Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens (original) (raw)
. Author manuscript; available in PMC: 2007 Jan 29.
Published in final edited form as: Nat Rev Immunol. 2006 Apr;6(4):318–328. doi: 10.1038/nri1810
Abstract
The central nervous system (CNS) regulates innate immune responses through hormonal and neuronal routes. The neuroendocrine stress response and the sympathetic and parasympathetic nervous systems generally inhibit innate immune responses at systemic and regional levels, whereas the peripheral nervous system tends to amplify local innate immune responses. These systems work together to first activate and amplify local inflammatory responses that contain or eliminate invading pathogens, and subsequently to terminate inflammation and restore host homeostasis. Here, I review these regulatory mechanisms and discuss the evidence indicating that the CNS can be considered as integral to acute-phase inflammatory responses to pathogens as the innate immune system.
The local acute-phase inflammatory response is characterized by_rubor_ (redness), dolor (pain) and_calor_ (heat). Although these are all cardinal clinical features of inflammation, their proximal triggers are neural in origin. Similarly, the systemic acute-phase response also involves key neural elements — fever and activation of the central hormonal-stress response — mediated by the effects of immune factors on the hypothalamus. The cellu lar and molecular components of the innate immune system provide the first line of defence against invading pathogens1, through recognition of pathogen-associated molecular patterns (PAMPs), and initial nonspecific cellular and humoral responses2. However, immune mediators and cytokines that are subsequently released by the innate immune system rapidly activate neural responses that both amplify local immune responses to clear pathogens and trigger systemic neuroendocrine and regional neural responses that eventually return the system to a resting state. Although this interplay constitutes an important feedback loop that optimizes innate inflammatory responses to invading pathogens, prolonged or inappropriate central nervous system (CNS) counter-regulatory responses might also predispose the host to excess inflammation (in the context of inadequate hormonal suppression) or uncontrolled infection (in the context of excess or prolonged anti-inflammatory hormonal responses). These can lead to pathological and lethal effects, including toxic shock, tissue damage and death.
In this Review, I describe how specific elements of the CNS and innate immune system interact, present evidence indicating that these two systems form a cohesive and integrated early host response to pathogens, and identify areas for future research efforts to fully elucidate this interaction.
General principles and areas of controversy
The nervous system has several characteristics that make it an ideal partner with the innate immune system in immediate nonspecific host defence. It reacts rapidly (in the order of milliseconds to minutes) to many types of nonspecific environmental stimuli. Neurotransmitters and neuropeptides often bind to G-protein-coupled receptors that activate the same secondary signalling pathways (such as those including protein kinase A, cyclic AMP and protein kinase C) as signals triggered by immune mediators. Immune mediators often interact with neuro-transmitter receptors3 and also modulate neural pathways that are integral to the acute-phase response, such as pain4,5. In turn, neuropeptides (such as substance P) trigger the release of pro-inflammatory mediators (such as histamine) that might amplify or facilitate inflammation by enhancing vasodilation, blood flow, vascular leakiness and leukocyte trafficking to sites of inflammation.
G-protein-coupled receptors —
Cell-surface receptors that are coupled to G-proteins, and have seven transmembrane-spanning domains. The acetylcholine, adrenergic and neuropeptide receptors are all members of this family. Typically, activation of the G-protein-coupled receptor produces a diffusible second messenger that, in turn, triggers various biochemical cascades.
The effects of neural factors on inflammatory responses, while rapid in onset, might vary over time, enhancing or dampening these responses6,7. Such variations in effects, coupled with the lower magnitude of neural compared to immune stimuli (2–3 fold compared to more than 1,000 fold, respectively) might be considered by some as evidence that neural regulation of inflammatory responses are of little biological relevance. In fact, this pattern and scale of neural effects are well suited for physiological modulators, because normal endogenous systems are characterized by tempered oscillations over time, rather than by large changes8. The nervous system is therefore ideally positioned to modulate the immediate nonspecific inflammatory response to immune stimuli and partner with it in a unified response to pathogens.
CNS-mediated regulation of immunity occurs through systemic, regional and local routes (FIG. 1). The peripheral nervous system provides a first line defence at local sites of inflammation through the release of neuropeptides that generally increase local inflammatory responses. The sympathetic (or adrenergic) nervous system (SNS), and the parasympathetic (or cholinergic) nervous system generally inhibit inflammation at a regional level, through innervation of immune organs. Neuroendocrine responses control inflammation at a systemic level by the hypothalamic–pituitary–adrenal (HPA) axis through the anti-inflammatory effects of glucocorticoids released from the adrenal cortex; by the hypothalamic–pituitary–gonadal (HPG) axis through sex hormones released from the ovaries and testes; and by the hypothalamic–pituitary–thyroid hormone axis through thyroid hormones released from the thyroid gland.
Figure 1. Schematic illustration of connections between the nervous and immune systems.
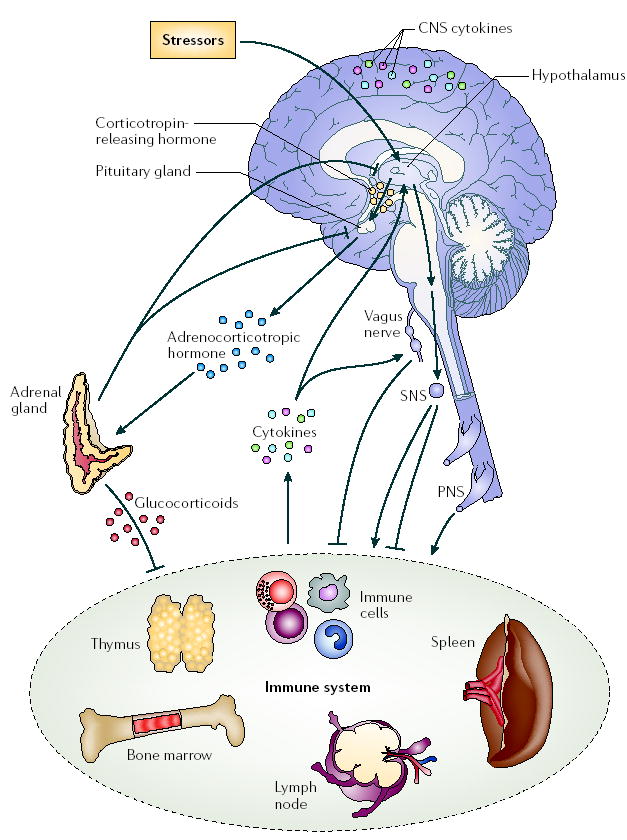
Signalling between the immune system and the central nervous system (CNS) through systemic routes, the vagus nerve, the hypothalamic–pituitary–adrenal (HPA) axis, the sympathetic nervous system (SNS) and the peripheral nervous system (PNS) are shown. Figure modified with permission from Molecular Psychiatry REF. 140 © (2005) Macmillan Magazines Ltd.
Sympathetic nervous system —
(SNS). A division of the autonomic nervous system that consists of fibres projecting from the central nervous system, through ganglia near the spinal cord, to innervate organs such as the heart, lungs, intestine, blood vessels and sweat glands. In general, sympathetic nerves dilate the pupils, constrict peripheral blood vessels and increase heart rate.
Parasympathetic nervous system —
A division of the autonomic nervous system that consists of nerve fibres projecting from the central nervous system and sacral portion of the spinal cord, which extend to nerve-cell clusters (ganglia) at specific sites, from which fibres are distributed to blood vessels, glands and other internal organs. Functions of parasympathetic nerves include slowing the heart rate; inducing the secretion of bile, insulin and digestive juices; dilating peripheral blood vessels; and contracting the bronchioles, pupils and oesophagus.
Extensive literature has shown that many immune cells contain the molecular machinery required to respond to neural factors, including receptors for neurotransmitters, neuropeptides and neurohormones, and the components of their signalling pathways. However, some debate still exists regarding the specific immune-cell types that express neural receptors9 and the biological significance of neural modulation of immune responses. This is, in part, because the effects of neural factors on some elements of innate immunity, such as macrophages and cytokine production, have been more extensively studied than others, such as dendritic cells (DCs) and Toll-like receptor (TLR) expression, which have been only superficially addressed. In addition, the mechanisms of the effects of neural factors on many immune responses have not been fully investigated. Also, neural factors might have varying effects over the time course of an immune response, or at different stages of immune-cell maturation10,11.
Similarly, although the effects of pathogen products, such as bacterial lipopolysaccharide (LPS), on CNS responses have been long established, detailed analyses of the mechanisms of these effects have only recently begun to be addressed. Therefore, issues such as the potential differences of various PAMPs on patterns of CNS activation have not been studied. It has only recently been shown that LPS can trigger CNS inflammation directly through TLR4, without the involvement of peripheral cytokines12; that microglia in the CNS express TLRs after exposure to various pathogens or pathogen products, such as LPS, peptidoglycans and CpG DNA; and that mice deficient in the TLR adaptor protein MyD88 (myeloid differentiation primary-response gene 88) show a decreased CNS-mediated inflammatory response to Streptococcus pneumoniae2,13,14.
HPA-axis-mediated regulation of immunity
The HPA axis provides an important physiological feedback loop of inflammation through the anti-inflammatory effects of glucocorticoids (BOX 1). Although the Nobel Prize was awarded for the use of glucocorticoids in the treatment of rheumatoid arthritis in 1950 (REF. 15), it was not until recently that the important physiological role of glucocorticoids in regulating multiple aspects of immune-cell function was recognized. For example, glucocorticoids can cause a shift in adaptive immune responses from a T helper 1 (TH1) type to a TH2 type, largely through inhibiting the production of the TH1-cell-inducing cytokine interleukin-12 (IL-12) by DCs and macrophages16, and in physiological concentrations, glucocorticoids can increase delayed-type hypersensitivity6. Moreover, as highlighted in this Review, they also regulate the innate immune response to bacterial and viral infection.
Delayed-type hypersensitivity —
A cellular immune response to antigen that develops over 24–72 hours with the infiltration of T cells and monocytes, and depends on the production of T helper 1-cell-specific cytokines.
Box 1 Components of the hypothalamic–pituitary–adrenal axis
The physiological feedback loop through which glucocorticoid release is regulated consists of a set of brain regions (the hypothalamus) and endocrine organs (the pituitary and cortex of the adrenal glands) known as the hypothalamic–pituitary–adrenal (HPA) axis (FIG. 1). Systemic exposure of the host to pro-inflammatory stimuli (such as bacterial lipopolysaccharide) as well as to physical or psychological stimuli, results in secretion of corticotropin-releasing hormone from cells of the paraventricular nucleus of the hypothalamus into the hypophyseal blood supply around the pituitary gland. This stimulates the release of adrenocorticotropic hormone from the anterior pituitary into the blood, which in turn stimulates the synthesis and release of endogenous glucocorticoids from the adrenal cortex (FIG. 1). This bi-directional communication between the immune system and the central nervous system (CNS)136,137, in which cytokines, including interleukin-1 (IL-1), IL-6 and tumour-necrosis factor (TNF), signal to the brain, and the brain responds by regulating the immune system, in part, through the anti-inflammatory effects of glucocorticoids, constitutes the main hormonal negative-feedback loop for CNS regulation of immunity. In addition to their role in regulating the immune system, glucocorticoids also downregulate the HPA axis itself, and are essential for the maintenance of several homeostatic mechanisms in the body, including the CNS and cardiovascular system, as well as for metabolic homeostasis.
Impaired HPA-axis function and disease
A blunted HPA axis is seen in a wide range of autoimmune and inflammatory diseases across several species, such as thyroiditis and scleroderma in chickens17; systemic lupus erythematosus (SLE) in some mouse models (such as MRL mice); inflammatory arthritis, experimental allergic encephalomyelitis and other autoimmune and inflammatory diseases in certain susceptible rat strains18; and rheumatoid arthritis, Sjogren’s syndrome, SLE, irritable bowel syndrome, chronic fatigue syndrome and fibromyalgia in humans19,20. Furthermore, interruptions of the HPA axis, through surgical interventions (adrenalectomy or hypophysectomy) or through pharmacological intervention with glucocorticoid antagonists (such as RU486), renders otherwise relatively inflammation-resistant animals highly susceptible to inflammation and increases mortality from septic shock following exposure to infectious or pro-inflammatory triggers, such as streptococcal cell walls21, Salmonella typhimurium22, cytomegalovirus23 and Shiga toxin24. Conversely, reconstitution of the HPA axis by treatment with exogenous glucocorticoids or by hypothalamic transplantation21 reverses this effect and prevents mortality in these animal models21–25.
Glucocorticoid resistance
Impaired glucocorticoid control of inflammation might also result from lack of glucocorticoid responsiveness, or glucocorticoid resistance, of target cells or tissues, and might contribute to autoimmune, inflammatory and allergic diseases26–31 (TABLE 1). Glucocorticoid receptors are members of a superfamily of nuclear hormone receptors (which includes the receptors for progesterone, oestrogen, androgen, mineralocorticoid and thyroid hormone) that reside in the cytoplasm or nucleus of almost all cells and, once bound to hormone, move to the nucleus and function as transcription factors32. Glucocorticoid resistance might result from mutations or polymorphisms of glucocorticoid receptors26,33,34 or impaired interactions of the receptor with one of the >200 cofactors, such as corticosterone-binding globulin35 and multidrug resistance-1 (REF. 36), that are required for glucocorticoid-receptor function. In addition, expression of glucocorticoid receptor-β, an inactive form that does not bind ligand or activate gene transcription37,38, is induced during chronic inflammation39, and might result in relative glucocorticoid resistance in such states.
Table 1.
Mechanisms of glucocorticoid resistance in autoimmune and inflammatory diseases
| Disease | Glucocorticoid receptor mutations and polymorphisms | Glucocorticoid receptor expression | Associated protein expression levels | Glucocorticoid sensitivity | Refs |
|---|---|---|---|---|---|
| Rheumatoid arthritis | Glucocorticoid receptor-β polymorphism: (A/G) TTTA | Increased stability of glucocorticoid receptor-β mRNA | ND | ND | 26 |
| Systemic lupus erythematosus | Glucocorticoid receptor polymorphism: (T/C) GGA | ND | Increased multidrug resistance-1 | ND | 33,36 |
| Systemic lupus erythematosus- associated nephritis | Glucocorticoid receptor mutation (frameshift mutation, resulting in early stop signal) | Decreased | ND | ND | 34 |
| Crohn’s disease | ND | Increased glucocorticoid receptor-β mRNA | Increased binding of cortisol to serum albumin | ND | 31,35 |
| Asthma | ND | Increased glucocorticoid receptor-β mRNA | ND | Decreased binding to glucocorticoid- response elements | 27,28 |
| Multiple sclerosis | ND | ND | ND | Decreased | 29,30 |
Finally, pathogens might themselves induce gluco-corticoid resistance. We have recently shown that Bacillus anthracis lethal toxin selectively and potently represses the activity of glucocorticoid receptor and other nuclear hormone receptors in a non-competitive fashion40, by preventing glucocorticoid-receptor binding to DNA through interactions with one or more of its cofactors or accessory proteins40. Should other bacterial products behave similarly, it could provide a mechanism by which invading pathogens might interfere with the host’s glucocorticoid-mediated negative-feedback control of inflammation, potentially predisposing the host to toxic shock. Indeed, although the precise mechanisms of this effect are unclear, we have shown that interference with glucocorticoid production, such as by adrenalectomy, render otherwise anthraxlethal-toxin-resistant mouse strains highly susceptible to rapid death from this toxin41.
Excess HPA-axis activity and illness
An excess of circulating glucocorticoids, which can occur as a result of chronic stress, is associated with increased susceptibility to viral infections, prolonged wound healing or decreased antibody production after vaccination42,43. Fluctuations in the levels of circulating glucocorticoids, such as circadian variations or fluctuations that occur during exercise, also suppress IL-1β and tumour-necrosis factor (TNF) production by leukocytes44,45.
Taken together, these studies indicate that a fine balance of glucocorticoids is required for the maintenance of immune homeostasis and to avoid excessive immunosuppression and death from overwhelming infection, or death from shock resulting from excessive cytokine and pro-inflammatory responses (FIG. 2).
Figure 2. Effects of glucocorticoids on immune-cell populations.
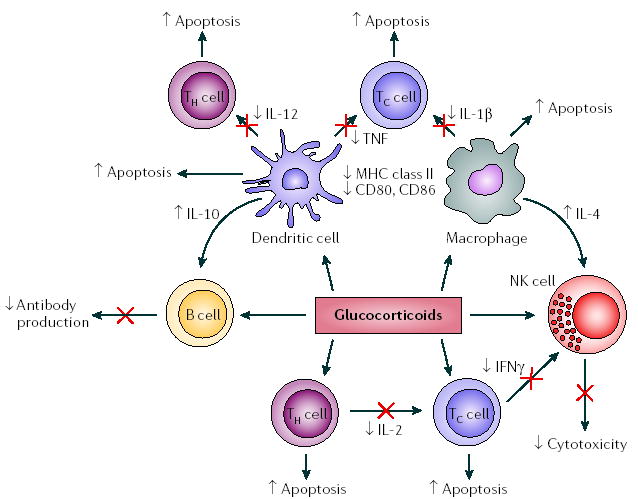
Glucocorticoids act on immune cells both directly and indirectly to suppress the induction of pro-inflammatory responses. They inhibit the production of pro-inflammatory cytokines, such as interleukin-1β (IL-1β) and tumour-necrosis factor (TNF), while promoting the production of anti-inflammatory cytokines, such as IL-10, by macrophages and dendritic cells. They also promote apoptosis of macrophages, dendritic cells and T cells, leading to inhibition of immune responses. IFNγ, interferon-γ; NK cell, natural killer cell; TC, cytotoxic T cell; TH, T helper cell.
Glucocorticoid effects on innate immune-cell function
In general, glucocorticoids suppress maturation, differentiation and proliferation of all immune cells, including DCs and macrophages (FIG. 2). Glucocorticoids inhibit DC differentiation depending on the stage of maturation46 and the DC subtype11. Glucocorticoids also reduce the capacity of DCs to promote allostimulatory responses and efficient activation of naive T cells in vitro, possibly owing to downregulation of expression of MHC class II and co-stimulatory molecules46,47. In vivo, dexa methasone (a synthetic glucocorticoid) impairs the ability of rat thymic DCs to produce IL-1β and TNF, but not IL-10 (REF. 48). Dexamethasone also inhibits IL-12p40 production by LPS-stimulated human monocytes by downregulation of activation of JUN N-terminal kinase (JNK), mitogen-activated protein kinase (MAPK), activator protein 1 (AP1) and nuclear factor-κB (NF-κB)49. In addition, in both mice that are transgenic for corticotropin-releasing hormone (CRH) (which chronically overproduce glucocorticoids) and in wild-type mice that are treated chronically with corticosterone50, splenic germinal centres fail to form, owing to impaired development of mature follicular DC networks.
Glucocorticoids regulate gene transcription by binding to hormone-response elements in the promoters of various genes, but they can also regulate gene expression through interaction with other transcription factors, such as NF-κB or AP1 (REF. 32). For example, NF-κB normally exists in an inactive state, bound to inhibitor of NF-κB (IκB). Upon cell activation, NF-κB is released from IκB and translocates to the nucleus, where it can bind to specific promoter sites and upregulate the expression of pro-inflammatory cytokines51 (FIG. 3). Glucocorticoids inhibit NF-κB by interacting with RelA (also known as p65, a subunit of NF-κB) and by inducing the expression of IκB51. Consistent with this inhibition of NF-κB, physiological doses and preparations of glucocorticoids (such as corticosterone and hydrocortisone) lead to decreased production of pro-inflammatory cytokines (such as IL-1, IL-6 and TNF)52.
Figure 3. Molecular mechanisms of neurotransmitter and glucocorticoid regulation of cytokine production.
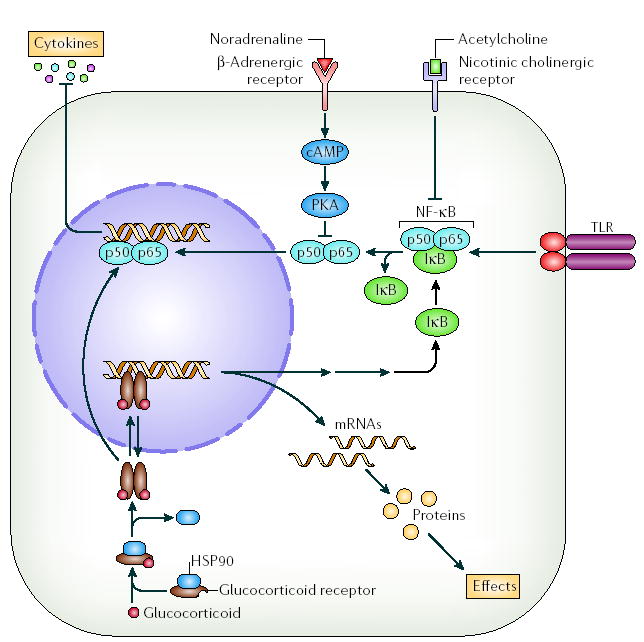
Glucocorticoids bind to glucocorticoid receptors in the cytosol, which displaces heat-shock protein 90 (HSP90) and allows receptor dimerization, movement into the nucleus and binding of the glucocorticoid–glucocorticoid-receptor complex to DNA. This leads to transcription and translation of proteins, including inhibitor of nuclear factor-κB (IκB). IκB then sequesters NF-κB, preventing it from activating transcription of pro-inflammatory cytokines. In addition, the glucocorticoid–glucocorticoid-receptor complex can interact with NF-κB directly to suppress cytokine production136. Noradrenaline binding to β-adrenergic receptors at the cell surface induces cyclic AMP (cAMP) and protein kinase A (PKA) activation, which inhibits cytokine production through inhibition of NF-κB. Acetylcholine binds to nicotinic cholinergic cell-surface receptors and inhibits cytokine production again through inhibition of NF-κB95. TLR, Toll-like receptor.
Glucocorticoid effects on immune-cell trafficking
Both endogenous and synthetic (dexamethasone) glucocorticoids inhibit the expression of many cell-adhesion molecules that are involved in cell trafficking, including intracellular adhesion molecule 1 (ICAM1), endothelial-leukocyte adhesion molecule 1 (ELAM1; also known as E-selectin)53 and vascular adhesion molecule 1 (VCAM1)54,55. Glucocorticoids also inhibit the production and secretion of chemokines, such as CC-chemokine ligand 2 (CCL2; also known as MCP1) and CXC-chemokine ligand 8 (CXCL8; also known as IL-8) by eosinophils56, as well as expression of the mRNA encoding the eosinophil chemoattractant IL-5 by mast cells and T cells57,58. Finally, dexamethasone downregulates T-cell expression of CCL8 (also known as MCP2) and CCL7 (also known as MCP3), which are chemotactic for many cell types including monocytes, DCs, and natural killer (NK) cells59.
Glucocorticoid effects on TLR expression
Although the effects of glucocorticoids on TLR expression by immune cells have not been addressed, recent studies indicate that glucocorticoids might alter TLR expression levels of non-immune cells. For example, in respiratory epithelial cells, dexamethasone upregulates TLR2 mRNA levels60, and might function synergistically with TNF and interferon-γ (IFNγ)60,61. Glucocorticoids can also upregulate expression of TLR2 mRNA by human epithelial cells in vitro, through upregulation of MAPK phosphatase 1 (MKP1)62. Interestingly, TLR2-deficient mice have an impaired corticosterone response to LPS, which is related to decreased amounts of IL-1, IL-6 and TNF in the blood and adrenal glands (REF. 63). These mice also have increased mortality rates when exposed to Mycobacterium tuberculosis and Staphylococcus aureus64,65. TLR2 and TLR4 have also been found to be expressed in cells of the human adrenal cortex63. Although their function in this location has not been defined, further clarification of interactions between immune cells, pathogens and TLRs expressed in the adrenal cortex could elucidate the role of this gland in pathogen defence. Indeed, recent studies indicate that during viral infection an increased IL-6 response might directly induce adrenal glucocorticoid responses that are independent of hypothalamic CRH66, indicating more of a direct role for the adrenal glands in host defence.
Together, these effects of glucocorticoids on innate immune responses tend to prevent immune-cell trafficking from immune organs to sites of inflammation, reduce activation of innate immune cells and cytokine production at sites of inflammation, and dampen the inflammatory response (TABLE 2).
Table 2.
Interaction of neural and neuroendocrine factors with components of the innate immune system
| Toll-like receptors | Dendritic cells | Macrophages | Natural killer cells | Innate cytokines (IL-1, IL-6, IL-12 and TNF) | Chemokines (CCL2 and CXCL8) | Chemokine receptors | |
|---|---|---|---|---|---|---|---|
| Glucocorticoid | + | − | − | − | − | − | ND |
| Noradrenaline | ND | ND | − | − | − | + | ND |
| Neuropeptide Y | ND | ND | − | ND | − | ND | ND |
| Acetylcholine | ND | ND | − | ND | − | + | ND |
| CRH | ND | ND | ND | + | + | ND | ND |
| CGRP | ND | − | ND | + | + | + | ND |
| Substance P | ND | ND | ND | ND | + | + | ND |
| α-MSH | − | ND | ND | ND | − | + | ND |
| Opioids | ND | ND | ND | ND | − | + | − |
| VIP | − | ND | − | − | − | − | ND |
| Oestrogen | ND | − | ND | ND | − | − | ND |
| Progesterone | ND | + | ND | ND | ND | − | ND |
Regional SNS control of immunity
The SNS includes a neuronal component that regulates immunity at a regional level through the innervation of immune organs and the release of noradrenaline, and a hormonal component that regulates immunity systemically through the release of adrenaline from the medulla of the adrenal glands (BOX 2).
Noradrenaline —
The primary neurotransmitter of the sympathetic nervous system. It is a biogenic amine derived from tyrosine and its metabolite dopamine, which is converted to noradrenaline by the enzyme β-hydroxylase.
Adrenaline —
A neurotransmitter of the sympathetic nervous system. It is a biogenic amine derived from tyrosine and its metabolite dopamine, which is converted to adrenaline from noradrenaline by the enzyme phenylethanolamine-_N_-methyl transferase.
Box 2 Components of the sympathetic nervous system
The neuronal component of the sympathetic nervous system consists of brain regions and noradrenergic sympathetic nerve fibres that release the neurotransmitter noradrenaline and connect central nervous system (CNS) adrenergic brainstem regions to primary and secondary lymphoid organs. Noradrenaline and related ligands bind to two forms of the adrenergic receptor, α and β, which are both classic seven-transmembrane domain G-protein-coupled receptors composed of two different subunits that assemble as heterodimers to form multiple subtypes138. Binding of noradrenaline to β2-adrenergic receptors on dendritic cells and macrophages leads to upregulation of cyclic AMP and activation of protein kinase A, which in turn leads to suppression of pro-inflammatory cytokine production, such as tumour-necrosis factor (TNF), interleukin-1 (IL-1), IL-6 and IL-12 through inhibition of nuclear factor-κB139.
Noradrenaline effects on innate immunity
Most studies addressing the direct effects of catecholamines, such as noradrenaline, and the SNS on immunity have examined their effects on adaptive immunity and antibody production67, and less is known about adrenergic effects on innate immunity. In vivo studies in which the SNS is ablated by chemical sympathectomy or interrupted surgically by cutting sympathetic nerve innervation of lymphoid organs, indicate that the SNS has an important role in regulating immunity at a regional level. Although some studies show that catecholamines and SNS activation increase immune and pro-inflammatory responses68,69, most studies indicate that SNS effects are inhibitory10. Activation of the SNS that occurs following stressful stimuli, such as exercise and psychological stress, is associated with decreased NK-cell activity70, which can be prevented by pretreatment with adrenergic receptor antagonists. Furthermore, excessive SNS-mediated signalling to the immune system caused by a large release of noradrenaline (for example, soon after chemical sympathectomy) can be immunosuppressive7,71.
In vitro, noradrenaline mediates its immunosuppressive effects on DCs and monocytes by inhibiting the production of pro-inflammatory cytokines, including TNF, IL-1, IL-6 and IL-12, while upregulating the production of anti-inflammatory cytokines, such as IL-10, from these cells10,72. Pharmacological blockade of adrenergic receptors has been shown to potentiate IL-6 secretion and inhibit IL-10 secretion in vivo73,74. Noradrenaline also increases secretion of the chemotactic chemokine CXCL8 by fibroblasts isolated from patients with rheumatoid arthritis and increases the migration of NK cells, monocytes and macrophages, but inhibits DC migration in vitro10,75,76. Noradrenaline inhibits the in vitro DC-chemotactic response to the chemokines CCL19 and CCL21 (which are important for DC migration from the site of antigen to regional lymph nodes), through upregulation of anti-inflammatory IL-10 production10. Taken together, it has been proposed that the effects of the SNS on DCs might not only have an anti-inflammatory role but might also contribute to the clearance of pathogens and modulation of the type and strength of the adaptive response10. However, as depletion of noradrenaline has been shown to reduce resistance to some bacterial infections76, the role of SNS responses in bacterial host defence remains to be determined.
Neuropeptide Y effects on immune cells
Neuropeptide Y (NPY) is synthesized and released simultaneously with noradrenaline from sympathetic neurons and other cells throughout the body, including cells in the adrenal medulla and possibly immune cells77.
NPY binds to any of five G-protein-coupled NPY receptors (Y1–5), which are differentially expressed in tissues78. The Y1 receptor is expressed widely throughout the body, including by leukocytes, whereas the Y2 and Y5 receptors are mainly expressed in the CNS. Exposure of macrophages to NPY inhibits IL-6 secretion, and when co-cultured with noradrenaline, its secretion is further inhibited76,79.
These studies indicate that the anatomical and molecular machinery exists to allow bi-directional signalling between the SNS and immune system. Activation of the innate inflammatory response by pathogens leads to the rapid release of innate cytokines (IL-1 and TNF) that, in turn, stimulate the SNS to release noradrenaline and related transmitters from sympathetic nerve endings in immune organs. This inhibits NK cells and macrophage pro-inflammatory cytokine production, providing negative-feedback inhibition to dampen the inflammatory response and restore homeostasis. By contrast, the noradrenaline-mediated stimulation of immune-cell migration and CXCL8 production indicates that, in some circumstances, local or regional release of noradrenaline might serve to perpetuate some aspects of local innate immune responses and inflammation, and could contribute to resolution of the response by promoting wound healing.
Adrenal medulla factor effects on immunity
Whereas noradrenaline and NPY released by sympathetic nerve endings provide regional regulation within immune organs, adrenaline and NPY released from the adrenal medulla regulate immunity systemically. Direct administration of adrenaline decreases circulating numbers of monocytes, B and T cells, and NK cells in vivo, by signalling through β-adrenergic receptors expressed by immune cells80,81. In animal studies, although adrenaline infusion has no effect on mortality from toxic shock, β-adrenergic-receptor blockade increases mortality, highlighting the role of the SNS in this syndrome81.
In addition to adrenaline, the adrenal medulla contains and releases large amounts of IL-6 and TNF in response to inflammatory stimuli, such as LPS, IL-1α and IL-1β82. The discovery of antimicrobial chromogranin peptides that are released simultaneously with adrenaline from chromaffin cells of the adrenal medulla83 coupled with the presence of TLRs on cells of the adrenal cortex raise the interesting possibility that the adrenal glands might have a more direct role in the response to pathogens, activation of innate immune responses and clearing of infectious agents than previously thought.
In summary, most of the systemic effects of the SNS are anti-inflammatory, although some effects increase inflammation, such as the induction of chemotaxis and chemotactic chemokines such as CXCL8 (TABLE 2).
Regional parasympathetic control of immunity
The parasympathetic nervous system modulates immune responses at a regional level through both the efferent and afferent fibres of the vagus nerve. The afferent fibres of the vagus nerve can signal the presence of peripheral inflammation to the brain, through IL-1 receptors expressed by paraganglia cells located in parasympathetic ganglia5. Therefore, during inflammation in the gut or peritoneum, IL-1 released by activated innate immune cells binds to paraganglia cells, activates afferent fibres of the vagus nerve and induces rapid activation of parasympathetic brainstem regions. This is the first step in the ‘inflammatory reflex’, which leads to the release of acetylcholine from efferent vagus nerve fibres and resultant negative-feedback control of inflammation84. Cutting the vagus nerve both prevents immune signalling to the brain with its concomitant activation of cholinergic brainstem regions and removes vagal control of inflammation and toxic shock5,84,85. There is some controversy in the literature regarding the cellular source of acetylcholine that regulates inflammation9. It has been suggested that the source of acetylcholine acting on macrophages is more likely to be immune-cell derived than released from nerve endings9.
Vagus nerve —
The main nerve trunk of the parasympathetic nervous system. It contains both afferent fibres that carry signals from the periphery to the brain, and efferent fibres that carry signals from the brain to the peripheral organs that it innervates.
Cholinergic effects on innate immunity
The primary parasympathetic neurotransmitter is acetylcholine, which binds to two general receptor subtypes, nicotinic and muscarinic cholinergic receptors, each of which consist of many different subunits that heterodimerize and provide cell and tissue specificity for cholinergic effects. Both of these receptors are found in immune cells, but nicotinic receptors specifically mediate cholinergic anti-inflammatory effects in macrophages.
The main cholinergic receptor expressed on macrophages is the α7 subunit of the nicotinic acetylcholine receptor86. Activation of this receptor on macrophages inhibits NF-κB signalling, thereby inhibiting pro-inflammatory cytokine production86 (FIG. 3). A recent report indicates that nicotine, a cholinergic agonist for the α7 subunit, inhibits macrophage cytokine production by recruiting JAK2 (Janus kinase 2) to the α7 subunit. This initiates the anti-inflammatory STAT3 (signal transducer and activator of transcription 3) and SOCS3 (suppressor of cytokine signalling 3) signalling cascade87. Interestingly, administration of nicotine promotes the survival of animals that are exposed to various inflammatory stimuli88,89, indicating that acetylcholine receptor α7-subunit agonists might be useful for treatment of inflammatory disease.
The cholinergic neuronal inflammatory reflex pathway also prevents increased serum concentrations of TNF during toxic shock, through the release of acetylcholine from the vagus nerve, indicating that this pathway has an important role in preventing excessive inflammatory responses. Accordingly, vagotomy exacerbates the TNF response to inflammatory stimuli and sensitizes animals to the lethal effects of endotoxin, whereas cholinergic agonists and vagus-nerve stimulation prevent these effects84. Acetylcholine also inhibits endotoxin-induced release of pro-inflammatory cytokines (IL-1, IL-6 and TNF) but not anti-inflammatory cytokines (IL-10) from macrophages86,90.
Additional support for a role for the parasympathetic nervous system in dampening inflammatory responses is provided by acetylcholine-mediated inhibition of high mobility group box 1 (HMGB1), a protein released by necrotic cells, and activated macrophages and somatic cells during severe sepsis. HMGB1, which is secreted later than the ‘classic’ pro-inflammatory cytokines (12–18 hours after administration of LPS_in vivo_ compared with TNF and IL-1, which peak at 2 hours and 4–6 hours, respectively)91, might signal through TLR2 and TLR4 to stimulate the production of TNF and IL-1. In turn, expression of HMGB1 is induced by activated macrophages, NK cells and mature DCs in response to IL-1, TNF and IFNγ. Consistent with a role for HMGB1 in sepsis, HMGB1-specific antibodies have been shown to prevent the lethal effects of endotoxin in animal models92,93.
The effects of the parasympathetic nervous system on leukocyte trafficking are less clear, as acetylcholine has been shown to increase the production of CCL2 by monocytes94, but both vagus-nerve stimulation and an acetylcholine agonist, acting through the α7 subunit, have been shown to inhibit leukocyte recruitment to endothelial cells by suppressing the expression of VCAM1 (REF. 95).
Therefore, as for glucocorticoid and SNS regulation of innate immunity, the parasympathetic nervous system is activated by cytokines released during activation of the innate immune system, and in turn provides a negative-feedback control of innate immune responses to restore homeostasis.
Local peripheral nervous system control
The peripheral nervous system regulates inflammation locally at inflammatory sites through the release of neuropeptides from sensory peripheral nerves that are involved in pain, touch and temperature perception. Peripheral neuropeptides that are known to regulate inflammation include CRH, substance P and calcitonin gene-related peptide (CGRP)96. Numerous studies have shown that these neuropeptides are expressed at sites of inflammation and are generally pro-inflammatory97,98, thereby favouring pathogen clearance (FIG. 4). The stimulation of peripheral nerves by innate immune mediators results in the characteristic features of local inflammation, including vasodilation, vascular leakiness, oedema and pain. Antagonists to these neuropeptides generally reduce inflammation at inflamed sites, and cutting peripheral nerves reduces inflammation that is distal to the site of the nerve lesion99.
Figure 4. Effects of the peripheral nervous system on immune-cell populations in lymphoid organs.
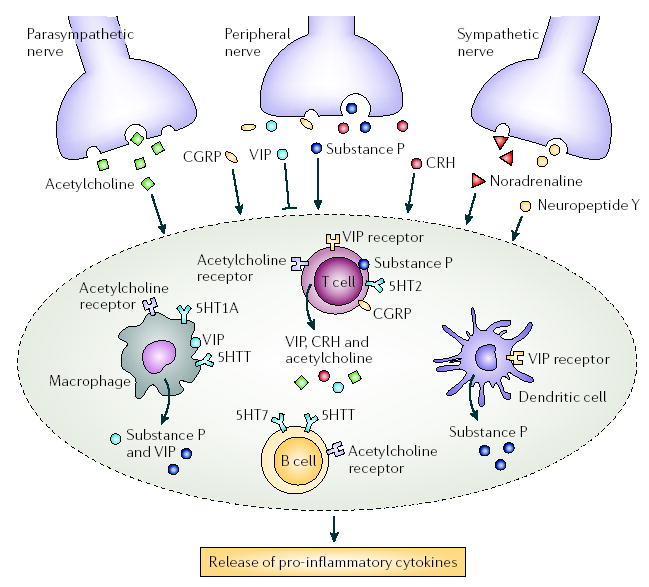
Neurotransmitters released through nerve terminals can both directly and indirectly affect immune responses. With the exception of vasoactive intestinal peptide (VIP), these neuropeptides stimulate the production of pro-inflammatory cytokines. In addition, some neuropeptides and their receptors are expressed by immune cells, including acetylcholine; VIP; calcitonin gene-related peptide (CGRP); substance P; muscarinic and nicotinic acetylcholine receptors; and serotonin receptors, such as 5HTT (5-hydroxytryptamine transporter), 5HT1A, 5HT2 and 5HT7. CRH, corticotropin-releasing hormone.
Corticotropin-releasing hormone
Although hypothalamic CRH has an anti-inflammatory effect through the induction of glucocorticoid release from the adrenal glands, it is pro-inflammatory when released locally from nerve endings at sites of inflammation97. Macrophages and B and T cells all express CRH receptors, and B and T cells also express CRH mRNA and release CRH protein100. Highlighting the biological significance of these effects are studies showing that the CRH receptor 1 antagonist, antalarmin, suppresses macrophage TNF, IL-1 and IL-6 production and secretion in vitro97 and reduces inflammation in vivo in a rat model of inflammatory arthritis101.
Calcitonin gene-related peptide
CGRP, which is a 37-amino-acid neuropeptide that mediates its effects through G-protein-coupled receptors102, is mainly expressed by sensory nerve fibres that innervate most organs in the body. CGRP has been reported to increase or suppress cytokine production by T cells and peripheral blood mononuclear cells (PBMCs)98,103, suppressing IL-2 production from T cells in vitro, but stimulating IL-6 and TNF release from human PBMCs after LPS administration_in vitro_98. Given these contradictory findings, more research is needed to elucidate the effects of CGRP on the innate immune system. CGRP increases the synthesis of CXCL8 by human epithelial cells, but has no effect on CCL2 (REF. 104), whereas in explants from human dental pulp, CGRP administration moderately increased CCL2 production while CXCL8 secretion remained unchanged105. CGRP has direct effects on DCs, inhibiting their activation, resulting in reduced expression of MHC class II and co-stimulatory molecules, and reduced production of IL-12, thereby impairing their ability to activate T cells103. In the thymus96, CGRP might be involved in inducing thymocyte apoptosis, possibly through the inhibition of NF-κB signalling pathways106. Upregulation of CGRP expression in certain brain regions after injury (such as induced by ischaemia) indicates that it might also have a role in the nervous system response to injury96.
Substance P
Substance P, which is a neuropeptide released by peripheral nerves that mediate pain perception, interacts with immune cells through two receptors, neurokinin-1 and neurokinin-2 (REF. 107). Substance P stimulates macrophage and eosinophil secretion of the pro-inflammatory cytokines TNF, IL-1β, IL-2 and IL-6 in vitro98,108; increases NK-cell activity and migration_in vitro_109,110; and stimulates the release of CXCL8 and CCL2 from leukocytes105,111. Furthermore, substance P induces the release of vaso-active mediators from mast cells, including histamine and serotonin112,113, and therefore contributes to vascular leakiness and oedema at sites of inflammation. Immune cells, including lymphocytes, monocytes and DCs, can also produce substance P75. Neonatal rats in which substance-P-containing primary afferent nerves are destroyed by capsaicin administration show a reduced capacity to mount a neurogenic inflammatory response114. This has led some researchers to suggest that substance P might have an important role in regulating the inflammatory response. Indeed, substance P antagonists have been shown to be effective as anti-inflammatory agents115.
Both glucocorticoids and catecholamines regulate CGRP and substance P. Glucocorticoids upregulate the expression and release of CGRP in dorsal root ganglion sensory neurons in vitro and noradrenaline inhibits the production and release of CGRP and substance P through α2-adrenoreceptors in dorsal root ganglion sensory neurons116. These interactions between the hormones and neurotransmitters of the HPA axis, and the sympathetic, parasympathetic and peripheral nervous systems indicate that modulation of immune responses by peripheral neuropeptides might occur not only through direct effects on immune cells but also indirectly through interactions of multiple neuronal and neuroendocrine pathways.
α -Melanocyte-stimulating hormone
The neuropeptide α-melanocyte-stimulating hormone (α-MSH) is a 13-amino-acid peptide derived from cleavage of the pro-opiomelanocortin gene product from which adrenocorticotropic hormone (ACTH) is also derived. α-MSH is released by pituitary cells117, neurons of the arcuate nucleus and the paraventricular nucleus in the hypothalamus118, and from peripheral nerves. It suppresses cytokine-mediated inflammation through induction of IL-10 production by monocytes, and reduces physiological markers of shock when injected systemically119. Furthermore, α-MSH inhibits LPS-induced stimulation of macrophages by blocking TLR4 signalling through an intracellular inhibitor IRAK-M (IL-1-receptor-associated kinase M)120. α-MSH inhibits the activation of transcription factors, such as NF-κB121, and inhibits the production of IL-1, IL-2, TNF and IFNγ119, but promotes CXCL8 mRNA expression and secretion by fibroblasts122.
Pro-opiomelanocortin —
(POMC). A 241-amino-acid precursor polypeptide that is synthesized in corticotrophin cells of the pituitary gland. Biologically active peptides derived from POMC include adrenocorticotropic hormone, enkephalins and α-melanocyte-stimulating hormone.
Arcuate nucleus —
A collection of neurons in the hypothalamus. It regulates the secretion of hormones through afferent dopaminergic projections to the pituitary.
Paraventricular nucleus —
(PVN). A collection of neurons in the hypothalamus that are adjacent to the third ventricle. It contains mainly neurosecretory neurons that secrete corticotrophin-releasing hormone, which stimulates pituitary corticotrophs. In addition, PVN neurons project to the sympathetic brainstem nuclei, parasympathetic brainstem pre-ganglionic neurons and spinal cord.
Opioids
Both synthetic opiates (morphine) and endogenous opioids (endorphins and enkephalins), which are pro-opiomelanocortin gene products that are released by pituitary cells together with ACTH, have been shown to be anti-inflammatory in vitro and in vivo3. Although endogenous opioids have previously been reported to stimulate chemotaxis of PBMCs123, more recent studies indicate that they inhibit chemokine-induced chemotaxis of human neutrophils and monocytes3. In addition, the endogenous opioid methionine-enkephalin desensitizes monocyte chemokine receptors by phosphorylating them124. Endogenous and exogenous opioids mediate their effects through three main membrane-bound opioid receptor subtypes, μ, δ and κ, which are expressed in the CNS, in dorsal root ganglia, by central and peripheral nerve terminals, and by lymphocytes and monocytes125. In addition to direct receptor-mediated effects on immune cells, opioids might mediate some of their anti-inflammatory effects indirectly, through glucocorticoids, as opioids are known to stimulate the HPA axis and induce glucocorticoid release126.
Endorphins —
Endogenous opioid peptides that are produced by the pituitary gland and the hypothalamus. They regulate feelings of pain and hunger.
Enkephalins —
Short five-amino-acid polypeptides that are members of endogenous opioid family and that bind to opiate receptors.
Vasoactive intestinal peptide
In contrast to the pro-inflammatory effects of neuropeptides described earlier, vasoactive intestinal peptide (VIP) released by peripheral nerves or microglia127, acting through its two G-protein-coupled receptors, VPAC1 and VPAC2, has been reported to inhibit inflammation. VIP inhibits the production of pro-inflammatory cytokines, such as IL-2, IL-6, IL-12 and TNF, by PBMCs, monocytes and T cells128–131; inhibits the expression of CXCL8 by monocytes in response to endotoxin132; inhibits NK-cell activity; prevents macrophage activation; promotes DC maturation133; stimulates macrophage production of the anti-inflammatory cytokine IL-10 (REF. 130); and decreases TLR2 and TLR4 mRNA expression by intestinal epithelial cells in vitro134. In addition to production by neurons, VIP is also produced by mast cells, leukocytes and neutrophils, and by lymphocytes in response to immune stimuli135. Therefore, the neuropeptide VIP can be considered as both a neuroeffector and a lymphocyte effector.
Taken together, these studies indicate that most neuropeptides, with the exception of VIP released from peripheral nerves, are pro-inflammatory (TABLE 2), and either directly or indirectly contribute to the vascular leakiness, leukocyte accumulation and immune activation at sites of inflammation.
Concluding remarks
Activation of the innate immune system in response to pathogen recognition provides both initial signals for the induction of inflammatory responses as well as signals for the activation of counter-regulatory CNS responses that terminate inflammation. Activation of the peripheral nervous system at local inflammatory sites serves to enhance innate immune responses and stimulate the release of vasoactive mediators that increase immune-cell recruitment and amplify local clearance of the pathogen. After pathogen clearance, activation of the HPA axis, SNS and parasympathetic nervous systems dampen inflammatory responses and restore host homeostasis. Removal of these inhibitory CNS pathways would allow uncontrolled activation of innate immune responses to the extent that they could become deleterious to the host.
For many years, the concept that the central and peripheral nervous system regulate immunity was considered to be highly controversial. However, as described in this Review, a wealth of data has now accumulated, indicating that the molecular machinery required to respond to neurotransmitters, neuropeptides and neurohormones are indeed present in immune cells. In addition, in some cases, immune cells produce and secrete neural mediators, supporting the importance of such mechanisms of immune regulation. However, the precise cellular source and maturation phase at which immune cells either produce such molecules or respond to them still requires clarification. In addition, the signalling mechanisms and physiological circumstances under which such regulation occurs largely remain unaddressed. And with most studies so far focusing on the effects of glucocorticoids on innate immune responses, evidence for a role of other neuropeptides and neurotransmitters in innate recognition systems, such as TLRs on immune cells, is scarce.
Research that focuses on these issues should provide further important insights into the molecular mechanisms and physiological role of the links between the immune system and the CNS that occur at systemic, regional and local levels, and will provide further evidence that these interactions have important physiological roles in health and disease.
Acknowledgments
The author would like to thank H. Gorby and C. Butts for their important contributions to this manuscript.
Footnotes
Competing interests statement
The author declares no competing financial interests.
References
- 1.Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the pathogenesis of human disease. Nature Immunol. 2004;5:975–979. doi: 10.1038/ni1116. [DOI] [PubMed] [Google Scholar]
- 2.Beutler B. The Toll-like receptors: analysis by forward genetic methods. Immunogenetics. 2005;57:385–392. doi: 10.1007/s00251-005-0011-3. A review of landmark studies on TLRs. [DOI] [PubMed] [Google Scholar]
- 3.Grimm MC, et al. Opiate inhibition of chemokine-induced chemotaxis. Ann N Y Acad Sci. 1998;840:9–20. doi: 10.1111/j.1749-6632.1998.tb09544.x. [DOI] [PubMed] [Google Scholar]
- 4.Milligan ED, et al. Controlling neuropathic pain by adeno-associated virus driven production of the anti-inflammatory cytokine, interleukin-10. Mol Pain. 2005;1:9. doi: 10.1186/1744-8069-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watkins LR, Maier SF. Implications of immune-to-brain communication for sickness and pain. Proc Natl Acad Sci USA. 1999;96:7710–7713. doi: 10.1073/pnas.96.14.7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dhabhar FS. Stress-induced enhancement of cell-mediated immunity. Ann N Y Acad Sci. 1998;840:359–372. doi: 10.1111/j.1749-6632.1998.tb09575.x. [DOI] [PubMed] [Google Scholar]
- 7.Madden KS, Felten SY, Felten DL, Sundaresan PR, Livnat S. Sympathetic neural modulation of the immune system. I. Depression of T cell immunity in vivo and vitro following chemical sympathectomy. Brain Behav Immun. 1989;3:72–89. doi: 10.1016/0889-1591(89)90007-x. [DOI] [PubMed] [Google Scholar]
- 8.Amaral LA, Diaz-Guilera A, Moreira AA, Goldberger AL, Lipsitz LA. Emergence of complex dynamics in a simple model of signalling networks. Proc Natl Acad Sci USA. 2004;101:15551–15555. doi: 10.1073/pnas.0404843101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawashima K, Fujii T. Expression of non-neuronal acetylcholine in lymphocytes and its contribution to the regulation of immune function. Front Biosci. 2004;9:2063–2085. doi: 10.2741/1390. A comprehensive review of non-neuronal cholinergic regulation of immunity. [DOI] [PubMed] [Google Scholar]
- 10.Maestroni GJ, Mazzola P. Langerhans cells β2-adrenoceptors: role in migration, cytokine production, Th priming and contact hypersensitivity. J Neuroimmunol. 2003;144:91–99. doi: 10.1016/j.jneuroim.2003.08.039. [DOI] [PubMed] [Google Scholar]
- 11.Woltman AM, Massacrier C, de Fijter JW, Caux C, van Kooten C. Corticosteroids prevent generation of CD34+-derived dermal dendritic cells but do not inhibit Langerhans cell development. J Immunol. 2002;168:6181–6188. doi: 10.4049/jimmunol.168.12.6181. [DOI] [PubMed] [Google Scholar]
- 12.Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J Neurosci. 2005;25:1788–1796. doi: 10.1523/JNEUROSCI.4268-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 14.Koedel U, et al. MyD88 is required for mounting a robust host immune response to Streptococcus pneumoniae in the CNS. Brain. 2004;127:1437–1445. doi: 10.1093/brain/awh171. [DOI] [PubMed] [Google Scholar]
- 15.Hench PS, Kendall EC, Slocumb CH, Polley HF. Effects of cortisone acetate and pituitary ACTH on rheumatoid arthritis, rheumatic fever and certain other conditions. Arch Med Interna. 1950;85:545–666. doi: 10.1001/archinte.1950.00230100002001. [DOI] [PubMed] [Google Scholar]
- 16.Agarwal SK, Marshall GD., Jr Dexamethasone promotes type 2 cytokine production primarily through inhibition of type 1 cytokines. J Interferon Cytokine Res. 2001;21:147–155. doi: 10.1089/107999001750133159. [DOI] [PubMed] [Google Scholar]
- 17.Wick G, et al. The obese strain of chickens: an animal model with spontaneous autoimmune thyroiditis. Adv Immunol. 1989;47:433–500. doi: 10.1016/s0065-2776(08)60666-5. [DOI] [PubMed] [Google Scholar]
- 18.Sternberg EM, et al. A central nervous system defect in biosynthesis of corticotropin-releasing hormone is associated with susceptibility to streptococcal cell wall-induced arthritis in Lewis rats. Proc Natl Acad Sci USA. 1989;86:4771–4775. doi: 10.1073/pnas.86.12.4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crofford LJ, et al. Hypothalamic-pituitary-adrenal axis perturbations in patients with fibromyalgia. Arthritis Rheum. 1994;37:1583–1592. doi: 10.1002/art.1780371105. [DOI] [PubMed] [Google Scholar]
- 20.Johnson EO, Vlachoyiannopoulos PG, Skopouli FN, Tzioufas AG, Moutsopoulos HM. Hypofunction of the stress axis in Sjogren’s syndrome. J Rheumatol. 1998;25:1508–1514. [PubMed] [Google Scholar]
- 21.Sternberg EM, et al. Inflammatory mediator-induced hypothalamic-pituitary-adrenal axis activation is defective in streptococcal cell wall arthritis-susceptible Lewis rats. Proc Natl Acad Sci USA. 1989;86:2374–2378. doi: 10.1073/pnas.86.7.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edwards CK, Yunger LM, Lorence RM, Dantzer R, Kelley KW. The pituitary gland is required for protection against lethal effects of Salmonella typhimurium. Proc Natl Acad Sci USA. 1991;88:2274–2277. doi: 10.1073/pnas.88.6.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruzek MC, Pearce BD, Miller AH, Biron CA. Endogenous glucocorticoids protect against cytokine-mediated lethality during viral infection. J Immunol. 1999;162:3527–3533. [PubMed] [Google Scholar]
- 24.Gomez SA, et al. Endogenous glucocorticoids attenuate Shiga toxin-2-induced toxicity in a mouse model of haemolytic uraemic syndrome. Clin Exp Immunol. 2003;131:217–224. doi: 10.1046/j.1365-2249.2003.02057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacPhee IA, Antoni FA, Mason DW. Spontaneous recovery of rats from experimental allergic encephalomyelitis is dependent on regulation of the immune system by endogenous adrenal corticosteroids. J Exp Med. 1989;169:431–445. doi: 10.1084/jem.169.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Derijk RH, et al. A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor β-isoform mRNA is associated with rheumatoid arthritis. J Rheumatol. 2001;28:2383–2388. [PubMed] [Google Scholar]
- 27.Leung DY, Szefler SJ. Diagnosis and management of steroid-resistant asthma. Clin Chest Med. 1997;18:611–625. doi: 10.1016/s0272-5231(05)70405-6. [DOI] [PubMed] [Google Scholar]
- 28.Adcock IM, et al. Differences in binding of glucocorticoid receptor to DNA in steroid-resistant asthma. J Immunol. 1995;154:3500–3505. [PubMed] [Google Scholar]
- 29.DeRijk RH, Eskandari F, Sternberg EM. Corticosteroid resistance in a subpopulation of multiple sclerosis patients as measured by ex vivo dexamethasone inhibition of LPS induced IL-6 production. J Neuroimmunol. 2004;151:180–188. doi: 10.1016/j.jneuroim.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 30.van Winsen LM, et al. Sensitivity to glucocorticoids is decreased in relapsing remitting multiple sclerosis. J Clin Endocrinol Metab. 2005;90:734–740. doi: 10.1210/jc.2004-0306. [DOI] [PubMed] [Google Scholar]
- 31.Towers R, et al. High levels of glucocorticoid receptors in patients with active Crohn’s disease may predict steroid resistance. Clin Exp Immunol. 2005;141:357–362. doi: 10.1111/j.1365-2249.2005.02846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ogawa S, et al. Molecular determinants of crosstalk between nuclear receptors and Toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. This study defines nuclear receptor–TLR interactions and clinical implications for glucocorticoid effects in different infections. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee YM, et al. A mutation of the glucocorticoid receptor gene in patients with systemic lupus erythematosus. Tohoku J Exp Med. 2004;203:69–76. doi: 10.1620/tjem.203.69. [DOI] [PubMed] [Google Scholar]
- 34.Jiang T, et al. The phase-shift mutation in the glucocorticoid receptor gene: potential etiologic significance of neuroendocrine mechanisms in lupus nephritis. Clin Chim Acta. 2001;313:113–117. doi: 10.1016/s0009-8981(01)00661-1. [DOI] [PubMed] [Google Scholar]
- 35.Mingrone G, et al. The steroid resistance of Crohn’s disease. J Investig Med. 1999;47:319–325. [PubMed] [Google Scholar]
- 36.Diaz-Borjon A, et al. Multidrug resistance-1 (MDR-1) in rheumatic autoimmune disorders. Part II: Increased P-glycoprotein activity in lymphocytes from systemic lupus erythematosus patients might affect steroid requirements for disease control. Joint Bone Spine. 2000;67:40–48. [PubMed] [Google Scholar]
- 37.DeRijk RH, Schaaf M, de Kloet ER. Glucocorticoid receptor variants: clinical implications. J Steroid Biochem Mol Biol. 2002;81:103–122. doi: 10.1016/s0960-0760(02)00062-6. A recent review describing the concept and clinical implications of glucocorticoid resistance. [DOI] [PubMed] [Google Scholar]
- 38.Oakley RH, Sar M, Cidlowski JA. The human glucocorticoid receptor β isoform. Expression, biochemical properties, and putative function. J Biol Chem. 1996;271:9550–9559. doi: 10.1074/jbc.271.16.9550. An interesting study describing the expression, biochemical properties, and function of the glucocorticoid receptor-β in humans. [DOI] [PubMed] [Google Scholar]
- 39.Leung DY, et al. Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor β. J Exp Med. 1997;186:1567–1574. doi: 10.1084/jem.186.9.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Webster JI, et al. Anthrax lethal factor represses glucocorticoid and progesterone receptor activity. Proc Natl Acad Sci USA. 2003;100:5706–5711. doi: 10.1073/pnas.1036973100. The first report of bacterial toxin repressing nuclear hormone receptor transactivation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moayeri M, Webster JI, Wiggins JF, Leppla SH, Sternberg EM. Endocrine perturbation increases susceptibility of mice to anthrax lethal toxin. Infect Immun. 2005;73:4238–4244. doi: 10.1128/IAI.73.7.4238-4244.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nature Rev Immunol. 2005;5:243–251. doi: 10.1038/nri1571. A comprehensive review of mechanisms of stress effects on immunity. [DOI] [PubMed] [Google Scholar]
- 43.Vedhara K, et al. Chronic stress in elderly carers of dementia patients and antibody response to influenza vaccination. Lancet. 1999;353:627–631. doi: 10.1016/S0140-6736(98)06098-X. [DOI] [PubMed] [Google Scholar]
- 44.DeRijk R, et al. Exercise and circadian rhythm-induced variations in plasma cortisol differentially regulate interleukin-1 β (IL-1 β), IL-6, and tumour necrosis factor-α (TNF α) production in humans: high sensitivity of TNFα and resistance of IL-6. J Clin Endocrinol Metab. 1997;82:2182–2191. doi: 10.1210/jcem.82.7.4041. [DOI] [PubMed] [Google Scholar]
- 45.Singh A, et al. Lymphocyte subset responses to exercise and glucocorticoid suppression in healthy men. Med Sci Sports Exerc. 1996;28:822–828. doi: 10.1097/00005768-199607000-00008. [DOI] [PubMed] [Google Scholar]
- 46.Matyszak MK, Citterio S, Rescigno M, Ricciardi-Castagnoli P. Differential effects of corticosteroids during different stages of dendritic cell maturation. Eur J Immunol. 2000;30:1233–1242. doi: 10.1002/(SICI)1521-4141(200004)30:4<1233::AID-IMMU1233>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 47.Moser M, et al. Glucocorticoids downregulate dendritic cell function in vitro and in vivo. Eur J Immunol. 1995;25:2818–2824. doi: 10.1002/eji.1830251016. [DOI] [PubMed] [Google Scholar]
- 48.Sacedon R, Vicente A, Varas A, Jimenez E, Zapata AG. Early differentiation of thymic dendritic cells in the absence of glucocorticoids. J Neuroimmunol. 1999;94:103–108. doi: 10.1016/s0165-5728(98)00231-8. [DOI] [PubMed] [Google Scholar]
- 49.Ma W, et al. Dexamethasone inhibits IL-12p40 production in lipopolysaccharide-stimulated human monocytic cells by downregulating the activity of c-Jun N-terminal kinase, the activation protein-1, and NF-κB transcription factors. J Immunol. 2004;172:318–330. doi: 10.4049/jimmunol.172.1.318. [DOI] [PubMed] [Google Scholar]
- 50.Murray SE, et al. Overproduction of corticotropin-releasing hormone blocks germinal center formation: role of corticosterone and impaired follicular dendritic cell networks. J Neuroimmunol. 2004;156:31–41. doi: 10.1016/j.jneuroim.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 51.Ghosh S, May MJ, Kopp EB. NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 52.Scheinman RI, Gualberto A, Jewell CM, Cidlowski JA, Baldwin AS., Jr Characterization of mechanisms involved in transrepression of NF-κ B by activated glucocorticoid receptors. Mol Cell Biol. 1995;15:943–953. doi: 10.1128/mcb.15.2.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cronstein BN, Kimmel SC, Levin RI, Martiniuk F, Weissmann G. A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc Natl Acad Sci USA. 1992;89:9991–9995. doi: 10.1073/pnas.89.21.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Atsuta J, Plitt J, Bochner BS, Schleimer RP. Inhibition of VCAM-1 expression in human bronchial epithelial cells by glucocorticoids. Am J Respir Cell Mol Biol. 1999;20:643–650. doi: 10.1165/ajrcmb.20.4.3265. [DOI] [PubMed] [Google Scholar]
- 55.Pitzalis C, et al. Corticosteroids inhibit lymphocyte binding to endothelium and intercellular adhesion: an additional mechanism for their anti-inflammatory and immunosuppressive effect. J Immunol. 1997;158:5007–5016. [PubMed] [Google Scholar]
- 56.Miyamasu M, et al. Glucocorticoids inhibit chemokine generation by human eosinophils. J Allergy Clin Immunol. 1998;101:75–83. doi: 10.1016/S0091-6749(98)70196-4. [DOI] [PubMed] [Google Scholar]
- 57.Sewell WA, Scurr LL, Orphanides H, Kinder S, Ludowyke RI. Induction of interleukin-4 and interleukin-5 expression in mast cells is inhibited by glucocorticoids. Clin Diagn Lab Immunol. 1998;5:18–23. doi: 10.1128/cdli.5.1.18-23.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Richards DF, Fernandez M, Caulfield J, Hawrylowicz CM. Glucocorticoids drive human CD8+T cell differentiation towards a phenotype with high IL-10 and reduced IL-4, IL-5 and IL-13 production. Eur J Immunol. 2000;30:2344–2354. doi: 10.1002/1521-4141(2000)30:8<2344::AID-IMMU2344>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 59.Pype JL, et al. Expression of monocyte chemotactic protein (MCP)-1, MCP-2, and MCP-3 by human airway smooth-muscle cells. Modulation by corticosteroids and T-helper 2 cytokines. Am J Respir Cell Mol Biol. 1999;21:528–536. doi: 10.1165/ajrcmb.21.4.3660. [DOI] [PubMed] [Google Scholar]
- 60.Homma T, et al. Corticosteroid and cytokines synergistically enhance Toll-like receptor 2 expression in respiratory epithelial cells. Am J Respir Cell Mol Biol. 2004;31:463–469. doi: 10.1165/rcmb.2004-0161OC. [DOI] [PubMed] [Google Scholar]
- 61.Hermoso MA, Matsuguchi T, Smoak K, Cidlowski JA. Glucocorticoids and tumour necrosis factor α cooperatively regulate Toll-like receptor 2 gene expression. Mol Cell Biol. 2004;24:4743–4756. doi: 10.1128/MCB.24.11.4743-4756.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shuto T, et al. Glucocorticoids synergistically enhance nontypeable Haemophilus influenzae-induced Toll-like receptor 2 expression via a negative cross-talk with p38 MAP kinase. J Biol Chem. 2002;277:17263–17270. doi: 10.1074/jbc.M112190200. [DOI] [PubMed] [Google Scholar]
- 63.Bornstein SR, et al. Impaired adrenal stress response in Toll-like receptor 2-deficient mice. Proc Natl Acad Sci USA. 2004;101:16695–16700. doi: 10.1073/pnas.0407550101. This paper provides evidence for a crucial role of TLR2 in adrenal glucocorticoid regulation in septicaemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 65.Drennan MB, et al. Toll-like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am J Pathol. 2004;164:49–57. doi: 10.1016/S0002-9440(10)63095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Silverman MN, Miller AH, Biron CA, Pearce BD. Characterization of an interleukin-6- and adrenocorticotropin-dependent, immune-to-adrenal pathway during viral infection. Endocrinology. 2004;145:3580–3589. doi: 10.1210/en.2003-1421. [DOI] [PubMed] [Google Scholar]
- 67.Sanders VM. Interdisciplinary research: noradrenergic regulation of adaptive immunity. Brain Behav Immun. 2005 11 Oct; doi: 10.1016/j.bbi.2005.07.004. (doi:10.1016/ j.bbi.2005.08.005) [DOI] [PubMed] [Google Scholar]
- 68.Johnson JD, et al. Adrenergic receptors mediate stress-induced elevations in extracellular Hsp72. J Appl Physiol. 2005;99:1789–1795. doi: 10.1152/japplphysiol.00390.2005. [DOI] [PubMed] [Google Scholar]
- 69.Madden KS, Sanders VM, Felten DL. Catecholamine influences and sympathetic neural modulation of immune responsiveness. Annu Rev Pharmacol Toxicol. 1995;35:417–448. doi: 10.1146/annurev.pa.35.040195.002221. [DOI] [PubMed] [Google Scholar]
- 70.Benschop RJ, et al. Effects of β-adrenergic blockade on immunologic and cardiovascular changes induced by mental stress. Circulation. 1994;89:762–769. doi: 10.1161/01.cir.89.2.762. [DOI] [PubMed] [Google Scholar]
- 71.Chelmicka-Schorr E, Checinski M, Arnason BG. Chemical sympathectomy augments the severity of experimental allergic encephalomyelitis. J Neuroimmunol. 1988;17:347–350. doi: 10.1016/0165-5728(88)90125-7. [DOI] [PubMed] [Google Scholar]
- 72.van der Poll T, Jansen J, Endert E, Sauerwein HP, van Deventer SJ. Noradrenaline inhibits lipopolysaccharide-induced tumour necrosis factor and interleukin 6 production in human whole blood. Infect Immun. 1994;62:2046–2050. doi: 10.1128/iai.62.5.2046-2050.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hasko G, Elenkov IJ, Kvetan V, Vizi ES. Differential effect of selective block of α 2-adrenoreceptors on plasma levels of tumour necrosis factor-α, interleukin-6 and corticosterone induced by bacterial lipopolysaccharide in mice. J Endocrinol. 1995;144:457–462. doi: 10.1677/joe.0.1440457. [DOI] [PubMed] [Google Scholar]
- 74.Woiciechowsky C, et al. Sympathetic activation triggers systemic interleukin-10 release in immunodepression induced by brain injury. Nature Med. 1998;4:808–813. doi: 10.1038/nm0798-808. [DOI] [PubMed] [Google Scholar]
- 75.Lang K, Drell TL, Niggemann B, Zanker KS, Entschladen F. Neurotransmitters regulate the migration and cytotoxicity in natural killer cells. Immunol Lett. 2003;90:165–172. doi: 10.1016/j.imlet.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 76.Straub RH, et al. Neurotransmitters of the sympathetic nerve terminal are powerful chemoattractants for monocytes. J Leukoc Biol. 2000;67:553–558. doi: 10.1002/jlb.67.4.553. [DOI] [PubMed] [Google Scholar]
- 77.Schwarz H, Villiger PM, von Kempis J, Lotz M. Neuropeptide Y is an inducible gene in the human immune system. J Neuroimmunol. 1994;51:53–61. doi: 10.1016/0165-5728(94)90128-7. [DOI] [PubMed] [Google Scholar]
- 78.Larhammar D. Structural diversity of receptors for neuropeptide Y, peptide YY and pancreatic polypeptide. Regul Pept. 1996;65:165–174. doi: 10.1016/0167-0115(96)00110-3. [DOI] [PubMed] [Google Scholar]
- 79.Straub RH, et al. Neuropeptide Y cotransmission with noradrenaline in the sympathetic nerve-macrophage interplay. J Neurochem. 2000;75:2464–2471. doi: 10.1046/j.1471-4159.2000.0752464.x. [DOI] [PubMed] [Google Scholar]
- 80.Jetschmann JU, et al. Expression and in-vivo modulation of α- and β-adrenoceptors on human natural killer (CD16+) cells. J Neuroimmunol. 1997;74:159–164. doi: 10.1016/s0165-5728(96)00221-4. [DOI] [PubMed] [Google Scholar]
- 81.Oberbeck R, et al. Adrenergic modulation of survival and cellular immune functions during polymicrobial sepsis. Neuroimmunomodulation. 2004;11:214–223. doi: 10.1159/000078439. [DOI] [PubMed] [Google Scholar]
- 82.Papanicolaou DA, Tsigos C, Oldfield EH, Chrousos GP. Acute glucocorticoid deficiency is associated with plasma elevations of interleukin-6: does the latter participate in the symptomatology of the steroid withdrawal syndrome and adrenal insufficiency? J Clin Endocrinol Metab. 1996;81:2303–2306. doi: 10.1210/jcem.81.6.8964868. [DOI] [PubMed] [Google Scholar]
- 83.Metz-Boutigue MH, Kieffer AE, Goumon Y, Aunis D. Innate immunity: involvement of new neuropeptides. Trends Microbiol. 2003;11:585–592. doi: 10.1016/j.tim.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 84.Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–859. doi: 10.1038/nature01321. A review of antifungal and antibacterial properties of some adrenal peptides. [DOI] [PubMed] [Google Scholar]
- 85.Laye S, et al. Subdiaphragmatic vagotomy blocks induction of IL-1 β mRNA in mice brain in response to peripheral LPS. Am J Physiol. 1995;268:R1327–R1331. doi: 10.1152/ajpregu.1995.268.5.R1327. [DOI] [PubMed] [Google Scholar]
- 86.Wang H, et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. This paper provides evidence for a role for nicotinic acetylcholine receptor in regulating inflammation. [DOI] [PubMed] [Google Scholar]
- 87.de Jonge WJ, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signalling pathway. Nature Immunol. 2005;6:844–851. doi: 10.1038/ni1229. The mechanism of action of non-pharmacological cholinergic anti-inflammatory intervention (vagus nerve stimulation) is described. [DOI] [PubMed] [Google Scholar]
- 88.Czura CJ, Tracey KJ. Autonomic neural regulation of immunity. J Intern Med. 2005;257:156–166. doi: 10.1111/j.1365-2796.2004.01442.x. [DOI] [PubMed] [Google Scholar]
- 89.van Westerloo DJ, et al. The cholinergic anti-inflammatory pathway regulates the host response during septic peritonitis. J Infect Dis. 2005;191:2138–2148. doi: 10.1086/430323. [DOI] [PubMed] [Google Scholar]
- 90.Borovikova LV, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 91.Yang H, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen G, et al. Suppression of HMGB1 release by stearoyl lysophosphatidylcholine:an additional mechanism for its therapeutic effects in experimental sepsis. J Lipid Res. 2005;46:623–627. doi: 10.1194/jlr.C400018-JLR200. [DOI] [PubMed] [Google Scholar]
- 93.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nature Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 94.Reyes-Reyna S, Stegall T, Krolick KA. Muscle responds to an antibody reactive with the acetylcholine receptor by upregulating monocyte chemoattractant protein 1: a chemokine with the potential to influence the severity and course of experimental myasthenia gravis. J Immunol. 2002;169:1579–1586. doi: 10.4049/jimmunol.169.3.1579. [DOI] [PubMed] [Google Scholar]
- 95.Saeed RW, et al. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J Exp Med. 2005;201:1113–1123. doi: 10.1084/jem.20040463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bulloch K, et al. Induction of calcitonin gene-related peptide-like immunoreactivity in hippocampal neurons following ischemia: a putative regional modulator of the CNS injury/immune response. Exp Neurol. 1998;150:195–205. doi: 10.1006/exnr.1997.6765. [DOI] [PubMed] [Google Scholar]
- 97.Agelaki S, Tsatsanis C, Gravanis A, Margioris AN. Corticotropin-releasing hormone augments proinflammatory cytokine production from macrophages in vitro and in lipopolysaccharide-induced endotoxin shock in mice. Infect Immun. 2002;70:6068–6074. doi: 10.1128/IAI.70.11.6068-6074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cuesta MC, Quintero L, Pons H, Suarez-Roca H. Substance P and calcitonin gene-related peptide increase IL-1β, IL-6 and TNFα secretion from human peripheral blood mononuclear cells. Neurochem Int. 2002;40:301–306. doi: 10.1016/s0197-0186(01)00094-8. [DOI] [PubMed] [Google Scholar]
- 99.Green PG, Luo J, Heller PH, Levine JD. Further substantiation of a significant role for the sympathetic nervous system in inflammation. Neuroscience. 1993;55:1037–1043. doi: 10.1016/0306-4522(93)90317-9. [DOI] [PubMed] [Google Scholar]
- 100.Baker C, Richards LJ, Dayan CM, Jessop DS. Corticotropin-releasing hormone immunoreactivity in human T and B cells and macrophages: co-localization with arginine vasopressin. J Neuroendocrinol. 2003;15:1070–1074. doi: 10.1046/j.1365-2826.2003.01099.x. [DOI] [PubMed] [Google Scholar]
- 101.Webster EL, et al. In vivo and in vitro characterization of antalarmin, a nonpeptide corticotropin-releasing hormone (CRH) receptor antagonist: suppression of pituitary ACTH release and peripheral inflammation. Endocrinology. 1996;137:5747–5750. doi: 10.1210/endo.137.12.8940412. [DOI] [PubMed] [Google Scholar]
- 102.Brain SD, Grant AD. Vascular actions of calcitonin gene-related peptide and adrenomedullin. Physiol Rev. 2004;84:903–934. doi: 10.1152/physrev.00037.2003. [DOI] [PubMed] [Google Scholar]
- 103.Carucci JA, et al. Calcitonin gene-related peptide decreases expression of HLA-DR and CD86 by human dendritic cells and dampens dendritic cell-driven T cell-proliferative responses via the type I calcitonin gene-related peptide receptor. J Immunol. 2000;164:3494–3499. doi: 10.4049/jimmunol.164.7.3494. [DOI] [PubMed] [Google Scholar]
- 104.Tran MT, Ritchie MH, Lausch RN, Oakes JE. Calcitonin gene-related peptide induces IL-8 synthesis in human corneal epithelial cells. J Immunol. 2000;164:4307–4312. doi: 10.4049/jimmunol.164.8.4307. [DOI] [PubMed] [Google Scholar]
- 105.Park SH, Hsiao GY, Huang GT. Role of substance P and calcitonin gene-related peptide in the regulation of interleukin-8 and monocyte chemotactic protein-1 expression in human dental pulp. Int Endod J. 2004;37:185–192. doi: 10.1111/j.0143-2885.2004.00782.x. [DOI] [PubMed] [Google Scholar]
- 106.Millet I, et al. Inhibition of NF-κB activity and enhancement of apoptosis by the neuropeptide calcitonin gene-related peptide. J Biol Chem. 2000;275:15114–15121. doi: 10.1074/jbc.275.20.15114. [DOI] [PubMed] [Google Scholar]
- 107.Mapp CE, et al. The distribution of neurokinin-1 and neurokinin-2 receptors in human central airways. Am J Respir Crit Care Med. 2000;161:207–215. doi: 10.1164/ajrccm.161.1.9903137. [DOI] [PubMed] [Google Scholar]
- 108.Lotz M, Vaughan JH, Carson DA. Effect of neuropeptides on production of inflammatory cytokines by human monocytes. Science. 1988;241:1218–1221. doi: 10.1126/science.2457950. [DOI] [PubMed] [Google Scholar]
- 109.Croitoru K, Ernst PB, Bienenstock J, Padol I, Stanisz AM. Selective modulation of the natural killer activity of murine intestinal intraepithelial leucocytes by the neuropeptide substance P. Immunology. 1990;71:196–201. [PMC free article] [PubMed] [Google Scholar]
- 110.Feistritzer C, et al. Natural killer cell functions mediated by the neuropeptide substance P. Regul Pept. 2003;116:119–126. doi: 10.1016/s0167-0115(03)00193-9. [DOI] [PubMed] [Google Scholar]
- 111.Serra MC, Calzetti F, Ceska M, Cassatella MA. Effect of substance P on superoxide anion and IL-8 production by human PMNL. Immunology. 1994;82:63–69. [PMC free article] [PubMed] [Google Scholar]
- 112.Guhl S, Lee HH, Babina M, Henz BM, Zuberbier T. Evidence for a restricted rather than generalized stimulatory response of skin-derived human mast cells to substance P. J Neuroimmunol. 2005;163:92–101. doi: 10.1016/j.jneuroim.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 113.Wang L, Stanisz AM, Wershil BK, Galli SJ, Perdue MH. Substance P induces ion secretion in mouse small intestine through effects on enteric nerves and mast cells. Am J Physiol. 1995;269:G85–G92. doi: 10.1152/ajpgi.1995.269.1.G85. [DOI] [PubMed] [Google Scholar]
- 114.Helme RD, Eglezos A, Andrews PV. The effects of capsaicin denervation on leucocyte and complement components of the inflammatory response. Clin Exp Neurol. 1987;24:207–211. [PubMed] [Google Scholar]
- 115.Duffy RA. Potential therapeutic targets for neurokinin-1 receptor antagonists. Expert Opin Emerg Drugs. 2004;9:9–21. doi: 10.1517/eoed.9.1.9.32956. [DOI] [PubMed] [Google Scholar]
- 116.Kawasaki H, Nuki C, Saito A, Takasaki K. Adrenergic modulation of calcitonin gene-related peptide (CGRP)-containing nerve-mediated vasodilation in the rat mesenteric resistance vessel. Brain Res. 1990;506:287–290. doi: 10.1016/0006-8993(90)91263-g. [DOI] [PubMed] [Google Scholar]
- 117.Lee TH, Lerner AB, Buettner-Janusch V. The isolation and structure of α- and β-melanocyte-stimulating hormones from monkey pituitary glands. J Biol Chem. 1961;236:1390–1394. [PubMed] [Google Scholar]
- 118.Roselli-Rehfuss L, et al. Identification of a receptor for γ melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc Natl Acad Sci USA. 1993;90:8856–8860. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lipton JM, Catania A. Anti-inflammatory actions of the neuroimmunomodulator α-MSH. Immunol Today. 1997;18:140–145. doi: 10.1016/s0167-5699(97)01009-8. [DOI] [PubMed] [Google Scholar]
- 120.Taylor AW. The immunomodulating neuropeptide α-melanocyte-stimulating hormone (α-MSH) suppresses LPS-stimulated TLR4 with IRAK-M in macrophages. J Neuroimmunol. 2005;162:43–50. doi: 10.1016/j.jneuroim.2005.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mandrika I, Muceniece R, Wikberg JE. Effects of melanocortin peptides on lipopolysaccharide/ interferon-γ-induced NF-κB DNA binding and nitric oxide production in macrophage-like RAW 264.7 cells: evidence for dual mechanisms of action. Biochem Pharmacol. 2001;61:613–621. doi: 10.1016/s0006-2952(00)00583-9. [DOI] [PubMed] [Google Scholar]
- 122.Kiss M, et al. Effects of the neuropeptides substance P, calcitonin gene-related peptide and α-melanocyte-stimulating hormone on the IL-8/IL-8 receptor system in a cultured human keratinocyte cell line and dermal fibroblasts. Inflammation. 1999;23:557–567. doi: 10.1023/a:1020294507767. [DOI] [PubMed] [Google Scholar]
- 123.van Epps DE, Saland L. β-endorphin and met-enkephalin stimulate human peripheral blood mononuclear cell chemotaxis. J Immunol. 1984;132:3046–3053. [PubMed] [Google Scholar]
- 124.Zhang N, Hodge D, Rogers TJ, Oppenheim JJ. Ca2+-independent protein kinase Cs mediate heterologous desensitization of leukocyte chemokine receptors by opioid receptors. J Biol Chem. 2003;278:12729–12736. doi: 10.1074/jbc.M300430200. Evidence for crosstalk between opioid receptors and chemokine receptors that are potentially relevant to pain mechanisms and therapy. [DOI] [PubMed] [Google Scholar]
- 125.Bidlack JM. Detection and function of opioid receptors on cells from the immune system. Clin Diagn Lab Immunol. 2000;7:719–723. doi: 10.1128/cdli.7.5.719-723.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mellon RD, Bayer BM. The effects of morphine, nicotine and epibatidine on lymphocyte activity and hypothalamic-pituitary-adrenal axis responses. J Pharmacol Exp Ther. 1999;288:635–642. [PubMed] [Google Scholar]
- 127.Blondel O, et al. A glia-derived signal regulating neuronal differentiation. J Neurosci. 2000;20:8012–8020. doi: 10.1523/JNEUROSCI.20-21-08012.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hernanz A, Tato E, De la Fuente M, de Miguel E, Arnalich F. Differential effects of gastrin-releasing peptide, neuropeptide Y, somatostatin and vasoactive intestinal peptide on interleukin-1 β, interleukin-6 and tumour necrosis factor-α production by whole blood cells from healthy young and old subjects. J Neuroimmunol. 1996;71:25–30. doi: 10.1016/s0165-5728(96)00118-x. [DOI] [PubMed] [Google Scholar]
- 129.Martinez C, et al. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide modulate endotoxin-induced IL-6 production by murine peritoneal macrophages. J Leukoc Biol. 1998;63:591–601. doi: 10.1002/jlb.63.5.591. [DOI] [PubMed] [Google Scholar]
- 130.Delgado M, Munoz-Elias EJ, Gomariz RP, Ganea D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide enhance IL-10 production by murine macrophages: in vitro and in vivo studies. J Immunol. 1999;162:1707–1716. [PubMed] [Google Scholar]
- 131.Ganea D. Regulatory effects of vasoactive intestinal peptide on cytokine production in central and peripheral lymphoid organs. Adv Neuroimmunol. 1996;6:61–74. doi: 10.1016/s0960-5428(96)00007-1. [DOI] [PubMed] [Google Scholar]
- 132.Delgado M, Ganea D. Vasoactive intestinal peptide inhibits IL-8 production in human monocytes. Biochem Biophys Res Commun. 2003;301:825–832. doi: 10.1016/s0006-291x(03)00059-7. [DOI] [PubMed] [Google Scholar]
- 133.Delneste Y, et al. Vasoactive intestinal peptide synergizes with TNF-α in inducing human dendritic cell maturation. J Immunol. 1999;163:3071–3075. [PubMed] [Google Scholar]
- 134.Gomariz RP, et al. Time-course expression of Toll-like receptors 2 and 4 in inflammatory bowel disease and homeostatic effect of VIP. J Leukoc Biol. 2005;78:491–502. doi: 10.1189/jlb.1004564. [DOI] [PubMed] [Google Scholar]
- 135.Delgado M, Pozo D, Ganea D. The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol Rev. 2004;56:249–290. doi: 10.1124/pr.56.2.7. [DOI] [PubMed] [Google Scholar]
- 136.Webster JI, Tonelli L, Sternberg EM. Neuroendocrine regulation of immunity. Annu Rev Immunol. 2002;20:125–163. doi: 10.1146/annurev.immunol.20.082401.104914. [DOI] [PubMed] [Google Scholar]
- 137.Steinman L. Elaborate interactions between the immune and nervous systems. Nature Immunol. 2004;5:575–581. doi: 10.1038/ni1078. [DOI] [PubMed] [Google Scholar]
- 138.Kobilka BK, et al. cDNA for the human β2-adrenergic receptor: a protein with multiple membrane-spanning domains and encoded by a gene whose chromosomal location is shared with that of the receptor for platelet-derived growth factor. Proc Natl Acad Sci USA. 1987;84:46–50. doi: 10.1073/pnas.84.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Le Tulzo Y, et al. Hemorrhage increases cytokine expression in lung mononuclear cells in mice: involvement of catecholamines in nuclear factor-κB regulation and cytokine expression. J Clin Invest. 1997;99:1516–1524. doi: 10.1172/JCI119314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Marques-Deak A, Cizza G, Sternberg E. Brain-immune interactions and disease susceptibility. Mol Psychiatry. 2005;10:239–250. doi: 10.1038/sj.mp.4001643. [DOI] [PubMed] [Google Scholar]