Modulation of ASIC channels in rat cerebellar purkinje neurons by ischaemia-related signals (original) (raw)
Abstract
Acid-sensing ion channels (ASICs), activated by a decrease of extracellular pH, are found in neurons throughout the nervous system. They have an amino acid sequence similar to that of ion channels activated by membrane stretch, and have been implicated in touch sensation. Here we characterize the pH-dependent activation of ASICs in cerebellar Purkinje cells and investigate how they are modulated by factors released in ischaemia. Lowering the external pH from 7.4 activated an inward current at −66 mV, carried largely by Na+ ions, which was half-maximal for a step to pH 6.4 and was blocked by amiloride and gadolinium. The H+-gated current desensitized within a few seconds, but approximately 30% of cells showed a sustained inward current (11% of the peak current) in response to the maintained presence of pH 6 solution. The peak H+-evoked current was potentiated by membrane stretch (which occurs in ischaemia when [K+]o rises) and by arachidonic acid (which is released when [Ca2+]i rises in ischaemia). Arachidonic acid increased to 77% the fraction of cells showing a sustained current evoked by acid pH. The ASIC currents were also potentiated by lactate (which is released when metabolism becomes anaerobic in ischaemia) and by FMRFamide (which may mimic the action of related mammalian RFamide transmitters). These data reinforce suggestions of a mechanosensory aspect to ASIC channel function, and show that the activation of ASICs reflects the integration of multiple signals which are present during ischaemia.
Acid-sensing ion channels (ASICs), which are activated by extracellular protons and blocked by amiloride, were first reported in rat trigeminal and dorsal root ganglion neurons (Krishtal & Pidoplichko, 1981), and have since been found in a range of central and peripheral neurons and glia (Akaike & Ueno, 1994; Bevan, 1998). The protein subunits making up these channels contain two membrane-spanning regions and exist in at least six different forms: ASIC1a (also known as BNaC2), ASIC1b, ASIC2a (also known as MDEG1 or BNaC1), ASIC2b (MDEG2), ASIC3 (DRASIC) and ASIC4. ASIC proteins are structurally related to epithelial sodium channels, and to channels activated by mechanical stimuli and by the snail neuropeptide FMRFamide (Lingueglia et al. 1995; Waldmann et al. 1997; Waldmann & Lazdunski, 1998; Mano & Driscoll, 1999). Mutations in C. elegans mechano-transduction channels lead to constitutive activation of these channels and thus to an excessive cation entry causing neuronal degeneration (Driscoll & Chalfie, 1991), so the channel superfamily which includes the ASICs is denoted DEG/ENaC, for _deg_enerin/_e_pithelial _Na c_hannel.
Knocking out ASIC2 or ASIC3 alters touch sensation, suggesting a mechanosensing role for these subunits (Price et al. 2000, 2001), although direct evidence of mechanosensing by ASICs is lacking. Furthermore, the pH-gating of ASIC1- and ASIC3-containing channels is potentiated by the neuropeptide FMRFamide (Askwith et al. 2000). This raises the possibility that the common evolutionary history of the DEG/ENaC channel superfamily has resulted in ASICs being able to integrate several different signals - acidity, mechanical stimulation and peptide transmitter binding.
The widespread expression of ASIC channels throughout the CNS, and the large membrane current that they can generate, suggests that they may have an important functional role. However, many ASIC channels require a large and rapid fall of pH to be activated and it is unclear when this might happen in the CNS (see Discussion). This suggests that a co-agonist or modulator may be needed, in addition to acid pH, to activate the channels under natural conditions, but little is known about the modulation of ASIC channels in central neurons. Here we characterize pH-gated currents in cerebellar Purkinje cells, which express ASIC subunits 1a, 2a and 2b (Garcia-Anoveros et al. 1997; Lingueglia et al. 1997), and determine how they are modulated by factors present during ischaemia. We focus on ischaemia for two reasons. First, the fall of extracellular pH by more than one pH unit during ischaemia (Kraig et al. 1983; Silver & Erecinska, 1992) might be expected to activate ASICs. Second, Purkinje cells are prone to dying necrotically in ischaemia (Cervos-Navarro & Diemer, 1991), despite their lack of the NMDA subtype of glutamate receptor which promotes ischaemic death in other neurons, and an ASIC-mediated cation influx might contribute to this death.
Methods
Cerebellar slices and acutely isolated Purkinje cells
Cerebellar slices (250 μm thick) were cut from the brains of 12- to 14-day-old rats (killed by cervical dislocation in accordance with the UK Animals (Scientific Procedures) Act, 1986), in a solution containing (mm): NaCl 120, KCl 2.5, MgCl2 2, CaCl2 2, NaH2PO4 1, NaHCO3 26, glucose 10, Na-kynurenate (to block glutamate receptors) 1, bubbled with 95 % O2-5 % CO2. Slices were dissociated by papain treatment followed by trituration with a fine glass pipette, as for isolated salamander Müller cells (Brew & Attwell, 1987) but with the tonicity of the solution raised to that for mammals by the addition of NaCl and with the solutions bubbled with 100 % O2. When the dissociated slice was plated into the experimental chamber, granule cells could be identified by their small spherical appearance (diameter < 8 μm), and Purkinje cells by their larger size (diameter > 20 μm) and the remains of axons and dendritic trees (Hockberger et al. 1994; Billups et al. 1998). Deep cerebellar nuclear cells also have a large diameter, but are present at only one tenth of the number of Purkinje cells (Caddy & Biscoe, 1979) and furthermore are rarely obtained by papain dissociation of cerebellum (Hockberger et al. 1994), suggesting that they did not significantly contaminate our sample of Purkinje cells.
Electrodes
Purkinje cells (isolated or in cerebellar slices) were whole-cell clamped using electrodes pulled from thin-walled borosilicate tube with a resistance in external solution of 2–4 MΩ. The series resistance in whole-cell clamp mode was ≈5 MΩ, leading to voltage errors of < 5 mV for peak ASIC currents of < 1 nA at a holding potential of −60 to −70 mV.
Solutions
Experiments were carried out at room temperature (23-26 °C). External solution contained (mm): NaCl 140, KCl 2.5, MgCl2 2, CaCl2 2.5, NaH2PO4 1, Hepes or Mes 10, glucose 10, pH adjusted with NaOH. Solutions were bubbled with 100 % O2 for slice experiments; the properties of isolated cells were not affected whether or not O2 was bubbled, presumably because sufficient O2 diffuses from the surface of the shallow bath solution for the metabolism of cells plated at low density, so bubbling was omitted. Hepes was used as the pH buffer for pH values > 6.7, and Mes for pH < 6.7 (at pH 6.7 the ASIC current amplitude was independent of the buffer used). NaH2PO4 was omitted from all solutions for experiments on gadolinium, which is chelated by phosphate (Caldwell et al. 1998). Most drugs (FMRFamide, GdCl3, lysophosphatidylinositol) were added to the external solution from a stock solution in water. Arachidonic acid was added from a stock solution of its sodium salt in water, or from a stock of the acid in ethanol - results with the two stocks were indistinguishable. Na-lactate was added as a replacement for NaCl. For experiments on cell swelling, 140 mm NaCl was replaced by 100 mm NaCl plus 40 mm choline-Cl, and the choline-Cl was omitted to produce 25 % hypotonic solution. None of these agents or manipulations affected the resting current at pH 7.4, apart from gadolinium and arachidonic acid which appeared to increase the resistance of the seal between the electrode and the cell. External solutions in different reservoirs were connected through a valve and tubing to a syringe needle, aimed at the whole-cell clamped cell. Solution was superfused through the needle at a rate of 11 ml min−1 into a bath of volume 1 ml. This flow rate gave a 10–90 % activation time of 77 ± 5 ms in 19 cells for ASIC currents activated by a switch (using manual valves) from pH 7.4 to pH 6. The pipette solution contained (mm): either CsF 110, CsCl 30, NaCl 4 and (_N_-methyl-d-glucamine)2EGTA 5, or KCl 140 and Na2EGTA 5, or NaCl 140 and Na2EGTA 5 (see text), in addition to CaCl2 0.5 and Hepes 10; pH was set to 7.0 using CsOH, KOH or NaOH respectively. When blockers of arachidonic acid metabolism were included in the internal solution (20 μM indomethacin to block cyclooxygenase, 20 μM nordihydroguiaretic acid (NDGA) to block lipoxygenase, and 20 μM 5,8,11,14-eicosatetraynoic acid (ETYA) to block epoxygense and lipoxygenase), we allowed > 8 min for them to diffuse into the cell before testing the effect of arachidonic acid. Electrode junction potentials (-6, −3 and 0 mV for the Cs+-, K+- and Na+-based internal solutions) were compensated for. ASIC currents were evoked by low pH solution at 1-1.5 min intervals to allow 96–99 % recovery from desensitization. All chemicals were from Sigma except FMRFamide, which was from Calbiochem.
Data analysis
Data are presented as means ± s.e.m. P values are from Student's two-tailed t test. Unless otherwise stated, drugs (or hypotonic solution) were applied for 2–3 min. Amplitudes of ASIC currents in the presence of drugs (or hypotonic solution) were measured as the average of two responses to acid pH solution applied after ≈1 and 2 min in the drug, and were normalized to the average of two control responses measured ≈1 and 2 min before the drug was applied and two responses measured ≈1 and 2 min after washing out the drug with control solution (to compensate for slow rundown of the ASIC currents). For the pH dose-response curve (Fig. 1_F_), responses to test pH solutions (applied in random order) were normalized to the average of bracketing responses to pH 5 solution; most cells were tested with five different pH solutions as well as pH 5.
Figure 1. ASIC currents in cerebellar Purkinje neurons.
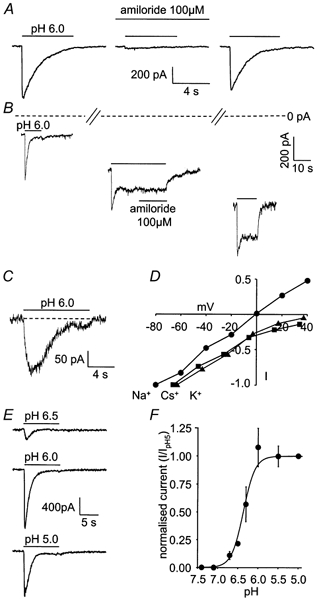
Membrane potential was −66 mV and internal solution contained Cs+ as the main cation, except where stated. A, response of an isolated Purkinje cell to pH 6.0 solution in control conditions, in the presence of 100 μM amiloride, and after washout of amiloride. B, increase of sustained component of response of an isolated Purkinje cell to pH 6.0 solution during deterioration of cell condition (shown by increase of holding current: distance below dashed line). Slashes on dashed line denote time intervals of ≈2 min between the responses shown. Middle trace shows that amiloride does not block the sustained current. C, response of a Purkinje cell in a cerebellar slice to pH 6.0 solution (solutions contained 10 μM NBQX, 40 μM bicuculline and 1 μM TTX, to block AMPA and GABAA receptors and Na+ action potentials). D, specimen I-V relations for the peak ASIC current evoked by pH 6.0 solution in three isolated cells whole-cell clamped with internal solutions in which the main cation was Na+ (•), Cs+ (▪) or K+ (▴); data were normalized to the inward current recorded at the most negative voltage tested. E, responses of an isolated cell to solutions of pH 6.5, 6.0 and 5.0. F, mean current-pH curve from 20 isolated cells. Smooth curve has the form [H+]o3/([H+]o3 + H0.53) with pH0.5 = -log10H0.5 = 6.4.
Results
Purkinje cells show an ASIC-mediated current
In isolated Purkinje cells voltage clamped at −66 mV, stepping the external pH from 7.4 to 6.0 always led to the activation of a large inward current (mean value 806 ± 71 pA in 79 cells) which then desensitized (Fig. 1_A_). The current was reduced reversibly by 93 ± 2 % by 100 μM amiloride in five cells (Fig. 1_A_), consistent with it being mediated by ASIC channels (Waldmann et al. 1997). During maintained application of pH 6.0 solution, the current desensitized with a time constant of 1.63 ± 0.10 s (in 20 cells). From the response to a second pulse of acid solution, the time constant of the current recovery from desensitization (assumed to be exponential) was 18.0 ± 1.5 s in three cells (data not shown) similar to data from other cell types (Bevan, 1998). In many cells desensitization was complete, while in 72 of 230 cells (31 %) with a healthy appearance and input resistance, to which pH 6 solution was applied, a small sustained inward current was present after the desensitization (Fig. 1_B_ and Fig. 4_C_). In cells showing a sustained current at the start of recording, the ratio of the sustained to the peak current evoked by pH 6 solution was 0.11 ± 0.01 (n = 72). The sustained current was not blocked by amiloride (5 cells, Fig. 1_B_), as reported previously in other preparations (Benson et al. 1999). During recordings in which the cell condition deteriorated, as assessed by a fall of input resistance, the sustained current tended to increase with time (Fig. 1_B_), suggesting that a sustained cation influx is more likely to be evoked by acid pH in cells with compromised health; these cells were excluded from further analysis.
Figure 4. Modulation by lactate and neurotransmitters of ASIC currents in isolated Purkinje cells.
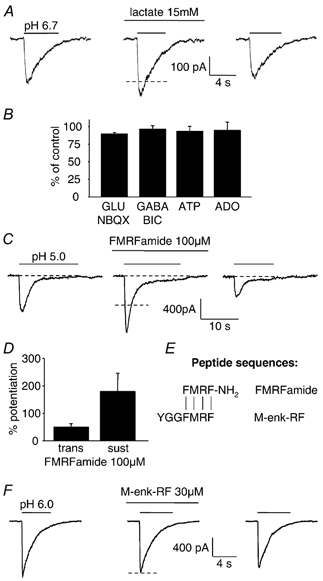
A, response of an isolated cell to pH 6.7 solution in control conditions, in 15 mm lactate, and after washing out the lactate. B, mean peak ASIC current (evoked by pH 6.0 solution) in the presence of 100 μM glutamate (GLU) + 10 μM NBQX (n = 5 cells), 100 μM GABA + 40 μM bicuculline (BIC, n = 5), 100 μM ATP (n = 5), and 100 μM adenosine (ADO, n = 3). C, response of a Purkinje cell to pH 5.0 solution in control conditions, in the presence of 100 μM FMRFamide, and after washout of the neuropeptide. D, mean potentiation by FMRFamide of the peak and sustained components of the ASIC current evoked by pH 5.0 solution, in 5 and 3 cells respectively. E, alignment of sequences of FMRFamide and met-enkephalin-arginine-phenylalanine (M-enk-RF); vertical lines show identical amino acids. F, M-enk-RF (30 μM) did not potentiate the ASIC current evoked by pH 6.0 solution. Cells were clamped at −66 mV with internal solution containing Cs+. Dashed lines show mean amplitude of control responses bracketing responses in lactate, FMRFamide and M-enk-RF.
Acid solution evoked a similar current in Purkinje cells in cerebellar slices (Fig. 1_C_, 6 cells) showing that the pH-activated current was not an artefact produced by cell isolation. In the slice, the activation kinetics of the current were slower and its amplitude was smaller than in isolated cells, presumably because the pH of the solution just outside the cell in the slice is altered more slowly by the superfusion solution, allowing more desensitization to occur during the current onset. All subsequent experiments were performed on isolated cells.
Consistent with previous work on ASIC-mediated currents, with Na+ as the main cation in the whole-cell pipette, the reversal potential of the peak acid-evoked current was at ≈0 mV, while with K+ or Cs+ as the main cation, the extrapolated reversal potential was more positive than +40 mV or +50 mV, respectively (Fig. 1_D_). This implies that Na+ is the main carrier of the peak ASIC current, but does not rule out a small Ca2+ component since, from the Goldman-Hodgkin-Katz equation, a permeability ratio of _P_Ca/_P_Na = 0.4 as reported for ASIC1a channels (Waldmann et al. 1997) would displace the reversal potential measured with Na+ as the main intracellular cation from 0 mV to only 0.35 mV. The sustained current component, when present, was too small to determine its ionic selectivity reliably.
The ASIC current was activated when the pH was lowered beyond 6.8, and larger acidifications produced a larger current, although in some cells the response to pH 5 solution was smaller than that to pH 6 (Fig. 1_E_). On average, the pH-dependence of the current could be fitted approximately by a Hill equation with a coefficient of 3 and a pH for half-maximal response of pH0.5 = 6.4 (Fig. 1_F_). This pH0.5 is consistent with the transient current being generated mainly by homomers of ASIC1a (which have pH0.5 = 6.2-6.6) or possibly heteromers of ASIC1a and ASIC2b, and rules out a major contribution from channels made from ASIC2a alone (pH0.5 = 4.1), ASIC2b alone (which does not form functional channels), ASIC1a and ASIC2a (pH0.5 = 4.8), or ASIC2a and ASIC2b (pH0.5 = 3.5) (Bassillana et al. 1997; Lingueglia et al. 1997; Waldmann et al. 1997; Escoubas et al. 2000).
Potentiation by cell swelling
During brain ischaemia the extracellular pH becomes acid (due to a switch to anaerobic respiration) at the same time as a rise in extracellular [K+] (produced by Na+-K+ pump inhibition) leads to cell swelling (Hansen, 1985; Walz et al. 1993). Since structural relatives of ASIC channels can sense membrane stretch (see Introduction), we investigated whether the Purkinje cell ASIC current showed one of the signatures of mechanosensitive channels, a sensitivity to gadolinium (Hamill & McBride, 1996), or was modulated by membrane stretch.
Gadolinium (100 μM) completely abolished the ASIC current in all three cells tested (Fig. 2_A_), while 1 μM gadolinium gave a 51 ± 8 % block in two cells. In contrast, 100 μM gadolinium produced only a 65 %, 40 % or 20 % block of channels formed by expression of ASIC2a + ASIC3, ASIC3 or ASIC2a subunits, respectively, in oocytes (Babinski et al. 2000). The complete block by 100 μM gadolinium of the Purkinje cell ASICs presumably reflects the presence of ASIC1a and/or ASIC2b (see above). External solution that was made 25 % hypotonic (at constant sodium concentration, see Methods) had no effect on the resting Purkinje cell membrane current (15 cells, data not shown), but potentiated the ASIC response to pH 6 solution by 20 ± 2 % (P = 2 × 10−7; Fig. 2_B_). Thus, the Purkinje cell ASIC current is potentiated by cell swelling.
Figure 2. Modulation of ASIC currents by gadolinium and cell swelling.
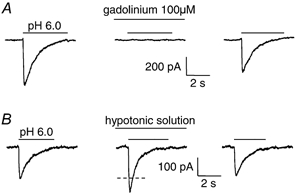
A, response of an isolated Purkinje cell to pH 6.0 solution in control conditions, after 1 min in the presence of 100 μM gadolinium, and 1 min after washout of the 2 min exposure to gadolinium. Solutions were phosphate-free (see Methods). B, response of an isolated Purkinje cell to pH 6.0 solution in normal external solution, after 1 min in 25 % hypotonic solution, and again in normal solution 1 min after the end of a 2 min exposure to hypotonic solution. Dashed line shows mean amplitude of control responses before and after hypotonic solution. Cells were clamped at −66 mV, with internal solution containing Cs+.
Potentiation by arachidonic acid
The rise of intracellular calcium concentration which occurs during brain ischaemia leads to an activation of phospholipase A2 and a massive release of arachidonic acid (Rehncrona et al. 1982). Arachidonic acid is known to have effects on channels similar to those produced by membrane stretch, for example potentiating the opening of NMDA receptor channels (Miller et al. 1992; Paoletti & Ascher, 1994; Casado & Ascher, 1998). Because it has a so-called ‘inverted cone’ shape, with a head group narrower than its lipid tail, insertion of arachidonic acid into the membrane is expected to mimic membrane stretch at the outer interface of the lipid phase with the extracellular water (Casado & Ascher, 1998). In agreement with this idea and with the potentiation of ASIC currents by swelling described above, application of arachidonic acid consistently potentiated the peak ASIC response (Fig. 3_A_; P = 3.4 × 10−7 for 10 μM arachidonic acid). The potentiation took ≈1 min to occur, and was roughly proportional to the concentration of arachidonic acid (Fig. 3_B_) as reported for the effect of arachidonic acid on mechanosensitive TREK-2 K+ channels (Bang et al. 2000), implying that the potentiation mechanism was not saturated by the doses of arachidonic acid we used. The fractional potentiation by arachidonic acid was not significantly different for activation by solutions with pH between 6.7 and 6.0 (Fig. 3_C_), implying that arachidonic acid did not alter the pH-dependence of activation of the ASIC current. Blocking the breakdown of arachidonic acid, by including enzyme blockers in the whole-cell pipette (see Methods), did not affect the potentiation by arachidonic acid. With the blockers present, 5 μM arachidonic acid potentiated the ASIC current evoked by pH 6 solution by 45 ± 10 % in three cells, which is not significantly different (P = 0.7) to the 41 ± 5 % potentiation in the absence of blockers shown in Fig. 3_B_. Thus, the potentiation is produced directly by arachidonic acid and not by its derivatives.
Figure 3. Modulation of ASIC currents in isolated Purkinje cells by arachidonic acid.
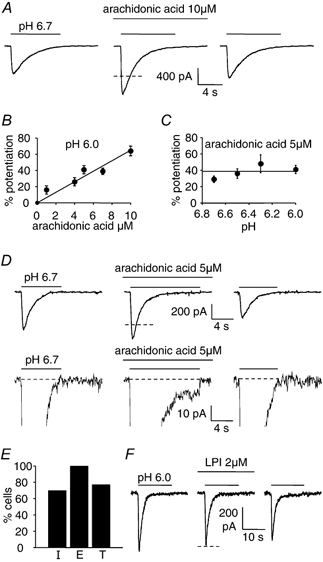
A, response of a Purkinje cell to pH 6.7 solution in the absence of arachidonic acid, in the presence of 10 μM arachidonic acid, and after washout of the arachidonic acid. B, potentiation of the peak current response to pH 6.0 solution as a function of arachidonic acid concentration (data from 6, 4, 4, 5 and 20 cells for 1, 4, 5, 7 and 10 μM arachidonic acid). C, potentiation by 5 μM arachidonic acid of the peak response to solutions of different pH (5, 3, 2 and 4 cells for pH 6.7, 6.5, 6.3 and 6.0). D, induction by 5 μM arachidonic acid of a sustained component to the ASIC response to pH 6.7 solution. Top traces show entire responses; bottom traces are at higher amplification to show the sustained component. E, percentage of 44 cells lacking a sustained component in control solution in which arachidonic acid induced (I) a sustained component, percentage of 13 cells showing a sustained component in control solution which arachidonic acid enhanced (E), and percentage of all 57 cells in which arachidonic acid induced or enhanced a sustained component (total, T). F, lysophosphatidylinositol (LPI, 2 μM) does not alter the Purkinje cell response to pH 6.0 solution (data typical of 7 cells). Cells were clamped at −66 mV, with internal solution containing Cs+. Dashed lines show mean amplitude of control responses bracketing responses in arachidonic acid and LPI.
In addition to potentiating the peak of the transient ASIC current, arachidonic acid either reversibly enhanced an existing sustained current component produced by acid solution, or reversibly induced a sustained component where one did not occur in control conditions (Fig. 3_D_). Of 44 cells showing no sustained component in control solution, arachidonic acid (1-10 μM) induced a sustained component in 31 (70 %), while in 13 cells showing a sustained component in control solution, arachidonic acid enhanced it in all 13 (100 %; Fig. 3_E_). In total, 77 % of cells had a sustained component induced or enhanced by arachidonic acid (Fig. 3_E_), and the fraction of cells showing a sustained component was thus increased from 23 % in control solution (13/57, not significantly different (P = 0.27) from the 72/230 quoted above) to 77 % with arachidonic acid present. For application of pH 6 solution, 1, 4, 5, 7 and 10 μM arachidonic acid produced an additional sustained current of 28.5 ± 7.8 pA (P = 0.035) in 4 of 5 cells (one cell showed no extra sustained current), 22.0 ± 2.5 pA (P = 0.003, in 4 of 4 cells), 27.6 ± 4.9 pA (P = 0.005, in 5 of 5 cells), 38.3 ± 3.9 pA (P = 0.0002, in 6 of 6 cells), and 34 pA (in 1 of 5 cells), respectively.
In the continuous presence of 10 μM arachidonic acid, 25 % hypotonic solution increased the peak ASIC current evoked by pH 6 solution by 17 ± 7 % in 6 cells, which is not significantly different (P = 0.58) from the 20 ± 2 % potentiation produced by hypotonic solution in the absence of arachidonic acid (Fig. 2_B_). The lack of occlusion by arachidonic acid of the potentiation produced by cell swelling is consistent with the potentiation induced by arachidonic acid being far from saturated (Fig. 3_B_), or with arachidonic acid and swelling acting via different mechanisms.
In contrast to arachidonic acid, the head group of lysophosphatidylinositol (LPI) is broader than its lipid tail, and when it inserts into the membrane it is expected to mimic membrane compression at the outer membrane- water interface (Casado & Ascher, 1998). However, whereas for NMDA receptor channels LPI had the opposite effect to arachidonic acid and membrane stretch, decreasing channel opening (Casado & Ascher, 1998), we found no effect of LPI (2 μM) on ASIC responses to pH 6 solution (Fig. 3_F_; in 7 cells the peak current was 93 ± 6 % of the control value, P = 0.26).
Potentiation by lactate
During brain ischaemia, the switch to anaerobic respiration leads to the production of lactate, which can rise from a resting value of 1–2 mm to reach a concentration of 12–20 mm in the extracellular space (Schurr & Rigor, 1997). As for cardiac ASICs (Immke & McCleskey, 2001), 15 mm lactate potentiated the current through Purkinje cell ASICs activated by pH 6.7 solution by 25 ± 2 % (P = 0.0002) in 6 cells (Fig. 4_A_). Analysis of the mechanism of this effect in cardiac ASICs has shown that lactate chelates Ca2+ and thereby increases the sensitivity to protons (Immke & McCleskey, 2001).
Modulation by neurotransmitters
The depolarization which occurs in ischaemia as a result of Na+-K+ pump inhibition leads to a rise in the extracellular concentration of most neurotransmitters (Phillis et al. 1991). We investigated whether the Purkinje cell ASIC current was modulated by the fast transmitters glutamate (100 μM, in the presence of 10 μM 2,3-dihydroxy-6-nitro-7-sulphamoyl-benzo(_f)_quinoxaline (NBQX) to block AMPA receptor currents, the pH-dependence of which could otherwise confound the results, 5 cells), GABA (100 μM, in the presence of 40 μM bicuculline to block pH-dependent GABAA receptor currents, 5 cells) and ATP (100 μM, 5 cells), or by adenosine (100 μM, 3 cells) which is also released in brain ischaemia (Phillis et al. 1991). None of these agents altered by more than 10 % the amplitude of the current evoked by a switch to pH 6 solution (Fig. 4_B_).
As reported (Askwith et al. 2000) for heterologously expressed and dorsal root ganglion ASIC currents, both the initial peak, and the sustained component of the pH-gated current were potentiated by the neuropeptide FMRFamide (100 μM, Fig. 4_C_). In five cells the peak current evoked by pH 5 solution was potentiated by 51 ± 13 % (P = 0.007), while in three cells showing a sustained response to pH 5 solution the sustained current was potentiated by 181 ± 67 % (P = 0.05; Fig. 4_D_). However the structurally-related endogenous opioid peptide met-enkephalin-arginine-phenylalanine, which lacks an amide group (Fig. 4_E_), did not affect the ASIC current (reduced by 3 ± 3 % in four cells, P = 0.34; Fig. 4_F_) suggesting that the amide group is obligatory for this potentiation. Although FMRFamide is not found in mammalian cerebellum, other RFamide peptides are, making them candidates for endogenous modulators of these channels (see Discussion).
Discussion
Cerebellar Purkinje neurons exhibit a transient inward cation current in response to a solution of acid pH (Fig. 1). The amiloride sensitivity of this current indicates that it is generated by ASIC channels (Waldmann et al. 1997). Purkinje cells express ASIC subunits 1a, 2a and 2b, but the dependence of their ASIC currents on pH suggests that the properties of their ASICs are dominated by the presence of ASIC1a subunits, possibly in combination with ASIC2b. Notably, we have shown that Purkinje cell ASIC currents are not just controlled by the extracellular pH, but are modulated by a number of factors which are produced during brain ischaemia.
Membrane stretch is produced when the extracellular fluid becomes hypotonic, or during ischaemia when the extracellular [K+] rises to 60 mm a few minutes after the start of a stroke (Hansen, 1985; Walz et al. 1993). Membrane stretch potentiated ASIC currents (Fig. 2), as did arachidonic acid (Fig. 3), which is released when the intracellular calcium concentration rises during brain ischaemia (Rehncrona et al. 1982). Arachidonic acid has been shown to mimic the effect of membrane stretch on NMDA receptor channels by distorting the membrane lipid bilayer (Casado & Ascher, 1998), and so may potentiate ASIC currents by the same mechanism as membrane stretch, but we cannot rule out an alternative mechanism of action. We found that 5–10 μM arachidonic acid produced a larger potentiation of the ASIC current than did 25 % hypotonicity. However, water movement to the whole-cell pipette will have reduced the membrane stretching effect of hypotonicity in our experiments, so membrane stretch may have a much larger effect in vivo. Arachidonic acid also induced or potentiated a sustained component to the response to acid solution (Fig. 3_D_ and E), which will lead to a sustained cation influx in conditions of ischaemia. This current is small in our experiments on cells lacking most of their dendritic tree, but since ASICs are expressed in the dendrites of Purkinje cells (Duggan et al. 2002) the sustained current may be much larger in intact cells.
Our data are consistent with a mechanosensory aspect to ASIC channel function. Membrane stretch or arachidonic acid alone did not activate the Purkinje cell ASICs, but touch sensation is affected by knocking out ASIC2 or ASIC3 (Price et al. 2000, 2001). This suggests that ASICs which have a different subunit composition to those in Purkinje cells, or are linked to other interacting proteins, may show direct activation by membrane deformation. Interestingly, in C. elegans, the mechanosensitivity of channels composed of MEC-4 or MEC-10 subunits is conferred by interaction with MEC-2 (Goodman et al. 2002), which is homologous to stomatin - a scaffolding protein expressed strongly in Purkinje cells (Mannsfeldt et al. 1999). Thus, the swelling sensitivity of the Purkinje cell ASICs may be mediated by an interaction with stomatin.
Lactate also potentiated Purkinje cell ASIC currents (Fig. 4_A_), probably as a result of lactate chelating extracellular Ca2+ (Immke & McCleskey, 2001). This makes lactate a potential modulator of ASIC channels during ischaemia when the extracellular lactate concentration can rise from 1–2 mm to 12–20 mm as a result of the switch to anaerobic respiration (Schurr & Rigor, 1997).
Of the neurotransmitters which we tested, only FMRFamide potentiated Purkinje cell ASIC currents (Fig. 4_C_ and D), as reported previously (Askwith et al. 2000) for currents mediated by ASIC1 and ASIC3 and for ASIC currents in dorsal root ganglion cells. The endogenous opioid peptide met-enkephalin-arginine-phenylalanine, which contains a C-terminal FMRF sequence but lacks the amide group, did not potentiate the Purkinje cell ASIC current, suggesting that the peptide binding site on ASICs recognizes in part the amide group. FMRFamide is not found in the vertebrate CNS, and its vertebrate analogue neuropeptide FF (which also potentiates ASIC currents (Askwith et al. 2000)) is only found at low levels in the cerebellum (Majane et al. 1989), but other RFamides have recently been discovered which are present in the cerebellum (Hinuma et al. 2000; Ukena & Tsutsui, 2001) and which may therefore be endogenous modulators of the Purkinje cell ASIC currents.
Activation of the Purkinje cell ASIC channels by a fall of pH in vivo may occur during synaptic transmission, as a result of the acid pH in presynaptic vesicles (about 5.5). When the vesicular contents are released during synaptic transmission a rapid acidification of the extracellular space may occur, and activation of ASIC channels (which are located in clusters in Purkinje cell dendrites (Duggan et al. 2002)) could contribute to postsynaptic currents. A rapid (though small) acidification during exocytosis has been detected both directly with a pH-sensitive dye (Krishtal et al. 1987) and indirectly from a modulatory effect on presynaptic calcium channels (DeVries, 2001). However, the idea that postsynaptic ASIC currents contribute significantly to excitatory or inhibitory synaptic transmission is apparently ruled out by the fact that glutamate or GABA receptor blockers, which we found to have little effect on the Purkinje cell ASIC current (Fig. 4_B_ and Fig. 1_C_), can essentially abolish postsynaptic currents in Purkinje cells (NBQX reduces AMPA receptor-mediated EPSCs by 97 % (Tempia et al. 1998); bicuculline reduces GABAA receptor mediated IPSCs by 96 % (Konnerth et al. 1990)).
During ischaemia, although the pH falls by up to one unit as a result of metabolism becoming anaerobic (Kraig et al. 1983; Silver & Erecinska, 1992), it takes several minutes to do so, which is too slow to evoke the large transient component of ASIC currents - channel desensitization occurring as the pH falls prevents a large current developing. Nevertheless some ASIC subunit combinations show a sustained current in low pH solution, and in Purkinje cells this was seen in ≈30 % of cells. Of the modulators that we tested, both arachidonic acid and FMRFamide significantly enhanced the sustained current component, and arachidonic acid increased to 77 % the fraction of cells exhibiting a sustained component. Thus, when arachidonic acid is released in ischaemia, at a time when the extracellular space has become acid, it will co-activate (with the pH change) a tonic inward cation current in most Purkinje cells. This may contribute to neuronal death in two ways. First, Na+ entry will tend to be followed by Cl− entry and an osmotic entry of water, which will produce cell swelling and possibly membrane rupture. Second, since ASIC1a subunits (possibly in combination with ASIC2b) appear to dominate the Purkinje cell ASIC current and ASIC1a homomeric channels are calcium-permeable (Waldmann et al. 1997), there may be an increased Ca2+ entry which can cause cell death by activating Ca2+-dependent proteases, lipases and nucleases. Testing the involvement of ASICs in ischaemic neuronal death is hindered at present by the fact that neither amiloride (Fig. 1_B_) nor toxin blockers of the channels block the sustained current component (Benson et al. 1999; Escoubas et al. 2000).
In summary, we have demonstrated that the activity of Purkinje cell ASIC channels reflects the integration of several signals which these channels sense: pH, membrane stretch, arachidonic acid, lactate, and peptide transmitters. Thus, the magnitude of ASIC currents and their contribution to neuronal depolarization or degeneration will reflect not just the pH of the extracellular solution but also the levels of the other signals sensed by these channels.
Acknowledgments
We thank Céline Auger, Liam Drew, Alasdair Gibb, Martine Hamann, Paikan Marcaggi, Paola Pedarzani, David Rossi and Angus Silver for comments on the manuscript. Supported by the Wellcome Trust and a Wolfson-Royal Society award to D.A. Nicola Allen is in the four year PhD Programme in Neuroscience at University College London.
References
- Akaike N, Ueno S. Proton-induced current in neuronal cells. Progress in Neurobiology. 1994;43:73–83. doi: 10.1016/0301-0082(94)90016-7. [DOI] [PubMed] [Google Scholar]
- Askwith CC, Cheng C, Ikuma M, Benson C, Price MP, Welsh MJ. Neuropeptide FF and FMRFamide potentiate acid-evoked currents from sensory neurons and proton-gated channels. Neuron. 2000;26:133–141. doi: 10.1016/s0896-6273(00)81144-7. [DOI] [PubMed] [Google Scholar]
- Babinski K, Catarsi S, Biagini G, Seguela P. Mammalian ASIC2a and ASIC3 subunits co-assemble into heteromeric proton-gated channels sensitive to Gd3+ Journal of Biological Chemistry. 2000;275:28519–28525. doi: 10.1074/jbc.M004114200. [DOI] [PubMed] [Google Scholar]
- Bang H, Kim Y, Kim D. TREK-2, a new member of the mechanosensitive tandem-pore K+ channel family. Journal of Biological Chemistry. 2000;275:17412–17419. doi: 10.1074/jbc.M000445200. [DOI] [PubMed] [Google Scholar]
- Bassillana F, Champigny G, Waldmann R, De Weille JR, Heurteaux C, Lazdunski M. The acid-sensitive ionic channel subunit ASIC and the mammalian degenerin MDEG form a heteromultimeric H+-gated Na+ channel with novel properties. Journal of Biological Chemistry. 1997;272:28819–28822. doi: 10.1074/jbc.272.46.28819. [DOI] [PubMed] [Google Scholar]
- Benson CJ, Eckert SP, McCleskey EW. Acid-evoked currents in cardiac sensory neurons. Circulation Research. 1999;84:921–928. doi: 10.1161/01.res.84.8.921. [DOI] [PubMed] [Google Scholar]
- Bevan S. Proton-gated ion channels in neurons. In: Kaila K, Ransom BR, editors. pH and Brain Function. New York: Wiley-Liss; 1998. pp. 447–475. [Google Scholar]
- Billups B, Rossi D, Oshima T, Warr O, Takahashi M, Sarantis M, Szatkowski M, Attwell D. Physiological and pathological operation of glutamate transporters. Progress in Brain Research. 1998;116:45–58. doi: 10.1016/s0079-6123(08)60429-x. [DOI] [PubMed] [Google Scholar]
- Brew H, Attwell D. Electrogenic glutamate uptake is a major current carrier in the membrane of axolotl retinal glial cells. Nature. 1987;327:707–709. doi: 10.1038/327707a0. [DOI] [PubMed] [Google Scholar]
- Caddy KW, Biscoe TJ. Structural and quantitative studies on the normal C3H and Lurcher mutant mouse. Philosophical Transactions of the Royal Society B. 1979;287:167–201. doi: 10.1098/rstb.1979.0055. [DOI] [PubMed] [Google Scholar]
- Caldwell RA, Clemo HF, Baumgarten CM. Using gadolinium to identify stretch-activated channels: technical considerations. American Journal of Physiology. 1998;275:C619–621. doi: 10.1152/ajpcell.1998.275.2.C619. [DOI] [PubMed] [Google Scholar]
- Casado M, Ascher P. Opposite modulation of NMDA receptors by lysophospholipids and arachidonic acid: common features with mechanosensitivity. Journal of Physiology. 1998;513:317–330. doi: 10.1111/j.1469-7793.1998.317bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervos-Navarro J, Diemer NH. Selective vulnerability in brain hypoxia. Critical Reviews in Neurobiology. 1991;6:149–182. [PubMed] [Google Scholar]
- Devries SH. Exocytosed protons feedback to suppress the Ca2+ current in mammalian cone photoreceptors. Neuron. 2001;32:1107–1117. doi: 10.1016/s0896-6273(01)00535-9. [DOI] [PubMed] [Google Scholar]
- Driscoll M, Chalfie M. The Mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature. 1991;349:588–593. doi: 10.1038/349588a0. [DOI] [PubMed] [Google Scholar]
- Duggan A, Garcia-Anoveros J, Corey DP. The PDZ domain protein PICK1 and the sodium channel BNaC1 interact and localize at mechanosensory terminals of DRG neurons and dendrites of central neurons. Journal of Biological Chemistry. 2002;277:5203–5208. doi: 10.1074/jbc.M104748200. [DOI] [PubMed] [Google Scholar]
- Escoubas P, De Weille JR, Lecoq A, Diochot S, Waldmann R, Champigny G, Moinier D, MeneZ A, Lazdunski M. Isolation of a tarantula toxin specific for a class of proton-gated Na+ channels. Journal of Biological Chemistry. 2000;275:25116–25121. doi: 10.1074/jbc.M003643200. [DOI] [PubMed] [Google Scholar]
- Garcia-Anoveros J, Derfler B, Neville-Golden J, Hyman BT, Corey DP. BNaC1 and BNaC2 constitute a new family of human neuronal sodium channels related to degenerins and epithelial sodium channels. Proceedings of the National Academy of Sciences of the USA. 1997;94:1459–1464. doi: 10.1073/pnas.94.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MB, Ernstrom GG, Chelur DS, O'Hagan R, Yao CA, Chalfie M. MEC-2 regulates C. elegans DEG/ENaC channels needed for mechanosensation. Nature. 2002;415:1039–1042. doi: 10.1038/4151039a. [DOI] [PubMed] [Google Scholar]
- Hamill OP, McBride DW., Jr The pharmacology of mechanogated membrane ion channels. Pharmacological Reviews. 1996;48:231–252. [PubMed] [Google Scholar]
- Hansen AJ. Effect of anoxia on ion distribution in the brain. Physiological Reviews. 1985;65:101–148. doi: 10.1152/physrev.1985.65.1.101. [DOI] [PubMed] [Google Scholar]
- Hinuma S, Shintani Y, Fukusumi S, Iijima N, Matsumoto Y, Hosoya M, Fujii R, Watanabe T, Kikuchi K, Terao Y, Yano T, Yamamoto T, Kawamata Y, Habata Y, Asada M, Kitada C, Kurokawa T, Onda H, Nishimura O, Tanaka M, Ibata Y, Fujino M. New neuropeptides containing carboxy-terminal RFamide and their receptor in mammals. Nature Cell Biology. 2000;2:703–708. doi: 10.1038/35036326. [DOI] [PubMed] [Google Scholar]
- Hockberger PE, Yousif L, Nam SC. Identification of acutely isolated cells from developing rat cerebellum. Neuroimage. 1994;1:276–287. doi: 10.1006/nimg.1994.1012. [DOI] [PubMed] [Google Scholar]
- Immke DC, McCleskey EW. Lactate enhances the acid-sensing Na+ channel on ischemia-sensing neurons. Nature Neuroscience. 2001;9:869–870. doi: 10.1038/nn0901-869. [DOI] [PubMed] [Google Scholar]
- Konnerth A, Llano I, Armstrong CM. Synaptic currents in cerebellar Purkinje cells. Proceedings of the National Academy of Sciences of the USA. 1990;87:2662–2665. doi: 10.1073/pnas.87.7.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraig RP, Ferreira-Filho CR, Nicholson C. Alkaline and acid transients in cerebellar microenvironment. Journal of Neurophysiology. 1983;49:831–850. doi: 10.1152/jn.1983.49.3.831. [DOI] [PubMed] [Google Scholar]
- Krishtal OA, Osipchuk YV, Shelest TN, Smirnoff SV. Rapid extracellular pH transients related to synaptic transmission in rat hippocampal slices. Brain Research. 1987;436:352–356. doi: 10.1016/0006-8993(87)91678-7. [DOI] [PubMed] [Google Scholar]
- Krishtal OA, Pidoplichko VI. Receptors for protons in the membrane of sensory neurons. Brain Research. 1981;214:150–154. doi: 10.1016/0006-8993(81)90446-7. [DOI] [PubMed] [Google Scholar]
- Lingueglia E, Champigny G, Lazdunski M, Barbry P. Cloning of the amiloride-sensitive FMRFamide peptide-gated sodium channel. Nature. 1995;378:730–733. doi: 10.1038/378730a0. [DOI] [PubMed] [Google Scholar]
- Lingueglia E, De Weille JR, Bassilana F, Heurteaux C, Sakai H, Waldmann R, Lazdunski M. A modulatory subunit of acid-sensing ion channels in brain and dorsal root ganglion cells. Journal of Biological Chemistry. 1997;272:29778–29783. doi: 10.1074/jbc.272.47.29778. [DOI] [PubMed] [Google Scholar]
- Majane EA, Panula P, Yang H-Y T. Rat brain regional distribution and spinal cord neuronal pathway of FLFQPQRF-NH2, a mammalian FMRF-NH2-like peptide. Brain Research. 1989;494:1–12. doi: 10.1016/0006-8993(89)90137-6. [DOI] [PubMed] [Google Scholar]
- Mannsfeldt AG, Carroll P, Stucky CL, Lewin GR. Stomatin, a MEC-2 like protein, is expressed by mammalian sensory neurons. Molecular and Cellular Neuroscience. 1999;13:391–404. doi: 10.1006/mcne.1999.0761. [DOI] [PubMed] [Google Scholar]
- Mano I, Driscoll M. DEG/ENaC channels: a touchy superfamily that watches its salt. Bioessays. 1999;21:568–578. doi: 10.1002/(SICI)1521-1878(199907)21:7<568::AID-BIES5>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Miller B, Sarantis M, Traynelis SF, Attwell D. Potentiation of NMDA receptor currents by arachidonic acid. Nature. 1992;355:722–725. doi: 10.1038/355722a0. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Ascher P. Mechanosensitivity of NMDA receptors in cultured mouse central neurons. Neuron. 1994;13:645–655. doi: 10.1016/0896-6273(94)90032-9. [DOI] [PubMed] [Google Scholar]
- Phillis JW, Walter JA, Simpson RE. Brain adenosine and transmitter amino acid release from the ischemic rat cerebral cortex: effects of the adenosine deaminase inhibitor deoxycoformin. Journal of Neurochemistry. 1991;56:644–650. doi: 10.1111/j.1471-4159.1991.tb08198.x. [DOI] [PubMed] [Google Scholar]
- Price MP, Lewin GR, McIlwrath SL, Cheng C, Xie J, Heppenstall PA, Stucky CL, Mannsfeldt AG, Brennan TJ, Drummond HA, Qiao J, Benson CJ, Tarr DE, Hrstka RF, Yang B, Williamson RA, Welsh MJ. The mammalian sodium channel BNC1 is required for normal touch sensation. Nature. 2000;407:1007–1011. doi: 10.1038/35039512. [DOI] [PubMed] [Google Scholar]
- Price MP, McIlwrath SL, Xie J, Cheng C, Qiao J, Tarr DE, Sluka KA, Brennan TJ, Lewin GR, Welsh MJ. The DRASIC cation channel contributes to the detection of cutaneous touch and acid stimuli in mice. Neuron. 2001;32:1071–1083. doi: 10.1016/s0896-6273(01)00547-5. [DOI] [PubMed] [Google Scholar]
- Rehncrona S, Westerberg E, Akesson B, Siesjo BK. Brain cortical fatty acids and phospholipids during and following complete and severe incomplete ischemia. Journal of Neurochemistry. 1982;38:84–93. doi: 10.1111/j.1471-4159.1982.tb10857.x. [DOI] [PubMed] [Google Scholar]
- Schurr A, Rigor BM. Brain anaerobic lactate production: a suicide note or a survival kit? Developmental Neuroscience. 1997;20:348–357. doi: 10.1159/000017330. [DOI] [PubMed] [Google Scholar]
- Silver IA, Erecinska M. Ion homeostasis in rat brain in vivo: intra- and extracellular [Ca2+] and [H+] in the hippocampus during recovery from short-term, transient ischemia. Journal of Cerebral Blood Flow and Metabolism. 1992;12:759–772. doi: 10.1038/jcbfm.1992.107. [DOI] [PubMed] [Google Scholar]
- Tempia F, Miniaci MC, Anchisi D, Strata P. Postsynaptic current mediated by metabotropic receptors in cerebellar Purkinje cells. Journal of Neurophysiology. 1998;80:520–528. doi: 10.1152/jn.1998.80.2.520. [DOI] [PubMed] [Google Scholar]
- Ukena K, Tsutsui K. Distribution of novel RFamide-related peptide-like immunoreactivity in the mouse central nervous system. Neuroscience Letters. 2001;300:153–156. doi: 10.1016/s0304-3940(01)01583-x. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated channel involved in acid-sensing. Nature. 1997;386:173–177. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Lazdunski M. H+-gated cation channels: neuronal acid sensors in the ENaC/DEG family of ion channels. Current Opinion in Neurobiology. 1998;8:418–424. doi: 10.1016/s0959-4388(98)80070-6. [DOI] [PubMed] [Google Scholar]
- Walz W, Klimaszewski A, Paterson IA. Glial swelling in ischemia: a hypothesis. Developmental Neuroscience. 1993;15:216–225. doi: 10.1159/000111337. [DOI] [PubMed] [Google Scholar]