RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement (original) (raw)
Abstract
Previous studies (Leadbetter, E.A., I.R. Rifkin, A.H. Hohlbaum, B. Beaudette, M.J. Shlomchik, and A. Marshak-Rothstein. 2002. Nature. 416:603–607; Viglianti, G.A., C.M. Lau, T.M. Hanley, B.A. Miko, M.J. Shlomchik, and A. Marshak-Rothstein. 2003. Immunity. 19:837–847) established the unique capacity of DNA and DNA-associated autoantigens to activate autoreactive B cells via sequential engagement of the B cell antigen receptor (BCR) and Toll-like receptor (TLR) 9. We demonstrate that this two-receptor paradigm can be extended to the BCR/TLR7 activation of autoreactive B cells by RNA and RNA-associated autoantigens. These data implicate TLR recognition of endogenous ligands in the response to both DNA- and RNA-associated autoantigens. Importantly, the response to RNA-associated autoantigens was markedly enhanced by IFN-α, a cytokine strongly linked to disease progression in patients with systemic lupus erythematosus (SLE). As further evidence that TLRs play a key role in autoantibody responses in SLE, we found that autoimmune-prone mice, lacking the TLR adaptor protein MyD88, had markedly reduced chromatin, Sm, and rheumatoid factor autoantibody titers.
Detection of pathogen-associated molecular patterns by members of the Toll-like receptor (TLR) gene family is a critical step in the activation of the antimicrobial immune response (1). TLRs can also recognize self-antigens released from stressed or damaged host tissues, and such self-recognition can potentially promote the development of autoimmune disease. As previously reported, immune complexes (ICs) consisting of IgG bound to mammalian chromatin have been shown to effectively activate transgenic rheumatoid factor (RF) B cells through a process that involves B cell antigen receptor (BCR) recognition of the IC and subsequent delivery of the DNA to TLR9 sequestered in an endosomal/lysosomal compartment (2, 3). The same low affinity RF B cells do not proliferate in response to protein-containing ICs. Similarly, chromatin ICs, but not protein ICs, stimulate myeloid and plasmacytoid DCs to secrete cytokines, in this case through coengagement of FcγRs and TLR9 (4, 5). Moreover, under the appropriate conditions, mammalian chromatin can also directly stimulate DNA-reactive cells, again through a TLR9-dependent process (6). Thus, it is not surprising that autoantibodies reactive with DNA or DNA-associated proteins are the earliest and most prevalent serological markers of SLE in both animal models and human disease (7).
RNA and RNA/protein macromolecules, such as Sm/RNP, constitute a second major category of autoantigen frequently targeted in systemic autoimmune diseases such as systemic lupus erythematosus (SLE). Sm/RNP particles consist of the uridine-rich U1 RNA bound by Sm and other associated proteins. In the course of recent studies that identified TLR7 and TLR8 as receptors for viral single-stranded (ss) RNA, uridine-rich ssRNA was found to be a particularly effective ligand (8, 9). These data raised the possibility that the BCR/TLR9 paradigm, previously established for chromatin/DNA activation of autoreactive B cells and DCs, would also apply to TLR7/8 involvement in the activation of B cells by RNA-associated autoantigens.
Results AND Discussion
RNA-containing ICs stimulate AM14 B cells through a TLR other than TLR9
The HEp2 staining patterns of representative mAbs reactive with RNA or RNA-associated proteins are depicted in Fig. 1 A. Y2, an mAb reactive with SmD (10), binds with a speckled nuclear pattern typical of Sm/RNP-reactive autoantibodies, in contrast to the homogeneous nuclear pattern typical of double-stranded DNA– or histone-reactive mAbs such as PL2-3 (11). BWR4, an mAb that directly binds RNA (12), stains with a predominantly cytoplasmic pattern. IgG2a-reactive B cells from the AM14 RF BCR transgenic line have proven an appropriate model for the study of autoantigen-driven B cell activation in that the BCR facilitates targeting of autoantigen to the RF B cell by means of a wide range of IgG2a mAbs (2, 13).
Figure 1.
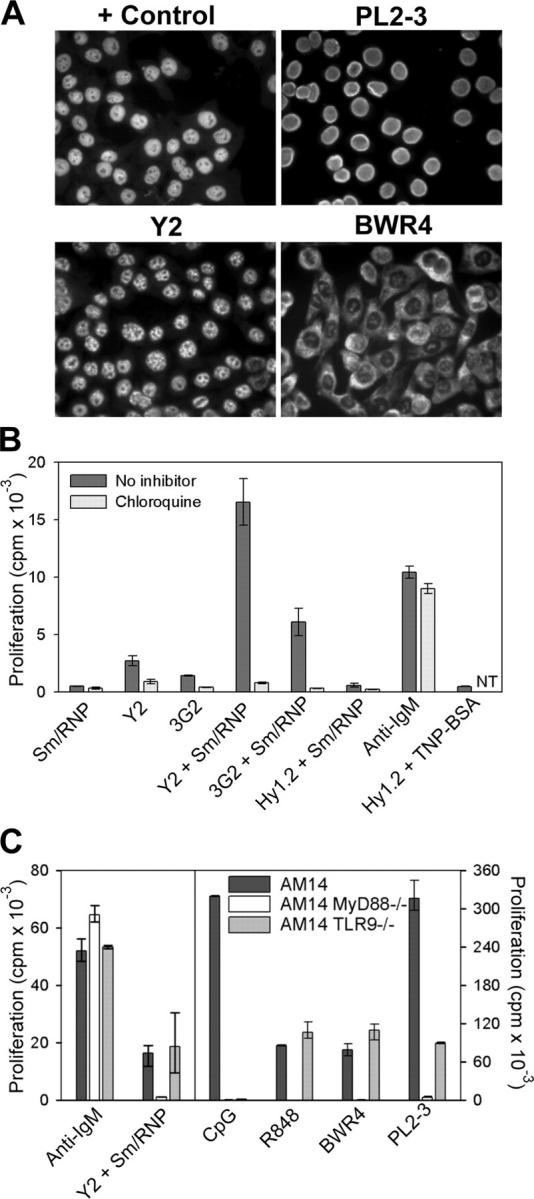
RNA-associated ICs activate AM14 B cells by a TLR-dependent mechanism. (A) HEp2 cells were stained with MRL/lpr serum (control) or the mAbs PL2-3, Y2, or BWR4. (B) AM14 B cells were stimulated with the anti-Sm mAbs Y2 and 3G2 or the anti-TNP mAb Hy1.2, alone or in combination with Sm/RNP antigen, with 5 μg/ml anti-IgM F(ab′)2 or with Hy1.2 + TNP BSA ICs in the presence or absence of chloroquine. Data shown are representative of between three and four experiments. NT, not tested. (C) AM14, AM14 MyD88-deficient, and AM14 TLR9-deficient B cells were stimulated with 5 μg/ml anti-IgM F(ab′)2, Sm/RNP IC, 1 μg/ml CpG s-ODN 1826, 10 ng/ml R848, BWR4, or PL2-3. Data shown are representative of at least two experiments per group. Values in B and C indicate means ± SD.
AM14 B cells routinely proliferated in response to ICs consisting of the IgG2a anti-Sm mAbs Y2 and 3G2 premixed with purified Sm/RNP antigen, whereas they failed to respond to ICs consisting of antihapten mAbs bound to haptenated proteins (Fig. 1 B). This response depended on IC formation, as Sm/RNP particles had no activity alone and also failed to enhance the response to uncomplexed IgG2a antihapten mAbs or to suboptimal concentrations of anti-IgM F(ab′)2 or to LPS (Fig. 1 B and not depicted). Stimulation with the anti-Sm mAbs alone consistently elicited a weak but detectable response, presumably caused by association with B cell–derived autoantigen present in the culture fluids (14). The addition of chloroquine or bafilomycin A to the cultures specifically inhibited both the relatively weak response to the directly added mAbs, as well as the more vigorous response to the preformed complexes, while having no effect on the response to anti-IgM F(ab′)2 or ligands for TLR2 or TLR4 (Fig. 1 B and not depicted). Chloroquine and bafilomycin A interfere with endosome/lysosome acidification and thereby block signaling events emanating from those TLRs located in intracellular compartments (15).
BWR4 effectively stimulated AM14 B cells in the absence of supplemental autoantigen (Fig. 1 C). As previously documented for chromatin IC activation of AM14 B cells, the BCR played a critical role in the recognition of RNA ICs, as both Y2 + Sm/RNP and BWR4 completely failed to stimulate nontransgenic B cells (unpublished data). AM14 B cells that lacked MyD88, an adaptor protein used by most TLR family members and essential for signal transduction through the TLR7 and TLR9 pathways (16), were not stimulated by either Y2 + Sm/RNP or BWR4 (Fig. 1 C). However, TLR9-deficient AM14 B cells responded to these ligands to the same extent as TLR9-sufficient cells. These data implicated TLR-derived signals as a critical factor in Sm/RNP IC activation of AM14 B cells. The data further indicated that the BWR4 mAb recognized RNA present in the culture wells and that the resulting ICs could activate AM14 B cells through a TLR other than TLR9. Therefore, its activity was not caused by cross-reactivity with DNA.
RNA IC stimulation of AM14 B cells is markedly enhanced by IFN-α
Accumulating data suggest an important role for IFN-α in the pathogenesis of human SLE (17). In addition, IFN-α has been found to have a direct effect on B cell activity by promoting plasmablast differentiation (18) and specifically up-regulating TLR7 and MyD88 expression (19). To determine whether IFN-α would influence the response of B cells to RNA-associated autoantigens, we precultured AM14 B cells with IFN-α for 2–3 h and then added either anti-Sm mAbs alone or preformed Sm/RNP ICs. Remarkably, IFN-α markedly enhanced the response of both AM14 and AM14 TLR9-deficient B cells to both spontaneous and preformed Sm/RNP ICs. This effect was limited to the autoantigen ICs; IFN-α did not induce a response to the Sm/RNP antigen alone (Fig. 2 A). A similar effect was seen in the response to BWR4 (Fig. 2 B). Over a series of 12 experiments, IFN-α increased the response to Sm/RNP ICs by an average of fivefold, had more moderate enhancing effects on the responses to R848 and CpG, and consistently suppressed the responses to anti-IgM F(ab′)2 and LPS (Fig. 2 C).
Figure 2.
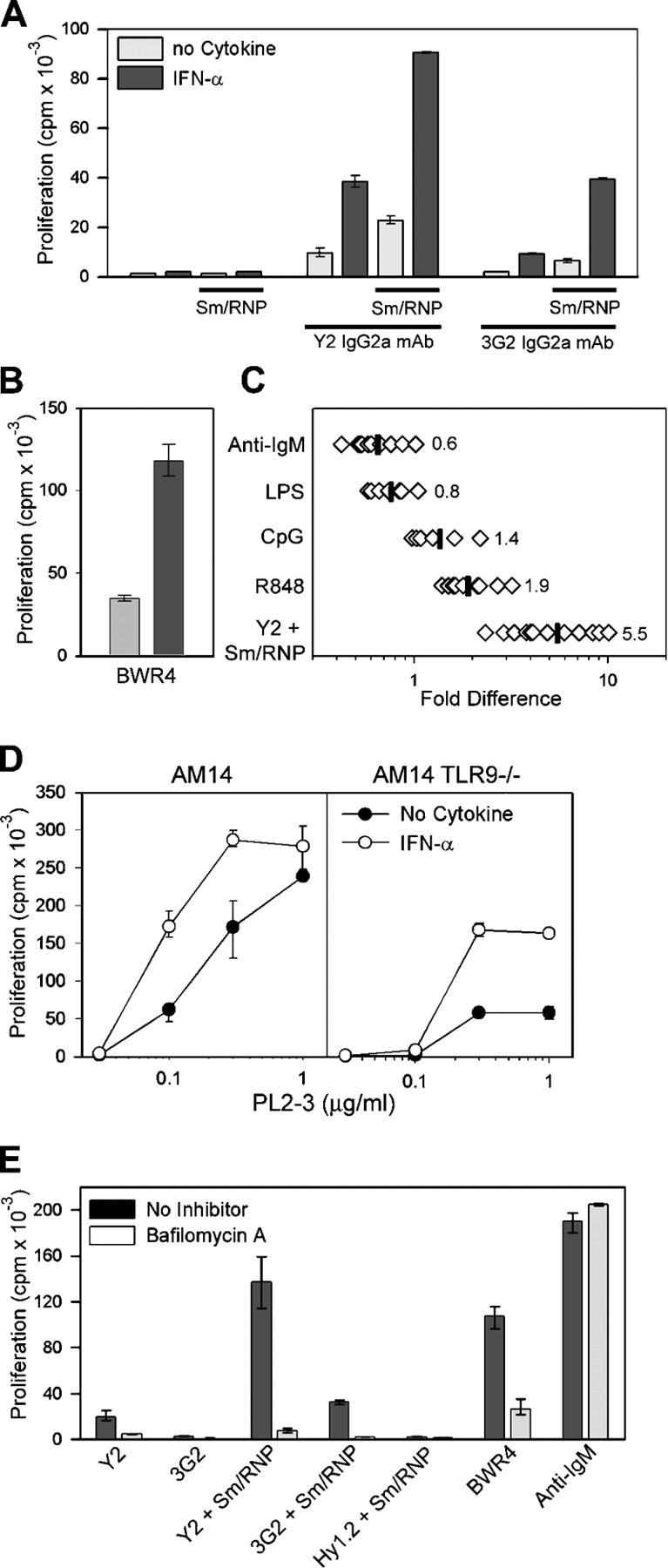
Sm/RNP IC and BWR4 activation of AM14 B cells is enhanced by IFN-α. Proliferative response of AM14 B cells to anti-Sm mAbs or Sm/RNP ICs (A) or BWR4 (B) in the presence or absence of 1,000 U/ml of IFN-α. Values indicate means ± SD. (C) Ratio of the response to anti-IgM F(ab′)2, LPS, CpG, R848, or Sm/RNP ICs stimulated in the presence or absence of IFN-α. Each point represents data from one experiment; data includes AM14 and AM14 TLR9-deficient responder populations. Mean values are indicated by vertical lines. (D) Response of AM14 and AM14 TLR9-deficient B cells to PL2-3 in the presence or absence of 1000 U/ml of IFN-α. (E) Stimulation of AM14 B cells by the indicated ligands after preincubation with IFN-α in the presence or absence of bafilomycin A. Values in D and E indicate means ± SD.
As previously reported, the response of TLR9-deficient AM14 B cells to PL2-3 is routinely 30–35% of the TLR9-sufficient AM14 B cell response (3). This residual activity was also dramatically enhanced by IFN-α, whereas the effect of IFN-α on the PL2-3 response of TLR9-sufficient cells was more modest (Fig. 2 D). By comparison, AM14 MyD88-deficient AM14 cells do not proliferate in response to PL2-3 (2, 3). Explanations for these data include the possibility that the histone dimers recognized by PL2-3 are bound directly to RNA or to apoptotic debris that incorporates RNA fragments. Evaluation of double-deficient TLR7/TLR9 AM14 B cells will help to address this issue, but these mice are not yet available because of the complicated breeding strategy required.
The IFN-α–enhanced response to Sm/RNP ICs and BWR4 was still suppressed by chloroquine and bafilomycin A, and IFN-α–primed MyD88-deficient cells failed to respond to these stimuli (Fig. 2 E and not depicted). Thus, IFN-α did not circumvent the TLR dependence of the RNA-associated IC response. These results establish a critical link between IFN-α and the activation of B cells specific for RNA-associated autoantigens.
Y2 + Sm/RNP and BWR4 activation of AM14 B cells is RNA dependent
To determine whether the stimulatory activity of the Sm/RNP complexes was dependent on the RNA component, the Sm/RNP antigen was preincubated with RNase A before the addition of mAb. RNase A markedly reduced the stimulatory activity of both the Sm/RNP ICs and BWR4 (Fig. 3 A). The effect was specific, as RNase had no effect on the B cell response to LPS. These data supported the idea that BCR recognition of RNA-containing ICs led to internalization of the complex and delivery of the RNA to an endosome/lysosome-associated RNA-sensitive TLR. TLR7 was a reasonable candidate based on its recently described capacity to recognize both microbial- and mammalian-derived ssRNA (8, 9, 20). AM14 TLR7-deficient B cells provided a further test of this premise. Both Y2 + Sm/RNP and BWR4 failed to stimulate TLR7-deficient AM14 B cells, as did the TLR7 ligand R848 (Fig. 3 B). This was not because of a global defect in TLR7-deficient B cells, as responses to anti-IgM, CpG DNA, and chromatin ICs (PL2-3 mAb) remained intact. Thus, the AM14 response to Y2 + Sm/RNP or BWR4 depends on BCR recognition and internalization of IgG2a ICs and subsequent engagement of the complexed mammalian RNA by TLR7.
Figure 3.

Sm/RNP IC and BWR4 activation of AM14 B cells depends on RNA activation of TLR7 and is blocked by s-ODN 2088. (A) AM14 B cells were stimulated with Y2 + Sm/RNP, BWR4, or LPS in the presence or absence of RNase A. Shown are the means of three experiment per group ± SD. (B) AM14 and AM14 TLR7-deficient B cells were stimulated with 1 μg/ml CpG, 10 ng/ml R848, PL2-3, or Y2 + Sm/RNP or BWR4. Data shown are representative of two experiments. (C) AM14 and AM14 TLR9-deficient B cells were stimulated with anti-IgM F(ab′)2, R848, CpG, Y2 + Sm/RNP, or BWR4 in the presence (○) or absence (•) of 4 μg/ml s-ODN 2088 and are representative of between four and seven experiments per group. In all cases, the Y2 + Sm/RNP– and BWR4-stimulated cells were preincubated with IFN-α. Values in B and C indicate means ± SD.
Intriguingly, the responses to Y2 + Sm/RNP and BWR4 could be blocked by s-ODN 2088 (Fig. 3 C). s-ODN 2088 was originally described as a specific inhibitor of TLR9 (21); however, it can also partially suppress the B cell response to the synthetic TLR7 ligand, R848, while having no effect on the response to LPS, anti-IgM F(ab′)2, or TLR2 ligands (Fig. 3 C) (2). Importantly, s-ODN 2088 can even suppress the Y2 + Sm/RNP response of AM14 TLR9-deficient cells (Fig. 3 C), proving that the inhibitory activity of s-ODN 2088 is completely independent of TLR9.
Reduced autoantibody production in MyD88-deficient autoimmune-prone mice
To confirm the relevance of our in vitro observations to the in vivo development of autoantibodies in SLE, we intercrossed autoimmune-prone MRL-lpr/gld mice with C57BL/6 mice deficient in the expression of MyD88. By 20–25 wk of age, sera from all the autoimmune lpr/lpr or gld/gld MyD88-sufficient offspring contained antinuclear antibodies (ANAs), as detected by HEp2 immunofluorescence. In contrast, none of the autoimmune MyD88-deficient offspring developed either a homogeneous nuclear or speckled nuclear staining pattern, indicative of a lack of antichromatin, anti-Sm, and any other ANA (Fig. 4 A). There was also no evidence for cytoplasmic RNA-associated antibodies.
Figure 4.

Absence of autoantibody production in MyD88-deficient mice. (A) ANA production by 20–25-wk-old autoimmune (lpr/lpr) MyD88-sufficient (MyD88+/− or MyD88+/+) and MyD88-deficient (MyD88−/−) mice. Intensity of staining of HEp2 cells was scored on a scale from 0 to 3. (right) Representative MyD88+/− autoimmune and MyD88−/− autoimmune sera, compared with standard MRL-lpr/gld and B6 serum samples, are shown. *, P < 0.00002 versus autoimmune MyD88−/− mice using Fisher's exact test. (B) Anti-Sm reactivity of 20–25-wk-old autoimmune (lpr/lpr or gld/gld) MyD88-sufficient (MyD88+/− or MyD88+/+) and MyD88-deficient (MyD88−/−) mice was scored by Western blotting as either negative (0) or positive, with positive reactivity being graded from 1+ (weaker reactivity) to 4+ (stronger reactivity). Distribution of Myd88−/− mice differs from autoimmune mice (P < 0.002 using χ2 test). (C and D) RF and IgG2a serum titers measured by ELISA. *, P < 0.01 versus autoimmune mice.
Under normal circumstances, almost all MRL-lpr and MRL-gld mice make antichromatin antibodies and ∼25% of MRL-lpr mice make anti-Sm antibodies (22). The resultant speckled anti-Sm reactivity pattern is often obscured in the HEp2 staining analysis by the presence of the homogeneously staining antichromatin antibodies. To prove that the intercrossed MyD88-sufficient autoimmune mice in this study could actually produce anti-Sm antibodies, the sera were further screened by Western blot analysis. In our sample population, 11 out of 24 autoimmune MyD88-sufficient mice made anti-SmD antibodies, whereas 2 out of the 28 autoimmune MyD88-deficient mice did so (Fig. 4 B). The MyD88-deficient mice also had very low RF titers, as predicted by the in vitro TLR-dependent response to chromatin and RNA-associated ICs (Fig. 4 C). Not only were the autoantibody titers reduced, but levels of total IgG2a were significantly lower in the MyD88-deficient autoimmune mice than in the MyD88-sufficient littermate controls (P < 0.01; Fig. 4 D). It is therefore likely that TLR9, TLR7, and/or other MyD88-dependent DNA/RNA-reactive receptors are involved in both the in vivo production of antichromatin and anti-Sm antibodies, as well as the subsequent IC-stimulation of RF B cells.
MyD88 is not only used in TLR signal propagation but also by the receptors for IL-1 and IL-18 (23), and we cannot formally exclude the possibility that the observed effects result from loss of signaling by these cytokines. However, in a mercuric chloride–induced lupus model, mice lacking IL-1β converting enzyme (and, thus, with defective production and release of bioactive IL-1β and IL-18) developed ANA titers comparable to WT mice (24). Moreover, Fas-deficient MRL mice lacking a functional IL-18 receptor still made anti-DNA antibodies (25), and Fas-deficient MRL mice treated with IL-1R antagonist showed no decrease in anti-DNA autoantibody titers (26). Therefore, the connection between MyD88 expression and autoantibody production is most likely TLR-dependent.
Elegant studies by Ronnblom et al. and Bave et al. have determined that similar kinds of chromatin- and RNA-containing ICs trigger the production of IFN-α by human plasmacytoid DCs (27, 28). Recent studies have shown that chromatin-containing ICs can induce cytokine production by murine BM-derived DCs through a mechanism involving FcγRIII and TLR9 (4, 5). Thus, the autoantibody production in vivo may well reflect direct effects of the ICs on B cells, as well as indirect effects mediated by IC activation of DCs and subsequent induction of cytokine production or priming of helper T cells. Additional studies will be necessary to evaluate the relative importance of DC and B cell intrinsic events.
Overall, these data establish a key role for TLR7, as well as TLR9, in the activation of autoreactive B cells. As reported recently, TLR9-deficient lpr/lpr mice failed to produce DNA-specific antibodies but still produced RNA and cardiolipin-reactive autoantibodies (29). Importantly, these mice developed clinical features of systemic autoimmune disease. Based on our study, it will be highly relevant to determine the disease profile of autoimmune-prone mice deficient in the expression of both TLR9 and TLR7. Simultaneous blockade of both receptors may prove to be a useful therapeutic strategy.
Materials and Methods
Mice
AM14, AM14 MyD88-deficient, and AM14 TLR9-deficient BCR transgenic mice have been described previously (3, 13) and were maintained at the Boston University School of Medicine (BUSM) Laboratory Animal Sciences Center. AM14 TLR7-deficient mice were derived from the intercross of AM14/MRL/lpr mice and TLR7-deficient mice (30) on the MRL/lpr background (BC5 or greater) at Yale University School of Medicine. TLR7-deficient mice were males with mutant X chromosomes, and these were compared with littermate control male or female mice with TLR7-intact X chromosomes. MyD88-deficient autoimmune mice were obtained at BUSM by intercrossing C57BL/6 MyD88-deficient mice (23) with MRL-lpr/gld mice. Offspring were screened for inheritance of both the lpr and gld mutations, and those mice typed as either lpr/lpr or gld/gld were considered autoimmune prone. The mice were given trimethoprim-sulfamethoxazole in the drinking water as prophylaxis against bacterial infection. All mice were bred and maintained in accordance with the regulations of the American Association for the Accreditation of Laboratory Animal Care.
mAbs
Hybridoma cell lines producing the IgG2a Sm-reactive mAbs Y2 and 3G2 (10) were provided by P. Cohen and R. Eisenberg (University of Pennsylvania, Philadelphia, PA), and the line producing the IgG2a RNA-reactive mAb BWR4 (12) was provided by D. Eilat (Hadassah University Hospital, Jerusalem, Israel). mAbs collected as culture fluids were affinity purified on protein A affinity columns. The mAbs Hy1.2 (anti-TNP) and PL2-3 (anti-H2a/H2b histone dimer) have been described previously (2). The isotype and specificity of these reagents were verified by either ELISA or Western blot analysis. All mAbs had <0.1 U endotoxin/ml as determined by limulus amoebocyte assay (Cambrex Bioscience).
In vitro stimulation
Sm/RNP ICs were preformed by mixing the anti-Sm mAbs with 0.628 mg/ml endotoxin-free Sm/RNP antigen (antibodies 260 nm/280 nm, >1.8; Arotec Diagnostics) for 1–2 h in serum-free medium. ICs were then added to magnetic bead–purified splenic AM14 B cells at a final concentration of 20 μg/ml mAb + 1:300−1,500 dilution of Sm/RNP. In some experiments, the Sm/RNP particles were pretreated with 80–160 μg/ml RNase A (USB) at 37°C for 1 h before IC formation. PL2-3 and BWR4 were used at final concentrations of 5 and 10 μg/ml, respectively, unless stated otherwise in the figure legends. For the BWR4 RNase experiments, the cells were preincubated with RNase A for 1 h at 37°C before the addition of mAb. The effect of IFN-α (PBL Biomed) was determined by preincubating the B cells with 1,000 U/ml of IFN-α for 2–3 h at 37°C before the addition of the other ligands. Culture conditions, additional inhibitors and ligands, and statistical parameters have been described previously (3, 6). Chloroquine, bafilomycin A, and s-ODN 2088 were used at final concentrations of 2 μg/ml, 7.5 nM/ml, and 4 μg/ml, respectively. Other ligands were used at suboptimal/optimal concentrations as follows: anti-IgM F(ab′)2, 5 μg/ml (suboptimal) or 15 μg/ml (optimal); CpG (s-ODN 1826), 0.3 μg/ml or 1.0 μg/ml; and R848, 10 ng/ml or 100 ng/ml.
Serological assays
ANAs were detected by indirect immunofluorescence using HEp-2 substrate slides (Antibodies Inc.). Serum RF and total IgG titers were determined by ELISA. Autoantibodies to Sm were measured by Western Blot analysis as previously described (10). Anti-Sm reactivity was scored as either negative or positive, with positive reactivity being graded from 1+ (weaker reactivity) to 4+ (stronger reactivity).
Acknowledgments
We thank Dr. A. Krieg for helpful discussions, J.R. Gee, Z. Ruhe, and J. Shupe for assistance with Western blotting and technical assistance, and Dr. M. Karow and her colleagues at Regeneron for derivation of the TLR7-deficient mice.
This work was supported by grants AR35230 (to A. Marshak-Rothstein), AR050256 (to A. Marshak-Rothstein, M.J. Shlomchik, G.A. Viglianti, and I.R. Rifkin), and AI36529 (to M.J. Mamula) from the National Institutes of Health; a grant from the National Kidney Foundation (to I.R. Rifkin); and a grant from the Lupus Research Institute (to I.R. Rifkin).
The authors have no conflicting financial interests.
References
- 1.Medzhitov, R., P. Preston-Hurlburt, and C.A. Janeway. 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 388:394–397. [DOI] [PubMed] [Google Scholar]
- 2.Leadbetter, E.A., I.R. Rifkin, A.H. Hohlbaum, B. Beaudette, M.J. Shlomchik, and A. Marshak-Rothstein. 2002. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 416:603–607. [DOI] [PubMed] [Google Scholar]
- 3.Marshak-Rothstein, A., L. Busconi, C.M. Lau, A.S. Tabor, E.A. Leadbetter, S. Akira, A.M. Krieg, G.B. Lipford, G.A. Viglianti, and I.R. Rifkin. 2004. Comparison of CpG s-ODNs, chromatin immune complexes, and dsDNA fragment immune complexes in the TLR9-dependent activation of rheumatoid factor B cells. J. Endotoxin. Res. 10:247–251. [DOI] [PubMed] [Google Scholar]
- 4.Boule, M.W., C. Broughton, F. Mackay, S. Akira, A. Marshak-Rothstein, and I.R. Rifkin. 2004. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin–immunoglobulin G complexes. J. Exp. Med. 199:1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Means, T.K., E. Latz, F. Hayashi, M.R. Murali, D.T. Golenbock, and A.D. Luster. 2005. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Invest. 115:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Viglianti, G.A., C.M. Lau, T.M. Hanley, B.A. Miko, M.J. Shlomchik, and A. Marshak-Rothstein. 2003. Activation of autoreactive B cells by CpG dsDNA. Immunity. 19:837–847. [DOI] [PubMed] [Google Scholar]
- 7.Burlingame, R., M.L. Boey, G. Starkebaum, and R. Rubin. 1994. The central role of chromatin in autoimmune responses to histones and DNA in systemic lupus erythematosus. J. Clin. Invest. 94:184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diebold, S.S., T. Kaisho, H. Hemmi, S. Akira, C. Reis, and E. Sousa. 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 303:1529–1531. [DOI] [PubMed] [Google Scholar]
- 9.Heil, F., H. Hemmi, H. Hochrein, F. Ampenberger, C. Kirschning, S. Akira, G. Lipford, H. Wagner, and S. Bauer. 2004. Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science. 303:1526–1529. [DOI] [PubMed] [Google Scholar]
- 10.Bloom, D.D., J.-L. Davignon, M.W. Retter, M.J. Shlomchik, D.S. Pisetsky, P.L. Cohen, R.A. Eisenberg, and S.H. Clarke. 1993. V region gene analysis of anti-Sm hybridomas from MRL/Mp-lpr/lpr mice. J. Immunol. 150:1591–1610. [PubMed] [Google Scholar]
- 11.Losman, M., T.M. Fasy, K.E. Novick, and M. Monestier. 1992. Monoclonal autoantibodies to subnucleosomes from a MRL/Mp-+/+ mouse. J. Immunol. 148:1561–1569. [PubMed] [Google Scholar]
- 12.Eilat, D., and R. Fischel. 1991. Recurrent utilization of genetic elements in V regions of antinucleic acid antibodies from autoimmune mice. J. Immunol. 147:361–368. [PubMed] [Google Scholar]
- 13.Wang, H., and M.J. Shlomchik. 1999. Autoantigen-specific B cell activation in Fas-deficient rheumatoid factor immunoglobulin transgenic mice. J. Exp. Med. 190:639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyer, C.M., R.A. Eisenberg, and P.L. Cohen. 1985. Quantitation of the Sm nuclear antigen in tissues and activated lymphocytes. Arthritis Rheum. 28:294–299. [DOI] [PubMed] [Google Scholar]
- 15.Hacker, H., H. Mischak, T. Miethke, S. Liptay, R. Schmid, T. Sparwasser, K. Heeg, G.B. Lipford, and H. Wagner. 1998. CpG-DNA-specific activation of antigen-presenting cells requires stress kinase activity and is preceded by non-specific endocytosis and endosomal maturation. EMBO J. 17:6230–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takeda, K., T. Kaisho, and S. Akira. 2003. Toll-like receptors. Annu. Rev. Immunol. 21:335–376. [DOI] [PubMed] [Google Scholar]
- 17.Baechler, E.C., P.K. Gregersen, and T.W. Behrens. 2004. The emerging role of interferon in human systemic lupus erythematosus. Curr. Opin. Immunol. 16:801–807. [DOI] [PubMed] [Google Scholar]
- 18.Jego, G., A.K. Palucka, J.P. Blanck, C. Chalouni, V. Pascual, and J. Banchereau. 2003. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 19:225–234. [DOI] [PubMed] [Google Scholar]
- 19.Bekeredjian-Ding, I.B., M. Wagner, V. Hornung, T. Giese, M. Schnurr, S. Endres, and G. Hartmann. 2005. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J. Immunol. 174:4043–4050. (published erratum appears in J. Immunol. 2005. 174:5884) [DOI] [PubMed] [Google Scholar]
- 20.Crozat, K., and B. Beutler. 2004. TLR7: a new sensor of viral infection. Proc. Natl. Acad. Sci. USA. 101:6835–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lenert, P., L. Stunz, A.-K. Yi, A.M. Krieg, and R.F. Ashman. 2001. CpG stimulation of primary mouse B cells is blocked by inhibitory oligodeoxyribonucleotides at a site proximal to NF-κB activation. Antisense Nucleic Acid Drug Dev. 11:247–256. [DOI] [PubMed] [Google Scholar]
- 22.Eisenberg, R.A., D.S. Pisetsky, S.Y. Craven, J.P. Grudier, M.A. O'Donnell, and P.L. Cohen. 1990. Regulation of anti-Sm autoantibody response in systemic lupus erythematosus mice by monoclonal anti-Sm antibodies. J. Clin. Invest. 85:86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adachi, O., T. Kawai, K. Takeda, M. Matsumoto, H. Tsutsui, M. Sakagami, K. Nakanishi, and S. Akira. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1 and IL-18-mediated function. Immunity. 9:143–150. [DOI] [PubMed] [Google Scholar]
- 24.Pollard, K.M., P. Hultman, and D.H. Kono. 2003. Using single-gene deletions to identify checkpoints in the progression of systemic autoimmunity. Ann. NY Acad. Sci. 987:236–239. [DOI] [PubMed] [Google Scholar]
- 25.Lin, L., and S.L. Peng. 2005. Interleukin-18 receptor signaling is not required for autoantibody production and end-organ disease in murine lupus. Arthritis Rheum. 52:984–986. [DOI] [PubMed] [Google Scholar]
- 26.Kiberd, B.A., and A.W. Stadnyk. 1995. Established murine lupus nephritis does not respond to exogenous interleukin-1 receptor antagonist; a role for the endogenous molecule? Immunopharmacology. 30:131–137. [DOI] [PubMed] [Google Scholar]
- 27.Ronnblom, L., and G.V. Alm. 2001. A pivotal role for the natural interferon α–producing cells (plasmacytoid dendritic cells) in the pathogenesis of lupus. J. Exp. Med. 194:F59–F63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bave, U., G.V. Alm, and L. Ronnblom. 2000. The combination of apoptotic U937 cells and lupus IgG is a potent IFN-alpha inducer. J. Immunol. 165:3519–3526. [DOI] [PubMed] [Google Scholar]
- 29.Christensen, S.R., M. Kashgarian, L. Alexopoulou, R.A. Flavell, S. Akira, and M.J. Shlomchik. 2005. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J. Exp. Med. 202:321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valenzuela, D.M., A.J. Murphy, D. Frendewey, N.W. Gale, A.N. Economides, W. Auerbach, W.T. Poueymirou, N.C. Adams, J. Rojas, J. Yasenchak, et al. 2003. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat. Biotechnol. 21:652–659. [DOI] [PubMed] [Google Scholar]