Passive Sexual Transmission of Human Immunodeficiency Virus Type 1 Variants and Adaptation in New Hosts (original) (raw)
Abstract
Human immunodeficiency virus type 1 (HIV-1) genetic diversity is a major obstacle for the design of a successful vaccine. Certain viral polymorphisms encode human leukocyte antigen (HLA)-associated immune escape, potentially overcoming limited vaccine protection. Although transmission of immune escape variants has been reported, the overall extent to which this phenomenon occurs in populations and the degree to which it contributes to HIV-1 viral evolution are unknown. Selection on the HIV-1 env gene at transmission favors neutralization-sensitive variants, but it is not known to what degree selection acts on the internal HIV-1 proteins to restrict or enhance the transmission of immune escape variants. Studies have suggested that HLA class I may determine susceptibility to HIV-1 infection, but a definitive role for HLA at transmission remains unproven. Comparing populations of acute seroconverters and chronically infected patients, we found no evidence of selection acting to restrict transmission of HIV-1 variants. We found that statistical associations previously reported in chronic infection between viral polymorphisms and HLA class I alleles are not present in acute infection, suggesting that the majority of viral polymorphisms in these patients are the result of transmission rather than de novo adaptation. Using four episodes of HIV-1 transmission in which the donors and recipients were both sampled very close to the time of infection we found that, despite a transmission bottleneck, genetic variants of HIV-1 infection are transmitted in a frequency-dependent manner. As HIV-1 infections are seeded by unique donor-adapted viral variants, each episode is a highly individual antigenic challenge. Host-specific, idiosyncratic HIV-1 antigenic diversity will seriously tax the efficacy of immunization based on consensus sequences.
The absence of an effective human immunodeficiency virus type 1 (HIV-1) vaccine is, in part, a consequence of the requirement to prime an immune response against extensive viral variation. The human leukocyte antigen (HLA) class I region shapes the cell-mediated arm of the HIV-1 immune response and is a determinant of plasma viral load (16) and clinical progression to AIDS (22). Studies have suggested a role for HLA class I in determining susceptibility to infection with HIV (7, 9, 13, 14), but a definitive role for HLA at the time of transmission is unproven. Viral diversity immediately after transmission is reduced (31, 32) and rare variants in the donor may be disproportionately represented in the recipient (29), but whether these findings are a consequence of any HLA-mediated selection is unknown. In recently infected individuals there is a greater loss of diversity in the HIV-1 envelope (env) gene than in gag, suggesting that selection at transmission is acting on env (31, 32); this is consistent with data showing that neutralization-sensitive env variants are preferentially transmitted (8). In addition, shortly after infection a high proportion of polymorphisms in the HIV-1 genome are located within cytotoxic T lymphocyte (CTL) epitopes (5). The processes governing transmission of the internal HIV-1 antigens which encode the majority of HLA class I-restricted epitopes recognized by CTLs (1) are less clear.
The selection pressure imposed by HLA class I on HIV-1 results in viral escape mutations that facilitate evasion of the immune response (25, 26) and may compromise vaccine efficacy (4). Recently, it has been shown that certain of these mutations can be transmitted both sexually (2, 12) and vertically (18). At the population level, the overall extent to which different escape mutations are transmitted, and also their impact on the ability of the new host to prime an immune response, is unknown.
Studies of genetic host-pathogen interactions are limited by the requirement for large numbers of patients. In a study of over 400 patients with chronic HIV-1 infection, 64 associations were described between specific amino acids in the HIV-1 RT gene and HLA class I molecules (of which 12 remained statistically significant after correction for multiple comparisons) (21), potentially shaping HIV-1 protein variation and driving HIV-1 viral evolution (19). The extent to which this is true is dependent on the dynamics of transmission and the rates of mutation reversion or accumulation (11, 18). Here, we present a permutation-based statistical approach for testing global host-pathogen genetic associations in populations and, in conjunction with detailed genetic analysis of transmission between pairs of individuals, we show that variants of HIV-1 internal proteins, including those with escape properties, are being transmitted passively with no evidence of limiting selection.
MATERIALS AND METHODS
Patients.
From 136 predominantly Caucasian male homosexual acute seroconverters sampled within 6 months of infection and recruited as part of a prospective study of acute HIV-1 infection at St. Mary's Hospital, London (20), we identified 101 patients infected with subtype B HIV-1. From the clinical cohort at St. Mary's Hospital, London, a subgroup of drug-naïve patients chronically infected with subtype B HIV-1 was identified (n = 62). Subtypes were confirmed from RT sequences, using LANL Treemaker software (http://hiv-web.lanl.gov) and subtype reference sequences, and double-checked with NCBI subtyping software (http://www.ncbi.nlm.nih.gov). Samples of plasma and genomic DNA from a second European cohort (n = 128) of HIV-1 subtype B chronically infected patients recruited as part of the SSITT therapeutic intervention trial (described in detail elsewhere [10]) and at centers around the United Kingdom were employed for further analysis of HLA class I-amino acid associations. From the acute cohort, four transmission pairs were identified by the clinicians and confirmed through close similarity of the RT sequence of infecting strains.
Diagnosis of acute HIV infection.
Criteria for acute HIV-1 infection were documented seronegative HIV-1 antibody test within the previous 6 months, acute symptomatic seroconversion illness, positive HIV-1 DNA PCR, or positive HIV-1 RNA quantification (Chiron 3.0; Chiron Inc., Emeryville, Calif.) in the absence of an antibody response. In order to confirm primary HIV-1 infection, sequential serum samples were later tested blindly with a “detuned” enzyme-linked immunosorbent assay (Abbott).
PCR amplification and sequencing.
For the analysis of the acute and chronic cohorts, viral RNA was extracted from patient plasma and the HIV-1 RT gene was sequenced using the HIV-1 genotyping kit (version 2; ABI) according to the manufacturer's instructions. For analysis of the four transmission pairs, sequences were derived from viral RNA, except for pair 3, for which only proviral DNA was available. HIV-1 virion RNA encoding gag, pol, and nef was reverse transcribed, amplified, cloned, and sequenced. Viral RNA was extracted using the NucleoSpin RNA virus kit (Macherey-Nagel GmbH). Peripheral blood mononuclear cells (PBMC) were obtained from heparinized blood using Ficoll gradient centrifugation, and DNA was extracted from a minimum of 1 × 106 cells using the Purigene DNA extraction kit (Gentra) according to the manufacturer's instructions. Viral RNA was reverse transcribed with the Reverse-iT 1st Strand synthesis blender (ABgene, United Kingdom) and antisense primers, described previously (23). These primers were also used for PCR amplification. Amplification from both cDNA and proviral DNA was achieved with two rounds of nested PCR (95°C for 2 min, followed by 95°C for 30 s, 50°C for 30 s, and 75°C for 1.5 to 3 min for 35 cycles, and finally 75°C for 10 min). PCR products were gel purified (gel purification kit; QIAGEN), ligated into the pCR4-TOPO vector (Invitrogen), and sequenced with M13 forward and reverse primers using ABI BigDye Terminator (v. 3.0; Applied Biosystems). Trace files were checked and assembled using the Staden software package (www.mrc-lmb.cam.ac.uk/pubseq/staden_home.html). Between 10 and 48 clones were sequenced for each time point.
Phylogenetics.
To illustrate the relationship between donor and recipient viruses in each of the transmission pairs, gag p24 nucleotide sequences obtained closest to the time of transmission were aligned manually using Se-Al and a neighbor-joining tree was constructed using PAUP*, assuming the HKY85 model of nucleotide substitution. The gag gene was used, as multiple clonal sequences for all eight members of the four transmission pairs were available, thereby facilitating a high-resolution phylogenetic comparison. The relationship between the four transmission pairs was confirmed using HIV-1 RT (data not shown). Subtype A and B reference sequences were obtained from the HIV Sequence Database (www.hiv.lanl.gov).
IFN-γ ELISPOT assay.
Wells of sterile 96-well microtiter plates containing polyvinylidene difluoride (Millipore) were coated with mouse anti-human gamma interferon (IFN-γ) monoclonal antibody 1-D1K (0.5 μg/ml; Mabtech). Frozen PBMC were added to the wells with peptide at a final concentration 10−5 to 10−9 M. Following overnight incubation, 0.5 μg/ml biotinylated anti-human IFN-γ monoclonal antibody clone 7-B6-1 (Mabtech) was added. Streptavidine-conjugated alkaline phosphatase was added (0.5 μg/ml) and, following incubation for 40 min, spots were developed using chromogenic alkaline phosphatase substrate (Bio-Rad) and counted using the AID version 2.9 ELISPOT plate reader. The number of spots in the negative control was subtracted, and the results were normalized to give the number of spot-forming cells per 106 PBMC.
Binding assay.
Thawed cryopreserved B-cell lines expressing the relevant HLA class I molecules were subjected to acid elution on ice using 0.263 M citric acid and 0.123 M Na2HPO4 in equal volumes. The cells were washed in RPMI-2% fetal calf serum and then incubated with fluorescein isothiocyanate-labeled high-affinity reference peptide (150 μM to 900 μM) and test peptide at threefold dilutions from 184 μM to 0.02 μM. Irrelevant peptides were included as negative controls. Following 24 h of incubation at 4°C, the cells were analyzed by flow cytometry. Percent inhibition of variant peptide against the fluorescent peptide was used as a measure of binding affinity.
Statistics.
Two-tailed Spearman's nonparametric correlation coefficients were used to compare the frequencies of variant alleles in the different patient cohorts. In order to test for associations between HLA class I molecules and polymorphic amino acids, the distribution of the number of mutations corresponding to the associated HLA type that would be likely to occur by chance was estimated by permutation. The amino acid sequences of the patients were assigned randomly among the two-digit HLA class I types of the cases, and a score was calculated as the total number of times that a mutation corresponding to the associated HLA type occurred within the population. This was repeated 19,999 times to estimate the distribution of this score expected under the null hypothesis of no association, and the observed score was compared to this distribution. Fisher's exact test was used for the determination of specific HLA-amino acid associations. To investigate the determinants of transmission in the four transmitter pairs, the median frequencies of transmitted and nontransmitted donor group epitope sequences were compared using the nonparametric Wilcoxon matched pairs test.
RESULTS
At the population level, HIV-1 retains a signature pattern of polymorphic variation.
The HIV-1 reverse transcriptase (RT) gene (amino acids [aa] 1 to 250) was sequenced from drug-naïve patients infected with HIV-1 subtype B strains in the acute (n = 101) and chronic (n = 62) London cohorts. The chronically infected European cohort (n = 128) (10) was sequenced in RT (amino acids 1 to 250) and typed for HLA class I. To maintain consistency across our experimental cohorts, amino acid variants were defined with reference to the HIV-1 subtype B reference strain, HXB2. Variant frequency at each site in the HIV-1 RT gene is shown in Fig. 1a and b for subtype B HIV-1-infected chronic and acute patients from the London cohort, respectively. Figure 1c shows the difference in the proportions of polymorphisms prevalent in the two populations. This figure reveals only two RT amino acid sites (aa 162 and aa 245) at which the proportion of variants in the chronic cohort varied significantly from that in the acute population. The graphical result of Fig. 1 is supported by statistical analysis. Using correlation coefficients, we found the frequency of amino acid variants in the two London cohorts (acute and chronic) to be very similar (Fig. 2a) (r = 0.680; P < 0.0001). On further comparison with chronically infected patients from the European cohort, these correlations remained statistically significant for both the acute and chronic cohorts from London (r = 0.74 and P < 0.0001 [acute]; r = 0.69 and P < 0.0001 [chronic]) (data not shown).
FIG. 1.

Population-level distribution of amino acid variation in RT in two patient cohorts. Variant frequency in the HIV-1 RT gene (amino acids 1 to 250) across a population of 62 drug-naïve chronically infected patients with subtype B HIV-1 recruited at St. Mary's Hospital, London (a) and a population of 101 individuals sampled shortly after infection recruited at St. Mary's Hospital, London (b). (c) Differences in polymorphism frequencies for the HIV-1 RT gene in acute and chronic patients. *, site at which the difference in proportions is statistically significant.
FIG. 2.
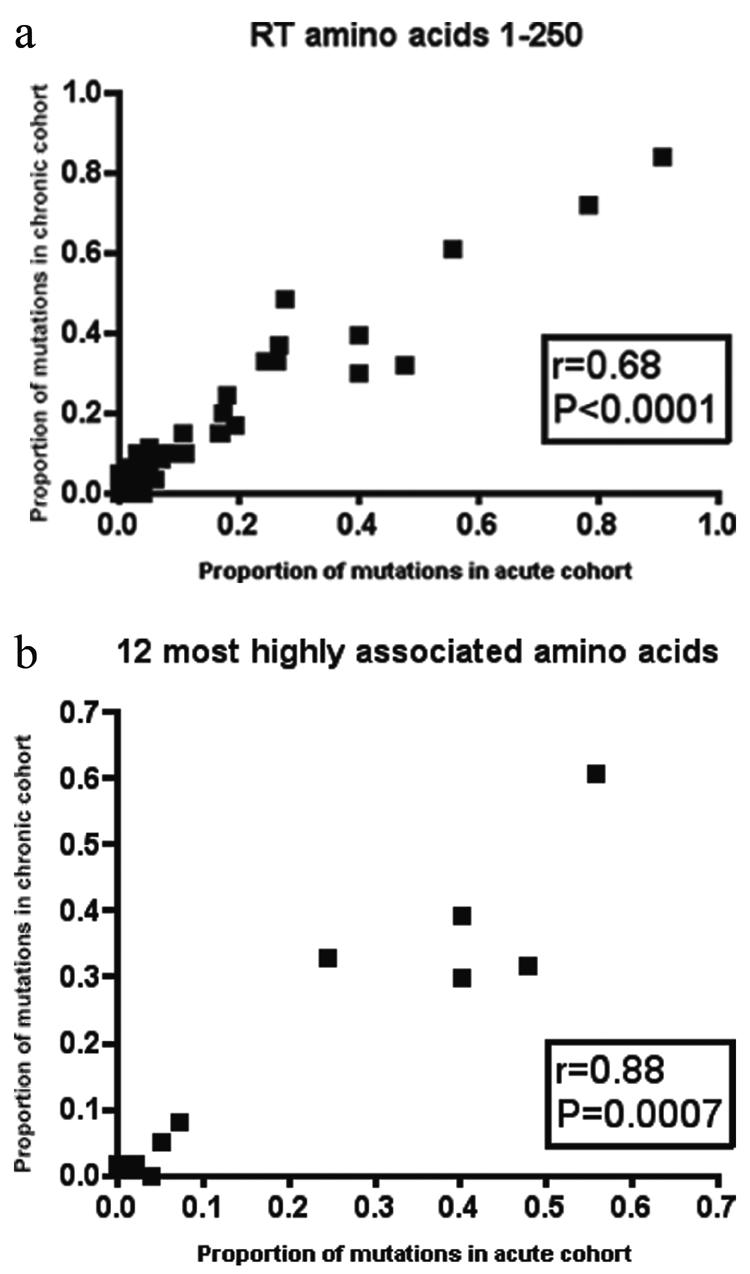
Analyses of variant frequencies in acute versus chronic patients using Spearman's nonparametric correlation coefficient for all polymorphic sites in RT (a) or only sites at which variation is associated with the presence of the restricting HLA class I allele (b). Correlations are shown between acute seroconverters from St. Mary's Hospital and chronically infected patients from St. Mary's Hospital.
These figures might merely demonstrate those amino acid sites within HIV-1 which are less structurally conserved. Evidence for statistically significant associations between specific HLA class I alleles and up to half of the polymorphic amino acids in RT (21) is suggestive that this conserved signature of HIV-1 variability may be the result of immune-mediated selection pressure. To exclude sites which might be polymorphic secondary to replicative plasticity rather than selection pressure, we focused on the 12 amino acids in RT for which the strongest associations with HLA class I, and thereby CTL selection pressure, have been reported following correction for multiple comparisons (21) (see Table S1 in the supplemental material). In these analyses the frequency of amino acid variants in the two London cohorts (acute and chronic) remained highly similar (r = 0.88; P = 0.0007) (Fig. 2b). It has been previously shown that HIV-1 env variants that have escaped antibody neutralization are selected against at transmission (8). Our data suggest that variants from an HIV-1 internal protein are transmitted without limiting selection.
Furthermore, one can also model the impact of HLA on the viral population by correcting for attributable mutations, according to reported odds ratios (ORs) (21). We adjusted the prevalence of the 12 sites in RT most strongly correlated with HLA class I to reflect an absence of selection (i.e., if the variability at these sites was random). We found that at the 12 tested sites, 15 out of 129 mutations (13%; 95% confidence interval, 7% to 18%) could be attributed to HLA class I associations. If we assumed an absence of HLA-mediated influence, the removal of these polymorphisms in chronic infection resulted in a significant reduction in the global number of polymorphisms at these sites (P = 0.03 by Fisher's exact test), although not at the individual amino acids. The polymorphism distributions in acute and chronic infections were highly similar prior to this correction and, if the polymorphism distribution in acute infection can be shown to be random (i.e., no associations with HLA class I), then the HIV-1 signature evident in chronic infection is contributed to by HLA-induced selection pressure.
In acute HIV-1 infection, the statistical associations between viral polymorphisms and HLA class I molecules are lost.
We, therefore, investigated polymorphisms in the acute seroconverters to see if associations with HLA class I molecules were present. The similarity between the frequency of HLA class I-adapted variants in the acute and chronic cohorts could reflect either the result of de novo adaptation of the virus to HLA in seroconverters or, alternatively, passive transmission of the HLA restriction pattern of the donor population. In the former case, associations between variation at a particular site and expression of a particular HLA class I allele in the recipient would be present in our acute infection cohort. To test this possibility, we again focused on the 12 amino acid-HLA class I associations in RT that have been identified in chronic infection (see Table S1 in the supplemental material) (21). For each acutely infected patient, we counted the number of times a variant was observed in association with expression of the appropriate HLA class I allele. We then summed the number of these observed associations for all individuals in the cohort to provide our test statistic, or score. For example, if each of the 12 associations was identified twice in the entire cohort, this would give a score of 2 × 12, or 24. The expected distribution of scores if there were no association between amino acid variation and HLA was estimated by randomly assigning the HLA allelic profiles of cases to the sequences in our data set and then recalculating the score. This process was repeated 19,999 times, creating a null distribution with a mean of 42.4, a standard deviation of 4.6, and an approximately Gaussian distribution (Fig. 3). The observed score for the seroconverter cohort was 46 (P = 0.38 for a two-tailed test). This analysis provides no evidence for a significant association between amino acid variation at these 12 selected sites and the HLA class I alleles expressed in acutely infected patients. This contrasts with the strong association that has been reported between HLA types and RT variation in chronically infected patients (21). If HLA associations of the strength reported in these chronically infected patients were present in our acute cohort, we would expect a score of 82.7, significantly different from the mean of the null distribution and the observed score of 46 (P < 0.0001). From this result, we conclude that the presence of HIV-1 variants in acute infection is not the result of de novo adaptation to the HLA alleles expressed by the new host.
FIG. 3.

HLA class I-independent variation in acutely infected patients. For each site known to adapt to the class I-restricted CTL response (see Table S1 in the supplemental material), the number of times a variant was observed in association with an HLA allele known to direct variation at that site was counted. The observed score for the seroconverter cohort was 46. Patient sequences were then randomly assigned to different HLA class I alleles expressed in the seroconverter cohort. Repetition of this process generated the null distribution of scores expected if the amino acid variants expressed by an infecting virus were unrelated to the HLA type of the patient. The mean for the null distribution was 42.4. The expected score for a cohort of previously described chronically infected patients (calculated using published odds ratios) (21) is 82.7, which is significantly different from the null distribution (P < 0.0001).
We looked for previously reported HLA class I -RT amino acid associations to see if they could be identified in either cohort. In the acutely infected cohort, only 1 of the 12 reported associations was present. This was at codon 135 at the carboxy terminal of the TAFTIPSI epitope (OR, 15.3; P = 0.001) restricted by HLA-B51, a known escape mutation (27). We then analyzed the European cohort of subtype B chronically infected patients to identify the number of associations that could be identified at this stage of infection and found that 3 of 12 were statistically significant, suggesting progressive adaptation to the new hosts over time (see Table S2 in the supplemental material). These were, again, in TAFTIPSI (OR, 20.7; P < 0.001) but also at codon 166 at the carboxy terminal in the A11-restricted epitope AIFQSSMTK (OR, 23.3; P < 0.001) and at amino acid position 7 of the B*07-restricted SPAIFQSSM (OR, 3.5; P = 0.027).
To examine this further we looked for variation in the 25 optimal epitopes previously documented in the fragment of the HIV-1 RT protein for which we had data (amino acids 29 to 225) (www.hiv.lanl.gov/content/immunology/maps/maps/html; see also Table S2 in the supplemental material). The second amino acid (position 2) and the carboxy-terminal amino acid are usually key to epitope binding in the HLA class I groove, and mutations at these sites frequently enable immune escape. We concentrated on these positions but also on any other intraepitopic sites at which there was significant variation. This analysis revealed four additional HLA class-amino acid associations (see Table S2). One of these was at the carboxy terminus of the HLA B*44-restricted EELRQHLLR epitope (aa 203 to 211). Interestingly, this association was only present in conjunction with HLA B*4403 and not other B*44 alleles. Three other associations at other amino acid sites were identified in the B*51-restricted epitope EI9 (aa 42 to 50), the B*35-restricted epitope VY10 (aa 118 to 127), and the B58-restricted epitope KL10 (aa 201 to 210) (see Table S2 in the supplemental material). Although these data support the previously published assertion that HLA class I is contributing to HIV-1 adaptation within human hosts, the extent of this selection appears less than that predicted from earlier reports (21).
In summary, the analysis of the population-level data in the acute and chronic cohorts suggests that transmission of HLA class I-restricted T-cell antigens is passive. In addition, statistical associations of HLA class I with specific viral polymorphisms are predominantly lost at acute seroconversion but become apparent in patients with chronic HIV-1 infection.
Viral diversity is decreased following transmission.
To examine the passive transmission of HIV-1 internal protein variants at greater resolution, we studied four HIV-1-infected transmitter pairs. In these four pairs, virus samples were obtained from both donor and recipient within 2 to 6 weeks (mean, 3.8 weeks) of the time when acute infection was diagnosed in the index case. HIV-1 virion RNA encoding gag, pol, and nef was reverse transcribed, amplified, cloned, and sequenced. To compare the viruses circulating in each individual, a neighbor-joining tree was constructed using gag p24 nucleotide sequences (Fig. 4a). Reference sequences from the HIV Sequence Database (www.hiv.lanl.gov) were included for comparison. For subtype B, these reference sequences were selected from patients sampled in London between 1993 and 2003 and closely represent the genetic diversity prevalent in our study population. For subtype A, reference sequences were obtained from the Los Alamos HIV-1 Subtype Reference Alignments Database. For pair 1 the transmitted virus clustered with subtype A sequences, whereas for pairs 2, 3, and 4 the sequences conformed to subtype B. The close similarity between the virus circulating in the peripheral blood of each pair, relative to the database sequences and virus in other pairs, confirms the authenticity of the transmission pairs. Compared to each donor, shorter branch lengths leading to the recipient sequences illustrate a substantial loss of diversity (P < 0.0001, paired two-sample t test), consistent with a genetic bottleneck at the time of transmission. The average reduction in nucleotide diversity, determined from the number of substitutions per site, was 81.3% for gag p17, 71.2% for gag p24, and 91.0% for nef (see Table S3 in the supplemental material).
FIG. 4.

(a) Neighbor-joining phylogenetic tree of gag p24 nucleotide sequences from donor-recipient pairs, assuming the HKY85 model of nucleotide substitution. Sequences were 843 bp long. The number of sequences for each patient was as follows: D1, 48; R1, 43; D2, 19; R2, 14; D3, 29; R3, 25; D4, 38; R4, 27. Subtype A and B reference sequences were obtained from the HIV Sequence Database (www.hiv.lanl.gov). Horizontal branch lengths are drawn to scale. Divergence is the number of substitutions per site, expressed as a percentage. (b) Retention of HIV-1 amino acid variants during sexual transmission. gag p24 from four donor-recipient pairs is shown. The variant frequency (proportion of amino acids at a particular site that differ from the subtype consensus sequence) is plotted on the vertical axis. Different-colored bars represent different amino acids at a given site. The horizontal axis represents the linear amino acid sequence from position 1 to 278. Regions of gag p24 restricted by the HLA alleles expressed in the infected patient are marked by red bars.
HIV-1 variation in the recipient closely resembles that in the donor.
We then concentrated on a single HIV-1 gene, gag p24, which contains many CTL epitopes, to identify the extent to which this bottleneck influenced the transmission of polymorphic variants from donor to new host. Graphical representation of amino acid variants encoded by gag p24 in viral clones from individual patients reveals a characteristic and idiosyncratic pattern for each virus (Fig. 4b). Transmission of these viral amino acid polymorphisms is illustrated by close similarity of the pattern of variant sites expressed in the donor and recipient of each pair. In individuals the gag gene variants that successfully propagate a new infection closely resemble those prevailing in the donor and are characteristic of each episode of transmission. This idiosyncrasy at the individual level is notable in view of the conserved population-level distribution of polymorphisms in different cohorts (Fig. 1).
Transmission of viral variants in HIV-1 internal proteins is frequency dependent with no evidence for selection.
To investigate more closely the determinants of transmission in HIV-1 internal proteins, we focused on CTL antigens restricted by the HLA class I alleles of each donor. For each polymorphic site within known CTL epitopes in the HIV-1 gag, pol, and nef genes, variant frequency in the donor close to the time of transmission was plotted against whether the variant was transmitted (as judged by detection in the recipient). We found that transmission of a variant was dependent on its frequency in the donor (P < 0.0001, two-tailed Mann-Whitney test; n = 53) with high-frequency variants being more likely to be transmitted (Fig. 5).
FIG. 5.
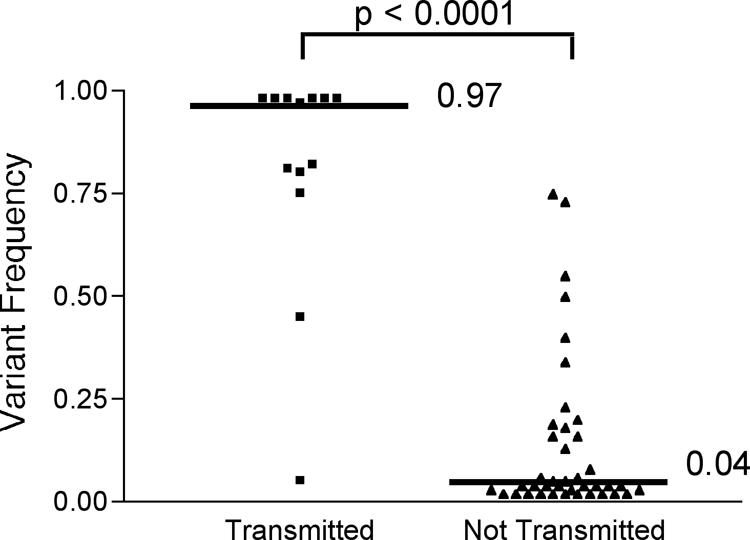
Frequency-dependent transmission of HLA class I antigenic variants, as shown by donor frequencies of transmission of all intraepitope, polymorphic sites. The median frequencies for the transmitted and nontransmitted variants are shown and were found to be significantly different (P < 0.0001, two-tailed Mann-Whitney test).
Variant CTL epitopes may have associated fitness costs (11, 18, 24, 30), which may reduce their chances of transmission. To assess further whether CTL escape has an influence on transmission, the immunological properties of HLA class I-restricted epitopes were characterized. At 20 epitopes identified in three of the transmitter pairs, 41 variants were immunologically characterized; 34 of these were classified as having escape properties (20). Immune escape was defined according to loss of binding affinity of peptide variants to HLA class I, using a fluorescent binding assay (15), or reduction in IFN-γ production in an ELISPOT assay. We found that when only escape variants were analyzed, their frequency in the donor was the main determinant of transmission (P < 0.0001, two-tailed Mann-Whitney test). This analysis might be biased by the inevitable transmission of variants that had reached fixation in the donor. It was, therefore, repeated using a more stringent approach, selecting only escape variants that had not reached fixation (n = 24). In this analysis, transmission remained dependent on donor frequency (P = 0.0029, two-tailed Mann-Whitney test). This finding of frequency-dependent transmission indicates that any coexisting selection is insufficient to impede transmission of the majority of epitope variants from immunogenic proteins, such as HIV-1 Gag, Pol, and Nef.
DISCUSSION
Adaptation of HIV-1 to the CTL immune response is documented at both individual and population levels. Whether this adaptation is likely to impact on viral evolution is dependent on whether mutants are transmitted and their fate following transmission. It is known that Env variants that escape humoral neutralization are selected against at transmission (8). Although transmission of CTL escape has been reported in individuals at specific well-characterized epitopes, this has not been investigated in populations, a prerequisite to understanding the full impact of CTL escape on HIV-1 evolution and antigenic variation.
In a large survey we identified a pattern of polymorphic variation which was conserved across different patient cohorts and, specifically, at sites associated with CTL selection pressure. This polymorphic signature is more than a reflection of functional constraint. This is supported by the contrasting idiosyncratic distribution of polymorphisms within infected individuals, the loss of similarity between acute and chronic patterns on eliminating known HLA class I-associated sites in the latter, and the previously reported finding of a high proportion of sites in RT under HLA-driven selection pressure (21).
We found that the frequency of variants present in acutely infected patients was closely correlated with that in chronic patients but that the reported statistical associations of variant sites with particular HLA alleles was absent. These data suggest that the majority of variants observed in the acutely infected patients were transmitted by the donor and were unlikely to have arisen de novo as a result of selection pressure in the new host.
Nevertheless, the rate at which new viral adaptation takes place at different loci is unlikely to be uniform but will reflect sites where selection pressures are strongest. The presence, in acute infection, of the strong association between HLA-B51 and RT amino acid 135 suggests that CTL-induced selection pressure is acting on at least one intraepitope site. This amino acid lies at the C-terminal end of the TAFTIPSI epitope, which is a recognized site for escape mutation (27). A limitation of this statistical approach is that this strong signal in acute infection could also be the result of rapid reversion to wild type in non-HLA-B51 patients, as well as reflecting early escape. Such early emergence of immune escape is comparable with the speed with which simian immunodeficiency virus can adapt to major histocompatibility complex class I in macaques, where new escape variants have been detected within 4 weeks of infection (3). Although the differential rate of reversion of transmitted escape mutations due to fitness costs has not yet been fully defined, rapid reversion has been observed (11, 18).
We identified a number of amino acid polymorphisms in known epitopes at which variation was associated with the restricting HLA class I molecule. However, we might have expected to find a greater number of reported associations in our chronic cohort. This might be because our cohort is smaller (n = 128 versus n = 473) and, although we could test previously reported sites, we were not powered to screen the HIV-1 genome for new HLA class I associations. In addition, we assumed that the distribution of HLA class I molecules among HIV-infected individuals in Perth—where the original study (21) was carried out—and London would be similar, and this is not certain.
From our observations of four episodes of sexual transmission in homosexual men, in which samples were available in all parties close to the time of infection, we found the successful transfer of CTL epitope variants was dependent on their frequency in the donor. Cytotoxic T-lymphocyte escape may impair viral replicative fitness (11, 18); however, when a broad spectrum of variants was analyzed we saw no evidence these viruses have diminished capacity for transmission. This contrasts with drug resistance mutations, which may also reduce the replicative capacity of HIV-1 but appear to be less likely to be transmitted (6, 17, 28).
We conclude that during the transmission of HLA class I-restricted antigens derived from immunogenic internal viral proteins, dominant variants present in the donor are able to establish infection in the recipient. This has important implications for viral pathogenesis, as many of the transmitted variants will encode CTL escape properties. This study has only looked at the variability of the transmitted virus. The fate of transmitted variants and the extent to which mutants revert following transmission are preserved or are acquired de novo in response to the new host will determine the significance of the passive transmission of internal protein variants. In HIV-infected populations the characteristic signature pattern of amino acid variation in RT is remarkably stable, but each infected individual selects an idiosyncratic pattern of variants. This variability constitutes the viral “challenge” to which naïve partners are exposed and places an enormous burden on any protective vaccine.
Supplementary Material
[Supplemental material]
Acknowledgments
We thank the patients and staff of St. Mary's Hospital in London for making the collection of these samples possible. We also thank A. McLean and C. Bangham for comments and suggestions.
The work was supported by the Medical Research Council (J.F.) and the Wellcome Trust (R.E.P., J.N.W., and P.J.G.), including the program grant SPARTAC. P.J.G. is supported by NIH grant AI046995-06.
We are indebted to the physicians and patients who contributed to the Spanish Swiss Intermittent Therapy Trial (SSITT).
Footnotes
REFERENCES
- 1.Addo, M. M., X. G. Yu, A. Rathod, D. Cohen, R. L. Eldridge, D. Strick, M. N. Johnston, C. Corcoran, A. G. Wurcel, C. A. Fitzpatrick, M. E. Feeney, W. R. Rodriguez, N. Basgoz, R. Draenert, D. R. Stone, C. Brander, P. J. Goulder, E. S. Rosenberg, M. Altfeld, and B. D. Walker. 2003. Comprehensive epitope analysis of human immunodeficiency virus type 1 (HIV-1)-specific T-cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load. J. Virol. 77**:**2081-2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen, T. M., M. Altfeld, X. G. Yu, K. M. O'Sullivan, M. Lichterfeld, S. Le Gall, M. John, B. R. Mothe, P. K. Lee, E. T. Kalife, D. E. Cohen, K. A. Freedberg, D. A. Strick, M. N. Johnston, A. Sette, E. S. Rosenberg, S. A. Mallal, P. J. Goulder, C. Brander, and B. D. Walker. 2004. Selection, transmission, and reversion of an antigen-processing cytotoxic T-lymphocyte escape mutation in human immunodeficiency virus type 1 infection. J. Virol. 78**:**7069-7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen, T. M., D. H. O'Connor, P. Jing, J. L. Dzuris, B. R. Mothe, T. U. Vogel, E. Dunphy, M. E. Liebl, C. Emerson, N. Wilson, K. J. Kunstman, X. Wang, D. B. Allison, A. L. Hughes, R. C. Desrosiers, J. D. Altman, S. M. Wolinsky, A. Sette, and D. I. Watkins. 2000. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407**:**386-390. [DOI] [PubMed] [Google Scholar]
- 4.Barouch, D. H., J. Kunstman, M. J. Kuroda, J. E. Schmitz, S. Santra, F. W. Peyerl, G. R. Krivulka, K. Beaudry, M. A. Lifton, D. A. Gorgone, D. C. Montefiori, M. G. Lewis, S. M. Wolinsky, and N. L. Letvin. 2002. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature 415**:**335-339. [DOI] [PubMed] [Google Scholar]
- 5.Bernardin, F., D. Kong, L. Peddada, L. A. Baxter-Lowe, and E. Delwart. 2005. Human immunodeficiency virus mutations during the first month of infection are preferentially found in known cytotoxic T-lymphocyte epitopes. J. Virol. 79**:**11523-111528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blower, S. M., A. N. Aschenbach, and J. O. Kahn. 2003. Predicting the transmission of drug-resistant HIV: comparing theory with data. Lancet Infect. Dis. 3**:**10-11. [DOI] [PubMed] [Google Scholar]
- 7.Cruse, J. M., M. N. Brackin, R. E. Lewis, W. Meeks, R. Nolan, and B. Brackin. 1991. HLA disease association and protection in HIV infection among African Americans and Caucasians. Pathobiology 59**:**324-328. [DOI] [PubMed] [Google Scholar]
- 8.Derdeyn, C. A., J. M. Decker, F. Bibollet-Ruche, J. L. Mokili, M. Muldoon, S. A. Denham, M. L. Heil, F. Kasolo, R. Musonda, B. H. Hahn, G. M. Shaw, B. T. Korber, S. Allen, and E. Hunter. 2004. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science 303**:**2019-2022. [DOI] [PubMed] [Google Scholar]
- 9.Fabio, G., R. S. Smeraldi, A. Gringeri, M. Marchini, P. Bonara, and P. M. Mannucci. 1990. Susceptibility to HIV infection and AIDS in Italian haemophiliacs is HLA associated. Br. J. Haematol. 75**:**531-536. [DOI] [PubMed] [Google Scholar]
- 10.Fagard, C., A. Oxenius, H. Gunthard, F. Garcia, M. Le Braz, G. Mestre, M. Battegay, H. Furrer, P. Vernazza, E. Bernasconi, A. Telenti, R. Weber, D. Leduc, S. Yerly, D. Price, S. J. Dawson, T. Klimkait, T. V. Perneger, A. McLean, B. Clotet, J. M. Gatell, L. Perrin, M. Plana, R. Phillips, and B. Hirschel. 2003. A prospective trial of structured treatment interruptions in human immunodeficiency virus infection. Arch. Intern. Med. 163**:**1220-1226. [DOI] [PubMed] [Google Scholar]
- 11.Friedrich, T. C., E. J. Dodds, L. J. Yant, L. Vojnov, R. Rudersdorf, C. Cullen, D. T. Evans, R. C. Desrosiers, B. R. Mothe, J. Sidney, A. Sette, K. Kunstman, S. Wolinsky, M. Piatak, J. Lifson, A. L. Hughes, N. Wilson, D. H. O'Connor, and D. I. Watkins. 2004. Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nat. Med. 10**:**275-281. [DOI] [PubMed] [Google Scholar]
- 12.Goulder, P. J., C. Brander, Y. Tang, C. Tremblay, R. A. Colbert, M. M. Addo, E. S. Rosenberg, T. Nguyen, R. Allen, A. Trocha, M. Altfeld, S. He, M. Bunce, R. Funkhouser, S. I. Pelton, S. K. Burchett, K. McIntosh, B. T. Korber, and B. D. Walker. 2001. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature 412**:**334-338. [DOI] [PubMed] [Google Scholar]
- 13.Greggio, N. A., M. Cameran, C. Giaquinto, F. Zacchello, D. Koroliuk, and V. Colizzi. 1993. DNA HLA-DRB1 analysis in children of positive mothers and estimated risk of vertical HIV transmission. Dis. Markers 11**:**29-35. [DOI] [PubMed] [Google Scholar]
- 14.Just, J., L. Louie, E. Abrams, S. W. Nicholas, D. Wara, Z. Stein, and M. C. King. 1992. Genetic risk factors for perinatally acquired HIV-1 infection. Paediatr. Perinat. Epidemiol. 6**:**215-224. [DOI] [PubMed] [Google Scholar]
- 15.Kessler, J. H., B. Mommaas, T. Mutis, I. Huijbers, D. Vissers, W. E. Benckhuijsen, G. M. Schreuder, R. Offringa, E. Goulmy, C. J. Melief, S. H. van der Burg, and J. W. Drijfhout. 2003. Competition-based cellular peptide binding assays for 13 prevalent HLA class I alleles using fluorescein-labeled synthetic peptides. Hum. Immunol. 64**:**245-255. [DOI] [PubMed] [Google Scholar]
- 16.Kiepiela, P., A. J. Leslie, I. Honeyborne, D. Ramduth, C. Thobakgale, S. Chetty, P. Rathnavalu, C. Moore, K. J. Pfafferott, L. Hilton, P. Zimbwa, S. Moore, T. Allen, C. Brander, M. M. Addo, M. Altfeld, I. James, S. Mallal, M. Bunce, L. D. Barber, J. Szinger, C. Day, P. Klenerman, J. Mullins, B. Korber, H. M. Coovadia, B. D. Walker, and P. J. Goulder. 2004. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 432**:**769-775. [DOI] [PubMed] [Google Scholar]
- 17.Leigh Brown, A. J., S. D. Frost, W. C. Mathews, K. Dawson, N. S. Hellmann, E. S. Daar, D. D. Richman, and S. J. Little. 2003. Transmission fitness of drug-resistant human immunodeficiency virus and the prevalence of resistance in the antiretroviral-treated population. J. Infect. Dis. 187**:**683-686. [DOI] [PubMed] [Google Scholar]
- 18.Leslie, A. J., K. J. Pfafferott, P. Chetty, R. Draenert, M. M. Addo, M. Feeney, Y. Tang, E. C. Holmes, T. Allen, J. G. Prado, M. Altfeld, C. Brander, C. Dixon, D. Ramduth, P. Jeena, S. A. Thomas, A. S. John, T. A. Roach, B. Kupfer, G. Luzzi, A. Edwards, G. Taylor, H. Lyall, G. Tudor-Williams, V. Novelli, J. Martinez-Picado, P. Kiepiela, B. D. Walker, and P. J. Goulder. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 10**:**282-289. [DOI] [PubMed] [Google Scholar]
- 19.McMichael, A., and P. Klenerman. 2002. HIV/AIDS. HLA leaves its footprints on HIV. Science 296**:**1410-1411. [DOI] [PubMed] [Google Scholar]
- 20.Milicic, A., C. Edwards, S. Hue, J. Fox, H. Brown, T. Pillay, J. W. Drijfhout, J. Weber, E. C. Holmes, H. T. Zhang, S. J. Fidler, and R. Phillips. 2005. Sexual transmission of single human immunodeficiency virus type 1 virions encoding highly polymorphic multisite cytotoxic T-lymphocyte escape variants. J. Virol. 79**:**13953-13962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore, C. B., M. John, I. R. James, F. T. Christiansen, C. S. Witt, and S. A. Mallal. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296**:**1439-1443. [DOI] [PubMed] [Google Scholar]
- 22.O'Brien, S. J., X. Gao, and M. Carrington. 2001. HLA and AIDS: a cautionary tale. Trends Mol. Med. 7**:**379-381. [DOI] [PubMed] [Google Scholar]
- 23.Oelrichs, R. B., V. A. Lawson, K. M. Coates, C. Chatfield, N. J. Deacon, and D. A. McPhee. 2000. Rapid full-length genomic sequencing of two cytopathically heterogeneous Australian primary HIV-1 isolates. J. Biomed. Sci. 7**:**128-135. [DOI] [PubMed] [Google Scholar]
- 24.Peyerl, F. W., D. H. Barouch, W. W. Yeh, H. S. Bazick, J. Kunstman, K. J. Kunstman, S. M. Wolinsky, and N. L. Letvin. 2003. Simian-human immunodeficiency virus escape from cytotoxic T-lymphocyte recognition at a structurally constrained epitope. J. Virol. 77**:**12572-12578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phillips, R. E., S. Rowland-Jones, D. F. Nixon, F. M. Gotch, J. P. Edwards, A. O. Ogunlesi, J. G. Elvin, J. A. Rothbard, C. R. Bangham, C. R. Rizza, et al. 1991. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354**:**453-459. [DOI] [PubMed] [Google Scholar]
- 26.Price, D. A., P. J. Goulder, P. Klenerman, A. K. Sewell, P. J. Easterbrook, M. Troop, C. R. Bangham, and R. E. Phillips. 1997. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. USA 94**:**1890-1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sipsas, N. V., S. A. Kalams, A. Trocha, S. He, W. A. Blattner, B. D. Walker, and R. P. Johnson. 1997. Identification of type-specific cytotoxic T lymphocyte responses to homologous viral proteins in laboratory workers accidentally infected with HIV-1. J. Clin. Investig. 99**:**752-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turner, D., B. Brenner, J. P. Routy, D. Moisi, Z. Rosberger, M. Roger, and M. A. Wainberg. 2004. Diminished representation of HIV-1 variants containing select drug resistance-conferring mutations in primary HIV-1 infection. J. Acquir. Immune Defic. Syndr. 37**:**1627-1631. [DOI] [PubMed] [Google Scholar]
- 29.Wolinsky, S. M., C. M. Wike, B. T. Korber, C. Hutto, W. P. Parks, L. L. Rosenblum, K. J. Kunstman, M. R. Furtado, and J. L. Munoz. 1992. Selective transmission of human immunodeficiency virus type-1 variants from mothers to infants. Science 255**:**1134-1137. [DOI] [PubMed] [Google Scholar]
- 30.Yang, O. O., P. T. Sarkis, A. Ali, J. D. Harlow, C. Brander, S. A. Kalams, and B. D. Walker. 2003. Determinant of HIV-1 mutational escape from cytotoxic T lymphocytes. J. Exp. Med. 197**:**1365-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang, L. Q., P. MacKenzie, A. Cleland, E. C. Holmes, A. J. Brown, and P. Simmonds. 1993. Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon primary infection. J. Virol. 67**:**3345-3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu, T., H. Mo, N. Wang, D. S. Nam, Y. Cao, R. A. Koup, and D. D. Ho. 1993. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science 261**:**1179-1181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental material]