Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome (original) (raw)
Abstract
The E2F family of transcription factors regulates basic cellular processes. Here, we take an unbiased approach towards identifying E2F1 target genes by examining localization of E2F1-binding sites using high-density oligonucleotide tiling arrays. To begin, we developed a statistically-based methodology for analysis of ChIP-chip data obtained from arrays that represent 30 Mb of the human genome. Using this methodology, we identified regions bound by E2F1, MYC, and RNA Polymerase II (POLR2A). We found a large number of binding sites for all three factors; extrapolation suggests there may be ∼20,000–30,000 E2F1- and MYC-binding sites and ∼12,000–17,000 active promoters in HeLa cells. In contrast to our results for MYC, we find that the majority of E2F1-binding sites (>80%) are located in core promoters and that 50% of the sites overlap transcription starts. Only a small fraction of E2F1 sites possess the canonical binding motif. Surprisingly, we found that ∼30% of genes in the 30-Mb region possessed an E2F1 binding site in a core promoter and E2F1 was bound near to 83% of POLR2A-bound sites. To determine if these results were representative of the entire human genome, we performed ChIP-chip analyses of ∼24,000 promoters and confirmed that greater than 20% of the promoters were bound by E2F1. Our results suggest that E2F1 is recruited to promoters via a method distinct from recognition of the known consensus site and point toward a new understanding of E2F1 as a factor that contributes to the regulation of a large fraction of human genes.
Members of the E2F family of transcription factors are present in all cell types and are conserved from plants to animals. There are eight known members of the E2F family in mammals. In general, E2Fs 1, 2, and 3 are classified as activators; E2Fs 4, 5, and 6 are classified as repressors; and E2F7 and E2F8 have not yet been studied well enough to be appropriately classified (Nevins 1998; Attwooll et al. 2004). However, it must be considered that the actual role that a specific E2F plays at a given promoter is defined by interactions with pocket proteins (i.e., Rb, p107, and p130) and histone-modifying complexes (Brehm et al. 1998; Dyson 1998; Luo et al. 1998; Magnaghi-Jaulin et al. 1998; Nielsen et al. 2001; Frolov and Dyson 2004). Because the first E2F target genes were shown to regulate basic cellular processes such as cell cycle progression and DNA repair, it was believed that the E2Fs played a critical, yet highly specific, role in cell biology. However, these first analyses of E2F target genes were highly biased toward certain classes of genes. Clearly, to understand the full role of E2F in the cell, an unbiased approach is required. Toward this goal, large sets of genes regulated by the E2F family were first identified using over- or underexpression of a particular family member, coupled with gene expression microarray analyses. Such studies suggested that perhaps 7% of the human genes were influenced by the E2F family (Muller et al. 2001). Interpretation of these studies, however, is complicated by the fact that the set of identified genes may include genes whose mRNA levels were simply responding to alterations in signal transduction cascades (leading to potential identification of indirect targets) and by probable functional redundancy created by the existence of multiple E2F gene family members. As a second approach to identifying sets of E2F target genes, two groups have used ChIP-chip analyses to identify E2F1-binding sites in either core promoter regions (Ren et al. 2002; Balciunaite et al. 2005) or in CpG islands (Weinmann and Farnham 2002; Oberley et al. 2003; Wells et al. 2003). Although these ChIP-chip experiments allowed a broader analysis of the binding patterns of E2F family members than previous one-gene-at-a-time approaches, they were restricted by the type of arrays used. The core promoter arrays, which allowed the analysis of 700 bp upstream and 200 bp downstream of the transcription start site of 13,000 human genes, provided an estimate that ∼2% of human promoters are bound by the E2Fs (Ren et al. 2002; Balciunaite et al. 2005). However, if E2F plays a role in transcriptional regulation that includes binding outside of core promoter regions, many binding sites (and thus many target genes) may have been missed. An alternative approach used microarrays representing CpG islands, which are present in both core promoter regions and other types of regulatory regions, allowing analysis of a set of regulatory regions that is probably more inclusive than core promoters. These microarrays again yielded estimates that ∼2% of human promoters are bound by E2Fs (Weinmann and Farnham 2002; Oberley et al. 2003; Wells et al. 2003). However, these arrays would, of course, be unable to identify binding sites in regulatory regions that were not CpG islands. Recent studies have suggested that binding sites for transcription factors are not necessarily near the start sites of genes or in CpG islands. For example, using an oligonucleotide tiling array that encompasses chromosomes 21 and 22, Cawley et al. (2004) found that only 18% of the MYC-binding sites are within 1 kb of a 5′-exon and only 24% are within 1 kb of a CpG island. If binding sites for the E2F family members are similarly located, then perhaps the majority of E2F target genes were missed using core promoter and/or CpG arrays.
In this study, we have used an unbiased approach to analyze the binding sites for E2F1 in the ENCODE regions. There are 44 ENCODE regions representing different sections of the human genome, each section spanning from 500 kb to 1.9 Mb, with the summed length being 30 Mb (1% of the human genome). A subset of the regions were selected manually because of the presence of extensively characterized genes or the availability of a large amount of comparative sequence data. The remaining targets were chosen at random using an algorithm that ensured that the complete set of targets represented the range of gene content and level of non-exonic conservation (relative to mouse) found in the human genome (see ENCODE Project Consortium 2004). Importantly, our analysis of 30 Mb of the human genome allows us to determine what percentage of E2F1-binding sites falls near core promoter regions, near CpG islands, or outside of either of these two types of regions. Unlike all of the previous ChIP-chip studies of E2Fs that used spotted microarrays, we have used high-density oligonucleotide arrays for our studies. The identification of binding sites on such arrays is not trivial, and therefore we began by developing and testing a statistically based methodology for analysis of ChIP-chip data on high-density tiling arrays. Using this methodology, we found a large number of E2F1-binding sites. As described below, our results suggest a possible new role for E2F1 as a transcription factor that may contribute to the regulation of a very large percentage of the genome.
Results
A statistical model for peak finding for ChIP-chip data obtained using high-density oligonucleotide arrays
We performed three independent ChIP assays (with ∼1 × 107 HeLa cells per immunoprecipitation) using a monoclonal antibody specific for E2F1 and a negative control IgG antibody; each of the three replicates (A, B, and C) represents a ChIP sample from cells that were grown, cross-linked, and assayed independently of the other two samples. As a positive control for the ChIP assays, primers specific for the MCM4 promoter (a known E2F target gene) were used. We found that, as expected, the MCM4 promoter was enriched in the E2F1 samples, but not in the IgG samples (Fig. 1A). As a negative control, we showed that a region of chromosome 21 was not enriched in either the E2F1 or in the IgG ChIP samples. Next, the E2F1 ChIP samples, the IgG ChIP samples, and a portion of total input DNA were blunt-ended, ligated to a unidirectional linker, and amplified to generate enough DNA to probe the microarrays. Before proceeding to the microarray step, the amplicons were analyzed to confirm that the PCR amplification step had retained the specificity of the starting ChIP samples (Fig. 1B); that is, the E2F target promoter was enriched in the E2F1 amplicons, but not in the IgG amplicons, whereas the negative control was not enriched in either amplicon. Similar control PCR reactions were performed for all ChIP samples used for ChIP-chip analysis in this study.
Figure 1.

Confirmation of ChIP and amplicons. Primers specific for the MCM4 promoter and for a region of chromosome 21 were used as positive and negative controls, respectively, for analysis of (A) ChIP or (B) amplicon samples that had been immunoprecipitated with an E2F1 antibody or a nonspecific rabbit IgG.
Having confirmed that the amplicons were good representations of the starting ChIP samples, the experimental E2F1 (or IgG) amplicons were then labeled with Cy5 dye, and the total input amplicons were labeled with Cy3 dye and cohybridized to high-density oligonucleotide tiling arrays representing the 44 ENCODE regions (ENCODE Project Consortium 2004). The nonrepetitive portions of the ENCODE regions were tiled at a density of one 50-mer every 38 bp, leading to a final total of ∼380,000 50-mers on the array. Previous investigations of microarrays have emphasized that, because of several factors, there is often significant variation in the range and distribution of amplitudes between arrays, even for biological replicates (Stekel 2003). In accord with this expectation, simple inspection of our data (Supplemental Fig. S1) also showed significant variation between arrays that were hybridized with samples from three independent experiments, with some arrays having more apparent “noise” than other arrays. In addition, we found that peaks corresponding to confirmed binding sites varied greatly in waveform, amplitude, and size (see Methods). Therefore, our first task was to develop an automated method by which peaks could be identified and selected for further consideration.
We sought an approach to peak detection that made minimal assumptions about the shape and amplitude of peaks representing true binding sites. The binding sites should appear in the data as runs of consecutive points (each point representing a 50-mer) with enhanced amplitude. Hence, a simple approach is to set a threshold for acceptability and then look for peaks of a certain width, as has previously been used (Kim et al. 2005). However, this leaves open the difficult question of setting an appropriate combination of threshold and width for each array. Clearly, a threshold requirement for an array that shows strong signals (e.g., array A of Supplemental Fig. S1A) should be very different than for an array that shows weaker signals (e.g., array B of Supplemental Fig. S1A). Therefore, for a threshold we use a percentile for each array (95th and 98th percentile) of log2 oligomer ratios. Use of this percentile “normalizes” the threshold values for each array to reflect both the amplitudes and distribution of signal in the arrays and, furthermore, presents a consistent, nonarbitrary way to set thresholds for different arrays. To determine the appropriate “run length” (or width) for a valid peak, we analogized this problem to a known statistical model in which the _P_-value for any run length can be calculated (Supplemental Figs. S1 and S2; see Methods for detailed explanation). Following this approach, for each threshold (95th percentile and 98th percentile), we use P < 0.0001 for a very stringent _P_-value (which requires six consecutive points above the 98th percentile or eight consecutive points above the 95th percentile) and P < 0.05 for a less stringent _P_-value cutoff (which requires four consecutive points above the 98th percentile or five consecutive points above the 95th percentile). Hence, our four conditions, in decreasing stringency, are 98th percentile threshold and P < 0.0001; 95th percentile threshold and P < 0.0001; 98th percentile threshold and P < 0.05; 95th percentile and P < 0.05. For clarity, we will refer to these as L1–L4, with L1 being the most stringent. It is important to note that as we lower stringency, we keep adding peaks to the set. So (with a few very rare exceptions—see Supplemental Methods), every L1 peak is present at L2; every L2 peak is present at L3, and so on. Hence, we distinguish “the set of L2 peaks” (meaning every peak present in the L2 set) from “peaks that first appear at L2” (which is “the set of L2 peaks” minus “the set of L1 peaks”).
Figure 2 displays the results of peak-calling on a single array trace (array C from Supplemental Fig. S1A). As stringency is decreased from L1 to L3, we see a small increase in the number of detected peaks and in the apparent size of the peaks (as shown by the width of the vertical lines). However, at L4, we see a large jump in the number of peaks. Similar results were seen in other ENCODE regions and with other array samples (e.g., Supplemental Table S2). An obvious concern, which is addressed below, is that lowering the stringency results in an increase in false positives in L4. Because a true binding site should be detected in multiple ChIP assays, we performed and analyzed three independent ChIP-chip arrays. The most common approach to combining array data from biological replicates has been to apply normalization methods to the separate data sets, combine all data into a single set, and then make predictions based on this single combined set (Fig. 3A, “combine-first”). While our peak prediction approach can be applied to a combined data set produced by this strategy, we chose to first predict peak locations for each array, then combine predictions by defining a binding site as a region that is called a peak on at least two of the three arrays (Fig. 3B, “peak-first”); rationale and details of this procedure are in the Supplemental Methods. This procedure is conservative in that it tends to produce long peaks with the prediction that there is at least one binding site in the area and may underestimate the number of binding sites if two or more sites are very closely spaced. However, given that the resolution of the ChIP experiments will not allow one to confirm the presence of two closely spaced binding sites, we chose the more conservative approach of calling these as one peak. We have analyzed our array data using the “peak-first” strategy and the more widely used “combine-first strategy” (Fig. 3D; see Methods for combination details). For each stringency level (L1–L4), our “peak-first” approach yielded fewer peaks, and the “peak-first” set was essentially a subset of the “combine-first set.” Hence, in this limited analysis, our “peak-first” approach appears to be more conservative than the “combine-first” approach, but more extensive testing and analysis using multiple data sets from multiple factors will be necessary to more clearly evaluate this issue.
Figure 2.

Peak-calling on a single array trace. The hybridization data for a portion of ENr232 are shown. Above the trace, four different levels of peak-calling stringency are indicated (L1–L4; see Results for explanation of levels). As the stringency is lowered to L4, more peaks are called.
Figure 3.

Combining array data sets. (A) A schematic illustration of the “combine-first” approach. (B) A schematic illustration of the “peak-first” approach. (C) An example of peak-calling and combination (at L1 for each array) on the same region from ENr223 from three independent arrays. The maximum bar height (log2) is 5.44 for “array A”; 2.10 for “array B”; and 4.18 for “array C”. The final derived peak is shown by the black rectangle on the top line and extends from chr6:74156406 to 74157748. All three arrays contributed significantly to the set of peaks; for the total set of 205 L1 peaks, 100 were found in all three arrays. (D) Comparison of binding site predictions (at L1) for peak-first and combine-first approaches for HeLa E2F1 data. Note that there are fewer “peak-first” predictions and that nearly all “peak-first” predictions (96%) are in the “combine-first” set. See Results and Supplemental Methods for additional description.
Analysis of peak predictions
A concern in ChIP experiments is that enriched DNA may be simply the result of nonspecific antibody binding, which is commonly tested using IgG as a control antibody. Hence, we analyzed the peaks produced from triplicate IgG ChIP samples from the same cross-linked cells as those used for the E2F1 ChIP experiments. For a rigorous test, we compared the entire set of the lowest stringency IgG peaks (L4), which led to the greatest number of peaks, to the set of genuine E2F1 peaks at L1–L4. In each case, we found that only ∼1% of predicted E2F1 binding sites overlapped IgG peaks. Hence, the peaks we have identified as E2F1-binding sites are not false positives because of nonspecific antibody/DNA interactions.
As another measure of analysis of our peak predictions, we examined E2F1 ChIP samples from another cell type, MCF7 breast cancer cells. Again, three biological replicates were analyzed, peaks were called, and a set of predicted binding sites appearing in at least two of three arrays was determined. We found that 75% of the MCF7 E2F1-binding sites (111/148; L1) were also identified as binding sites in HeLa cells (Fig. 4). Hence, a completely separate set of results using a second cell type lends support to our predictions.
Figure 4.

Most E2F1-binding sites are found in both HeLa and MCF7 cells. (A) Raw array data (one array for each cell type shown) and predicted E2F1-binding sites (L1) from ENr324 (chrX) in HeLa and MCF7 cells. Predicted sites (indicated as HeLa TFBS and MCF7 TFBS) are the results of using our standard analysis approach on three arrays from each cell type (see Results). (B) Pie chart showing that 75% of MCF7 sites overlap a HeLa site. (TFBS) Transcription factor binding site.
As a more direct test of our predictions, we performed standard PCR analyses of the predicted peaks from the E2F1 set. For these confirmations, we used amplicons from the same samples that were hybridized to the arrays (Supplemental Table S1). We examined predictions in both HeLa and MCF7 cells; in a subset of cases, the same peak was examined in both cell types. After analyzing 82 individual array predictions and 29 peak predictions (20 distinct peaks; nine peaks were examined in both HeLa and MCF7 cells), we have, in the L1 set, a 95% confirmation rate of individual array predictions and 100% confirmation rate of peak predictions. Hence, this L1 level appears to provide excellent specificity of predictions. Sparser testing of peaks first appearing at the L2 and L3 levels provided support that peaks appearing at these lower stringencies are also genuine sites. In contrast, only 1/5 of peaks first appearing at L4 were confirmed, and on a single array level, only 6/11 of single array predictions first appearing at L4 were confirmed. Hence, it appears that the array and peak predictions at L1 are very highly reliable; limited evidence suggests peaks and array predictions first appearing at L2 and L3 are reliable, and the evidence points toward the additional peaks at L4 being mostly artifactual.
In summary, we produce three lines of evidence to support our L1 peak predictions: (1) the peak predictions have minimal overlap with a set of nonspecific (IgG) binding events, (2) the predictions are verified at a high rate by PCR, and (3) the peak predictions are largely held in common in a second cell type. More limited but similar results apply to L2 and L3, but at L4, these tests fail. Hence, we suggest that the parameters at L1 provide a very high specificity (very low false positives, but some false negatives), L2 and L3 provide good specificity but greater sensitivity (some false positives but fewer false negatives), but that L4 is not appropriate (too many false positives).
Genomic distribution of E2F1-binding sites
Having established a reliable approach to E2F1-binding site identification, we next investigated general themes revealed by this unbiased survey. Unless otherwise indicated, we used the conservative L1 set of E2F1-binding sites for all analyses. However, our conclusions drawn using L1 were qualitatively and quantitatively quite similar to those when the L2 or L3 set was used (Supplemental Table S4). For E2F1, we found a range of 0–25 E2F1-binding sites per ENCODE region at L1, 0–27 at L2, 0–38 at L3, and 1–69 at L4 (Supplemental Table S2). If we examine the randomly chosen ENCODE regions alone (which are each the same length, 500 kb), we find 0–11, 0–13, 0–18, and 0–41 peaks per region at L1–L4, respectively. Note that, for most regions, particularly in the random set, (1) there is a small increase in the number of binding sites from L1 to L2; (2) L2 and L3 have quite similar numbers of binding sites; and (3) there is a large jump in the number of predicted binding sites from L3 to L4. We noted that the great majority of E2F1-binding sites were spaced far from each other. However, occasionally, there were small chains (typically two to three peaks/chain) of binding sites spaced <1 kb from each other. We considered it likely that these small, relatively closely spaced chains of E2F-binding sites were probably in the same regulatory region. Others (Cawley et al. 2004) have also clustered binding sites that are separated by <1 kb. Hence, to provide an estimate of the number of regulatory regions, we considered E2F1-binding sites that were <1 kb from each other to be in the same regulatory region. For L1, the 205 total E2F1-binding sites produce 170 separate regulatory regions by this criterion; the number of separate regulatory regions at L2, L3, and L4 is 237, 253, and 460, respectively. Using these data and the fact that the ENCODE regions represent 1% of the human genome, we roughly and approximately estimate that there are between 205 × 100 = 20,500 (L1) and 337 × 100 = 33,700 (L3) E2F1-binding sites in the human genome.
We performed the same analysis of MYC-binding sites by performing triplicate ChIP-chip experiments and calling peaks as described in Figure 3. Interestingly, we found a similar number of MYC- and E2F1-binding sites in the ENCODE regions (Supplemental Table S2). For example, at L1 we found 172 (hg17; see Methods) MYC-binding sites (as compared to 205 E2F1-binding sites), at L2 we found 332, and at L3 we found 354. Extrapolation to the entire human genome suggests that there are ∼17,000–33,000 MYC-binding sites. Although we found similar numbers of MYC- and E2F1-binding sites, these sites clustered somewhat differently in the different ENCODE regions (Supplemental Table S2), suggesting that a different set of genes was regulated by the two factors (see below for a more detailed analysis of this issue).
We sought to examine whether our experimentally determined E2F1-binding sites possess the well-known strong consensus motif for E2F1 sites, which is TTTSSCGC with S = C or G (Tao et al. 1997). To do so, we mapped the location of TTTSSCGC on both the forward and reverse strands for all the ENCODE regions, producing a total of 511 instances of this octamer. However, only 25/205 of high-stringency L1 E2F1-binding sites contained the consensus motif (Table 1; see also Supplemental Fig. S4), and there were only a total of 27 instances of this motif in these 205 binding sites (two of the experimentally determined binding sites had two TTTSSCGC hits each). Clearly, the small overlap between E2F1-binding sites and TTTSSCGC locations indicates that using the presence of TTTSSCGC as a marker for identifying E2F1-binding sites would not be appropriate. It is possible that a larger percentage of experimentally and computationally determined E2F1-binding sites would overlap if the motif were allowed to contain a 1-bp mismatch from the E2F consensus. However, when the location of all the 1-bp mismatch sites was mapped, we found that there are 37,750 such motifs in the ENCODE regions, or >1 per kilobase. Clearly, this number of sites is too large to be of predictive value. Others have observed that E2F1 can bind to sites that resemble the consensus but are mismatched in one of the three Ts (Tao et al. 1997), and a structural study (Zheng et al. 1999) has emphasized the importance of the central SSCGC for E2F binding. Therefore, it may be more biologically relevant to allow a mismatch only in the T stretch. If the single mismatch from consensus is allowed to vary only in the three Ts, there are still 5450 sites in the ENCODE region, or ∼1 site per 5.5 kb. Again, the high frequency of this motif obviates any predictive value. These results are in accord with recent studies emphasizing that the presence of a canonical motif is a poor guide to finding actual binding sites and that many experimentally determined binding sites lack canonical motifs (Cawley et al. 2004; Wasserman and Sandelin 2004). CpG arrays have previously been successfully used to find E2F-binding sites, suggesting that using CpG islands as a criterion for E2F1-binding sites might be appropriate. However, it has never been examined if a significant portion of E2F1 sites lie outside of CpG islands. We found that 27% of the CpG islands in the ENCODE region were bound by E2F1 and the great majority (>80%) of E2F1-binding sites overlapped CpG islands (Table 1). Although a significant fraction of E2F1-binding sites are outside of these regions, CpG islands clearly have better predictive value than do E2F consensus motifs.
Table 1.
Characteristics of E2F1-binding sites

Previous work on E2F1 has revealed binding sites near transcription start sites (Kel et al. 2001). However, these previous studies only examined core promoter regions, and therefore the frequency with which E2F1 bound near a start site could not be estimated. By analyzing 30 Mb of the human genome, we can now determine the preferred location of E2F1-binding sites with respect to transcription units. We used the gene annotations provided by the GENCODE project (GENCODE; http://genome.imim.es/gencode/), a subproject within ENCODE that has been investigating genes and transcripts within the ENCODE region in detail. As recommended by the GENCODE project, for a high-confidence set of protein coding genes, we used annotations categorized as “Known” and “Novel_CDS,” which includes all confirmed protein-coding genes (we refer to as “protein-coding set”). In addition, we examined a more loose set comprising every category except “Artifact” (we refer to as “NotArtifact set”). We display data and quantitation using the “protein-coding set” (except for analysis of E2F1 with respect to non-coding transcripts); however, results using either set were very similar. Also, for comparison, we performed the same analyses with the “Known Genes” set (Known Genes, March 2004 set; http://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=61274972&g=knownGene) and derived very similar results (data not shown). To begin, we quantified the percentage of experimentally determined E2F1-binding sites within various distances of a transcription start site and, for comparison, of 3′ transcription end sites (Table 2; see also Supplemental Table S4). We found that 51% of the L1 E2F1-binding sites were localized at the transcription start site, while only 5% overlapped the 3′-end of the transcript. Furthermore, 82% of E2F1-binding sites were found within 1 kb of the transcription start site. A minority (8%) of E2F1-binding sites were found >10 kb from the transcription start site of known genes. It is possible that many of the E2F1 sites that appear to be far from core promoter regions are, in fact, near start sites of as yet undiscovered genes or transcripts. Interestingly, a subpopulation of E2F1 sites is close to the 5′-ends of novel and/or non-coding transcripts (derived using the “NotArtifact” set of GENCODE annotations) (Supplemental Table S3).
Table 2.
Comparative statistics for E2F1-, POLII-, and MYC-binding sites
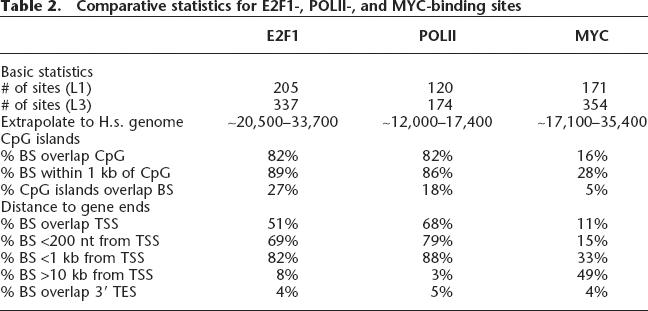
As a comparison to the E2F1 analysis, we also analyzed the location of the MYC-binding sites (Table 2). We found that only 11% of the MYC-binding sites overlap a transcription start site and only 33% are within 1 kb of the start site. This binding pattern is very different from that of E2F1. Although several groups have published ChIP-chip studies examining MYC target promoters in mammalian and Drosophila cells using promoter or cDNA-based arrays (Fernandez et al. 2003; Li et al. 2003; Mao et al. 2003; Orian et al. 2003), only one group has used genomic tiling arrays, which allow an unbiased location analysis of MYC binding to be performed. Our results are similar to this previous study that reported that only 18% of the MYC-binding sites on chromosomes 21 and 22 were within 1 kb of a 5′-exon (Cawley et al. 2004). In addition, while we found that most E2F1 sites are near or overlap CpG islands (82% overlap; 89% within 1 kb), only 16% of MYC sites that we identified overlapped CpG islands, and only ∼28% of MYC sites were within 1 kb of a CpG island. In accord with these findings, Cawley et al. (2004) found ∼24% of MYC sites within 1 kb of a CpG island.
E2F1 is recruited to a large fraction of human genes
A comparison of the position of the E2F1-binding sites to the location of known genes revealed that many of the genes in the ENCODE regions have E2F1 bound near the 5′-end. To quantify this relationship throughout all the ENCODE regions, we considered an E2F-binding site that is located within 1 kb of a transcription start site to be in the core promoter region of that gene. Here, it is important to note that many genes have several transcription start sites because of alternative first exon usage; hence, if a gene had an E2F1-binding site within the core promoter region of at least one start site, we considered that gene to be an E2F1 target gene. Strikingly, a large fraction (∼35%) of genes in the ENCODE region possessed an E2F1-binding site within the promoter region of at least one transcript (Table 1). Use of the “Known Genes” set of transcripts (Known Genes, March 2004 set; http://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=61274972&g=knownGene), which includes a smaller set of transcripts (but a larger number of predicted genes), yielded a similar percentage (28.3%). Using the GENCODE analysis and extrapolating to the entire human genome, we arrive at a rough estimate that 0.35 × 25,000 = ∼8750 human genes may be regulated by E2F1. Genes commonly produce several transcripts. We were interested in determining the fraction of this set of total possible transcripts that may be regulated by E2F1. Annotation of all possible transcripts from a gene is difficult for many reasons, including the fact that there are no current highly reliable computational models predicting all possible transcripts and the fact that some transcripts may be restricted to rare, small subsets of cells and/or only produced in rare cellular states. However, the GENCODE project provides an estimate of the number of transcripts for the ENCODE regions. Hence, we asked what fraction of this set of total possible transcripts might be regulated by E2F1, using the identification of an E2F1-binding site within 1 kb of the 5′-end of a transcript to indicate possible regulation. We found that ∼26% of all transcripts in the ENCODE regions were possibly regulated by E2F1 (Table 1). Use of the “Known Genes” set of transcripts (Known Genes, March 2004 set; http://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=61274972&g=knownGene) yielded a very similar percentage (26.9%).
The ENCODE regions represent only a small (1%) portion of the human genome and were not chosen to reflect the functional diversity of human genes. Hence, extrapolation from the ENCODE regions to the entire genome may not be appropriate. To directly test whether E2F1 is bound to a large fraction of human promoters, we used ChIP-chip to examine E2F1 localization in an array design containing from −1300 to +200 surrounding the mapped 5′-ends of ∼24,000 human promoters, each represented by 15 probes with an average probe spacing of 100 nt. Because our ENCODE array data indicate that E2F1 often binds very close to the transcription start site (Table 2), this array design is appropriate for investigating E2F1 binding. It is important to note that these ∼24,000 5′-ends do not necessarily reflect all the promoters in the genome. In addition, owing to lack of definitive information on exact locations of actual transcription start sites (and thus the promoter regions) for most genes, the 1500 bp on the array may actually represent sequences farther 5′ or 3′ than −1300 to +200. However, this array design does provide a large-scale data set for analysis of E2F1 binding. As described above, data from the ENCODE arrays indicate that E2F1 could possibly regulate ∼26% of transcripts (see above). Hence, simple extrapolation from our ENCODE data would imply that ∼6200 promoters of the ∼24,000 would be bound.
We sought to develop a nonarbitrary approach to calling a positive versus negative target promoter. Our analysis approach used for the ENCODE arrays (described above) is based on a model in which actual oligomers in genuine E2F1-binding sites are only a very small fraction of total oligomers. For example, on the ENCODE arrays (which tile coding sequence, introns, and intergenic regions), we find that ∼1 in every 200–300 oligomers is bound by E2F1. For transcription factors that bind a relatively small fraction of the promoters in the promoter array design, our analysis approach developed for ENCODE arrays is appropriate and leads to verifiable predictions (M.C. Bieda, S.R. Krig, H. O’Geen, and P.J. Farnham, unpubl.). However, if E2F1 binds to several thousand promoters in the promoter array, potentially one in every 12 points (or more) would be positive (see Supplemental Methods for calculation), indicating that another analysis approach is necessary. Therefore, to guide our analysis of the promoter array data, we examined E2F1 target promoters identified from the ENCODE arrays that also appeared on the human promoter array. We found a set of 25 regions on the promoter array that were at least 60% covered by an E2F1 hit from the ENCODE arrays (L1 stringency). The median values (of the normalized log2 ratios of antibody/total for all 15 oligomers for each promoter) for these 25 promoters varied from 0.025 to 2.5, but there was a sharp division in that 20% of the hits were <0.33 and the remaining 80% of hits were >0.707. Hence, we used a median value of 0.707 as a cutoff value. Using this cutoff value, we found that 6183 promoters were bound by E2F1 in HeLa cells. We performed PCR analysis of 10 randomly selected promoters from this set of 6183, and all confirmed to be bound by E2F1 (Supplemental Fig. S3). These results suggest that numerical extrapolation from results from the ENCODE arrays provides a reasonable representation of the percentage of promoters bound by E2F1 in the entire human genome. As another indication of the robustness of the promoter ChIP-chip data, we examined the median values of the log2 ratios for nine known E2F target genes that were identified in our previous ChIP-chip studies using CpG island arrays (Supplemental Fig. S3). Importantly, all nine of these promoters have been previously confirmed to be bound by E2F1 using PCR analysis of ChIP samples (Weinmann and Farnham 2002; Oberley et al. 2003; Wells et al. 2003). Additionally, we examined the Dhfr promoter, the first, and perhaps most-studied, E2F target gene (Blake and Azizkhan 1989; Means et al. 1992; Slansky and Farnham 1993; Slansky et al. 1993; Fry et al. 1999) and the E2F1 promoter, another well-characterized E2F1 target promoter (Hsiao et al. 1994; Johnson et al. 1994). The median log2 ratio for these 11 known target genes ranged from 0.792 (Dhfr) to 2.98 (CBX5), and thus each was called as positive with the log2 cutoff of 0.707. Because the median value for a promoter region is calculated based on the signals from all 15 oligomers (many of which are far from the binding site), the actual maximum enrichment found in each promoter region is higher than the median (Supplemental Fig. S3). For example, for MAGEF1, the median value (log2) of all 15 probes was 1.9, but the maximum value was 3.0; similarly for RAD51, the median value (log2) of all probes was 1.0, but the maximum value was 2.5.
As a final test to determine if the E2F1 promoter array data were reliable, we performed another ChIP-chip analysis using a biologically independent set of E2F1 amplicons. To gain an estimate of the number of promoters bound by E2F1 in this second sample, we examined the same 25 promoters using the logic described above. For this second sample, there was a less-clear cutoff; however, we find that conservative thresholds that would capture 64%–76% of this set of 25 yielded an estimate of 5554–7352 promoters bound by E2F1, similar to the 6183 estimated above. For the analysis of the ENCODE arrays, we had found that only 49% of the called peaks were found in all three of the biologically independent E2F1 ChIP samples that were analyzed in ChIP-chip experiments. The source of the variation could be due to differences in cell culture conditions from one experiment to the next, to differences created during amplicon preparation, or to inconsistencies in array hybridization or washing; we are currently investigating each of these possibilities as part of a larger group of investigators. Regardless, the results of the ENCODE arrays suggest that about half of the promoters called as a target on one array would also be called as targets in all subsequent promoter ChIP-chip experiments. A comparison of the top 6000 ranked promoters in our two promoter ChIP-chip experiments indicated that 45% are in common. Thus, the promoter arrays and the ENCODE arrays give similar results, providing support for our suggestion that E2F1 binds to a large fraction of the promoters in the human genome.
The fact that E2F1 binds to almost 30% of the promoters in the human genome is striking. However, these data do not reveal whether E2F1 contributes to the regulation of each of the promoters to which it is bound. Clearly, analysis of the regulation of these promoters on an individual basis is not practical. However, as a measure of the relationship between E2F1 and promoter activity, we have compared the binding pattern of E2F1 to that of POLR2A (assuming that most promoters that are bound by POLR2A are transcriptionally active in that cell type). To perform this comparison, we performed triplicate ChIP-chip experiments using an antibody to POLR2A and hybridized to the ENCODE arrays, used our standard binding site determination method (as in Fig. 3) and thus identified the set of active promoters in the ENCODE regions in HeLa cells (Table 2). We found that 94 known genes were bound by POLR2A in the ENCODE regions in HeLa cells. Interestingly, E2F1 was found within 1 kb of 83% of bound POLR2A (Table 1), and 90% of the active genes (as defined by having a promoter region occupied by POLR2A) were bound by E2F1. Thus, there is a very strong correlation between the binding of POLR2A and the binding of E2F1 to a promoter.
Discussion
The most surprising finding of this report is that E2F1 appears to be recruited to the promoter regions of a large fraction (at least 25%–35%) of human genes and is closely associated with active genes (as defined by a bound POLR2A), suggesting that E2F1 plays a much more widespread role in the human genome than expected from previous work. Our findings were based on analyses of ENCODE region high-density tiling arrays and the human promoter array. To derive binding site information from the ENCODE arrays, we developed and tested a new analysis approach. Using these binding site predictions along with other data sets for the ENCODE regions, we found that: (1) most E2F1-binding sites are within 1 kb of a transcription start site; (2) the great majority of E2F1 sites lack the canonical E2F motif, and the great majority of canonical E2F1 motifs are outside of E2F1 binding sites; (3) most POLR2A-binding sites are within 1 kb of an E2F1-binding site; and (4) the majority of E2F1-binding sites are shared between at least two cell types. Analysis of the large-scale (∼24,000 promoters) human promoter tiling array supported the finding of a large number of E2F1-binding sites and suggested that a large fraction of genes may be regulated by E2F1.
Analysis approach
We present and test a new, simple, and minimal statistically based analysis methodology for high-density ChIP tiling arrays and apply it to find E2F1-, MYC-, and POLR2A-binding sites in the ENCODE regions. Our approach bears some resemblance to that used by Kim et al. (2005). However, we differ from that approach in that we put the determination of width on a statistical basis, we do not use a triangular filter, and we explore several ranges of parameters. In addition, our approach differs from previous approaches in that we choose to determine candidate binding sites in each of three separate biological samples, then combine the predictions (“peak-first”), as opposed to combining the arrays and then making a single determination of binding sites (“combine-first”).
Several lines of evidence support our approach: (1) Most importantly, PCR testing supports our peak predictions and argues strongly against use of our lowest stringency set (L4). (2) Our results fit very well with expectations from previous work on E2F1; for example, others have shown that CpG arrays are effective in identifying E2F target genes, and we found that 74%–82% of the identified E2F1 (L1 stringency) binding sites are in CpG islands. (3) We found that most of the identified HeLa binding sites were also identified in MCF7 cells. (4) We find that control IgG experiments show that only two of the 205 E2F1-binding sites (L1 stringency) are due to nonspecific immunoprecipitation. (5) We find that using our analysis approach to identify the preferred location of MYC-binding sites with respect to core promoter regions produces results that closely match the location analysis of Myc-binding sites from a previous study using a different array platform (Affymetrix Chr21 + 22 tiling arrays) and a different analysis approach (Cawley et al. 2004). Hence, our procedure produces a reproducible, heavily verifiable population of E2F1-binding sites. One concern is that our approach might be biased toward only producing a very stringent set of binding sites and might therefore suffer from low sensitivity. However, in testing areas in which no peak was predicted, we find that only rarely do we detect a binding site. In addition, we find that the peaks first appearing at our lowest stringency (L4) are often not confirmable, indicating that at this level, we have sacrificed specificity for sensitivity. Hence, our stringency range of L1–L4 seems to “bracket” the ranges of high-specificity/lower sensitivity (i.e., L1) to lower specificity/higher sensitivity (L4).
However, there are several weaknesses to our method. First, it estimates the random rate by looking at the rate of points above a certain threshold. Because a significant number of these points are real data points, we are actually estimating a too high rate of random noise and hence are being too conservative. Second, our method depends on high sensitivity and a single below-threshold oligomer can stop detection of a peak. Again, this would bias our approach toward being conservative. One way to decrease this problem would be to use a three-point median filter to smooth the data before peak detection, as has been used previously (Kim et al. 2005). Third, our choice of _P_-values and threshold values, although based on a reasonable set of parameters that are similar to those used in previous work, is somewhat arbitrary. It is important to note here that all of these factors would point toward our analysis approach being conservative; hence, these points argue that our extrapolations to the whole genome may also provide low estimates of the number of E2F1-binding sites. In addition, our calculation of _P_-values assumes statistical independence of points on the array (an assumption made by other ChIP-chip peak-finding approaches; e.g., Kim et al. [2005 and Cawley et al. [2004]). Clearly, given both the nature of the array (overlapping tiles) and, more importantly, the fact that the sheared DNA fragments cover many consecutive tiles on the array, this is an oversimplification, and corrections to the statistical calculations used in this field must be developed in the future to address these dependencies.
E2F1 and MYC may bind to thousands of genomic locations
Our unbiased sampling of the human genome using the ENCODE arrays points toward a large number of E2F1 sites in the genome (we estimate ∼20,500–34,000). This estimate is a crude extrapolation from the number of experimentally determined E2F1 sites in the ENCODE regions, and fundamentally assumes that multiplying values derived from the ENCODE regions (which in total length are 1% of the genome) by 100 will yield a good approximation to the entire genome. We view this extrapolated number of sites as an order-of-magnitude approximation; to gain a better approximation, it will be necessary to examine whole genome arrays. Although the estimated number of E2F1-binding sites is large, it is similar to the number of estimated MYC sites in a previous study (Cawley et al. 2004), and it is comforting to note that our extrapolated number of MYC sites obtained using ENCODE region data (17,100–33,000) is similar to the previous study’s estimate (25,000). Several factors indicate that our estimates, both numerically and of percentages, may be low, including (1) we estimate based on one cell type (HeLa), yet our preliminary investigation of MCF7 cells reveals an additional population of sites; and (2) our analysis approach (at L1 level in particular) tends to favor specificity over sensitivity, indicating that we are not detecting all sites (see above discussion). However, our use of HeLa cells raises the possibility that many of the identified E2F1 sites may primarily be occupied only in tumor cells, perhaps because of higher than normal E2F1 production or, potentially, abnormal chromatin modifications unmasking normally obscured binding sites. Therefore, it will be important for future studies to assay E2F1 binding sites in normal cells. It would be very interesting if oncogenic transformation led to a enhanced role for E2F1 in regulating the genome.
Localization of E2F1-binding sites
Our results concerning the localization of E2F1 in reference to transcription start sites and to sites bound by POLR2A were robust across our analysis stringency levels of L1–L3 (e.g., Tables 1 and 2) and with several sets of gene annotations (“Known Genes” and two subsets of GENCODE annotations; see Results for details). We found that the great majority of E2F1 sites were in a core promoter region (i.e., within 1 kb of a transcription start site), a marked contrast with results concerning other transcription factors studied in an unbiased manner (Sp1, p53, NF-kb, or MYC) (Cawley et al. 2004; Carroll et al. 2005; this study). In fact, we find that the presence of an E2F1-binding site is as predictive of the presence of a core promoter region as is the presence of a TAF1-binding site (Kim et al. 2005). In addition, we found that 83% of POLR2A sites had a bound E2F1 within 1 kb and that 90% of active genes (as defined by POLR2A in the core promoter region) had a bound E2F1 within 1 kb of a transcription start site. However, it is important to note that there is a significant set of E2F1 sites that are far from POLR2A sites. Many of these E2F1-binding sites that are far from POLR2A sites are in fact localized near the 5′-end of a gene. It is interesting to speculate that these E2F1 sites may be sites with partially pre-assembled pre-initiation complexes and may represent promoters that are active in other cell types. In addition, we find that a small set of E2F1 sites that are far from the 5′-ends of known genes are near the 5′-ends of novel and/or non-coding transcripts, as defined by the GENCODE annotations (Supplemental Table S3). This raises the interesting possibility that E2F1 may play a significant role in regulating non-coding transcripts. In all, our results suggest that discovery of an E2F1-binding site far from an annotated transcript would imply that the region around that E2F1-binding site should be investigated for transcriptional activity in a variety of cell types, without regard to the type of transcript that may be produced from that genomic location.
Canonical motif localization
We find that very few E2F1 sites possess the canonical binding site motif (TTTSSCGC), and, conversely, very few of the canonical binding site motifs found in the ENCODE regions are actually within a binding site. One basic conclusion from these results is that using the presence or absence of a canonical binding site motif as a marker for E2F1 binding is not a good strategy, a finding in line with a growing consensus on this issue (Cawley et al. 2004; Wasserman and Sandelin 2004). In addition, we “relaxed” the binding site motif to allow either one mismatch at any position or one mismatch in only the initial TTT stretch. In both cases, we found a very large number of these variant motifs in the ENCODE regions (37,500 and 5450, respectively), which obviated any predictive value that they might have. However, it is important to note that this merely demonstrates that finding these variant motifs has little predictive value. It is still possible that E2F1 actually uses these degenerate motifs to bind to DNA, but that other factors are required to assist E2F1 binding to a small subset of the sites and/or to “veto” E2F1 binding to the majority of the sites. This latter possibility would be consistent with another level of control of binding site accessibility, such as larger-scale chromatin modifications (e.g., histone modification). In addition, E2F1 could bind DNA via a completely different motif. Our preliminary investigations have not revealed any clear candidates for this potential second binding motif (M.C. Bieda and P.J. Farnham, unpubl.). Clearly, further investigation of the mechanisms by which E2F1 is recruited to chromatin in living cells is warranted.
Comparison with other array studies
Previous ChIP-chip experiments using CpG arrays and core promoter arrays have also been successful in identifying E2F-binding sites. These previous studies detected E2F1 binding at between 1% and 3% of the promoters on the array (Ren et al. 2002; Weinmann et al. 2002; Oberley et al. 2003; Wells et al. 2003; Cam et al. 2004). We find that, even with conservative analysis parameters, probably 25%–35% of the promoters in the ENCODE regions are bound by E2F1. Our results suggest that the previous ChIP-chip studies were detecting only a small fraction of the total number of E2F1-bound promoters. Although one might assume that the genomic tiling arrays detected a larger number of E2F1 target promoters because they contained a larger amount of upstream sequences (in this study, each region that was analyzed ranged from 500,000 to 1,900,000 bp, whereas only 1–2-kb promoter regions were used in earlier experiments), this is not the case, because 80% of the E2F1-binding sites were within 1 kb of the transcription start site. Thus, our present study strongly suggests that the sensitivity of the high-density genomic tiling arrays used in our current studies is much greater than that of spotted arrays. Recent data in which we have compared the number of E2F4 target promoters in HeLa cells identified with spotted CpG arrays versus high-density oligonucleotide tiling arrays support this conclusion (M.C. Bieda, X. Xu, and P.J. Farnham, unpubl.).
A speculative model for E2F1 action
We find that only ∼12% of the experimentally determined E2F1-binding sites possess the canonical motif. It is likely, but not yet proven, that E2F1 is recruited to this set of target promoters via binding to the consensus site (Fig. 5A). We also found that ∼50% of the experimentally determined E2F1-binding sites possess a derivative of the consensus that lacks one of the Ts. Others have shown that E2F family members can cooperate with certain site-specific DNA-binding factors to regulate transcription (Schlisio et al. 2002; Giangrande et al. 2003). Perhaps E2F1 is recruited to a significant number of target promoters via cooperative interactions (Fig. 5B). However, our finding that 50% of the E2F1-binding sites actually overlap the start site of transcription suggests an intriguing possibility that E2F1 may, in many cases, be recruited to promoters via interaction with components of the general transcriptional machinery (Fig. 5C). Previous studies have shown that the E2F1 transactivation domain can interact with TBP and TFIIH (Pearson and Greenblatt 1997; Fry et al. 1999). It has been assumed that binding of E2F1 to a promoter occurs first and then the interactions between the E2F1 transactivation domain and TBP or TFIIH help to recruit the transcription pre-initiation complex to the DNA. However, it is possible that E2F1 recruitment to some promoters occurs as a consequence, and not a cause, of transcription complex formation. This could explain the very high correlation we see between the localization of E2F1 and RNA Polymerase II in HeLa cells. Interestingly, using a highly artificial model system, a previous study (Blau et al. 1996) has suggested that E2F1 falls into a class of transcription factors that can stimulate both pre-initiation and post-initiation events. Perhaps E2F1 enhances pre-initiation complex formation at a set of promoters to which it directly binds to DNA and plays a role in promoter clearance at a set of promoters to which it is recruited by the transcriptional machinery. In light of the fact that 83% of the sites that are bound by RNA Polymerase II in HeLa cells are also bound by E2F1, the concept that a site-specific transcription factor such as E2F1 may play a general role in transcriptional regulation requires further investigation.
Figure 5.

A speculative model for E2F1 recruitment. (A) Classical E2F1 sites. At a subset of sites (mostly associated with cell cycle and DNA repair genes), E2F1 plays a major role in controlling transcriptional output by binding directly to a consensus site and helping to recruit the transcriptional machinery. (B) Cooperative E2F1 sites. At these sites, E2F1 binds cooperatively with other DNA-bound factors to sites that resemble the E2F consensus motif. (C) Inverted recruitment of E2F1. Here, E2F1 does not directly bind DNA, but perhaps is recruited to the promoter via interaction of the transactivation domain with general transcription factors such as TBP or TFIIH to play a role after pre-initiation complex formation. See Discussion for description.
Methods
ChIP-chip assays
HeLa cells were grown and cross-linked with formaldehyde as previously described (Weinmann et al. 2001). A complete protocol can be found on our Web site at http://genomics.ucdavis.edu/farnham/ and in Oberley et al. (2004). A mixed monoclonal antibody against E2F1 (KH20/KH95) was purchased from Upstate Biotechnology; a rabbit polyclonal antibody against MYC (N-202; cat# sc-764x) was purchased from Santa Cruz Biotechnology; a POLR2A antibody (N-20, cat# sc-899) was purchased from Santa Cruz Biotechnology; rabbit IgG (cat# 210-561-9515) was purchased from Alpha Diagnostic; and the secondary rabbit anti-mouse IgG (cat# 55,436) was purchased from MP Biomedicals. For analysis of the ChIP samples prior to amplicon generation, immunoprecipitates were dissolved in 50 μL of water, except for input samples, which were dissolved in 100 μL. Standard PCR reactions using 2 μL of the immunoprecipitated DNA were performed. PCR products were separated by electrophoresis through 1.5% agarose gels and visualized by ethidium bromide intercalation. For details concerning the generation of amplicons from ChIP samples, see http://genomics.ucdavis.edu/farnham/ and Oberley et al. (2004). High-density ENCODE oligonucleotide arrays were created by NimbleGen Systems and contained ∼380,000 50-mer probes per array, tiled every 38 bp. The regions included on the arrays encompassed the 30 Mb of the repeat masked ENCODE sequences, representing ∼1% of the human genome. The arrays were hybridized, and the data were extracted according to standard operating procedures by NimbleGen Systems Inc. Confirmation of the predicted binding sites was performed using standard PCR analysis of the amplicons that were applied to the arrays (quantitation methods are presented in Supplemental Methods). The primers used for all PCR reactions will be provided upon request.
Array analysis
To normalize array data, the log2 of the ratio of Cy5 (E2F1 ChIP amplicons) to Cy3 (Total DNA amplicons) for each point was calculated. Then, the biweight mean of this log2 ratio was subtracted from each point; this procedure is approximately equivalent to mean-normalization of each channel. For our peak detection method, we use a percentile for each array (95th and 98th percentile) of log2 oligomer ratios; further details and rationale are presented in Results. For a given threshold, we recode the array with points above the threshold as 1, points below as 0, and missing regions (due primarily to repeat-masking) as X (Supplemental Fig. S2). Because we have observed that some confirmed binding sites display peak-like waveforms that are “interrupted” by missing points, we ignore the X points for our analysis. This results in a long string of ∼380,000 1s and 0s to represent the full set of oligomers for the ENCODE regions on the array. Genuine binding sites should be represented as a series of points of elevated amplitude; this is equivalent to a run of 1s. However, just by chance, there will occasionally be two or more consecutive points of high amplitude that will be coded as 1s. Given the sequence length (e.g., ∼380,000 for our ENCODE arrays) and the probability of getting a 1 at any position, we can calculate the longest run of 1s expected purely by chance using the well-known Erdos-Renyi Law (Erdos and Renyi 1970). Importantly, this has been extended to exact results enabling calculation of the actual probabilities (_P_-values) associated with each run length (Waterman et al. 1987). We use the following six relations (Waterman et al. 1987) to calculate _P_-values for a run of 1s, where w is the length of the run; L is the length of the sequence (∼380,000 for these ENCODE arrays); p is the probability of having any given point be a 1 [for the 98th percentile threshold, this value is (1 − 0.98) = 0.02]; SD denotes the standard deviation; z is the _z_-score; and P is the final _P_-value reflecting the probability of getting this run length of 1s:
- mean Rn = log(1/p) L + (0.577)/Θ − 0.5
- variance Rn = π2/(6Θ2) + 1/12
- Θ = ln 1/p
- SD = (variance)1/2
- z = (w − mean Rn)/SD
- P(Z > z) = 1 − exp[−exp(−1.2825_z_ − 0.577)]
Following Waterman et al. (1987), the first two of these indicate the mean and variance for the maximum run length; the third is a convenient definition; the fourth and fifth are listed for clarity and reflect well-known formulas for the standard deviation and _z_-score; the last indicates the equation for conversion of _z_-score to _P_-value (based on extreme-value distribution). To ensure that we are not calling false peaks because of random consecutive high amplitude points, we use P < 0.0001 for a very stringent _P_-value and P < 0.05 for a less stringent _P_-value cutoff. See Results for actual parameters for L1–L4 and Figure 2 for an example of array peak-calling. Additional explication is presented in Supplemental Methods.
Data analysis and visualization
All data coordinates reference the July 2003 build of the human genome (hg16). However, hg17 (May 2004) ENCODE arrays were used for MYC, and the human promoter array is also in hg17 coordinates. To process MYC hg17 data, we derived MYC-binding sites using our standard methods (Fig. 3) using the hg17 data, then used the liftOver tool from Jim Kent (freely available at http://hgdownload.cse.ucsc.edu/downloads.html#liftover) to convert hg17 coordinates to hg16 coordinates. liftOver was able to map 171/172 (>99%) of MYC-binding sites to hg16. Note that the Supplemental Material has all the files in the original coordinate system (i.e., hg16 for all files except for MYC files and promoter array data, which are all hg17). Files used for comparison to binding site localization were downloaded from the UCSC genome browser in July and August 2005 (all hg16; CpG islands, GENCODE genes, Known Genes II, Known Genes tracks). For the “peak-first” experiments, custom programs written in Perl 5.8.3 and shell (bash) were produced. These are available from the authors upon request. Custom programs were written in Perl 5.8.3 to calculate overlap statistics and to output the sets of overlapping and non-overlapping hits. For the “combine-first” experiments, we used the track downloaded from the UCSC browser. To produce this track, the three array replicates were combined by taking the Tukey Bi-Weight mean at each point. Some displayed figures were images derived from the UCSC genome browser (http://genome.ucsc.edu). SignalMap 1.7–1.8 (NimbleGen Inc.) was used to visualize raw data sets for exploratory data analysis and some figure production. The total set of locations of TTTSSCGC (and variants) was mapped in the ENCODE regions using the emboss program fuzznuc (Rice et al. 2000), with both forward and reverse strands searched.
Acknowledgments
This work was supported in part by Public Health Service grants CA45250 and HG003129. We thank Sharon Squazzo and Alina Rabinovich for providing the E2F1-binding data for MCF7 cells, Catherine Gordon for work on PCR confirmations of predicted human promoter array targets, and the Farnham laboratory for helpful discussions. M.B. acknowledges the previous financial support of the Santa Fe Institute that allowed study of the sequence analysis statistics used in this report.
Footnotes
References
- Attwooll C., Denchi L.E., Helin K. The E2F family: Specific functions and overlapping interests. EMBO J. 2004;23:4709–4716. doi: 10.1038/sj.emboj.7600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balciunaite E., Spektor A., Lents N.H., Cam H., te Riele H., Scime A., Rudnicki M.A., Young R., Dynlacht B.D. Pocket protein complexes are recruited to distinct targets in quiescent and proliferating cells. Mol. Cell. Biol. 2005;25:8166–8178. doi: 10.1128/MCB.25.18.8166-8178.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake M.C., Azizkhan J.C. Transcription factor E2F is required for efficient expression of the hamster dihydrofolate reductase gene in vitro and in vivo. Mol. Cell. Biol. 1989;9:4994–5002. doi: 10.1128/mcb.9.11.4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blau J., Xiao H., McCracken S., O’Hare P., Greenblatt J., Bentley D. Three functional classes of transcriptional activation domains. Mol. Cell. Biol. 1996;16:2044–2055. doi: 10.1128/mcb.16.5.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm A., Miska E.A., McCance D.J., Reid J.L., Bannister A.J., Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- Cam H., Balciunaite E., Blais A., Spektor A., Scarpulla R.C., Young R., Kluger Y., Dynlacht B.D. A common set of gene regulatory networks links metabolism and growth inhibition. Mol. Cell. 2004;16:399–411. doi: 10.1016/j.molcel.2004.09.037. [DOI] [PubMed] [Google Scholar]
- Carroll J.S., Liu X.S., Brodsky A.S., Li W., Meyer C.A., Szary A.J., Eeckhoute J., Shao W., Hestermann E.V., Geistlinger T.R., et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Cawley S., Bekiranov S., Ng H.H., Kapranov P., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. doi: 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- Dyson N. The regulation of E2F by pRB-family proteins. Genes & Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- Erdos P., Renyi A. On a new law of large numbers. J. Anal. Math. 1970;23:103–111. [Google Scholar]
- Fernandez P.C., Frank S.R., Wang L., Schroeder S., Liu S., Green J., Cocito A., Amati B. Genomic targets of the human c-Myc protein. Genes & Dev. 2003;17:1115–1129. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov M.V., Dyson N.J. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J. Cell Sci. 2004;117:2173–2181. doi: 10.1242/jcs.01227. [DOI] [PubMed] [Google Scholar]
- Fry C.J., Pearson A., Malinowski E., Bartley S.M., Greenblatt J., Farnham P.J. Activation of the murine dihydrofolate reductase promoter by E2F1: A requirement for CBP recruitment. J. Biol. Chem. 1999;274:15883–15891. doi: 10.1074/jbc.274.22.15883. [DOI] [PubMed] [Google Scholar]
- Giangrande P.H., Hallstrom T.C., Tunyaplin C., Calame K., Nevins J.R. Identification of E-box factor TFE3 as a functional partner for the E2F3 transcription factor. Mol. Cell. Biol. 2003;23:3707–3720. doi: 10.1128/MCB.23.11.3707-3720.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K.-M., McMahon S.L., Farnham P.J. Multiple DNA elements are required for the growth regulation of the mouse E2F1 promoter. Genes & Dev. 1994;8:1526–1537. doi: 10.1101/gad.8.13.1526. [DOI] [PubMed] [Google Scholar]
- Johnson D.G., Ohtani K., Nevins J.R. Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes & Dev. 1994;8:1514–1525. doi: 10.1101/gad.8.13.1514. [DOI] [PubMed] [Google Scholar]
- Kel A.E., Kel-Margoulis O.V., Farnham P.J., Bartley S.M., Wingender E., Zhang M.Q. Computer-assisted identification of cell cycle-related genes: New targets for E2F transcription factors. J. Mol. Biol. 2001;309:99–120. doi: 10.1006/jmbi.2001.4650. [DOI] [PubMed] [Google Scholar]
- Kim T.H., Barrera L.O., Zheng M., Qu C., Singer M.A., Richmond T.A., Wu Y., Green R.D., Ren B. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Van Calcar S., Qu C., Cavenee W.K., Zhang M.Q., Ren B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc. Natl. Acad. Sci. 2003;100:8164–8169. doi: 10.1073/pnas.1332764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R.X., Postigo A.A., Dean D.C. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–473. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L., Groisman R., Naguibneva I., Robin P., Lorain S., Villain J.P., Troalen F., Trouche D., Harel-Bellan A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- Mao D.Y.L., Watson J.D., Yan P.S., Barsyte-Lovejoy D., Khosravi F., Wong W.W.-L., Farnham P.J., Huang T.H.-M., Penn L.Z. Analysis of Myc bound loci identified by CpG island arrays shows that Max is essential for Myc-dependent repression. Curr. Biol. 2003;13:882–886. doi: 10.1016/s0960-9822(03)00297-5. [DOI] [PubMed] [Google Scholar]
- Means A.L., Slansky J.E., McMahon S.L., Knuth M.W., Farnham P.J. The HIP1 binding site is required for growth regulation of the dihydrofolate reductase gene promoter. Mol. Cell. Biol. 1992;12:1054–1063. doi: 10.1128/mcb.12.3.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H., Bracken A.P., Vernell R., Moroni M.C., Christians F., Grassilli E., Prosperini E., Vigo E., Oliner J.D., Helin K. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes & Dev. 2001;15:267–285. doi: 10.1101/gad.864201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins J.R. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 1998;9:585–593. [PubMed] [Google Scholar]
- Nielsen S.J., Schneider R., Bauer U.-M., Bannister A.J., Morrison A., O’Carroll D., Firestein R., Cleary M., Jenuwein T., Herrera R.E., et al. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412:561–565. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- Oberley M.J., Inman D., Farnham P.J. E2F6 negatively regulates BRCA1 in human cancer cells without methylation of histone H3 on lysine 9. J. Biol. Chem. 2003;278:42466–42476. doi: 10.1074/jbc.M307733200. [DOI] [PubMed] [Google Scholar]
- Oberley M.J., Tsao J., Yau P., Farnham P.J. High throughput screening of chromatin immunoprecipitates using CpG island microarrays. Methods in Enzymol. 2004;376:316–335. doi: 10.1016/S0076-6879(03)76021-2. [DOI] [PubMed] [Google Scholar]
- Orian A., van Steensel B., Delrow J., Bussemaker H.J., Li L., Sawado T., Williams E., Loo L.W., Cowley S.M., Yost C., et al. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes & Dev. 2003;17:1101–1114. doi: 10.1101/gad.1066903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson A., Greenblatt J. Modular organization of the E2F1 activation domain and its interaction with general transcription factors TBP and TFIIH. Oncogene. 1997;15:2643–2658. doi: 10.1038/sj.onc.1201451. [DOI] [PubMed] [Google Scholar]
- Ren B., Cam H., Takahashi Y., Volkert T., Terragni J., Young R.A., Dynlacht B.D. E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes & Dev. 2002;16:245–256. doi: 10.1101/gad.949802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P., Longden I., Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Schlisio S., Halperin T., Vidal M., Nevins J.R. Interaction of YY1 with E2Fs, mediated by RYBP, provides a mechanism for specificity of E2F function. EMBO J. 2002;21:5775–5786. doi: 10.1093/emboj/cdf577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slansky J.E., Farnham P.J. The role of the transcription factor E2F in the growth regulation of DHFR. Plenum Press; New York.: 1993. [Google Scholar]
- Slansky J.E., Li Y., Kaelin W.G., Farnham P.J. 1993. A protein synthesis-dependent increase in E2F1 mRNA correlates with growth regulation of the dihydrofolate reductase promoter. Mol. Cell. Biol. 13 1610 1618 [author’s correction: 13 7201]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stekel D. Microarray bioinformatics. Cambridge University Press; Cambridge, UK.: 2003. [Google Scholar]
- Tao Y., Kassatly R., Cress W.D., Horowitz J.M. Subunit composition determines E2F DNA-binding site specificity. Mol. Cell. Biol. 1997;17:6994–7007. doi: 10.1128/mcb.17.12.6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman W., Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 2004;5:267–287. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- Waterman M.S., Gordon L., Arratia R. Phase transitions in sequence matches and nucleic acid structure. Proc. Natl. Acad. Sci. 1987;84:1239–1243. doi: 10.1073/pnas.84.5.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinmann A.S., Farnham P.J., Identification of unknown target genes of human transcription factors through the use of chromatin immunoprecipitation. Methods. 2002;26:37–47. doi: 10.1016/S1046-2023(02)00006-3. [DOI] [PubMed] [Google Scholar]
- Weinmann A.S., Bartley S.M., Zhang M.Q., Zhang T., Farnham P.J. The use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol. Cell. Biol. 2001;21:6820–6832. doi: 10.1128/MCB.21.20.6820-6832.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinmann A.S., Yan P.S., Oberley M.J., Huang T.H.-M., Farnham P.J. Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes & Dev. 2002;16:235–244. doi: 10.1101/gad.943102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells J., Yan P.S., Cechvala M., Huang T., Farnham P.J. Identification of novel pRb binding sites using CpG microarrays suggests that E2F recruits pRb to specific genomic sties during S phase. Oncogene. 2003;22:1445–1460. doi: 10.1038/sj.onc.1206264. [DOI] [PubMed] [Google Scholar]
- Zheng N., Fraenkel E., Pabo C.O., Pavletich N.P. Structural basis of DNA recognition by the heterodimeric cell cycle transcription factor E2F-DP. Genes & Dev. 1999;13:666–674. doi: 10.1101/gad.13.6.666. [DOI] [PMC free article] [PubMed] [Google Scholar]