Selecting for Functional Alternative Splices in ESTs (original) (raw)
Abstract
The expressed sequence tag (EST) collection in dbEST provides an extensive resource for detecting alternative splicing on a genomic scale. Using genomically aligned ESTs, a computational tool (TAP) was used to identify alternative splice patterns for 6400 known human genes from the RefSeq database. With sufficient EST coverage, one or more alternatively spliced forms could be detected for nearly all genes examined. To identify high (>95%) confidence observations of alternative splicing, splice variants were clustered on the basis of having mutually exclusive structures, and sample statistics were then applied. Through this selection, alternative splices expected at a frequency of >5% within their respective clusters were seen for only 17%–28% of genes. Although intron retention events (potentially unspliced messages) had been seen for 36% of the genes overall, the same statistical selection yielded reliable cases of intron retention for <5% of genes. For high-confidence alternative splices in the human ESTs, we also noted significantly higher rates both of cross-species conservation in mouse ESTs and of validation in the GenBank mRNA collection. We suggest quantitative analytical approaches such as these can aid in selecting useful targets for further experimental characterization and in so doing may help elucidate the mechanisms and biological implications of alternative splicing.
Alternative splicing of eukaryotic pre-mRNAs is a mechanism for generating potentially many transcript isoforms from a single gene. It is known to play important regulatory functions. A classic example is the Drosophila sex-determination pathway, in which alternative splicing acts as a sex-specific genetic switch that forms the basis of a regulatory hierarchy (Boggs et al. 1987; Baker 1989; Lopez 1999). Another intriguing example was found in the inner ear of the chicken, where differential distribution of splice variants for the calcium-activated potassium channel gene slo may form a tonotopic gradient and attune sensory hair cells to the detection of different sound frequencies (Black 1998; Ramanathan et al. 1999; Graveley 2001). Alternative splicing is also implicated in human diseases. For example, the neurodegenerative disease FTDP-17 has been associated with mutations that affect the alternative splicing of tau pre-mRNAs (Goedert et al. 2000; Jiang et al. 2000).
Initial sequencing and analysis of the human genome has placed further attention on the role of alternative splicing. The surprising finding that the genome contains some 30,000 protein-coding genes—significantly less than previously estimated—led to the proposal that alternative splicing contributes greatly to functional diversity (Ewing and Green 2000; Lander et al. 2001; Venter et al. 2001). Based on analysis of expressed sequence tags (ESTs), a number of studies have, indeed, shown that alternative splicing is prevalent in human as well as mouse genes (Mironov et al. 1999; Brett et al. 2000; Croft et al. 2000; Kan et al. 2001; Modrek et al. 2001; Kochiwa et al. 2002). These results indicate that a vast number of splice variants have yet to be discovered and characterized.
ESTs provide a tremendous resource for transcript analysis at the sequence level. More than 4.5 million human ESTs are available in the public domain dbEST database (Boguski et al. 1993), representing an in-depth sampling of the transcriptome. Methods have been developed to discover splice variants using the approach of EST self-clustering (Burke et al. 1998; Brett et al. 2000). With the availability of a nearly complete sequence of the human genome (Lander et al. 2001), aligning ESTs to the genomic sequence has become a practical strategy. A number of methods and resources based on this strategy have been developed, enabling large-scale or genome-wide surveys of alternative splicing (Mironov et al. 1999; Hide et al. 2001; Kan et al. 2001; Modrek et al. 2001; Brett et al. 2002) and heterogeneity of polyadenylation (Kan et al. 2000).
In a relatively short time, significant discoveries have been made through the combination of bioinformatics methodology and genomic resources. However, the sheer volume of predictions churned out by genomic-scale studies can create a bottleneck in terms of experimental validation (Modrek and Lee 2001). Concerns have also been raised about the reliability of the EST data (Modrek and Lee 2001), which could have an impact on the reliability of any dependent predictions. For example, many so-called splice variants in the ESTs may actually represent incompletely spliced heteronuclear RNA (hnRNA) or oligo(dT)-primed genomic DNA contaminants of cDNA library constructions. Furthermore, the splicing apparatus is known to make errors, resulting in aberrant transcripts that are degraded by the mRNA surveillance system and amount to little that is of functional importance (Maquat and Carmichael 2001). Consequently, the mere presence of a transcript isoform in the ESTs cannot establish a functional role for it. Because alternative splicing appears so pervasively for human genes, strategies are needed with which to screen targets for their potential significance, prior to embarking on downstream experimental verification.
We describe here a method of first identifying alternative splice patterns in transcript sequence data and then performing a statistical analysis of their frequencies of occurrence, in order to identify reliable observations of alternative splicing. The first step has been implemented in the software package TAP (Kan et al. 2001; http://sapiens.wustl.edu/∼zkan/TIP/), which can be used through a Web-based interface or downloaded for local installation. TAP was used here to delineate alternative splice patterns from 3.5 million genomically aligned ESTs for 6400 known human genes. The results confirm and extend previous observations of widespread alternative splicing. In the second step, we estimated with confidence values the relative abundance of splice variants, as compared to each variant's mutually exclusive spliced forms, using their observed frequencies in the EST resource. Subsets of splice variants were selected by their relative frequency and an associated confidence level. We then examined the alternative splices by more traditional functional tests, which included cross-species conservation and reproducibility in GenBank mRNAs. The statistical selection for reliable observations of alternative splicing was seen to comprise an effective selection for functional spliced forms, as well. We suggest that quantitative approaches such as these may be useful for directing experimental efforts toward specific subsets of the observed splice forms that would be the most promising for further study.
RESULTS
Identification of Alternative Splice Patterns
For a gene of interest, TAP identified alternative splice patterns by comparing a reference gene structure with genomically aligned transcript sequences (Fig. 1; see Methods). In this study, RefSeq mRNAs, each representing a unique gene locus, served as references. The following procedures were used to extract alternative splice information from dbEST:
(1)
Mapping and alignment. The known gene structures were obtained by mapping and aligning RefSeq mRNA sequences to the human genomic contigs. Once the genomic locus for a transcript was identified, the genomic sequence surrounding it was extracted. This template sequence was searched against dbEST, and high-scoring ESTs were aligned to the genomic template.
(2)
Delineation of mutually exclusive splice patterns. Genomically aligned ESTs with near identity were used. A “splice” refers to the donor/acceptor pair of splice sites flanking an intron. In the context of genomic alignment, a splice pattern is a series of coordinates denoting the boundaries of exons and introns for a given gene. ESTs exhibiting the same splice pattern were cataloged as a single, possibly partial, alternative gene structure. Structures inferred from EST alignments were compared with the reference gene structure to identify mutually exclusive relationships (Fig. 2). Each mutually exclusive splice pattern was therefore represented by a potential cluster of EST alignments exhibiting the same exon/intron pattern.
(3)
Reconstruction of splice variants. Mutually exclusive gene structures might result from intronic genes (genes contained within introns of other genes), oligo(dT)-primed genomic DNA contamination, or intron contamination by unprocessed or incompletely spliced pre-mRNAs (Wolfsberg and Landsman 1997). For these reasons, the working definition of “alternative splice patterns” was further refined in our study, by discarding from consideration two types of mutually exclusive gene structures, namely, “intronic exon” and “intron retention.” For the splice variants that remained, hypothetical gene structures were created by substituting the alternative splice for the corresponding splice pattern in the reference gene structure (see example in Fig. 3).
Figure 1.

Overview of alignment procedures. (a) A known mRNA sequence was searched against the human genome contig sequences to locate a genomic template. (b) The genomic sequence was RepeatMasked and searched against dbEST. (c) High-scoring EST hits were retrieved and aligned to the genomic sequence. (d) TAP identified alternative splicing events based on a comparison of genomic EST alignments with the known gene structure. (e) All splices were searched against mouse ESTs to assess conservation. In this example, the numbers of EST observations for splices S1–S4 are, respectively, 4, 2, 4, and 3. S2 and S3 are mutually exclusive. S2 is an alternative splice because its observed frequency is lower than that of S3 (_N_ASP = 2, _N_Others = 4, _N_Total = 6). The others are predominant splices.
Figure 2.

Mutually exclusive patterns. “Mutually exclusive” means that two exon/intron structures cannot belong to the same transcript. In this study, a mutually exclusive relationship was called if an intron in one gene structure overlapped an exon of another gene structure. We observed the following basic types of mutually exclusive relationships: exon insertion, exon skipping, splice-site mismatch, intron insertion, intron retention, alternative first exon, alternative last exon, partial exon insertion, partial intron retention, and intronic exon. Complex splice patterns may be composed of multiple basic patterns. Note that some of these relationships are reciprocal. For example, “exon skipping” is the reciprocal of “exon insertion.” The choice of which to use depends on whether the long form or the short form is selected as the reference. For two types of mutually exclusive patterns, the underlying biological processes may not involve alternative splicing at all. Firstly, “intron retention” may be representative of contamination caused by partially spliced or unprocessed pre-mRNAs. Secondly, “intronic exons” may be indicative of intronic genes (genes within genes) rather than alternative splicing.
Figure 3.

Full-length reconstruction of splice variant. Shown here is the graphic output of TAP that illustrates the reconstruction results for NM_000382 (ALDH3A2, aldehyde dehydrogenase 3 family, member A2). The known gene structure based on the RefSeq sequence is shown at the first level. Two GenBank mRNA sequences, AK025677 and AK000658, that exhibit alternative splice patterns are shown below the known gene structure. The gene structures labeled as ALT+# are hypothetical full-length gene structures, each combining an EST-inferred alternative splice pattern (green) with the known gene structure (yellow). Arrows indicate patterns of alternative splicing. Note that the alternative splice pattern in AK000658 was also identified in dbEST as ALT+4. In addition, the complete gene structure of AK000658 is accurately reconstructed.
Alternative Splicing Appears to Be Universal
We mapped 6400 RefSeq transcripts onto the existing genomic contig sequences. Using the EST resource, TAP identified 11,011 alternative splice patterns in 4032 genes (63%). Furthermore, the more ESTs that were found for a given gene, the more likely it was that an alternative splice pattern was detected for that gene. For example, in the subset of 3972 genes with >40 EST hits, 80% exhibited alternative splice patterns. This is consistent with a previously noted correlation between depth of EST coverage and observed extent of alternative splicing (Hide et al. 2001; Kan et al. 2001). Presumably the EST collection missed a substantial fraction of splice variants with rare expression patterns or expressed at low levels. If this kind of sampling bias were not accounted for, the prevalence of alternative splicing in human genes would be underestimated. To reduce sampling bias, we focused on genes with high EST coverage. Surprisingly, we found evidence of alternative splicing for 99% of the 152 genes with >700 EST hits. This result indicates that the overall prevalence of alternative splicing assessed through EST analysis may exceed present estimates by a wide margin, as EST data collection continues. It even seems possible that alternative splice patterns may be observed for all genes that undergo splicing.
Our observations extend previous studies that established pervasiveness of alternative splicing for human genes (for review, see Modrek et al. 2001). Almost all of these studies used EST resources alone to make their estimates, with the attendant potential for misaligning the sequences. We have therefore tried to refine the EST data by using genomically aligned ESTs. The possibility of contamination was minimized by discarding those mutually exclusive splice patterns that could have arisen from intronic genes or incompletely processed pre-mRNAs. Nevertheless, it remains possible that a fraction of the splice variants retained in our study were merely by-products of the gene expression process. Studies have established the presence of an mRNA surveillance system that degrades aberrant transcripts, such as those containing splice errors (for review, see Maquat and Carmichael 2001). Spurious transcript isoforms may occasionally be produced by the cell, but may not exist long enough to have any functional impact. Because EST/cDNA sequences provide a snapshot of the transcriptome, the detection of a particular isoform only establishes its presence, but is insufficient to prove sustained expression or functional importance. Based on these considerations, we recognized the need to identify a subset of the alternative splice patterns that are less likely to be spurious. This problem seems especially relevant at present, as the volume of targets generated through genome-wide surveys exceeds the capacity of present experimental methods and resources for validation and characterization.
Quantitative Analysis of Observed Frequencies
Here we introduce a statistical approach for analyzing the frequencies of alternative splice events. In producing the dbEST database, many EST libraries were sampled to varying degrees. In a complex manner, the proportion of cDNA clones from a given gene represented in dbEST is related to the overall abundance of the corresponding mRNA species under a variety of in vivo conditions. Cloning biases and normalization procedures will alter the relative abundance of different genes in the library (Soares et al. 1994). However, the effect of normalization on the relative abundance of splice variants from the same gene may be minimal. Transcript isoforms will often share significant stretches of identical sequence, which makes them more likely to be treated equally by procedures based on reassociation kinetics. The resultant cDNA clones were randomly selected for end-sequencing to generate the ESTs. A significant fraction of ESTs cover internal splice junctions in the canonical mRNA sequences. For example, 5′-ESTs often sample different internal regions of an mRNA because 5′-ends of cDNA inserts tend to be truncated (Kan et al. 2001). As a result, each EST may be thought of as covering a randomly selected set of splice junctions in a randomly selected transcript. More specifically, for our statistical analyses, we made the simplifying assumption that any two alternatively spliced transcripts for a gene were subject to the same cloning and sequencing biases. This is a reasonable assumption, because we did not dissect the dbEST into its diverse, constituent cDNA libraries, but instead treated the database as one large sample of the human transcriptome. We further assumed that any two alternative splice patterns had the same chance of being covered by EST sequences. This latter assumption seems reasonable, because the alternative splice patterns in our study tended to occupy similar positions along the transcript.
The concept of observed frequency of an alternative splice pattern is illustrated in the following scenario. Suppose a gene is alternatively expressed using two different 3′ (acceptor) splice sites, X and Y. With perfect knowledge about events underlying our actual observations, we might see that the splicing machinery tends to choose the X isoform 10:1 over the Y isoform. We would then say that the events X and Y have, respectively, 91% and 9% (relative) frequencies of occurrence. Then let us say a particular cDNA cloning experiment yielded 900 clones of isoform X and 95 clones of isoform Y from the EST library. Subsequently, among the subset of clones selected for sequencing, 130 X and 15 Y clones were present, which yielded 35 ESTs exhibiting X and 5 ESTs exhibiting Y. The sample size is then 40, yielding observed frequencies of 87.5% and 12.5% for X and Y, respectively. Note that only ESTs with a mutually exclusive relationship (either X or Y in this example) may help us in our analysis. ESTs from the same gene but not covering the same region of interest are not informative.
When two mutually exclusive outcomes were observed in our study, as in the foregoing example, the classical binomial distribution was applied to estimating the likelihood of observing various relative frequencies for X and Y. For some genes, more than two alternative splice patterns were observed, in which case the binomial distribution remained applicable, by classifying outcomes as being Y or non-Y for each pattern. In essence, we evaluated the frequencies of each pattern independently. We did not evaluate alternative splicing for each group of mutually exclusive patterns as a whole, as would be done with a multinomial distribution.
In this study, we compared all individual splices, whether from known gene structures or identified in the ESTs, against each other. Each splice was assigned to a cluster of one or more splices that were mutually exclusive to each other. We further defined an “alternative splice” as one having a lower observed frequency than at least one of its mutually exclusive splices; and the “predominant splice” as the one having the highest frequency—or no mutually exclusive relationship at all. Hence, for a given alternative splice, there was always a predominant splice that was observed more frequently. We identified a total of 13,352 alternative splices of which 86% were derived from ESTs. Only 6% of the 53,273 predominant splices were novel to the ESTs, with the vast majority being observed in the known gene structures.
Values for _N_ASP, _N_Others, and _N_Total were obtained for all alternative splices, where _N_ASP is the count of ESTs exhibiting a particular alternative splice, _N_Others is the count of ESTs showing mutually exclusive splices relative to the alternative, and _N_Total is the sum of _N_ASP and _N_Others, or the total size of the cluster. In the case of exon insertions (Fig. 2), _N_ASP represents the number of times the 3′-most splice was observed. Conditioned on an arbitrarily chosen threshold probability, p, that a given alternative splice Y might be made over all others in its cluster, we computed the binomial probability, P(‖_Y_‖ ≥ _N_ASP‖_N_Total, p), of observing Y at least _N_ASP times in _N_Total trials (see Methods). P < 0.05 was interpreted to mean that _Y_ would be expected with high (>95%) confidence to appear more frequently than the chosen threshold.
The 13,352 alternative splices were evaluated for their ability to satisfy the binomial test at the 95% confidence level using several values for the threshold probability. For example, we found 4951 alternative splices for 2518 genes (39%) likely to occur at >1% frequency. Considering only alternative splices that passed the binomial test at the 1% threshold, we noticed that the fraction of genes exhibiting alternative splicing was essentially independent of EST coverage and typically ranged from 45% to 50% (Fig. 4A). Above a coverage level of 700, nearly all genes exhibited alternative splices, but only 47% of these genes exhibited alternative splices passing the 1% frequency test. This indicates that the high prevalence of alternative splicing observed for genes with high EST coverage was caused by increased sensitivity at detecting infrequent alternative splice events at larger sample sizes.
Figure 4.


Prevalence of alternative splicing. (A) Shown here is the prevalence of alternative splicing (AS) versus the minimum available EST coverage for the genes. EST coverage refers to the total number of ESTs that mapped to a given gene. (% genes) The fraction of the 6400 genes for which the indicated EST coverage was minimally available; (% genes AS) the fraction of genes for which one or more alternative splice patterns were identified in dbEST; (p = x) means that only those genes associated with at least one pattern of alternative splicing that passed the binomial test at the threshold frequency x (95% C.L.) were counted when calculating % genes AS. For (p = 0), no frequency threshold was imposed, such that all genes exhibiting any degree of alternative splicing were counted. % genes AS (p = 0) increased with EST coverage and approached 100% at a coverage of 700 or more. In contrast, when a threshold frequency was imposed, the values of % genes AS varied little over a wide range of EST coverage. (B) The prevalence of intron retention (IR) events versus the minimum available EST coverage. In general, intron retention events were common for human genes, but relatively few genes exhibited them with high frequency.
We performed a similar statistical analysis on intron retention events, that is, clusters of unspliced EST alignments that contained intronic sequences. Such ESTs are believed to derive largely from unspliced or partially spliced pre-mRNAs. The prevalence of intron retention events was 36% overall and increased to 76% at EST coverage of 700, similar to the behavior seen for alternative splicing but with a lower plateau. Applying the binomial test significantly reduced the prevalence of these events, as well. In fact, rather than the statistical test merely flattening the curves as was seen for alternative splicing (Fig. 4A, p > 0), the rate of intron retention events exhibits an almost steady decline with respect to increasing EST coverage (Fig. 4B). Less than 5% of all genes (and only 2% at high EST coverage) exhibited intron retentions passing the 5% frequency threshold, whereas the prevalence of alternative splicing above the same threshold was 17% overall and hovered between 24% and 28% at moderate to high EST coverage. Observed frequencies might be helpful then in discriminating alternative splicing from spurious or artifactual events, such as intron contamination, owing to the fact that these different classes of events were seen here to occur at distinct ranges of frequencies.
Conservation of Alternative Splicing
Human–mouse conservation of alternative splicing was examined because conservation is indicative of a functional selection. An alternative splice is likely to be conserved if its homolog is identifiable in mouse ESTs. Two 50-nt exonic sequences flanking the donor and acceptor splice sites were joined to make a 100-nt sequence probe specifically representing a given splice isoform. These sequence probes were searched against mouse ESTs using BLASTN. A splice was “conserved” if the search yielded an HSP alignment that had >70% identity and spanned the midpoint (splice junction) of the sequence probe (Kan et al. 2001).
Overall, only 8% of alternative splices collected from the human data were observed in the mouse, compared with 61% of predominant splices. We also noticed that conserved alternative splices tended to appear at higher relative frequencies compared with their predominant splice counterparts (Fig. 5). At a threshold frequency of 5%, 15% of alternative splice isoforms had _z_-scores >2, whereas 41% of conserved alternative splices had _z_-scores >2. (A _z_-score >2 from the binomial test would mean that the splice is expected with 98% confidence to occur at a frequency greater than the chosen threshold.) With an odds ratio of 2.7:1 (41%/15%), an alternative splice from human with a _z_-score >2 was nearly three times more likely to be found conserved in mouse ESTs. As shown in Figure 5, the odds of observing cross-species conservation increased with _z_-score. The correlation indicates that alternative splices that are more reliably observed in a population of ESTs are more likely to play a functional role, as well.
Figure 5.

Greater observed frequency is associated with functional indicators. (All) refers to all alternative splices identified in the ESTs; (conserved) refers to the subset of alternative splices detected as being conserved in mouse ESTs; (mRNA verified) refers to the subset of alternative splices also identified in the GenBank mRNA collection. The _z_-score is interconvertible with the binomial probability P(‖_Y_‖ ≥ _k_‖ n, p), which was calculated for a threshold frequency of p = 0.05 (see Methods). (A) A significant difference between the _z_-score distribution for all alternative splices and the distribution for conserved alternative splices is seen, which indicates that conserved events tend to occur at higher frequencies. (B) A significant difference between the overall _z_-score distribution and the distribution for mRNA verified alternative splices is also seen.
When assessing the reliability of observations made of stochastic processes, one may need to take sampling bias into account. Observations of spliced forms in a population of ESTs constitute such a case. Suppose two random samples are taken from a given population of cDNAs. A particular spliced form observed in one sample may fail to be detected in a second sample from the same population. The chance of repeated detection depends both on the underlying frequency of the spliced form within the population and the sample size. When sampling ESTs from two different populations (human and mouse ESTs in our case), an observed difference may simply be caused by this random variation and not necessarily by any real difference between the human and mouse transcriptomes. Sampling bias in our case may arise from the fact that the mouse ESTs are fewer in number (a smaller sample) than the human ESTs, or that the mouse ESTs are derived from different tissues and developmental stages than the human ESTs.
We sought to limit sample bias differences between human and mouse ESTs by focusing on alternative splices likely to occur at a frequency greater than a minimum detection level in mouse ESTs. The minimum detection level was set at 1/m, where m is the _N_Total for the mouse ESTs. A splice with P(‖_Y_‖ ≥ k_‖_n, 1/m) < 0.05 in human would be expected to be seen in the mouse ESTs, if the frequency of the alternative splice in mouse is at least as great as the frequency in human. In total, we found 434 splices expected by these criteria to be detectable, and 184 (42%) of them were seen to be conserved. In comparison, the predominant splices were conserved in 5083 (99%) out of 5139 cases.
These results are consistent with the observation in genome comparison studies that gene structures are generally conserved between human and mouse (Batzoglou et al. 2000). Moreover, the extent of conservation for alternative splice patterns may be significantly more common than what it appears to be from the ESTs. Hence, variation in frequency and sample size in part explains why so few alternative splice patterns could be identified in mouse ESTs.
Reproducibility of Alternative Splicing
A biological observation is less likely to be spurious if it can be independently verified. We have identified alternative splice patterns for the same 6400 genes using an additional source of transcript data—the GenBank mRNA collection. The public domain EST collection was based on a relatively small number of large-scale sequencing projects that focused on gene discovery. On the other hand, the GenBank mRNA collection was based on many individual studies, each producing a small amount of sequence data. These resources represent two largely independent sampling studies of the human transcriptome. A total of 1957 alternative splices were identified for 1229 genes from the GenBank mRNA collection, and 830 (42%) of these were also found in the ESTs. More than 90% of the alternative splice patterns found in the ESTs could not be detected in the GenBank mRNA collection, possibly owing to the fact that the EST resource represents a much deeper sampling of the transcriptome than does the GenBank mRNA collection. On the other hand, the majority of alternative splices found in the GenBank mRNAs were not identified in the ESTs. This is an indication that EST-based sampling is still a long way from revealing the full extent of alternative splicing in human genes.
The subset of alternative splice patterns identified in both ESTs and GenBank mRNAs were considered independently reproducible observations. We found that these patterns also tended to have greater observed frequencies than the rest (Fig. 5). The results indicate that observed frequency is a good predictor of cross-species conservation and reproducibility, which are generally regarded as evidences of function. In our study, however, the vast majority of alternative splice patterns captured in the ESTs could not be verified either through the conservation or reproducibility tests. Hence, quantitative analysis of observed frequency, a source of information already encoded in the EST resource, may serve as a high-throughput method for prioritizing alternative splicing events for experimental validation.
DISCUSSION
We have developed a method for delineating alternative splice patterns by comparing a reference gene structure with genomically aligned EST/cDNA sequences. The method was implemented in the software tool TAP, which is readily accessible to the research community (http://sapiens.wustl.edu/∼zkan/TIP/). TAP was used to identify alternative splice variants for 6400 known human genes using ESTs. Stringent criteria were applied to remove potential contamination. The proportion of genes that exhibited alternative splicing was found to increase with EST coverage and reached 99% for the subset of genes with 700 or more EST hits. We introduced a novel statistical approach for estimating the frequency of alternative splicing events based on their observed frequencies in ESTs. From our analysis, the increasing prevalence of alternative splicing at higher EST coverage may simply be attributable to an increased chance of detecting low-probability events with continued sampling. Moreover, we found that alternative splice patterns generally appeared in the ESTs at greater frequencies than did potential intron contamination. We also established a correlation between frequency of observation of alternative splicing and other traditional indicators of biological function, namely, cross-species conservation and reproducibility.
Biological processes have inherent error rates that are difficult to quantify and could be highly variable. If errors by the splicing apparatus generate splice variants even at low frequency, it seems plausible that some degree of alternative splicing may be detected for all spliced genes. Theoretically two types of alternative splicing events might exist, one generated randomly and one generated through regulated processes. Spurious events are expected to occur at lower frequencies than regulated events; otherwise, cellular systems would be overwhelmed by the task of synthesizing and removing aberrant transcripts. However, EST sequencing may detect these rare events through in-depth sampling.
The chance of detecting any background noise in the EST library certainly increases with sample size, that is, with the number of ESTs sampled for a given gene. Suppose a gene has 6 introns and is sampled by 100 ESTs covering all splice junctions. If the frequency of spurious variation was merely 1% at each junction, the probability of detecting among the 100 ESTs at least one spurious splice for a single junction would be 0.63 (given by the Poisson approximation to binomial probability, 1 − _e_−100*0.01). Spurious variation would almost certainly be detected then for at least one of the 6 introns in the gene. Furthermore, the probability of detecting variation in two or more ESTs under these conditions would be 0.84. Unless the error rate of splicing is very low, one can not ignore the possibility that a significant fraction of splice variation captured in ESTs may be spurious. As illustrated in our study, sample statistics may then be essential to identifying reliable spliced structures amid a large population of ESTs.
We have presented the view of alternative splice events as being discrete outcomes of a stochastic process. Constitutive splicing, alternative splicing, and spurious splice variation represent three classes of events that occur at different frequencies. From an evolutionary perspective, it is difficult to imagine how the present diversity of transcript isoforms was achieved without a significant element of trial-and-error. And although many of the low-frequency events we observed may indeed be functional, it seems likely that many are not. In any case, the stochastic nature of alternative splicing may serve to open for exploration more possibilities for gene expression and may work synergistically with random mutational processes to promote molecular evolution.
METHODS
Data Sources
A total of 10,238 human mRNA sequences were derived from the RefSeq database released on December 2000 (Maglott et al. 2000; http://www.ncbi.nlm.nih.gov/LocusLink/refseq.html). The EST sequences were derived from dbEST release 060201 (http://www.ncbi.nlm.nih.gov/dbEST/, consisting of 3.5 million human ESTs and 2 million mouse ESTs. The UCSC assembly of the working draft of the human genome (Kent and Haussler 2001; http://genome.ucsc.edu/), December 2000 release, was used. The GenBank mRNA collection consists of 68,975 sequences from the nr database as of October 2001 (http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html).
Construction of Genomic EST Alignments
The RefSeq sequences were searched against the human genomic contigs using WU-BLASTN 2.0 (W. Gish 1996–2002, unpubl., http://blast.wustl.edu/). The high-scoring sequences were aligned to the matching contig sequences using sim4 1.0 (Florea et al. 1998). A locus was found if the overall percent identity of the genomic alignment was >98%. In the case in which one mRNA sequence mapped to multiple loci, the locus with the highest percent identity was chosen. In 126 cases (2%) in which multiple sequences were mapped to the same locus, one sequence was randomly selected. For each gene, the genomic sequence including the genic region and up to 40-kb extensions at both ends was extracted and set to the same orientation as the mRNA sequence. This process produced 6400 known gene structures. Known repetitive elements were masked using a combination of RepeatMasker (A. Smit and P. Green, unpubl.; http://ftp.genome.washington.edu/) in its most sensitive setting and MaskerAid (Bedell et al. 2000). The masked genomic templates were searched against human ESTs using WU-BLASTN. The high-scoring ESTs were aligned to the template using sim4. EST alignments with >93% identity overall were used for transcript reconstruction. Poorly aligned or tiny terminal exons (<20 nt) were removed from the alignments.
Delineating Alternative Splicing Events
The algorithm for inferring the predominant gene structure from genomic EST alignments was previously described (Kan et al. 2001). Here we focus on the method for delineating patterns of alternative splicing. EST alignments were partitioned into strand-specific sets based on EST labels, alignment orientation, and splice-site motifs. Splice patterns were extracted from EST alignment data. Each splice consists of a pair of donor and acceptor splice sites flanking an intron. It must have the consensus GT · AG motif or be observed in >2 ESTs. Each EST alignment is represented by its splice pattern. A connectivity matrix M(i,j) was built to record the EST-based connectivity between adjacent splices i and j. All splices are sorted by their 5′ coordinates. The connectivity between any two splices is classified into four mutually exclusive types: conflicting, contiguous, transitive, or gapped. A conflicting connection arises when two splices i and j overlap with one another but have different coordinates, and is penalized by a score of −∞. The connection is contiguous when one or more EST alignments carry i and j in adjacent positions, yielding a positive score equal to the number of such ESTs. When there is no contiguous connection, the program checks for EST overlaps. If there is overlapping coverage, the connection is “transitive,” and M(i,j) becomes 1. Otherwise, the connection is “gapped,” and M(i,j) becomes 0. A second matrix N was also built such that each cell, N(i,j), describes the long-range connectivity between splices i and j, set to the number of EST alignments carrying both splices.
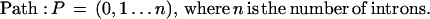
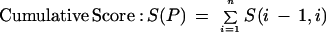
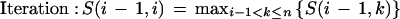
To identify the predominant gene structure, the program essentially finds a path through the matrix that maximizes the cumulative score. This is accomplished using a greedy strategy that follows the maximum connectivity score at each elongation step. The program uses a modified algorithm to identify alternative gene structures. First, splices from the known gene structure are stored into the connectivity matrix. It now consists of two types of splices, those that are known and those that are novel. The filled matrix is recursively traced to enumerate all paths containing novel splice patterns. However, the tracing process is “local” rather than “global.” Instead of assembling a complete path, the algorithm terminates paths at any gapped connection with a zero score. In addition, each elongation step requires a positive value of long-range connectivity, N(k,j), where k is any splice in the existing path and j is the next splice. This effectively requires that each path be represented by at least one EST alignment. Finally, these partial paths are reported as exon/intron structures. If an alternative gene structure consists of mutually exclusive splices and overlapped known exons, it is called an alternative splice pattern.
Statistical Analysis
_N_ASP is the count of ESTs showing a particular alternative splice, _N_Others is the count of ESTs showing mutually exclusive splice patterns, and _N_Total = _N_ASP + _N_Others. We would like to know if, in repeated samplings, the observed frequency of an alternative splice relative to _N_Total would be likely to exceed a given threshold frequency, p. To this end, the binomial probability P(‖_Y_‖ ≥ _k_‖ n, p) was calculated such that the alternative splice Y occurs at least k times in n trials. Here, k is _N_ASP and n is _N_Total. If P(‖_Y_‖ ≥ _k_‖ n, p) is below 0.05, then the observed frequency of the alternative splice is expected to exceed the given threshold with high (>95%) confidence in repeated samplings. The following formulas were used:
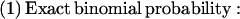
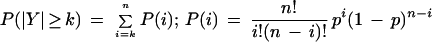
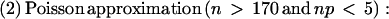
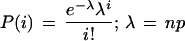
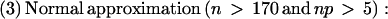
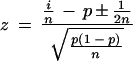
_z_-scores are interconvertible with probabilities by the following formulas:
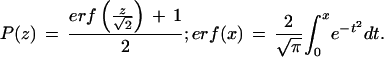
WEB SITE REFERENCES
http://blast.wustl.edu/; WU-BLAST 2.0.
http://ftp.genome.washington.edu/; RepeatMasker.
http://genome.ucsc.edu/; UCSC assembly of the working draft of the human genome, December 2000 release.
http://sapiens.wustl.edu/∼zkan/TIP/; additional data available at Z.K.'s Web site.
http://www.ncbi.nlm.nih.gov/dbEST/; the EST sequences were derived from dbEST release 060201.
http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html; GenBank mRNA from nr database October 2001..
http://www.ncbi.nlm.nih.gov/LocusLink/refseq.html; RefSeq at NCBI.
Acknowledgments
We sincerely thank Compaq Computer Corp. for providing access to their BioCluster supercomputing facility and HP/Intel for hardware support through the Itanium Processor Family (IPF) University Grants Program. This work was supported in part by grants from the National Institutes of Health (R01-HG01391) and the Department of Energy (DE-FG02-94ER61910). W.G. was funded by NIH/NHGRI U01-HG02042 and U01-HG02155.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL gish@watson.wustl.edu; FAX (314) 286-1810.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.764102.
REFERENCES
- Baker BS. Sex in flies: The splice of life. Nature. 1989;340:521–524. doi: 10.1038/340521a0. [DOI] [PubMed] [Google Scholar]
- Batzoglou S, Pachter L, Mesirov JP, Berger B, Lander E. Human and mouse gene structure: Comparative analysis and application to exon prediction. Genome Res. 2000;10:950–958. doi: 10.1101/gr.10.7.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedell JA, Korf I, Gish W. MaskerAid: A performance enhancement to RepeatMasker. Bioinformatics. 2000;16:1040–1041. doi: 10.1093/bioinformatics/16.11.1040. [DOI] [PubMed] [Google Scholar]
- Black DL. Splicing in the inner ear: A familiar tune, but what are the instruments? Neuron. 1998;20:165–168. doi: 10.1016/s0896-6273(00)80444-4. [DOI] [PubMed] [Google Scholar]
- Boggs RT, Gregor P, Idriss S, Belote JM, McKeown M. Regulation of sexual differentiation in D. melanogaster via alternative splicing of RNA from the transformer gene. Cell. 1987;50:739–747. doi: 10.1016/0092-8674(87)90332-1. [DOI] [PubMed] [Google Scholar]
- Boguski MS, Lowe TM, Tolstoshev CM. dbEST—Database for “expressed sequence tags.”. Nat Genet. 1993;4:332–333. doi: 10.1038/ng0893-332. [DOI] [PubMed] [Google Scholar]
- Brett D, Hanke J, Lehmann G, Hasse S, Delbruck S, Krueger S, Reich J, Bork P. EST comparison indicates 38% of human mRNAs contain possible alternative splice forms. FEBS Lett. 2000;47:83–86. doi: 10.1016/s0014-5793(00)01581-7. [DOI] [PubMed] [Google Scholar]
- Brett D, Pospisil H, Valcarcel J, Reich J, Bork P. Alternative splicing and genome complexity. Nat Genet. 2002;1:29–30. doi: 10.1038/ng803. [DOI] [PubMed] [Google Scholar]
- Burke J, Wang H, Hide W, Davison DB. Alternative gene form discovery and candidate gene selection from gene indexing projects. Genome Res. 1998;8:276–290. doi: 10.1101/gr.8.3.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft L, Schandorff S, Clark F, Burrage K, Arctander P, Mattick JS. ISIS, the intron information system, reveals the high frequency of alternative splicing in the human genome. Nat Genet. 2000;24:340–341. doi: 10.1038/74153. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Analysis of expressed sequence tags indicates 35,000 human genes. Nat Genet. 2000;25:232–234. doi: 10.1038/76115. [DOI] [PubMed] [Google Scholar]
- Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–974. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Ghetti B, Spillantini MG. τ gene mutations in frontotemporal dementia and Parkinsonism linked to chromosome 17 (FTDP-17). Their relevance for understanding the neurogenerative process. Ann NY Acad Sci. 2000;920:74–83. doi: 10.1111/j.1749-6632.2000.tb06907.x. [DOI] [PubMed] [Google Scholar]
- Graveley BR. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001;17:100–107. doi: 10.1016/s0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- Hide WA, Babenko VN, Van Heusden PA, Seoighe C, Kelso JF. The contribution of exon-skipping events on chromosome 22 to protein coding diversity. Genome Res. 2001;11:1848–1853. doi: 10.1101/gr.188001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Cote J, Kwon JM, Goate AM, Wu JY. Aberrant splicing of τ pre-mRNA caused by intronic mutations associated with the inherited dementia frontotemporal dementia with Parkinsonism linked to chromosome 17. Mol Cell Biol. 2000;20:4036–4048. doi: 10.1128/mcb.20.11.4036-4048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z, Gish W, Rouchka E, Glasscock J, States D. UTR reconstruction and analysis using genomically aligned EST Sequences. Intell Syst Mol Biol. 2000;8:218–227. [PubMed] [Google Scholar]
- Kan Z, Rouchka EC, Gish WR, States DJ. Gene structure prediction and alternative splicing analysis using genomically aligned ESTs. Genome Res. 2001;11:875–888. doi: 10.1101/gr.155001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent JW, Haussler D. Assembly of the working draft of the human genome with GigAssembler. Genome Res. 2001;11:1541–1548. doi: 10.1101/gr.183201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochiwa H, Suzuki R, Washio T, Saito R, Bono H, Carninci P, Okazaki Y, Miki R, Hayashizaki Y, Tomita M The RIKEN Genome Exploration Research Group Phase II Team. Inferring alternative splicing patterns in mouse from full-length cDNA library and microarray data. Genome Res. 2002;12:1286–1293. doi: 10.1101/gr.220302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lopez AJ. Alternative splicing of pre-mRNA: Developmental consequences and mechanisms of regulation. Annu Rev Genet. 1999;32:279–305. doi: 10.1146/annurev.genet.32.1.279. [DOI] [PubMed] [Google Scholar]
- Maglott DR, Katz KS, Sicotte H, Pruitt KD. NCBI's LocusLink and RefSeq. Nucleic Acids Res. 2000;28:126–128. doi: 10.1093/nar/28.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE, Carmichael GG. Quality control of mRNA function. Cell. 2001;104:173–176. doi: 10.1016/s0092-8674(01)00202-1. [DOI] [PubMed] [Google Scholar]
- Mironov AA, Fickett JW, Gelfand MS. Frequent alternative splicing of human genes. Genome Res. 1999;9:1288–1293. doi: 10.1101/gr.9.12.1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrek B, Lee C. A genomic view of alternative splicing. Nat Genet. 2001;30:13–19. doi: 10.1038/ng0102-13. [DOI] [PubMed] [Google Scholar]
- Modrek B, Resch A, Grasso C, Lee C. Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res. 2001;29:2850–2859. doi: 10.1093/nar/29.13.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan K, Michael TH, Jiang GJ, Hiel H, Fuchs PA. A molecular mechanism for electrical tuning of cochlear hair cells. Science. 1999;283:215–217. doi: 10.1126/science.283.5399.215. [DOI] [PubMed] [Google Scholar]
- Soares MB, Bonaldo MF, Jelene P, Su L, Lawton L, Efstratiadis A. Construction and characterization of a normalized cDNA library. Proc Natl Acad Sci. 1994;91:9228–9232. doi: 10.1073/pnas.91.20.9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Halt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Wolfsberg TG, Landsman D. A comparison of expressed sequence tags (ESTs) to human genomic sequences. Nucleic Acids Res. 1997;25:1626–1632. doi: 10.1093/nar/25.8.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]