Tuberculosis in children and adults: two distinct genetic diseases (original) (raw)
Abstract
Disseminated disease in children and pulmonary disease in adults constitute two major epidemiological and clinical forms of tuberculosis. Paradoxically, only a small fraction of infected individuals develop clinical tuberculosis, typically one form of the disease or the other. Mendelian and complex genetic predispositions to tuberculosis were reported recently in children and adults, respectively. Here, we argue that tuberculosis and its clinical expression largely reflect the underlying human genetic background.
The burden of tuberculosis is not equally shared
Tuberculosis, an infectious disease caused by Mycobacterium tuberculosis, remains a leading public health problem worldwide. The global incidence of tuberculosis is rising, with ∼8.8 million new cases and 2 million deaths each year (1). However, not all individuals exposed to M. tuberculosis become chronically infected. Epidemiological studies of tuberculosis in highly endemic countries indicate a consistent pattern of host stratification, with ∼20% of individuals retaining negative tuberculin skin tests throughout life, despite repeated exposure to the bacteria (2). As an intrinsic, global impairment of delayed type hypersensitivity is improbable in all nonresponders, a substantial fraction of these individuals are likely to be naturally resistant to tuberculous infection. Moreover, progression to clinical tuberculosis is far from being an inevitable consequence of persistent infection by M. tuberculosis, as only an estimated 10% of infected individuals develop clinical disease. Another level of interindividual variability also exists; there are two major clinical forms of tuberculosis in endemic areas, which correspond to the two age-dependent epidemiological peaks of incidence (Fig. 1). In young children, tuberculosis is often disseminated due to early, hematogenous spread of the mycobacterium after primary pulmonary infection. In adults, the infection is often limited to the lungs and reflects the reactivation of latent tuberculosis from a silent primary infection.
Figure 1.

Mortality rates for disseminated tuberculosis (blue bars) and chronic pulmonary tuberculosis (red bars) per 100,000 untreated persons of various ages living in Bavaria in 1905 (adapted from references 7 and 28). Note that there were too few deaths to accurately plot cases of pulmonary tuberculosis before the age of 20 and disseminated tuberculosis between the ages of 5 and 20 yr. These data relate to the natural history of tuberculosis in an endemic area before BCG vaccines and antimycobacterial antibiotics were available. Vaccination against and early diagnosis and treatment of tuberculosis in children may now have blurred the corresponding clinical phenotypes of tuberculosis, but not the underlying genotype. There are other forms of tuberculosis, with primary infection in adults and reactivation in late childhood. There is, however, clearly a “golden age” between the ages of 4 and 13 yr (reference 6).
Predisposition to tuberculosis is largely inherited
Before Pasteur's groundbreaking microbial theory of disease, tuberculosis was suspected to reflect an intrinsic host disorder, often echoing familial predisposition. As Benjamin Marten described in 1720, “‘Some persons are of such an happy constitution that if any of one of the inimical animals that causes consumption [of the lungs], happen to get into their bodies, they may likewise be quickly forced out again, through some of the emunctories, before they are produced into life; or else wholly destroyed’”(3). This “familial” hypothesis, suggested by the familial clustering of cases, gained favor by the end of the 18th century and dominated medical thinking for most of the 19th century (3, 4). However, Pasteur's microbial theory and Koch's subsequent identification of M. tuberculosis overturned such theories, which were based on anecdotal observations in the absence of epidemiological or experimental evidence. It was not until 1933 that rigorous genetic epidemiological studies provided strong evidence for the contribution of genetic factors to tuberculosis, with higher concordance rates of tuberculosis observed among monozygotic than dizygotic twin pairs (4, 5). In addition, although it was long known that the incidence of tuberculosis was particularly high in newly exposed populations (6), this observation was poorly understood. A genetic interpretation of this observation was provided when the ancestors of susceptible individuals, unlike those of resistant individuals living in the same environment, were found to be more likely to have originated from tuberculosis-free areas (7).
Immunodeficiencies favor the development of tuberculosis
The first molecular evidence that a predisposition to tuberculosis might reflect inborn errors of immunity was provided by the occurrence of overwhelming tuberculosis in children with rare, severe primary immunodeficiencies (PIDs) (Table I) (8, 9). Disseminated disease in children with PIDs is often caused by widespread, weakly virulent mycobacteria, such as bacillus Calmette-Guerin (BCG) vaccines and environmental mycobacteria (EM). In contrast, bona fide tuberculosis caused by virulent M. tuberculosis has been reported more rarely in these children. To date, one child with severe combined immunodeficiency, two children with X-linked hyper IgM syndrome, and one child with anhidrotic ectodermal dysplasia with immunodeficiency have been diagnosed with tuberculosis (Table I) (8). The rarity of these patients may be a mere consequence of the fortuitous occurrence of tuberculosis and immunodeficiency, but more probably reflects the lower levels of exposure to M. tuberculosis. Supporting this view, a large proportion of children (∼35%) with chronic granulomatous disease (CGD), a less severe PID, were diagnosed with severe tuberculosis in two endemic areas (10, 11). The high predisposition to tuberculosis conferred by CGD established a causal link between human genes and tuberculosis. This suggested that predisposition to tuberculosis in other children may also reflect Mendelian inborn errors of immunity. However, this issue is complicated by the fact that children with these PIDs also suffer from multiple opportunistic infectious diseases, unlike most other children with the common form of tuberculosis.
Table I.
Tuberculosis in children with Mendelian inborn errors of immunity
| Inherited defect | No.of patients | Age atdisease onset | Countryof origin | Reference | |
|---|---|---|---|---|---|
| PID | SCID | 1 | NA | Australia | cited in (8) |
| HIGM | 2 | 15,33 | NA, Japan | cited in (8) | |
| EDA-ID | 1 | 1.7 | NA | cited in (8) | |
| CGD | 6 | 1.5–7.5 | Hong Kong | Lau et al. (10) | |
| CGD | 13 | NA | Iran | Movahedi et al. (11) | |
| MSMD | complete IFN-γR1 | 1 | 12 | Turkey | Dorman et al. (29) |
| partial IFN-γR1 | 1 | 3 | Japan | Sasaki et al. (30) | |
| IL-12p40 | 1 | 2.5 | Turkey | Picard et al. (31) | |
| MST | partial IFN-γR1 | 1 | 3 | Portugal | Jouanguy et al. (17) |
| IL-12Rβ1 | 1 | 18 | Morocco | Altare et al. (14) | |
| IL-12Rβ1 | 2 | 5,12 | Spain | Caragol et al. (15) | |
| IL-12Rβ1 | 1 | 11 | Turkey | Ozbek et al. (16) |
The syndrome of Mendelian susceptibility to mycobacterial diseases
Further progress in the understanding of the genetic basis of tuberculosis was achieved from 1996 onwards, with studies of the syndrome known as Mendelian susceptibility to mycobacterial diseases (MSMD) (12). Patients with MSMD are particularly susceptible to weakly virulent mycobacteria (BCG and EM) but are resistant to most other infectious agents, with the exception of Salmonella. In the last decade, germline mutations have been found in five MSMD-causing genes (IFNGR1, IFNGR2, STAT1, IL12B, IL12RB1). These genes encode proteins that are involved in interleukin (IL)-12/23–dependent, interferon (IFN)-γ–mediated immunity. Extensive allelic heterogeneity at these loci accounts for the existence of twelve distinct genetic disorders responsible for MSMD, which were diagnosed in >300 patients worldwide in <10 yr, mostly in nonendemic areas. Three MSMD patients with BCG- and/or EM-induced disease, living in endemic areas, also developed bona fide tuberculosis. These patients, who had deficiencies in IFN-γR1 or IL-12p40 expression, developed tuberculosis between the ages of 2.5 and 12 yr (Table I). As these patients suffered from both tuberculosis and mycobacterial disease caused by the weakly virulent BCG or EM species, it remained unclear whether common cases of tuberculosis alone may also be attributable to a Mendelian predisposition.
Mendelian susceptibility to tuberculosis in children: a novel concept
Intriguingly, IL-12Rβ1 deficiency showed incomplete penetrance for the case-definition phenotype of MSMD (13), as not all individuals with this deficiency were susceptible to BCG and EM disease. Moreover, in children from three unrelated families, IL-12Rβ1 deficiency was found to be associated with culture-proven severe tuberculosis as the sole infectious phenotype (14–16) (Table I). In one family, an IL-12Rβ1–deficient patient developed abdominal tuberculosis (14). She had been vaccinated three times with BCG with no adverse effects, whereas her brother, who was also deficient for IL-12Rβ1, developed BCG disease after immunization. In another family, an IL-12Rβ1–deficient girl developed disseminated tuberculosis (15). Her IL-12Rβ1–deficient sister had a history of nontyphoidal extraintestinal salmonellosis and, despite prophylactic treatment with isoniazid, also developed tuberculosis. Finally, a girl with IL-12Rβ1 deficiency, but no relevant personal or familial history of mycobacteriosis or salmonellosis, developed disseminated tuberculosis (16). These cases of severe tuberculosis in children with IL-12Rβ1 deficiency provided proof-of-principle that tuberculosis can be a Mendelian disease. Mendelian predisposition to tuberculosis is not limited to IL-12Rβ1 deficiency, however, as one child with a partial IFN-γR1 deficiency suffered from tuberculosis (17). These observations raise the possibility that a substantial proportion of children worldwide who have disseminated tuberculosis have a Mendelian predisposition to disease (18).
Bayesian estimation of the frequency of Mendelian tuberculosis in endemic areas
In an attempt to calculate the frequency of Mendelian tuberculosis, we estimated the expected proportion of Mendelian cases of disseminated tuberculosis among children: P(Mendel/cTB). Direct application of Bayes' theorem for calculating conditional probabilities gives the following equation:
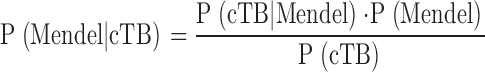
where P(cTB) is the cumulative incidence of disseminated tuberculosis by the age of 14 yr, P(Mendel) is the frequency of individuals carrying the relevant Mendelian mutations (all mutations predisposing to Mendelian tuberculosis), and P(cTB/Mendel) is the cumulative incidence of disseminated tuberculosis among these individuals (the cumulative penetrance). Based on epidemiological data, P(cTB) can be estimated at 2 × 10−4. We varied P(Mendel) from 10−5 to 10−4 and P(cTB/Mendel) from 0.5 to 0.9, based on past genetic reports (12, 13). Remarkably, the estimated proportion of Mendelian cases of disseminated tuberculosis (P[Mendel/cTB]) ranges from 3 to 45% (Fig. 2). It may seem provocative to suggest that almost half the children with disseminated tuberculosis might display a Mendelian predisposition. However, this high estimate is based on realistic assumptions: P(Mendel) = 10−4 and P(cTB|Mendel) = 0.9. It therefore seems likely that a substantial fraction of children suffering from tuberculosis have a Mendelian predisposition. This prediction is experimentally testable, by investigating groups of children with tuberculosis living in endemic areas.
Figure 2.

Bayesian estimation of the rate of Mendelian predisposition to disseminated tuberculosis in childhood in endemic areas. The x axis corresponds to P(Mendel), the y axis corresponds to P(cTB/Mendel), and the z axis corresponds to P(Mendel/cTB) (see “Bayesian estimation of the frequency…”). As an example, assuming P(Mendel) = 0.0001 and P(cTB/Mendel) = 0.5, the estimated proportion of Mendelian cases among children with disseminated tuberculosis is P(Mendel/cTB) = 0.25.
Complex genetic predisposition to tuberculosis in adults
Pediatric and adult tuberculosis differ markedly in epidemiological features (two distinct peaks of incidence), clinical appearance (disseminated versus pulmonary disease), and pathogenesis (primary infection vs. reactivation). These differences probably reflect differences in immunological and genetic control (Fig. 1). More genetic studies have focused on adult than on childhood tuberculosis but with less success. No adults with Mendelian tuberculosis have yet been reported, and no major susceptibility locus has been identified by genome-wide linkage studies (19). In addition, few studies reporting associations between human genes and adult tuberculosis have been reproduced (for review see reference 12). The most convincing associations to date were obtained with the natural resistance-associated macrophage protein 1 (NRAMP1, alias SLC11A1) gene, the human orthologue of the murine Nramp1 gene. This gene encodes a membrane transporter protein that depletes phagosomes of divalent cations that are essential for the intraphagosomal survival of some mycobacterial species. The NRAMP1 region was also found to be linked with tuberculosis during an outbreak in a Canadian aboriginal community (20). However, although this and other studies suggest that complex human genetic factors (NRAMP1 alleles in particular) may be involved in susceptibility to pulmonary tuberculosis in adults, the associations are weak, and causal relationships between genotypes and phenotypes have not been demonstrated.
A common MCP1 allele confers a high attributable risk
A study published in this issue reveals an important new genetic link to pulmonary tuberculosis in adults (21). In this study, Flores-Villanueva et al. identify a polymorphism in the promoter region of the gene encoding the chemokine monocyte chemoattractant protein–1 (MCP-1) that confers susceptibility to pulmonary tuberculosis in Mexican adults. This polymorphism is characterized by an adenine (A) to guanine (G) change at position −2518 of the MCP1 promoter. The results of the study are reported in terms of odds ratio (OR), which is a valid estimate of the relative risk (the risk of developing tuberculosis according to genotype) when the disease prevalence is low (typically <10%). The ORs of tuberculosis among individuals with AG and GG versus AA genotypes were estimated at 2.3 and 5.4, respectively. This means that individuals homozygous for the G allele were approximately five times more likely to develop tuberculosis than those homozygous for the A allele. The GG genotype is associated with an increase in MCP-1 production, which leads to decreased production of IL-12, a key antimycobacterial cytokine (12), providing a possible explanation for the observed increased susceptibility to tuberculosis. The MCP1 susceptibility allele is very common (occurring in ∼50% of the Mexican population), giving an estimated attributable risk as high as 64% in this population (22). In other words, if the −2518G MCP1 polymorphism were the only risk factor for tuberculosis among exposed individuals in Mexico (which is unlikely), the incidence of disease would be 64% lower in the absence of the G allele. This association was also found in a South Korean population (21). However, this MCP1 allele was previously reported not to be associated with tuberculosis in a Brazilian cohort (23). Nevertheless, this study reports the most substantial impact ever described of a human allele on adult tuberculosis at the population level.
From complex to Mendelian inheritance (and back again)
Common alleles that predispose adults to pulmonary tuberculosis provide candidate genes (such as MCP1) for the study of Mendelian tuberculosis in childhood. Conversely, MSMD-causing genes (such as IL12RB1) are candidate genes for complex predisposition to tuberculosis in adults. Allelic heterogeneity (allelic variations at a given locus) may account for the age-dependent clinical heterogeneity of tuberculosis. Alternatively, nonallelic heterogeneity may be involved, as the pools of genes that govern immunity to primary and latent mycobacterial infections may not overlap. In any event, disseminated tuberculosis seems to reflect Mendelian predispositions in a fraction of children, whereas adult pulmonary disease seems to reflect a more complex genetic predisposition. However, these modes of inheritance are artificially separated because modifier genes have a profound impact on the expression of Mendelian traits. In addition, major genes, which are genes whose common polymorphisms exert an effect strong enough to be detected in segregation studies and/or genome-wide linkage scans, may exert an almost Mendelian impact (9). Moreover, Mendelian and complex predispositions are not mutually exclusive in either age group, as suggested by the recent description of NRAMP1 susceptibility alleles in children with tuberculosis (24). Johann Gregor Mendel, the father of genetics, and Francis Galton, the father of biometrics, were ignorant of each other's work, although both were born in 1822 and lived in Europe (25). Most barriers of communication have since fallen, and research on tuberculosis has benefited from the combined efforts of molecular and population geneticists.
From the microbial to the genetic paradigm
The idea that even seemingly noncontagious diseases may be infectious, as highlighted by the relationship between Helicobacter pylori and gastric ulcers, has become a dominant paradigm in medicine. However, microbes, including M. tuberculosis, are necessary but not sufficient for the development of infectious disease. This novel idea was clearly expressed by Louis Pasteur himself when he described the two diseases of silk worms (pebrine and flacherie) that provided the experimental basis for his microbial theory (26). He found that the pebrine disease was apparently purely infectious, whereas flacherie was both infectious and hereditary, with only certain silkworm strains being vulnerable. Pasteur, who was also born in 1822, never heard of Mendel but explicitly spoke of “flacherie héréditaire” and added that “‘it is not the microbe that is transmitted from the parents to the offspring, but the predisposition to disease’” (26). The considerable success of Pasteur's microbial theory overshadowed his own audacious conception of the ecology and heredity of infectious diseases (26, 27). The genetic theory of infectious diseases has recently benefited from interactions between different disciplines (18). There is a continued need to reconsider infectious diseases to ensure the timely development of novel preventive and curative treatments (http://www.nap.edu/catalog/11471.html). Vaccines that specifically protect genetically predisposed individuals against infection and immunomodulatory drugs that restore impaired immunity are needed to circumvent the inevitable spread of antibiotic-resistant pathogens, including M. tuberculosis.
Acknowledgments
We thank E. Schurr, J. El Baghdadi, S. Dupuis-Boisson, D. Nolan, N. Remus and all members of the laboratory of Human Genetics of Infectious Diseases for helpful discussions.
The laboratory is supported by grants from the INSERM, Agence Nationale de la Recherche, March of Dimes, and Foundation BNP-Paribas. A.A. is supported in part by a grant from Assistance-Publique-Hôpitaux de Paris. J.-L.C. is an International Scholar of the Howard Hughes Medical Institute.
A.A., C.F., L.A., and J.-L.C. contributed equally to this work.
References
- 1.World Health Organization. 2004. Global tuberculosis control. Surveillance, planning, financing. WHO report.
- 2.Kassim, S., P. Zuber, S.Z. Wiktor, F.V. Diomande, I.M. Coulibaly, D. Coulibaly, A. Kadio, A. Yapi, K.C. Toure, P.B. Blekou, et al. 2000. Tuberculin skin testing to assess the occupational risk of Mycobacterium tuberculosis infection among health care workers in Abidjan, Cote d'Ivoire. Int. J. Tuberc. Lung Dis. 4:321–326. [PubMed] [Google Scholar]
- 3.Bernier, J. 2005. L'interprétation de la phtisie pulmonaire au XVIIIe siècle [The interpretation of pulmonary tuberculosis in the 18th century]. Can. Bull. Med. Hist. 22:35–56. [DOI] [PubMed] [Google Scholar]
- 4.Puffer, R. 1944. Familial susceptibility to tuberculosis; Its importance as a public health problem. Harvard University Press, Cambridge. 106 pp.
- 5.Diehl, K., and O.v. Verschuer. 1933. Zwillingstuberkulose, Zwillings-forschung und Tuberkulose-disposition. Gustav Fischer, Jena. 500 pp.
- 6.Dubos, R.J., and J. Dubos. 1952. The White Plague; Tuberculosis, Man, and Society. Little Brown, Boston, viii. 277 pp.
- 7.Stead, W.W. 1992. Genetics and resistance to tuberculosis. Could resistance be enhanced by genetic engineering? Ann. Intern. Med. 116:937–941. [DOI] [PubMed] [Google Scholar]
- 8.Reichenbach, J., S. Rosenzweig, R. Doffinger, S. Dupuis, S.M. Holland, and J.L. Casanova. 2001. Mycobacterial diseases in primary immunodeficiencies. Curr. Opin. Allergy Clin. Immunol. 1:503–511. [DOI] [PubMed] [Google Scholar]
- 9.Abel, L., and J.L. Casanova. 2000. Genetic predisposition to clinical tuberculosis: bridging the gap between simple and complex inheritance. Am. J. Hum. Genet. 67:274–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lau, Y.L., G.C. Chan, S.Y. Ha, Y.F. Hui, and K.Y. Yuen. 1998. The role of phagocytic respiratory burst in host defense against Mycobacterium tuberculosis. Clin. Infect. Dis. 26:226–227. [DOI] [PubMed] [Google Scholar]
- 11.Movahedi, M., A. Aghamohammadi, N. Rezaei, N. Shahnavaz, A.B. Jandaghi, A. Farhoudi, Z. Pourpak, M. Moin, M. Gharagozlou, and D. Mansouri. 2004. Chronic granulomatous disease: a clinical survey of 41 patients from the Iranian primary immunodeficiency registry. Int. Arch. Allergy Immunol. 134:253–259. [DOI] [PubMed] [Google Scholar]
- 12.Casanova, J.L., and L. Abel. 2002. Genetic dissection of immunity to mycobacteria: the human model. Annu. Rev. Immunol. 20:581–620. [DOI] [PubMed] [Google Scholar]
- 13.Fieschi, C., S. Dupuis, E. Catherinot, J. Feinberg, J. Bustamante, A. Breiman, F. Altare, R. Baretto, F. Le Deist, S. Kayal, et al. 2003. Low penetrance, broad resistance, and favorable outcome of interleukin 12 receptor beta1 deficiency: medical and immunological implications. J. Exp. Med. 197:527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altare, F., A. Ensser, A. Breiman, J. Reichenbach, J.E. Baghdadi, A. Fischer, J.F. Emile, J.L. Gaillard, E. Meinl, and J.L. Casanova. 2001. Interleukin-12 receptor β1 deficiency in a patient with abdominal tuberculosis. J. Infect. Dis. 184:231–236. [DOI] [PubMed] [Google Scholar]
- 15.Caragol, I., M. Raspall, C. Fieschi, J. Feinberg, M.N. Larrosa, M. Hernandez, C. Figueras, J.M. Bertran, J.L. Casanova, and T. Espanol. 2003. Clinical tuberculosis in 2 of 3 siblings with interleukin-12 receptor β1 deficiency. Clin. Infect. Dis. 37:302–306. [DOI] [PubMed] [Google Scholar]
- 16.Özbek, N., C. Fieschi, B.T. Yilmaz, L. De Beaucoudrey, Y.E. Bikmaz, J. Feinberg, and J.L. Casanova. 2005. Interleukin-12 receptor β 1 chain deficiency in a child with disseminated tuberculosis. Clin. Infect. Dis. 40:e55–e58. [DOI] [PubMed] [Google Scholar]
- 17.Jouanguy, E., S. Lamhamedi-Cherradi, F. Altare, M.C. Fondaneche, D. Tuerlinckx, S. Blanche, J.F. Emile, J.L. Gaillard, R. Schreiber, M. Levin, et al. 1997. Partial interferon-γ receptor 1 deficiency in a child with tuberculoid bacillus Calmette-Guerin infection and a sibling with clinical tuberculosis. J. Clin. Invest. 100:2658–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casanova, J.L., and L. Abel. 2005. Inborn errors of immunity to infection: the rule rather than the exception. J. Exp. Med. 202:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bellamy, R., N. Beyers, K.P. McAdam, C. Ruwende, R. Gie, P. Samaai, D. Bester, M. Meyer, T. Corrah, M. Collin, et al. 2000. Genetic susceptibility to tuberculosis in Africans: a genome-wide scan. Proc. Natl. Acad. Sci. USA. 97:8005–8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenwood, C.M., T.M. Fujiwara, L.J. Boothroyd, M.A. Miller, D. Frappier, E.A. Fanning, E. Schurr, and K. Morgan. 2000. Linkage of tuberculosis to chromosome 2q35 loci, including NRAMP1, in a large aboriginal Canadian family. Am. J. Hum. Genet. 67:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flores-Villanueva, P., J. Ruiz-Morales, C.-H. Song, L. Flores, E. Jo, M. Montano, P. Barnes, M. Selman, and J. Granados. 2005. A functional promoter polymorphism in monocyte chemoattractant protein-1 is associated with increased susceptibility to pulmonary tuberculosis. J. Exp. Med. 202:1649–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldgar, D.E. 2002. Population aspects of cancer genetics. Biochimie. 84:19–25. [DOI] [PubMed] [Google Scholar]
- 23.Jamieson, S.E., E.N. Miller, G.F. Black, C.S. Peacock, H.J. Cordell, J.M. Howson, M.A. Shaw, D. Burgner, W. Xu, Z. Lins-Lainson, et al. 2004. Evidence for a cluster of genes on chromosome 17q11-q21 controlling susceptibility to tuberculosis and leprosy in Brazilians. Genes Immun. 5:46–57. [DOI] [PubMed] [Google Scholar]
- 24.Malik, S., L. Abel, H. Tooker, A. Poon, L. Simkin, M. Girard, G.J. Adams, J.R. Starke, K.C. Smith, E.A. Graviss, et al. 2005. Alleles of the NRAMP1 gene are risk factors for pediatric tuberculosis disease. Proc. Natl. Acad. Sci. USA. 102:12183–12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunn, L. 1965. A Short History of Genetics. Iowa State University Press, Ames. 253 pp.
- 26.Pasteur, L. 1870. Etudes sur la maladie des vers a soie. La pébrine et la flacherie (tome I). Gauthiers-Villars, Paris, 282 pp.
- 27.Dubos, R.J. 1950. Louis Pasteur, Free Lance of Science. Little Brown, Boston, xii. 418 pp.
- 28.Ranke, K. 1910. Diagnose und epidemiologie der lungentuberculose des kindes. Arch. Kinderheilkd. 54:279–306. [Google Scholar]
- 29.Dorman, S.E., C. Picard, D. Lammas, K. Heyne, J.T. van Dissel, R. Baretto, S.D. Rosenzweig, M. Newport, M. Levin, J. Roesler, et al. 2004. Clinical features of dominant and recessive interferon γ receptor 1 deficiencies. Lancet. 364:2113–2121. [DOI] [PubMed] [Google Scholar]
- 30.Sasaki, Y., A. Nomura, K. Kusuhara, H. Takada, S. Ahmed, K. Obinata, K. Hamada, Y. Okimoto, and T. Hara. 2002. Genetic basis of patients with bacille Calmette-Guerin osteomyelitis in Japan: identification of dominant partial interferon-γ receptor 1 deficiency as a predominant type. J. Infect. Dis. 185:706–709. [DOI] [PubMed] [Google Scholar]
- 31.Picard, C., C. Fieschi, F. Altare, S. Al-Jumaah, S. Al-Hajjar, J. Feinberg, S. Dupuis, C. Soudais, I.Z. Al-Mohsen, E. Genin, et al. 2002. Inherited interleukin-12 deficiency: IL12B genotype and clinical phenotype of 13 patients from six kindreds. Am. J. Hum. Genet. 70:336–348. [DOI] [PMC free article] [PubMed] [Google Scholar]