., and W. R. Lieb. 1994. Molecular and cellular mechanisms of general-anesthesia. Nature. 367:607–614. [DOI] [PubMed] [Google Scholar]
- Franks, N. P., and W. R. Lieb. 1998. Which molecular targets are most relevant to general anaesthesia? Toxicol. Lett. 101:1–8. [DOI] [PubMed] [Google Scholar]
- Gratkowski, H., J. D. Lear, and W. F. DeGrado. 2001. Polar side chains drive the association of model transmembrane peptides. Proc. Natl. Acad. Sci. USA. 98:880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauet, N., F. Artzner, F. Boucher, C. Grabielle-Madelmont, I. Cloutier, G. Keller, P. Lesieur, D. Durand, and M. Paternostre. 2003. Interaction between artificial membranes and enflurane, a general volatile anesthetic: DPPC-enflurane interaction. Biophys. J. 84:3123–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm, C. A., P. Tippmannkrayer, H. Mohwald, J. Als-Nielsen, and K. Kjaer. 1991. Phases of phosphatidyl ethanolamine monolayers studied by synchrotron x-ray scattering. Biophys. J. 60:1457–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y. X., A. Lee, J. Y. Chen, V. Ruta, M. Cadene, B. T. Chait, and R. MacKinnon. 2003. X-ray structure of a voltage-dependent K+ channel. Nature. 423:33–41. [DOI] [PubMed] [Google Scholar]
- Johansson, J. S. 2001. Synthetic _α_-helical bundles as tools for defining the structural features of volatile general anesthetic binding sites on protein targets. Recent Res. Dev. Biophys. Chem. 2:37–52. [Google Scholar]
- Johansson, J. S. 2003. Noninactivating tandem pore domain potassium channels as attractive targets for general anesthetics. Anesth. Analg. 96:1248–1250. [DOI] [PubMed] [Google Scholar]
- Johansson, J. S., B. R. Gibney, F. Rabanal, K. S. Reddy, and P. L. Dutton. 1998. A designed cavity in the hydrophobic core of a four-_α_-helix bundle improves volatile anesthetic binding affinity. Biochemistry. 37:1421–1429. [DOI] [PubMed] [Google Scholar]
- Johansson, J. S., D. Scharf, L. A. Davies, K. S. Reddy, and R. G. Eckenhoff. 2000. A designed four-_α_-helix bundle that binds the volatile general anesthetic halothane with high affinity. Biophys. J. 78:982–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson, J. S., and R. G. Eckenhoff. 1996. Minimum structural requirement for an inhalational anesthetic binding site on a protein target. Biochim. Biophys. Acta. 1290:63–68. [DOI] [PubMed] [Google Scholar]
- Johansson, J. S., R. G. Eckenhoff, and P. L. Dutton. 1995. Binding of halothane to serum albumin demonstrated by using tryptophan fluorescence. Anesthesiology. 83:316–324. [DOI] [PubMed] [Google Scholar]
- Johansson, J. S., F. Rabanal, and P. L. Dutton. 1996. Binding of the volatile anesthetic halothane to the hydrophobic core of a tetra-_α_-helix-bundle protein. J. Pharmacol. Exp. Ther. 279:56–61. [PubMed] [Google Scholar]
- Krasowski, M. D., and N. L. Harrison. 1999. General anaesthetic actions on ligand-gated ion channels. Cell. Mol. Life Sci. 55:1278–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lear, J. D., Z. R. Wasserman, and W. F. Degrado. 1988. Synthetic amphiphilic peptide models for protein ion channels. Science. 240:1177–1181. [DOI] [PubMed] [Google Scholar]
- Lösche, M., M. Piepenstock, A. Diederich, T. Grunewald, K. Kjaer, and D. Vaknin. 1993. Influence of surface-chemistry on the structural organization of monomolecular protein layers adsorbed to functionalized aqueous interfaces. Biophys. J. 65:2160–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, P. Z., and R. L. Baldwin. 1997. Mechanism of helix induction by trifluoroethanol: a framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry. 36:8413–8421. [DOI] [PubMed] [Google Scholar]
- Manderson, G. A., and J. S. Johansson. 2002. Role of aromatic side chains in the binding of volatile general anesthetics to a four-_α_-helix bundle. Biochemistry. 41:4080–4087. [DOI] [PubMed] [Google Scholar]
- Marvin, J. S., E. E. Corcoran, N. A. Hattangadi, J. V. Zhang, S. A. Gere, and H. W. Hellinga. 1997. The rational design of allosteric interactions in a monomeric protein and its applications to the construction of biosensors. Proc. Natl. Acad. Sci. USA. 94:4366–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy, D., J. R. Calhoun, and J. D. Lear. 2003. Direct analysis of protein sedimentation equilibrium in detergent solutions without density matching. Anal. Biochem. 320:185–192. [DOI] [PubMed] [Google Scholar]
- Opella, S. J., F. M. Marassi, J. J. Gesell, A. P. Valente, Y. Kim, M. Oblatt-Montal, and M. Montal. 1999. Structures of the M2 channel-lining segments from nicotinic acetylcholine and NMDA receptors by NMR spectroscopy. Nat. Struct. Biol. 6:374–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popot, J. L., and D. M. Engelman. 2000. Helical membrane protein folding, stability, and evolution. Annu. Rev. Biochem. 69:881–922. [DOI] [PubMed] [Google Scholar]
- Richards, F. M. 1974. The interpretation of protein structures: total volume, group volume distributions and packing density. J. Mol. Biol. 82:1–14. [DOI] [PubMed] [Google Scholar]
- Sixma, T. K., and A. B. Smit. 2003. Acetylcholine binding protein (AChBP): a secreted glial protein that provides a high-resolution model for the extracellular domain of pentameric ligand-gated ion channels. Annu. Rev. Biophys. Biomembr. 32:311–334. [DOI] [PubMed] [Google Scholar]
- Skalicky, J. J., B. R. Gibney, F. Rabanal, R. J. B. Urbauer, P. L. Dutton, and A. J. Wand. 1999. Solution structure of a designed four-_α_-helix bundle maquette scaffold. J. Am. Chem. Soc. 121:4941–4951. [Google Scholar]
- Spencer, R. H., and D. C. Rees. 2002. The _α_-helix and the organization and gating of channels. Annu. Rev. Biophys. Biomembr. 31:207–233. [DOI] [PubMed] [Google Scholar]
- Strzalka, J. W., X. Chen, C. C. Moser, P. L. Dutton, B. M. Ocko, and J. K. Blasie. 2000. X-ray scattering studies of maquette peptide monolayers. 1. Reflectivity and grazing incidence diffraction at the air/water interface. Langmuir. 16:10404–10418. [Google Scholar]
- Strzalka, J., E. DiMasi, I. Kuzmenko, T. Gog, and J. K. Blasie. 2004a. Resonant x-ray reflectivity from a bromine-labeled fatty acid Langmuir monolayer. Phys. Rev. E. In press. [DOI] [PubMed]
- Strzalka, J., B. R. Gibney, S. Satija, and J. K. Blasie. 2004b. Specular neutron reflectivity and the structure of artificial protein maquettes vectorially oriented at interfaces. Phys. Rev. E. In press. [DOI] [PubMed]
- Tang, P., P. K. Mandal, and Y. Xu. 2002. NMR structures of the second transmembrane domain of the human glycine receptor _α_1 subunit: model of pore architecture and channel gating. Biophys. J. 83:252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian, S. A., and L. K. Tamm. 2000. Secondary structure, orientation, oligomerization, and lipid interactions of the transmembrane domain of influenza hemagglutinin. Biochemistry. 39:496–507. [DOI] [PubMed] [Google Scholar]
- Ulmschneider, M. B., and M. S. P. Sansom. 2001. Amino acid distributions in integral membrane protein structures. BBA Biomembr. 1512:1–14. [DOI] [PubMed] [Google Scholar]
- Unwin, N. 1995. Acetylcholine-receptor channel imaged in the open state. Nature. 373:37–43. [DOI] [PubMed] [Google Scholar]
- Xu, Y., T. Seto, P. Tang, and L. Firestone. 2000. NMR study of volatile anesthetic binding to nicotinic acetylcholine receptors. Biophys. J. 78:746–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, S., J. W. Strzalka, B. M. Discher, D. Noy, S. Zheng, P. L. Dutton, and J. K. Blasie. 2004. Amphiphilic 4-helix bundles designed for biomolecular materials applications. Langmuir. 20:5897–5904. [DOI] [PubMed] [Google Scholar]
- Zheng, S., J. Strzalka, D. H. Jones, S. J. Opella, and J. K. Blasie. 2003. Comparative structural studies of VPU peptides in phospholipid monolayers by x-ray scattering. Biophys. J. 84:2393–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]