Genetic and Immunohistochemical Analysis of Pancreatic Acinar Cell Carcinoma: Frequent Allelic Loss on Chromosome 11p and Alterations in the APC/β-Catenin Pathway (original) (raw)
Abstract
Acinar cell carcinomas (ACCs) are rare malignant tumors of the exocrine pancreas. The specific molecular alterations that characterize ACCs have not yet been elucidated. ACCs are morphologically and genetically distinct from the more common pancreatic ductal adenocarcinomas. Instead, the morphological, immunohistochemical, and clinical features of ACCs overlap with those of another rare pancreatic neoplasm, pancreatoblastoma. We have recently demonstrated a high frequency of allelic loss on chromosome arm 11p and mutations in the APC/β-catenin pathway in pancreatoblastomas, suggesting that similar alterations might also play a role in the pathogenesis of some ACCs. We analyzed a series of 21 ACCs for somatic alterations in the APC/β-catenin pathway and for allelic loss on chromosome 11p. In addition, we evaluated the ACCs for alterations in p53 and Dpc4 expression using immunohistochemistry, and for microsatellite instability (MSI) using polymerase chain amplification of a panel of microsatellite markers. Allelic loss on chromosome 11p was the most common genetic alteration in ACCs, present in 50% (6 of 12 informative cases). Molecular alterations in the APC/β-catenin pathway were detected in 23.5% (4 of 17) of the carcinomas, including one ACC with an activating mutation of the β-catenin oncogene and three ACCs with truncating APC mutations. One ACC (1 of 13, 7.6%) showed allelic shifts in four of the five markers tested (MSI-high), two (15.4%) showed an allelic shift in only one of the five markers tested (MSI-low), and no shifts were detected in the remaining 10 cases. The MSI-high ACC showed medullary histological features. In contrast, no loss of Dpc4 protein expression or p53 accumulation was detected. These results indicate that ACCs are genetically distinct from pancreatic ductal adenocarcinomas, but some cases contain genetic alterations common to histologically similar pancreatoblastomas.
Acinar cell carcinomas (ACCs) are rare neoplasms of the exocrine pancreas, comprising less than 1% of primary pancreatic tumors. 1,2 ACCs are distinct from the more common pancreatic ductal adenocarcinomas. Histologically and immunohistochemically, ACCs recapitulate the growth pattern and secretory products of nonneoplastic pancreatic acini, including frequent production of the digestive enzymes trypsin, lipase, chymotrypsin, and, less commonly, amylase. 1,3-5 In some patients the overproduction of lipase by the neoplasm produces a distinctive syndrome of subcutaneous fat necrosis and polyarthralgia, in contrast to the more frequent jaundice in patients with pancreatic ductal adenocarcinomas. 1,6-10 In addition, although the prognosis of ACC in adults is poor, with the majority of patients showing evidence of metastatic disease either at or subsequent to diagnosis, 1,11 the reported mean survival of 18 months and the occasional long-term survival of patients with ACC contrasts with the significantly worse prognosis for patients with pancreatic ductal adenocarcinomas. 1,4,12
Several studies have examined genetic alterations in ACCs, including evaluation for mutations in the K-ras oncogene and DPC4 and p53 tumor suppressor genes that characterize the stepwise molecular and histological progression of pancreatic ductal neoplasms. 13-19 Not surprisingly, given the clinicopathological differences between ductal adenocarcinoma and ACC, alterations of these genes have either been absent or only rarely present in ACCs. 17,20-24 Indeed, aside from the presence of aneuploidy 25 and the recent report of ACC allelotype, 24 specific molecular alterations characterizing ACCs have not yet been identified.
We have recently studied genetic alterations in another rare pancreatic neoplasm, pancreatoblastoma, and found frequent involvement of the APC/β-catenin pathway and allelic loss on chromosome arm 11p in these neoplasms. 26 In pancreatoblastomas, the rationale for molecular evaluation of the APC/β-catenin pathway and for chromosome 11p loss lies in their occasional occurrence in patients with familial adenomatous polyposis (FAP) and Beckwith-Wiedemann syndrome, respectively. 26 Beckwith-Wiedemann syndrome, a maldevelopmental disorder with tissue overgrowth and increased neoplastic risk, is characterized by dysregulation of cell-cycle genes on a heavily imprinted chromosomal region on 11p15.5. FAP, caused by germline mutation of the APC gene on chromosome 5q, imparts a markedly increased risk for colonic and extra-colonic neoplasms through second-hit alterations of APC (either intragenic mutation or allelic loss of 5q). In turn, the sporadic variants of FAP-associated neoplasms also frequently involve the APC/β-catenin pathway, either through bi-allelic APC inactivation or by activating mutations in the β-catenin oncogene.
Several clinicopathological similarities exist between ACC and pancreatoblastoma. Both these tumors are characterized histologically by variably sheet-like, trabecular, and acinar growth patterns, both consistently show acinar differentiation as detected by immunohistochemical labeling, 1,3,21,27,28 both may contain varying proportions of endocrine cells, 1,27,29-33 and both may produce α-fetoprotein. 1,4,11,27,34-38 We therefore undertook a molecular characterization of a series of ACCs. The alterations that characterize pancreatoblastomas, 26 including allelic loss on chromosome 11p and mutations in the APC/β-catenin pathway were examined, as were alterations in the p53 and DPC4 tumor suppressor genes that characterize the more common adult pancreatic ductal adenocarcinomas.
Materials and Methods
Case Selection
The study population consisted of 21 patients with pancreatic ACC who underwent biopsy (4 cases) or surgical resection (17 cases) between 1983 and 2001. Five cases were from The Johns Hopkins Hospital and 16 cases were from Memorial Sloan-Kettering Cancer Center. ACCs were diagnosed based on characteristic histological and immunohistochemical features that included varying proportions of sheet-like, trabecular, and acinar growth, as well as the absence of squamoid corpuscles, which distinguish pancreatoblastomas from ACCs (Figure 1) ▶ . 1,27
Figure 1.
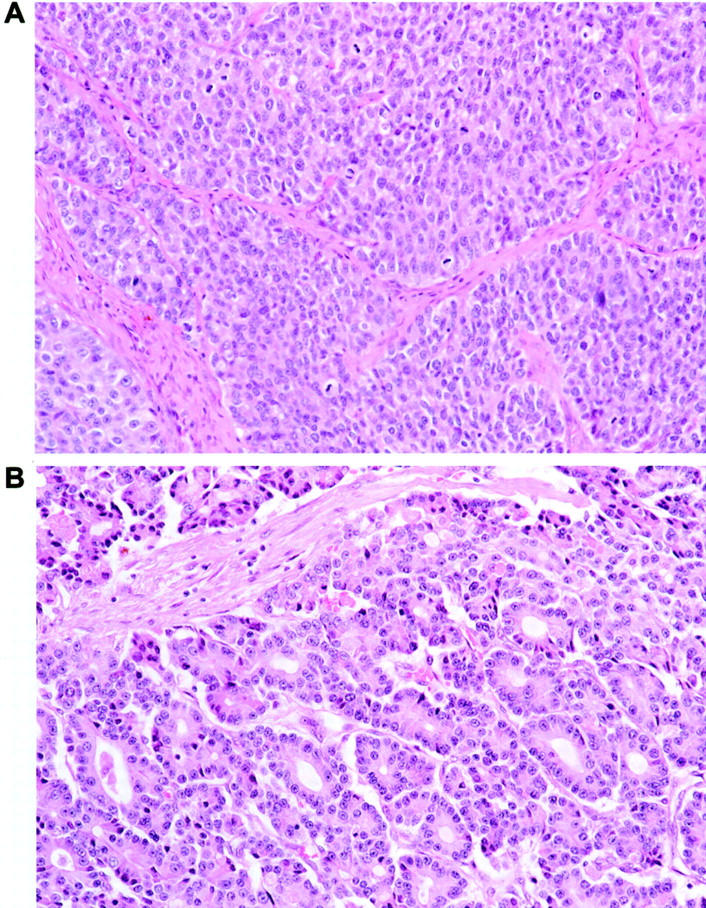
Histopathological appearance of pancreatic ACC. The neoplastic epithelial cells show variable growth patterns including areas of sheet-like growth divided by bands of fibrous tissue (A) and areas of acinar differentiation (B). Identical growth patterns can be present in ACCs and pancreatoblastomas, but characteristic squamoid corpuscles are not seen in the former.
Immunohistochemistry for β-Catenin, p53, and Dpc4
Immunohistochemical labeling using diaminobenzidine as the chromogen was performed on the Techmate 1000 automatic labeling system (BioTek Solutions, Tucson, AZ). Deparaffinized sections of formalin-fixed tissue at 5-μm thickness were labeled with β-catenin antibody (1:500 dilution, mouse monoclonal; Becton Dickinson Transduction Laboratories, Lexington, KY), p53 antibody (1:100 dilution, mouse monoclonal clone D07; DAKO, Carpinteria, CA), and Dpc4 antibody (1:100 dilution, monoclonal clone B8; Santa Cruz Biotechnology, Santa Cruz, CA). Heat-induced antigen retrieval using steam for 20 minutes at 80°C was used before incubation with all three antibodies.
For β-catenin, immunohistochemical labeling was evaluated for the presence of nuclear, cytoplasmic, and membranous β-catenin accumulation in both the ACCs and any normal surrounding tissues. Nuclear and cytoplasmic accumulation of β-catenin in ACCs was graded according to the percentage of neoplastic cells with strong immunolabeling. For p53, the percentage of positively labeled nuclei was recorded; we considered strong nuclear labeling in ≥30% of neoplastic cells as the cutoff for positivity. 39 For Dpc4, ACCs were classified as showing intact Dpc4 expression if they showed the normal pattern of strong, diffuse cytoplasmic labeling and labeling of scattered nuclei. ACCs were classified as showing loss of normal Dpc4 expression if they showed a complete loss of cytoplasmic and nuclear Dpc4 labeling. 40
DNA Extraction
Microdissection of ACCs for DNA extraction was performed from formalin-fixed, paraffin-embedded specimens. A 27 1/2-guage-needle tip was used for microdissection of routinely processed, 5-μm hematoxylin and eosin-stained slides under a low-power (×4) objective. Genomic DNA was extracted as described previously. 41 Corresponding normal control DNA was available in 16 cases and was extracted from adjacent nonneoplastic tissue (adjacent pancreatic acini and/or stroma in 11 cases, duodenum in 3 cases, liver in 1 case, and colon in 1 case).
Mutation Analysis of the β-Catenin Gene
Genomic DNA from each sample was amplified by polymerase chain reaction (PCR) using the primer pair: 5′-ATGGAACCAGACAGAAAAGC-3′ (sense) and 5′-GCTACTTGTTCTGAGTGAAG-3′ (anti-sense). These amplified a 200-bp fragment of exon 3 of the β-catenin gene that encompasses the region for GSK-3β phosphorylation. PCR reactions were performed under standard conditions in a 50-μl volume containing 38 μl of Platinum PCR SuperMix (Life Technologies, Inc., Rockville, MD), 5 μl of both 5′ and 3′ oligonucleotides (final concentration of 1 μmol/L), and 2 μl (∼ 50 ng) of genomic DNA. PCR conditions consisted of an initial denaturation at 94°C for 3 minutes, 40 cycles of 94°C for 1 minute, 58°C for 1 minute, and 72°C for 2 minutes, and a final extension at 72°C for 7 minutes. PCR products were purified with spin columns using QIAquick PCR purification kit (Qiagen, Inc., Valencia, CA) before sequencing. Automated sequencing of purified PCR products was performed on an ABI Prism 3700 DNA Analyzer (Applied Biosystems, Inc., Foster City, CA) using the internal primers: 5′-AAAGCGGCTGTTAGTCACTGG-3′ (sense) and 5′-CCTGTTCCCACTCATACAGG-3′ (anti-sense), and the resulting sequence data were analyzed with the Sequencher analysis program (Gene Codes, Ann Arbor, MI). Mutations were verified in both sense and anti-sense directions on independent PCR products.
Mutation Analysis of the APC Gene
Four sets of oligonucleotide primers (A1: 5′-CAGACTTATTGTGTAGAAGA-3′ and A2: 5′-CTCCTGAAGAAAATTCAACA-3′ for codons 1260 to 1359; B1: 5′-AGGGTTCTAGTTTATCTTCA-3′ and B2: 5′-TCTGCTTGGTGGCATGGTTT-3′ for codons 1339 to 1436; C1: 5′-GGCATTATAAGCCCCAGTGA-3′ and C2: 5′-AAATGGCTCATCGAGGCTCA-3′ for codons 1417 to 1516; D1: 5′-ACTCCAGATGGATTTTCTTG-3′ and D2: 5′-GGCTGGCTTTTTTGCTTTAC-3′ for codons 1497 to 1596) were used to amplify the mutation cluster region of the APC gene. 42 PCR reactions were performed in 50-μl volumes using the reaction mixture described above. PCR conditions consisted of an initial denaturation step of 94°C for 3 minutes, 40 cycles (94°C for 1 minute, 55°C for 1 minute, and 68°C for 1.5 minutes for APC-B, APC-C, and APC-D primer pairs and 94°C for 1 minute, 52°C for 1 minute, and 68°C for 1.5 minutes for APC-A), followed by a final extension at 72°C for 7 minutes. PCR products were purified and sequenced as described above using the same primers as for genomic DNA amplification. All mutations were verified in both sense and anti-sense directions on independent PCR products.
Allelic Loss on Chromosome 5q
Loss of heterozygosity (LOH) on 5q was evaluated in the ACCs for which nonneoplastic control tissue was available. LOH was assessed by microsatellite assays using PCR amplification of three microsatellite markers (D5S82, D5S299, and D5S346) as previously described. 43 Assays were performed in 96-well plates in 10-μl volumes, each containing 5 μl of PCR Master (Boehringer Mannheim, Mannheim, Germany), 3.5 μl of water, 1 μl of genomic DNA, 0.06 μl of 3′ oligonucleotide, and 0.4 μl of end-labeled 5′oligonucleotide. The 5′ oligonucleotide was end-labeled with (γ-32P)-ATP (NEN DuPont, Boston, MA) using T4 polynucleotide kinase (New England Biolabs, Beverly, MA). For D5S82 and D5S299, 38 cycles of 95°C for 30 seconds, 55°C for 30 seconds, and 72°C for 1 minute were performed, and for D5S346, 38 cycles of 95°C for 30 seconds, 58°C for 30 seconds, and 72°C for 1 minute were performed. PCR products were separated on 6% denaturing polyacrylamide gels and the gels were subjected to autoradiography. LOH was considered to be present when there was complete or near-complete disappearance of a heterozygous band in the ACC as compared with nonneoplastic control tissue in at least one informative marker.
Allelic Loss on Chromosome 11p
LOH on 11p was evaluated in the ACCs for which nonneoplastic control tissue was available, using the microsatellite markers TH (a tetranucleotide repeat polymorphism on 11p15.5-p15) and D11S1984 (a dinucleotide repeat on 11p15.5). Assays were performed and interpreted as described above using annealing temperatures of 62°C for TH and 55°C for D11S1984.
Microsatellite Instability (MSI) Analysis
MSI was evaluated in the ACCs for which nonneoplastic control tissue was available. MSI testing was performed using the five microsatellite loci (D5S346, as described above for 5q LOH, plus D2S123, D17S250, Bat-25, and Bat-26) recommended by the 1997 National Cancer Institute (NCI)-sponsored consensus conference. 44 Assays were performed as described above for 5q LOH analysis using annealing temperatures of 55°C for D2S123, D17S250, Bat-25, and Bat-26. The resultant bands on autoradiographs were interpreted according to the criteria described in detail by Berg and colleagues. 45 MSI-high (MSI-H) was considered to be present when at least two of the five microsatellite loci showed shifting, MSI-low (MSI-L) when only one locus was shifted, and microsatellite stable (MSS) when none of the loci were shifted, as per the NCI criteria. 44
Results
A summary of the clinicopathological and molecular findings in the 21 ACCs (designated A1 to A21) is presented in Table 1 ▶ .
Table 1.
Genetic Alterations in Pancreatic Acinar Cell Carcinomas
| Case | Age/sex | p53 accumulation | Dpc4 loss | MSI | Nuclear β-catenin | β-catenin mutation | APC mutation | 11p LOH |
|---|---|---|---|---|---|---|---|---|
| A1 | 15 /F | — | — | MSS | — | Wild-type | Wild-type | − |
| A2 | 21 /F | — | — | MSS | — | Wild-type | 1444X | − |
| A3 | 32 /F | — | — | MSS | — | Wild-type | Wild-type | − |
| A4 | 73 /F | — | — | MSI-L | — | Wild-type | Wild-type | + |
| A5 | 51 /M | — | — | MSI-L | 50% | Wild-type | Wild-type | − |
| A6 | 55 /F | — | — | MSS | — | Wild-type | Wild-type | N/I |
| A7 | 2 /F | — | — | MSS | — | Wild-type | Wild-type | + |
| A8 | 70 /M | — | — | MSS | — | Wild-type | Wild-type | − |
| A9 | 76 /M | — | — | MSS | — | Wild-type | Wild-type | + |
| A10 | 58 /M | — | — | MSS | — | Wild-type | Wild-type | − |
| A11 | 31 /M | — | — | MSS | — | Wild-type | Wild-type | + |
| A12 | 80 /M | — | — | MSS | — | Wild-type | Wild-type | + |
| A13 | 31 /M | — | — | N/N | 20% | T41I | Wild-type | N/N |
| A14 | 61 /M | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| A15 | 63 /M | — | — | N/A | — | N/A | N/A | N/A |
| A16 | 76 /M | — | — | N/N | 60% | Wild-type | 1554–1556FS | N/N |
| A17 | 54 /M | — | — | N/A | — | N/A | N/A | N/A |
| A18 | 60 /M | — | — | N/N | — | Wild-type | 1554–1556FS | N/N |
| A19 | 75 /M | — | — | MSI-H | — | Wild-type | Wild-type | + |
| A20 | 74 /M | — | — | N/A | — | N/A | N/A | N/A |
| A21 | 79 /M | — | — | N/N | — | Wild-type | Wild-type | N/N |
ClinicopathologicalCharacteristics
Nineteen of the ACCs arose in adults ranging from 21 to 80 years (mean, 59 years) and two cases were in pediatric patients aged 2 and 15 years. Six patients (29%) were female and 15 (71%) were male.
Alterations in the APC/β-Catenin Pathway
Amplifiable DNA for mutation analysis of β-catenin and APC was obtained from 17 of the 21 ACCs, and β-catenin or APC gene mutations were detected in a total of four (23.5%) of the 17 ACCs. One ACC (5.8%, case A13) contained a β-catenin gene mutation, a 1-bp C → T missense mutation at threonine codon 41 that would be predicted to result in β-catenin activation because of alteration of a presumptive residue for glycogen synthase kinase-3β (GSK-3β) phosphorylation. A mixture of both the wild-type and mutant peaks was present on DNA sequencing of this neoplasm, corresponding to the dominant nature of β-catenin gene alterations (Figure 2) ▶ . Three additional ACCs (17.6%, cases A2, A16, and A18) contained APC gene mutations, each of which would be predicted to result in APC inactivation because of premature protein truncation. In cases A16 and A18, frameshifts from insertion of a base A into a 6-base poly(A) tract spanning codons 1554 to 1556 were present. Although normal tissue was not available in either A16 or A18 for 5q LOH analysis, both cases demonstrated loss of the wild-type allele on DNA sequencing of the region of the mutated poly(A) tract, indicative of bi-allelic APC inactivation (Figure 2) ▶ . In case A2, a 1-bp C → T substitution at codon 1444 resulted in the formation of a premature stop codon (Figure 2) ▶ . Normal tissue was available for analysis in this case, and the somatic nature of this APC mutation was confirmed by the presence of only wild-type APC in the nonneoplastic tissue from this patient. When DNA from this ACC was sequenced, both the mutant and wild-type peaks were present, and no 5q LOH was present on allelic loss analysis. Bi-allelic APC inactivation therefore could not be demonstrated in this case. The 13 ACCs for which normal tissue was available were analyzed for 5q LOH, and allelic loss was present in two separate ACCs (15%, cases A8 and A9) for which no corresponding intragenic APC gene mutations were detected.
Figure 2.
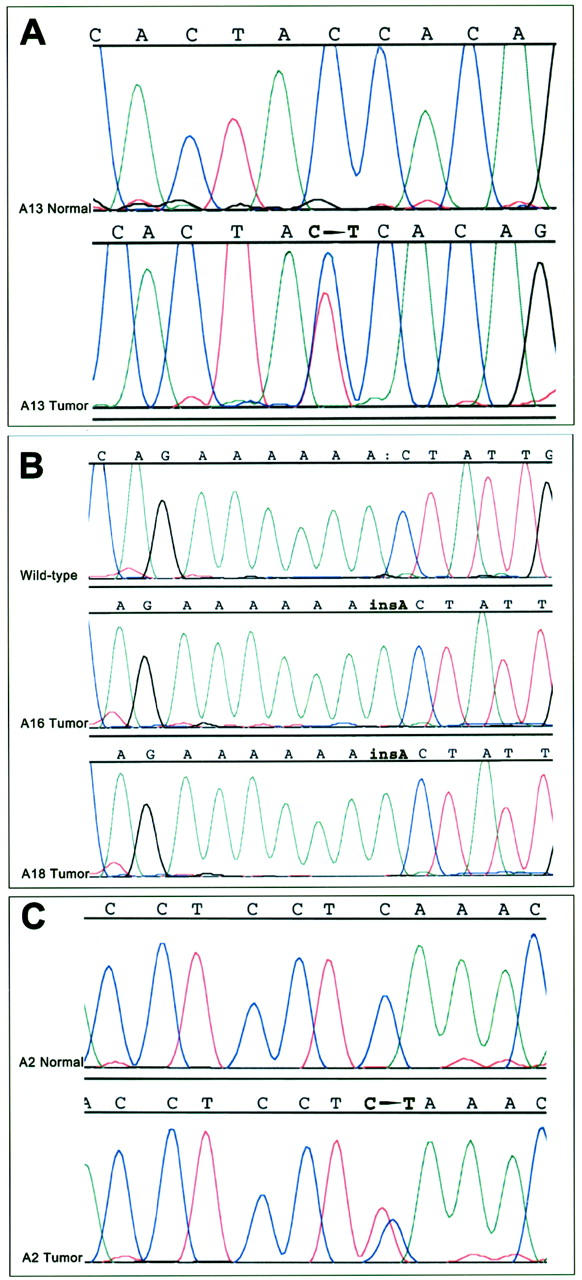
Alterations in the APC/β-catenin pathway in ACCs. A: DNA sequencing from case A13, showing an ACC (threonine) → ATC (isoleucine) mutation at codon 41 of the β-catenin gene. Sequencing of nonneoplastic tissue from this patient shows only wild-type β-catenin, confirming the somatic nature of the β-catenin mutation. B: Frameshift APC mutations in cases A16 and A18. DNA sequencing shows a 1-bp insertion A into a 6 base poly(A) tract spanning codons 1554 to 1556 in each case. The corresponding wild-type APC sequence from a different ACC is shown above. C: Nonsense APC mutation in case A2. DNA sequencing shows a CAA (glycine) → TAA (stop) mutation at codon 1444 that is present in the ACC but not in the nonneoplastic tissue from this patient.
Immunohistochemical labeling for β-catenin protein revealed strong nuclear and cytoplasmic accumulation in three (15%) ACCs (cases A5, A13, and A16). The labeling was patchy in nature in all three cases, ranging from 20 to 60% of the neoplastic cells (Figure 3) ▶ . Nonneoplastic pancreatic acini, ducts, and gastrointestinal epithelial cells showed the expected membranous and faint cytoplasmic labeling, but no nuclear or strong cytoplasmic β-catenin. Stromal cells in fibrous tissue between lobules of neoplastic epithelial cells did not show β-catenin accumulation.
Figure 3.

Immunohistochemical labeling for β-catenin in an ACC. Nuclear and cytoplasmic accumulation of β-catenin in neoplastic epithelial cells is present in this example (case A16), which also showed a truncating APC gene mutation. Stromal cells in the intervening fibrous bands show membranous β-catenin labeling, but are negative for nuclear and cytoplasmic accumulation.
Only a moderate degree of correlation was present between the detection of β-catenin or APC gene mutation within an ACC and β-catenin protein accumulation by immunohistochemistry. Of the four ACCs with β-catenin or APC mutations by sequencing, two (50%, cases A13 and A16) demonstrated patchy nuclear accumulation of β-catenin in 20% and 60% of neoplastic cells, respectively, whereas the other two did not. One additional ACC (case A5) demonstrated nuclear β-catenin in 50% of neoplastic cells but did not contain detectable β-catenin or APC mutation.
Allelic Loss on 11p
Allelic loss on 11p15.5 was present in 6 of 12 (50%) ACCs that contained amplifiable DNA and were informative in one or both 11p microsatellite markers (Figure 4) ▶ . All three cases that were informative at both TH and D11S1984 showed LOH on both markers in the ACCs.
Figure 4.
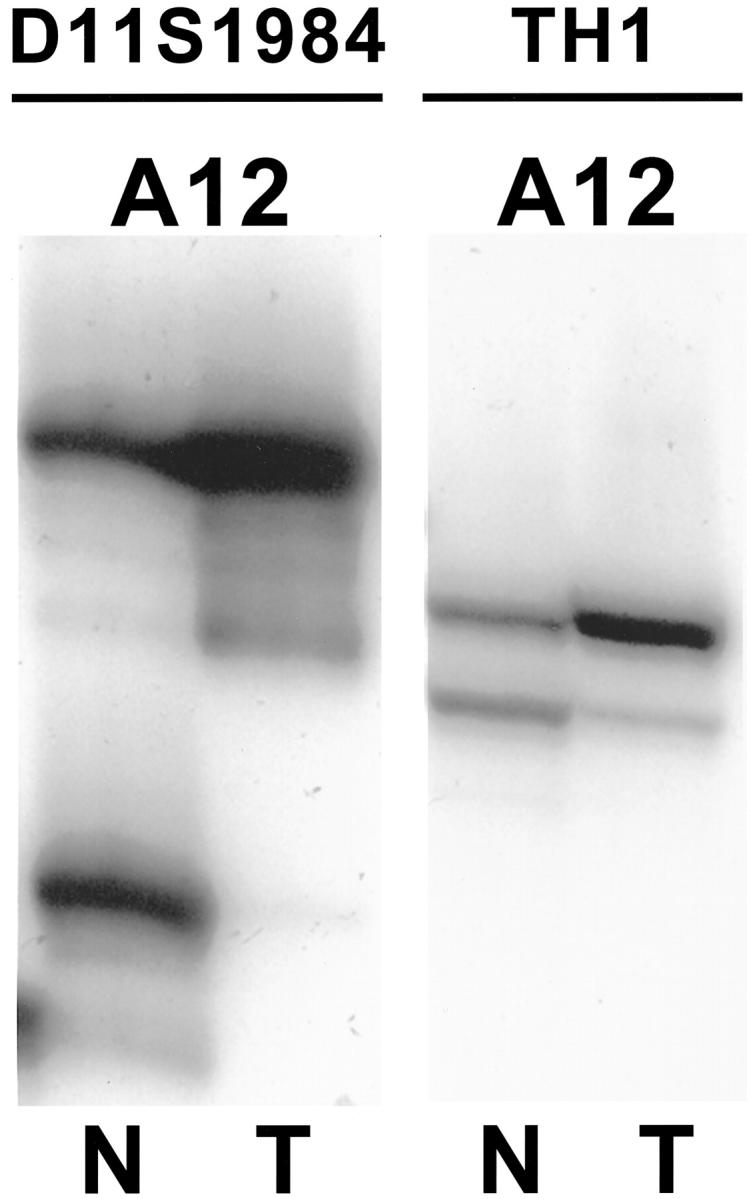
Allelic loss on 11p15.5 in ACC. LOH is present with both the D11S1984 and TH1 microsatellite markers for case A12, which was informative for both markers. N, normal; T, ACC.
MSI
MSI was present in 3 of 13 ACCs (23%) that contained amplifiable DNA and for which normal control tissue was available. One ACC (case A19) showed MSI-high, with allelic shifts in four of five NCI microsatellite markers (D5S346, D17S250, Bat-25, and Bat-26) as well as in D5S299 and D11S1984. Two ACCs showed MSI-low, with allelic shifts only in one marker (D17S250 in case A4 and Bat-26 in case A5). Correlation of the histopathological features in these neoplasms revealed that the ACC with MSI-high (A19) showed an interesting pattern of sharp demarcation between the neoplasm and the nonneoplastic pancreas, poor differentiation, and areas of syncytial growth of the neoplastic epithelial cells, features that have previously been described in medullary carcinomas of the pancreas. 46,47 There was not a pronounced inflammatory infiltrate in this ACC, but this feature is also lacking in the reported pancreatic medullary carcinomas. 46,47 However, unlike the other previously reported cases of medullary carcinoma of the pancreas, histological foci of clear-cut acini were present in this neoplasm (Figure 5) ▶ , and acinar differentiation of the neoplastic epithelial cells as evidenced by diffuse immunohistochemical expression of trypsin, chymotrypsin, and lipase was also present.
Figure 5.

MSI in an ACC. A: Case A19 demonstrates MSI-high, with allelic shifts in the microsatellite markers D5S346, D17S250, Bat-25, and Bat-26. Only D2S123 is stable. In contrast, no allelic shifts are present in case A12, which is microsatellite stable. B: The histopathological features of case A19 overlapped with those of medullary carcinoma of the pancreas, with areas of poor differentiation and syncytial growth. C: Unlike cases of previously described medullary carcinoma, foci of clear-cut acinar differentiation were also present.
Alterations in DPC and p53
Normal Dpc4 protein expression was preserved in all 20 ACCs. No significant p53 accumulation was detected by immunohistochemistry in any of the 20 ACCs (immunohistochemistry failed in one case).
Discussion
Studies of pancreatic ACCs to date have demonstrated the lack or rarity of genetic alterations commonly present in ductal adenocarcinomas, including mutations in the K-ras oncogene and p53 and DPC4 tumor suppressor genes. 17,20-23 The specific molecular alterations that do characterize ACCs have remained obscure. 24
Both the histopathological and immunohistochemical features of ACCs overlap with those of pancreatoblastomas, another rare but distinctive pancreatic malignancy. Although pancreatoblastomas occur predominantly in the pediatric population and ACCs predominantly in adults, occasional cases of ACCs in children (2 in this series of 21 ACCs) 1,48,49 and pancreatoblastomas in adults 27,50-54 are encountered, and some investigators regard pancreatoblastomas as the pediatric counterpart of ACCs. 27 We have recently characterized genetic alterations in pancreatoblastomas and have demonstrated high frequencies of both allelic loss on chromosome 11p and mutations in the APC/β-catenin pathway, a molecular genotype that is distinct from that of pancreatic ductal adenocarcinoma. 26 The histological and clinicopathological overlap between ACCs and pancreatoblastomas suggests that ACCs and pancreatoblastomas might share similar genetic alterations.
The most common molecular alteration we identified in ACCs was allelic loss on chromosome 11p. Fifty percent of ACCs in this series (6 of 12 informative cases) showed LOH for TH1 and D11S1984, microsatellite markers near the WT-2 locus on 11p15.5. This frequency is somewhat higher than the 25% reported by Rigaud and colleagues 24 in the only previous reported allelotype of pancreatic ACC. However, that study of eight informative ACCs used different 11p microsatellite markers (including one of two markers on 11p15.3), 24 making direct comparison of results difficult. The WT-2 locus on 11p15.5 is a heavily imprinted area containing growth- and cell cycle-regulatory genes. 55,56 Congenital disruption of this locus is associated with Beckwith-Wiedemann syndrome, a maldevelopment syndrome of tissue overgrowth and organomegaly that is characterized by an increased risk for embryonal malignancies including hepatoblastoma, Wilm’s tumor, and rhabdomyosarcoma. 55,56 Several cases of pancreatoblastomas in patients with Beckwith-Wiedemann syndrome have also been reported, 57,58 and indeed, among pancreatoblastomas studied by us, 86% also showed allelic loss on 11p15.5. 26 The somewhat lower rate of 11p LOH in ACCs compared with pancreatoblastomas mirrors the relationship between another typically adult tumor—hepatocellular carcinoma (HCC)—and its embryonal or pediatric counterpart—hepatoblastoma. Allelic loss on 11p (and in particular, loss of the maternal allele) is reported in up to 75% of hepatoblastomas, 59-63 but at lower rates in HCCs. 64-68
The second most common genetic alteration we identified in ACCs was mutation in the APC/β-catenin pathway in 23.5% (4 of 17) cases. One ACC contained a β-catenin gene mutation at threonine codon 41 that would be predicted to disrupt the APC/β-catenin pathway by constitutive β-catenin protein activation, and the other three ACCs contained APC gene mutations predicted to result in APC protein truncation. In comparison, we previously found a much higher APC/β-catenin mutation frequency of 67% in pancreatoblastomas. 26 Interestingly, both of the adult pancreatoblastoma cases in that study demonstrated targeting of this pathway, 26 whereas neither of the two pediatric ACC cases in the current series contained APC or β-catenin mutations. One of the adult pancreatoblastoma cases in our previous study occurred in a patient with familial adenomatous polyposis, 26 although none of the ACCs in the current study did.
The lower rate of APC/β-catenin targeting in ACCs in comparison with pancreatoblastomas again mirrors the molecular findings in adult HCCs and childhood hepatoblastomas. Among hepatoblastomas, alterations in the APC/β-catenin pathway are frequently present. 69-75 These most commonly take the form of activating β-catenin gene mutations, reported in one-half to two-thirds of hepatoblastomas in larger series. 71,73 In contrast, β-catenin mutations are found on average only in approximately one-fifth of HCCs. 76-82 Additionally, whereas most hepatoblastomas demonstrate strong nuclear accumulation of β-catenin protein on immunohistochemistry, the frequency of β-catenin labeling in HCCs is lower and the correlation between β-catenin mutation and immunolabeling is less clear; some HCCs contain β-catenin mutations without the expected nuclear accumulation of the stabilized protein, and some show β-catenin immunolabeling without corresponding mutation. 78,79,81,82 Similarly, we found that whereas the majority of pancreatoblastomas demonstrate strong nuclear β-catenin with good correlation between labeling and mutation status, 26 only 15% of ACCs show nuclear β-catenin accumulation, and only moderate correlation exists between the immunolabeling and mutation results.
The etiology of the discrepancy between β-catenin immunohistochemistry and APC/β-catenin mutation analysis in this study is not clear. However, in addition to some HCCs, 78,79,81,82 multiple other human organ systems have also been noted to share this discrepancy between nuclear β-catenin labeling and APC/β-catenin gene mutations. In colorectal polyps, thyroid carcinomas, uterine and ovarian carcinomas, and soft tissue sarcomas, some neoplasms harbor mutations in the APC/β-catenin pathway without the expected nuclear accumulation of β-catenin protein, whereas others show nuclear β-catenin labeling without demonstrable mutations. 83-88 In colonic adenomas, tumor size has been reported as a factor in nuclear β-catenin accumulation, 84 whereas proliferative activity is more closely correlated with nuclear β-catenin expression in high-grade sarcomas. 88
One ACC (7.6%) in the current series was suspected to harbor MSI based on the finding of allelic shifts in the 11p and 5q allelic loss assays, and this neoplasm subsequently demonstrated high-level MSI with the NCI microsatellite markers. 44 The MSI phenotype has recently been described in a subset of poorly differentiated carcinomas of the pancreas that show distinctive histological features common to medullary carcinomas of the colon (including a syncytial growth pattern, poor differentiation, and a pushing rather than infiltrative border, but interestingly only uncommonly a prominent lymphocytic infiltrate) as well as distinctive molecular features (frequent wild-type K-ras genes). 46,47 Histopathologically, the ACC with MSI-H in this study was characterized by areas of syncytial growth, poor differentiation, and a pushing border (although this latter feature is also common to ACCs in general), but areas of well-defined acinar differentiation both histologically and by immunohistochemistry were also present. Although the medullary carcinomas reported by Goggins and colleagues 46 and Wilentz and colleagues 47 were considered to represent poorly differentiated adenocarcinomas, the finding of MSI in an ACC in the present series suggests that MSI might constitute an alternative molecular pathway of neoplastic progression in a fraction of ACCs as well.
In contrast to the above genetic alterations in ACCs, we found no evidence for involvement of p53 or DPC4 genes in the molecular pathogenesis of ACC. Our findings are in agreement with previous investigations that have shown no or only rare alterations of the K-ras, DPC4, and p53 genes in pancreatic ACCs. 17,20-24 Among conventional pancreatic ductal adenocarcinomas, early mutational activation of the K-ras oncogene occurs in nearly all neoplasms, 13,14 inactivation of the DPC4 tumor suppressor gene occurs in slightly more than one-half, 18,19 and late inactivation of p53 occurs in up to 70%. 13,15-17 The findings in this study underscore the sharp clinicopathological and genetic contrast between pancreatic ACCs and ductal adenocarcinomas, and suggest that at least a subset of ACCs share both pathological and molecular genetic features with pancreatoblastomas.
Footnotes
Address reprint requests to Susan Abraham, M.D., Division of GI/Liver Pathology, Dept. of Pathology, Ross Bldg., Room 632, The Johns Hopkins University School of Medicine, 720 Rutland Ave., Baltimore, MD 21205-2196. E-mail: sabraham@jhmi.edu.
Supported by a National Cancer Institute SPORE grant in gastrointestinal cancer (grant P50-CA62924).
References
- 1.Klimstra DS, Heffess CS, Oertel JE, Rosai J: Acinar cell carcinoma of the pancreas. A clinicopathologic study of 28 cases. Am J Surg Pathol 1992, 16:815–837 [DOI] [PubMed]
- 2.Chen J, Baithun SI, Ramsay MA: Histogenesis of pancreatic carcinomas: a study based on 248 cases. J Pathol 1985, 146:65-76 [DOI] [PubMed] [Google Scholar]
- 3.Morohoshi T, Kanda M, Horie A, Chott A, Dreyer T, Kloppel G, Heitz PU: Immunocytochemical markers of uncommon pancreatic tumors. Acinar cell carcinoma, pancreatoblastoma, and solid cystic (papillary-cystic) tumor. Cancer 1987, 59:739-747 [DOI] [PubMed] [Google Scholar]
- 4.Shinagawa T, Tadokoro M, Maeyama S, Maeda C, Yamaguchi S, Morohoshi T, Ishikawa E: Alpha fetoprotein-producing acinar cell carcinoma of the pancreas showing multiple lines of differentiation. Virchows Arch 1995, 426:419-423 [DOI] [PubMed] [Google Scholar]
- 5.Caruso RA, Inferrera A, Tuccari G, Barresi G: Acinar cell carcinoma of the pancreas. A histologic, immunocytochemical and ultrastructural study. Histol Histopathol 1994, 9:53-58 [PubMed] [Google Scholar]
- 6.Foulet A, Copin MC, Jaillard S, Wurtz A, Gosselin B: Acinar cell carcinoma of the pancreas revealed by Weber-Christian syndrome. Ann Pathol 1995, 15:438-442 [PubMed] [Google Scholar]
- 7.Kuerer H, Shim H, Pertsemlidis D, Unger P: Functioning pancreatic acinar cell carcinoma: immunohistochemical and ultrastructural analyses. Am J Clin Oncol 1997, 20:101-107 [DOI] [PubMed] [Google Scholar]
- 8.MacMahon HE, Brown PA, Shen EM: Acinar cell carcinoma of the pancreas with subcutaneous fat necrosis. Gastroenterology 1965, 49:555-559 [PubMed] [Google Scholar]
- 9.Burns WA, Matthews MJ, Hamosh M, Weide GV, Blum R, Johnson FB: Lipase-secreting acinar cell carcinoma of the pancreas with polyarthropathy. A light and electron microscopic, histochemical, and biochemical study. Cancer 1974, 33:1002-1009 [DOI] [PubMed] [Google Scholar]
- 10.Radin DR, Colletti PM, Forrester DM, Tang WW: Pancreatic acinar cell carcinoma with subcutaneous and intraosseous fat necrosis. Radiology 1986, 158:67-68 [DOI] [PubMed] [Google Scholar]
- 11.Kawamoto S, Hiraoka T, Kanemitsu K, Kimura M, Miyauchi Y, Takeya M: Alpha-fetoprotein-producing pancreatic cancer—a case report and review of 28 cases. Hepatogastroenterology 1992, 39:282-286 [PubMed] [Google Scholar]
- 12.Kuopio T, Ekfors TO, Nikkanen V, Nevalainen TJ: Acinar cell carcinoma of the pancreas. Report of three cases. APMIS 1995, 103:69-78 [DOI] [PubMed] [Google Scholar]
- 13.Hruban RH, Goggins M, Parsons J, Kern SE: Progression model for pancreatic cancer. Clin Cancer Res 2000, 6:2969-2972 [PubMed] [Google Scholar]
- 14.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M: Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53:549-554 [DOI] [PubMed] [Google Scholar]
- 15.Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CJ, Kern SE: p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res 1994, 54:3025-3033 [PubMed] [Google Scholar]
- 16.DiGiuseppi JA, Hruban RH, Goodman SN, Polak M, van den Berg FM, Allsion DC, Cameron JL, Offerhaus GJA: Overexpression of p53 protein in adenocarcinoma of the pancreas. Am J Clin Pathol 1994, 101:684-688 [DOI] [PubMed] [Google Scholar]
- 17.Moore PS, Orlandini S, Zamboni G, Capelli P, Rigaud G, Falconi M, Bassi C, Lemoine NR, Scarpa A: Pancreatic tumours: molecular pathways implicated in ductal cancer are involved in ampullary but not in exocrine nonductal or endocrine tumorigenesis. Br J Cancer 2001, 84:253-262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM, Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H, Sidransky D, Casero Jr RA, Meltzer PS, Hahn SA, Kern SE: DPC4 gene in various tumor types. Cancer Res 1996, 56:2527–2530 [PubMed]
- 19.Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE: DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271:350-353 [DOI] [PubMed] [Google Scholar]
- 20.Terhune PG, Heffess CS, Longnecker DS: Only wild-type c-Ki-ras codons 12, 13, and 61 in human pancreatic acinar cell carcinomas. Mol Carcinog 1994, 10:110-114 [DOI] [PubMed] [Google Scholar]
- 21.Hoorens A, Lemoine NR, McLellan E, Morohoshi T, Kamisawa T, Heitz PU, Stamm B, Ruschoff J, Wiedenmann B, Kloppel G: Pancreatic acinar cell carcinoma. An analysis of cell lineage markers, p53 expression, and Ki-ras mutation. Am J Pathol 1993, 143:685-698 [PMC free article] [PubMed] [Google Scholar]
- 22.Pellegata NS, Sessa F, Renault B, Bonato M, Leone BE, Solcia E, Ranzani GN: K-ras and p53 gene mutations in pancreatic cancer: ductal and nonductal tumors progress through different genetic lesions. Cancer Res 1994, 54:1556-1560 [PubMed] [Google Scholar]
- 23.Longnecker DS: Molecular pathology of invasive carcinoma. Ann NY Acad Sci 1999, 880:74-82 [DOI] [PubMed] [Google Scholar]
- 24.Rigaud G, Moore PS, Zamboni G, Orlandini S, Taruscio D, Paradisi S, Lemoine NR, Kloppel G, Scarpa A: Allelotype of pancreatic acinar cell carcinoma. Int J Cancer 2000, 88:772-777 [DOI] [PubMed] [Google Scholar]
- 25.Hsueh C, Kuo TT: Acinar cell carcinoma of the pancreas. Report of two cases with complex histomorphologic features causing diagnostic problems. Int J Pancreatol 1992, 12:305-313 [PubMed] [Google Scholar]
- 26.Abraham SC, Wu TT, Klimstra DS, Finn L, Lee JH, Yeo CJ, Cameron JL, Hruban RH: Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas: frequent alterations in the APC/β-catenin pathway and chromosome 11p. Am J Pathol 2001, 159:1619-1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klimstra DS, Wenig BM, Adair CF, Heffess CS: Pancreatoblastoma. A clinicopathologic study and review of the literature. Am J Surg Pathol 1995, 19:1371-1389 [DOI] [PubMed] [Google Scholar]
- 28.Lonardo F, Cubilla AL, Klimstra DS: Microadenocarcinoma of the pancreas—morphologic pattern or pathologic entity? A reevaluation of the original series. Am J Surg Pathol 1996, 20:1385-1393 [DOI] [PubMed] [Google Scholar]
- 29.Klimstra DS, Rosai J, Heffess CS: Mixed acinar-endocrine carcinomas of the pancreas. Am J Surg Pathol 1994, 18:765-778 [DOI] [PubMed] [Google Scholar]
- 30.Kloppel G: Mixed exocrine-endocrine tumors of the pancreas. Semin Diagn Pathol 2000, 17:104-108 [PubMed] [Google Scholar]
- 31.Muramatsu T, Kijima H, Tsuchida T, Konagaya M, Matsubayashi H, Tada N, Nakamura M, Ueyama Y: Acinar-islet cell tumor of the pancreas: report of a malignant pancreatic composite tumor. J Clin Gastroenterol 2000, 31:175-178 [DOI] [PubMed] [Google Scholar]
- 32.Cho KJ, Kim JY, Lee SS, Khang SK, Kim CW: Mixed acinar-endocrine carcinoma of the pancreas—a case report. J Korean Med Sci 1996, 11:188-192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ordonez NG, Mackay B: Acinar cell carcinoma of the pancreas. Ultrastruct Pathol 2000, 24:227-241 [DOI] [PubMed] [Google Scholar]
- 34.Cingolani N, Shaco-Levy R, Farruggio A, Klimstra DS, Rosai J: Alpha-fetoprotein production by pancreatic tumors exhibiting acinar cell differentiation: study of five cases, one arising in a mediastinal teratoma. Hum Pathol 2000, 31:938-944 [DOI] [PubMed] [Google Scholar]
- 35.Nojima T, Kojima T, Kato H, Sato T, Koito K, Nagashima K: Alpha-fetoprotein-producing acinar cell carcinoma of the pancreas. Hum Pathol 1992, 23:828-830 [DOI] [PubMed] [Google Scholar]
- 36.Ono J, Sakamoto H, Sakoda K, Yagi Y, Hagio S, Sato E, Katsuki T: Acinar cell carcinoma of the pancreas with elevated serum alpha-fetoprotein. Int Surg 1984, 69:361-364 [PubMed] [Google Scholar]
- 37.Balian A, Hammel P, Terris B, Sauvanet A, Belghiti J, Ruszniewski P, Bernades P: Increase of serum alpha-fetoprotein in a patient with acinar cell carcinoma of the pancreas. Gastroenterol Clin Biol 1997, 21:231-232 [PubMed] [Google Scholar]
- 38.Ishizaki A, Koito K, Namieno T, Nagakawa T, Murashima Y, Suga T: Acinar cell carcinoma of the pancreas: a rare case of an alpha-fetoprotein-producing cystic tumor. Eur J Radiol 1995, 21:58-60 [DOI] [PubMed] [Google Scholar]
- 39.Baas IO, Mulder JW, Offerhaus GJ, Vogelstein B, Hamilton SR: An evaluation of six antibodies for immunohistochemistry of mutant p53 gene product in archival colorectal neoplasms. J Pathol 1994, 172:5-12 [DOI] [PubMed] [Google Scholar]
- 40.Wilentz RE, Su GH, Dai JL, Sparks AB, Argani P, Sohn TA, Yeo CJ, Kern SE, Rhuban RH: Immunohistochemical labeling for Dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol 2000, 156:37-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moskaluk CA, Kern SE: Microdissection and polymerase chain reaction amplification of genomic DNA from histological tissue sections. Am J Pathol 1997, 150:1547-1552 [PMC free article] [PubMed] [Google Scholar]
- 42.Yashima K, Nakamori S, Murakami Y, Yamaguchi A, Hayashi K, Ishikawa O, Konishi Y, Sekiya T: Mutations of the adenomatous polyposis coli gene in the mutation cluster region: comparison of human pancreatic and colorectal cancers. Int J Cancer 1994, 59:43-47 [DOI] [PubMed] [Google Scholar]
- 43.Wu TT, Watanabe T, Heitmiller R, Zahrak M, Forastiere AA, Hamilton SR: Genetic alterations in Barrett esophagus and adenocarcinomas of the esophagus and esophagogastric junction region. Am J Pathol 1998, 153:287-294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S: A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998, 58:5248-5257 [PubMed] [Google Scholar]
- 45.Berg KD, Glaser CL, Thompson RE, Hamilton SR, Griffin CA, Eshleman JR: Detection of microsatellite instability by fluorescence multiplex polymerase chain reaction. J Mol Diagn 2000, 2:20-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goggins M, Offerhaus GJ, Hilgers W, Griffin CA, Shekher M, Tang D, Sohn TA, Yeo CJ, Kern SE, Hruban RH: Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am J Pathol 1998, 152:1501-1507 [PMC free article] [PubMed] [Google Scholar]
- 47.Wilentz RE, Goggins M, Redston M, Marcus VA, Adsay NV, Sohn TA, Kadkol SS, Yeo CJ, Choti M, Zahurak M, Johnson K, Tascilar M, Offerhaus GJ, Hruban RH, Kern SE: Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas. A newly described and characterized entity. Am J Pathol 2000, 156:1641-1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osborne BM, Culbert SJ, Cangir A, MacKay B: Acinar cell carcinoma of the pancreas in a 9-year-old child: case report with electron microscopic observations. South Med J 1977, 70:370-372 [DOI] [PubMed] [Google Scholar]
- 49.Mah PT, Loo DC, Tock EP: Pancreatic acinar cell carcinoma in childhood. Am J Dis Child 1974, 128:101-104 [DOI] [PubMed] [Google Scholar]
- 50.Robin E, Terris B, Valverde A, Molas G, Belghiti J, Bernades P, Ruszniewski P: Pancreatoblastoma in adults. Gastroenterol Clin Biol 1997, 21:880-883 [PubMed] [Google Scholar]
- 51.Palosaari D, Clayton F, Seaman J: Pancreatoblastoma in an adult. Arch Pathol Lab Med 1986, 110:650-652 [PubMed] [Google Scholar]
- 52.Dunn JL, Longnecker DS: Pancreatoblastoma in an older adult. Arch Pathol Lab Med 1995, 119:547-551 [PubMed] [Google Scholar]
- 53.Hoorens A, Gebhard F, Kraft K, Lemoine NR, Kloppel G: Pancreatoblastoma in an adult: its separation from acinar cell carcinoma. Virchows Arch 1994, 424:485-490 [DOI] [PubMed] [Google Scholar]
- 54.Levey JM, Banner BF: Adult pancreatoblastoma: a case report and review of the literature. Am J Gastroenterol 1996, 91:1841-1844 [PubMed] [Google Scholar]
- 55.Li M, Squire JA, Weksberg R: Molecular genetics of Beckwith-Wiedemann syndrome. Curr Opin Pediatr 1997, 9:623-629 [DOI] [PubMed] [Google Scholar]
- 56.Li M, Squire JA, Weksberg R: Molecular genetics of Wiedemann-Beckwith syndrome. Am J Med Genet 1998, 79:253-259 [PubMed] [Google Scholar]
- 57.Kohda E, Iseki M, Ikawa H, Endoh M, Yokoyama J, Mukai M, Hata J, Yamazaki H, Miyauchi J, Saeki M: Pancreatoblastoma. Three original cases and review of the literature. Acta Radiol 2000, 41:334-337 [DOI] [PubMed] [Google Scholar]
- 58.Koh TH, Cooper JE, Newman CL, Walker TM, Kiely EM, Hoffmann EB: Pancreatoblastoma in a neonate with Wiedemann-Beckwith syndrome. Eur J Pediatr 1986, 145:435-438 [DOI] [PubMed] [Google Scholar]
- 59.Albrecht S, von Schweinitz D, Waha A, Kraus JA, von Deimling A, Pietsch T: Loss of maternal alleles on chromosome arm 11p in hepatoblastoma. Cancer Res 1994, 54:5041-5044 [PubMed] [Google Scholar]
- 60.Byrne JA, Simms LA, Little MH, Algar EM, Smith PJ: Three non-overlapping regions of chromosome arm 11p allele loss identified in infantile tumors of adrenal and liver. Genes Chromosom Cancer 1993, 8:104-111 [DOI] [PubMed] [Google Scholar]
- 61.Kiechle-Schwarz M, Scherer G, Kovacs G: Cytogenetic and molecular studies on six sporadic hepatoblastomas. Cancer Genet Cytogenet 1989, 41:286 [Google Scholar]
- 62.Koufos A, Hansen MF, Copeland NG, Jenkins NA, Lampkin BC, Cavenee WK: Loss of heterozygosity in three embryonal tumours suggests a common pathogenetic mechanism. Nature 1985, 316:330-334 [DOI] [PubMed] [Google Scholar]
- 63.Blaker H, Hofmann WJ, Reiker RJ, Penzel R, Graf M, Otto HF: Beta-catenin accumulation and mutation of the CTNNB1 gene in hepatoblastoma. Genes Chromosom Cancer 1999, 25:399-402 [PubMed] [Google Scholar]
- 64.Sheu JC, Lin YW, Chou HC, Huang GT, Lee HS, Lin YH, Huang SY, Chen CH, Wang JT, Lee PH, Lin JT, Lu FJ, Chen DS: Loss of heterozygosity and microsatellite instability in hepatocellular carcinoma in Taiwan. Br J Cancer 1999, 80:468-476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yumoto Y, Hanafusa T, Hada H, Morita T, Ooguchi S, Shinji N, Mitani T, Hamaya K, Koide N, Tsuji T: Loss of heterozygosity and analysis of mutations of p53 in hepatocellular carcinoma. J Gastroenterol Hepatol 1995, 10:179-185 [DOI] [PubMed] [Google Scholar]
- 66.Nishida N, Fukuda Y, Kokuryu H, Sadamoto T, Isowa G, Honda K, Yamaoka Y, Ikenaga M, Imura H, Ishizaki K: Accumulation of allelic loss on arms of chromosomes 13q, 16q and 17p in the advanced stages of human hepatocellular carcinoma. Int J Cancer 1992, 51:862-868 [DOI] [PubMed] [Google Scholar]
- 67.Kiechle-Schwarz M, Scherer G, Kovacs G: No evidence for loss of alleles at 11p in HBV negative hepatocellular carcinomas. Genes Chromosom Cancer 1990, 1:312-314 [DOI] [PubMed] [Google Scholar]
- 68.Zhang WD, Hirohashi S, Tsuda H, Shimosato Y, Yokota J, Terada M, Sugimura T: Frequent loss of heterozygosity on chromosomes 16 and 4 in human hepatocellular carcinoma. Jpn J Cancer Res 1990, 81:108-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kurahashi H, Takami K, Oue T, Kusafuka T, Okada A, Tawa A, Okada S, Nishisho I: Biallelic inactivation of the APC gene in hepatoblastoma. Cancer Res 1995, 55:5007-5011 [PubMed] [Google Scholar]
- 70.Oda H, Imai Y, Nakatsuru Y, Hata J, Ishikawa T: Somatic mutations of the APC gene in sporadic hepatoblastomas. Cancer Res 1996, 56:3320-3323 [PubMed] [Google Scholar]
- 71.Koch A, Denkhaus D, Albrecht S, Leuschner I, von Schweinitz D, Pietsch T: Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer Res 1999, 59:269-273 [PubMed] [Google Scholar]
- 72.Park WS, Oh RR, Park JY, Kim PJ, Shin MS, Lee JH, Kim HS, Lee SH, Kim SY, Park YG, An WG, Kim HS, Jang JJ, Yoo NJ, Lee JY: Nuclear localization of beta-catenin is an important prognostic factor in hepatoblastoma. J Pathol 2001, 193:483-490 [DOI] [PubMed] [Google Scholar]
- 73.Wei Y, Fabre M, Branchereau S, Gauthier F, Perilongo G, Buendia MA: Activation of beta-catenin in epithelial and mesenchymal hepatoblastomas. Oncogene 2000, 19:498-504 [DOI] [PubMed] [Google Scholar]
- 74.Jeng YM, Wu MZ, Mao TL, Chang MH, Hsu HC: Somatic mutations of beat-catenin play a crucial role in the tumorigenesis of sporadic hepatoblastoma. Cancer Lett 2000, 152:45-51 [DOI] [PubMed] [Google Scholar]
- 75.Takayasu H, Horie H, Hiyama E, Matsunaga T, Hayashi Y, Watanabe Y, Suita S, Kaneko M, Sasaki F, Hashizume K, Ozaki T, Furuuchi K, Tada M, Ohnuma N, Nakagawara A: Frequent deletions and mutations of the beta-catenin gene are associated with overexpression of cyclin D1 and fibronectin and poorly differentiated histology in childhood hepatoblastoma. Clin Cancer Res 2001, 7:901-908 [PubMed] [Google Scholar]
- 76.Legoix P, Bluteau O, Bayer J, Perret C, Balabaud C, Belghiti J, Franco D, Thomas G, Laurent-Puig P, Zucman-Rossi J: Beta-catenin mutations in hepatocellular carcinoma correlate with a low rate of loss of heterozygosity. Oncogene 1999, 18:4044-4046 [DOI] [PubMed] [Google Scholar]
- 77.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A, Perret C: Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci USA 1998, 95:8847-8851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Van Nhieu JT, Renard CA, Wei Y, Cherqui D, Zafrani ES, Buendia MA: Nuclear accumulation of mutated β-catenin in hepatocellular carcinoma is associated with increased cell proliferation. Am J Pathol 1999, 155:703-710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hsu HC, Jeng YM, Mao TL, Chu JS, Lai PL, Peng SY: β-catenin mutations are associated with a subset of low-stage hepatocellular carcinoma negative for hepatitis B virus and with favorable prognosis. Am J Pathol 2000, 157:763-770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Miyoshi Y, Iwao K, Nagasawa Y, Aihara T, Sasaki Y, Imaoka S, Murata M, Shimano T, Nakamura Y: Activation of the β-catenin gene in primary hepatocellular carcinomas by somatic alterations involving exon 3. Cancer Res 1998, 58:2524-2527 [PubMed] [Google Scholar]
- 81.Terris B, Pineau P, Bregeaud L, Valla D, Belghiti J, Tiollais P, Degott C, Dejean A: Close correlation between β-catenin gene alterations and nuclear accumulation of the protein in human hepatocellular carcinomas. Oncogene 1999, 18:6583-6588 [DOI] [PubMed] [Google Scholar]
- 82.Huang H, Fujii H, Sankila A, Mahler-Araujo BM, Matsuda M, Cathomas G, Ohgaki H: β-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C infection. Am J Pathol 1999, 155:1795-1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Valizadeh A, Karayiannakis AJ, El-Hariry I, Kmiot W, Pignatelli M: Expression of E-cadherin-associated molecules (α-, β-, and γ-catenins and p120) in colorectal polyps. Am J Pathol 1997, 150:1977-1984 [PMC free article] [PubMed] [Google Scholar]
- 84.Brabletz T, Herrmann K, Jung A, Faller G, Kirchner T: Expression of nuclear β-catenin and c-myc is correlated with tumor size but not with proliferative activity of colorectal adenomas. Am J Pathol 2000, 156:865-870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Garcia-Rostan G, Tallini G, Herrero A, D’Aquila TG, Carcangiu ML, Rimm DL: Frequent mutation and nuclear localization of β-catenin in anaplastic thyroid carcinoma. Cancer Res 1999, 59:1811-1815 [PubMed] [Google Scholar]
- 86.Fukuchi T, Sakamoto M, Tsuda H, Maruyama K, Nozawa S, Hirohashi S: β-catenin mutation in carcinoma of the uterine endometrium. Cancer Res 1998, 58:3526-3528 [PubMed] [Google Scholar]
- 87.Palacios J, Gamallo C: Mutations in the β-catenin gene (CTNNB1) in endometrioid ovarian carcinomas. Cancer Res 1998, 58:1344-1347 [PubMed] [Google Scholar]
- 88.Kuhnen C, Herter P, Muller O, Muehlberger T, Krause L, Homann H, Steinau HU, Muller KM: β-catenin in soft tissue sarcomas: expression is related to proliferative activity in high-grade sarcomas. Mod Pathol 2000, 13:1005-1013 [DOI] [PubMed] [Google Scholar]