Microenvironmental regulation of cancer development (original) (raw)
. Author manuscript; available in PMC: 2009 Feb 1.
Published in final edited form as: Curr Opin Genet Dev. 2008 Feb 20;18(1):27–34. doi: 10.1016/j.gde.2007.12.006
Abstract
Numerous studies have demonstrated that the tumor microenvironment not only responds to and supports carcinogenesis, but actively contributes to tumor initiation, progression, and metastasis. During tumor progression all cells composing the tumor undergo phenotypic and epigenetic changes. Paracrine signaling between epithelial and stromal cells is important for the regulation of the proliferation, invasive, angiogenic, and metastatic behavior of cancer cells. Better understanding the molecular mechanisms by which stromal cells exert these effects may open up new venues for cancer therapeutic and preventative interventions.
Introduction
The “seed and soil” hypothesis of tumor growth dates back as early as 1889 [1], but during the past decades advances in identifying aberrances in oncogenes and tumor suppressor genes within tumor epithelial cells resulted in the ignorance of the role of the microenvironment in tumorigenesis. However, a tumor is much more than clusters of transformed cells standing alone. Tumor epithelial cells can only thrive in an aberrant microenvironment composed of altered extracellular matrix (ECM) and various non-transformed cells (e.g., fibroblasts, myofibroblasts, leukocytes, and myoepithelial and endothelial cells) that play a role in the initiation and progression of neoplasms [2–5]. Cross-talk between epithelial and stromal cells is known to be essential for differentiation and development of normal organs and tissues as well as for the growth and progression of tumors [2,6,7]. Recent work has begun to address the importance of the microenvironment in supporting malignant growth and the molecular mechanisms by which stromal cells may contribute to tumorigenesis. In this review, we focus on the phenotypic, genetic, and epigenetic alterations found in cells composing the tumor microenvironment, the role of these cells in tumor progression, and the clinical implications of these findings.
The tumor microenvironment: from reactive neighborhood to active contributor
It was noted long time ago that changes in the microenvironment accompany tumor formation [8–10]. Increased fibroblast proliferation and ECM remodeling are often found adjacent to cancer cells. The tumor stroma in many aspects resembles wound healing and chronic inflammatory conditions, except that normal physiologic controls are not maintained [11]. Furthermore, even fibroblasts outside of the immediate vicinity of neoplastic lesions can demonstrate phenotypic changes. For example, skin fibroblasts from a significant portion of patients with breast cancer were described to display altered migratory behavior due to the expression of a soluble migration-stimulating factor not made by normal adult cells [12].
Large amounts of data support the hypothesis that the stroma is not just a passive bystander that simply reacts to the transformed cells. Rather, interactions between mesenchymal and epithelial cells result in reciprocal influences, and the microenvironment is an active participant throughout cancer initiation, progression, and metastasis.
First, phenotypic and genotypic abnormalities in cancer epithelial cells cannot fully delineate tumor phenotypes and clinical behavior. cDNA microarray profiling and hierarchical clustering analyses have been utilized to classify breast cancer subtypes and predict clinical outcome [13,14]. However, comprehensive gene expression and genetic profiling studies comparing in situ, invasive, and metastatic breast carcinomas have failed to identify tumor stage–specific gene signatures [15–19], implying that besides the intrinsic malignant properties of tumor epithelial cells, other factors such as microenvironmental changes may regulate progression to invasion and metastasis.
Second, normal cellular microenvironment can inhibit tumor cell proliferation and cancer formation. This was first and most dramatically demonstrated by the lack of tumor development in chick embryos infected with the Rous sarcoma virus expressing a potent oncogene [20]. Normal myoepithelial cells have been shown able to suppress the growth, invasion, and angiogenesis of multiple different breast cancer cell types [21–23], whereas the effects of normal fibroblasts are more dependent on the ratio of stromal and epithelial cells and the degree of malignancy of the tumor cells [24].
Third, stromal cells can create a permissive microenvironment for tumorigenesis. In contrast to normal myoepithelial cells, cancer-associated fibroblasts and myofibroblasts have been demonstrated to promote tumorigenesis via enhancing angiogenesis, and proliferation, survival, invasion, and metastatic spread of tumor epithelial cells [2–5]. Bone marrow-derived cells were found to colonize at sites of distant metastases prior to the arrival of the tumor cells, establishing a “pre-metastatic niche” that facilitates tumor cell growth in distant organs [25••,26–29]. Disruption of the normal microenvironment due to chronic inflammation or hereditary changes in genes regulating immune reactions and tissue remodeling may play similar “preparing the soil” roles in tumor initiation. Human epidemiologic data as well as experiments in model systems strongly support this hypothesis [30].
Fourth, the stroma may be a crucial target of carcinogens. In one study, neoplastic transformation of rat mammary epithelial cells occurred only when the mammary fat pad stroma was exposed to chemical carcinogens, regardless of whether or not the epithelial cells were treated [31]. However, similar experiments by another group failed to reproduce these results and pointed out that carcinogen treatment of the epithelial cells is required for tumor formation [32].
Finally, targeting the tumor microenvironment may be a feasible therapeutic approach for cancer treatment and prevention. In contrast to genetically unstable tumor cells, the genetically stable cells composing the microenvironment are less plastic and less likely to acquire drug resistance, and therefore, potentially make better targets for cancer therapy. In line with this, anti-angiogenic drugs have been developed to target tumor endothelial cells as a means of cancer treatment [33]. Elimination of carcinoma-associated macrophages in murine models of metastatic breast, colon, and non-small cell lung cancers decreased tumor angiogenesis, growth, and metastasis [34]. Similarly, targeting tumor-associated fibroblasts using a vaccine-based approach in multi-drug resistant murine colon and breast carcinomas suppressed primary tumor growth and metastasis, decreased the expression of collagen type I, and increased the uptake of chemotherapeutic drugs [35]. The results of these studies demonstrate the feasibility and success of combined targeting of cancer cells and their microenvironment using various immuno- and chemotherapeutic approaches.
Phenotypic and molecular alterations in the tumor microenvironment
To define the molecular basis underlying microenvironmental changes in tumorigenesis, Allinen et al. characterized the comprehensive gene expression profiles of several major cell types from normal human breast tissue, ductal carcinoma in situ (DCIS), and invasive breast carcinomas, using SAGE (Serial Analysis of Gene Expression) [36]. The results of this study demonstrated that dramatic gene expression changes occur in all cell types, including tumor epithelial, endothelial, and myoepithelial cells, myofibroblasts, fibroblasts, and leukocytes, during breast tumor progression. Similarly, gene expression [37] and enzyme activity [38] profiles of human breast cancer cell lines grown in three different ways: (1) in vitro cultures, (2) primary tumors in the mammary fat pad, and (3) distant metastases in different organs, showed changes in both epithelial cells and neighboring host stroma, suggesting reciprocal interactions during tumor formation and metastasis.
Despite the dramatic and universal changes of gene expression in all cell types during tumor progression, clonally selected genetic alterations were restricted in tumor epithelial cells and could not be found in any of the stromal cells analyzed by aCGH (array comparative genomic hybridization) and SNP (Single Nucleotide Polymorphism) arrays [36]. Controversy arises when data from other groups suggested that genetic aberrances also happen in breast tumor stroma, including gene copy number changes, LOH (loss of heterozygosity), microsatellite instability (MSI) and point mutations in tumor suppressor genes and oncogenes [39–45]. Because the analysis of small numbers of cells from archived tissue samples using PCR based methods is prone to yield erroneous results, determining if these findings are reproducible using fresh tissue samples and alternative approaches would be imperative. Indeed, recent results obtained using fresh or frozen tissue samples, and primary cultured cells indicate that LOH and copy number alterations are extremely rare in breast and ovarian carcinoma-associated fibroblasts (Qiu et al., unpublished data).
The presence of gene expression alterations, but lack of genetic abnormalities point to epigenetic changes including DNA methylation and chromatin modification being responsible for the relative stability of the abnormal phenotypes of cancer-associated stromal cells. To explore this possibility, Hu et al. characterized the comprehensive DNA methylation profiles of epithelial and myoepithelial cells, and fibroblasts isolated from normal and neoplastic breast tissues using MSDK (Methylation-Specific Digital Karyotyping) [46••]. DNA methylation changes were detected in all three cell types in DCIS and invasive tumors, supporting the hypothesis that the phenotypic changes observed in tumor stromal cells are at least in part due to epigenetic modifications. Studies in HER2+ breast cancer [47] and prostate tumors [48] also demonstrated distinct methylation patterns of selected genes in tumor epithelial and surrounding stromal cells. A significant fraction of genes aberrantly methylated in cancer cells (both epithelium and others) encode transcription factors with known function in development and differentiation [46]. It is proposed that tumor-associated myofibroblasts and fibroblasts develop from bone marrow-derived mesenchymal stem cells recruited to the stroma of developing tumors [49]. Thus, their abnormal phenotypes and epigenetic profiles may reflect their abnormal differentiation due to factors present in tumors.
Signaling networks between the microenvironment and tumor epithelial cells
Stromal cells influence epithelial cell behavior by secreting various ECM proteins, chemokines, cytokines, growth factors, proteases, and protease inhibitors. A large proportion of genes differentially expressed in epithelial and stromal cells during breast tumor progression encode for secreted proteins and cell surface receptors [36]. An extensive network of cross-talks between cancer cells and the host was identified including regulations of cell-ECM interaction and growth factor signaling using a lung adenocarcinoma xenograft model [50]. Cell adhesion is an important regulator of neoplastic behavior, particularly that of invasion and metastasis [51,52]. Thus, the modulation of the expression of genes involved in cell adhesion may be another mechanism by which stromal cells regulate tumor cell invasion and metastasis.
Co-injection of lethally irradiated fibroblasts with human cancer cells increased the tumorigenic potential of prostate, bladder, and breast cancer cell lines, and cells derived from the ascites fluids of patients with metastatic renal or prostate cancers [53], supporting a role for paracrine signaling between fibroblasts and tumor epithelial cells. Injection of fibroblast-conditioned medium at the inoculation site of tumor cells enhanced tumor growth, suggesting the involvement of soluble factors secreted by fibroblasts [54]. Inhibiting TGF-β signaling in fibroblasts by specifically deleting the TGF-β type II receptor in these cells, enhances the growth and oncogenic potential of adjacent epithelia via activation of TGF-α, MSP (macrophage-stimulating protein), and HGF (hepatocyte growth factor) pathways [55,56••,57]. Similarly, the CXCL14 and CXCL12 (CXC motif chemokine ligand 14 and 12) chemokines, overexpressed in DCIS myoepithelial cells and myofibroblasts, respectively, promote tumor cell proliferation, migration, invasion, angiogenesis, and metastasis [36,58,59].
Clinical observations have indicated that tumor cells metastasize to distant organs in a non-random manner, although they scatter ubiquitously [60]. To investigate the molecular mechanisms underlying homing and preferential tumor cell growth at certain sites, the gene expression profiles of variants of the MDA-MB-231 breast cancer cell line with elevated metastatic activity to particular organs such as bone or lung were characterized [61,62•,63]. Distinct groups of secreted and membrane proteins are found to be associated with organ preference for metastatic colonization and growth, suggesting the involvement of tumor-host interactions in these processes in part mediated by chemokines/cytokines and their receptors. In a separate study, co-injection of bone marrow-derived mesenchymal stem cells enhanced the metastatic potency of MDA-MB-231 human breast carcinoma cells growing at subcutaneous sites via upregulating CCL5-CCR5 signaling [64•]. Another study on the other hand proposed that bone marrow-derived stem cells are mobilized due to tumor growth and arrive to sites of future distant metastasis prior to tumor epithelial cells [25–29]. Thus, the local microenvironment created by these circulating stem cells prepares a “niche” for metastases. Therefore, paracrine signaling between tumor epithelial cells and the host/organ microenvironment appears to have major influence on tumor progression to metastatic disease (Fig. 1). Overall, cells composing the microenvironment can regulate epithelial cell behavior multiple different ways including direct and indirect mechanisms.
Figure 1. The dual role of bone marrow derived mesenchymal stem cells (BMD-MSCs) in cancer metastasis.
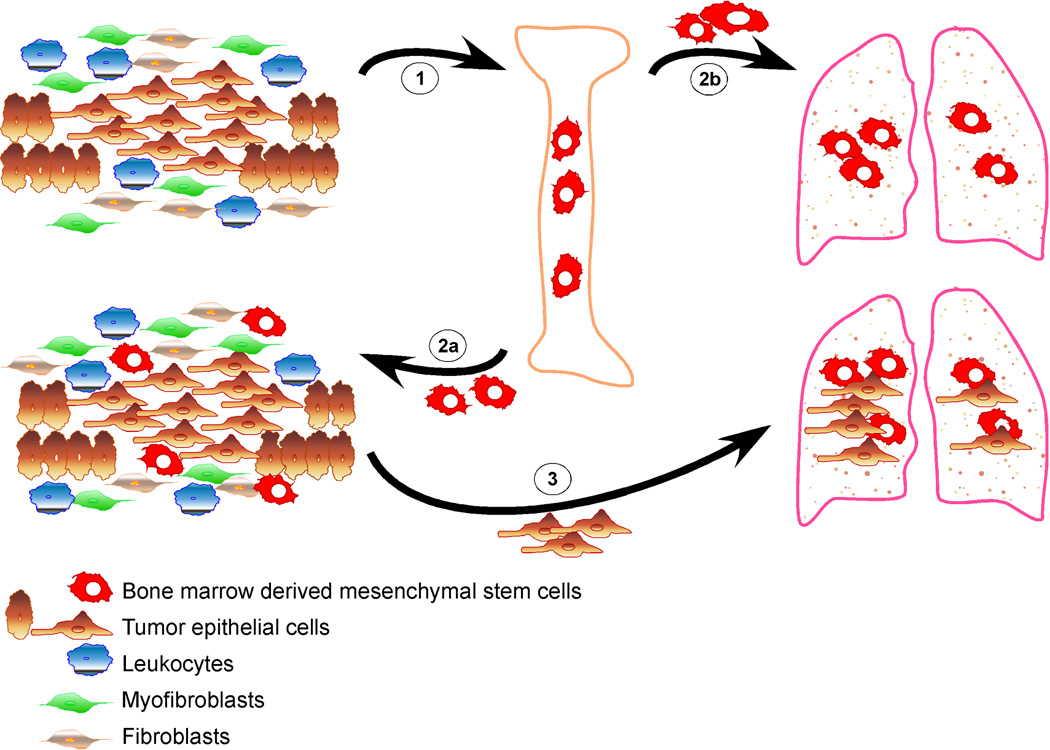
1. Primary tumor cells send signals to the bone marrow mobilizing BMD-MSCs. 2. BMD-MSCs are recruited to the primary tumor (2a), and also mobilized to other organs such as lungs (2b), creating a pre-metastatic niche prior to the dissemination of the cancer cells. 3. The interaction between cancer cells and BMD-MSCs within the primary tumor enhances the motility, invasive and metastatic capacity of tumor cells via paracrine interactions (e.g., CCL5-CCR5). In addition, BMD-MSCs can differentiate into myofibroblasts and other stromal cell types that further support the growth and progression of the tumor. Furthermore, disseminated cancer cells preferentially grow at sites where BMD-MSCs are localized forming distant metastases.
Role of stromal cells in the _in situ_-to-invasive carcinoma progression
The transition of in situ to invasive carcinoma is a key event in breast tumor progression that is poorly understood. Phenotypic, genetic and epigenetic changes have been detected in tumor epithelial cells during this transition step yet stage-specific molecular signatures could not been identified [16–19,36,46]. Meanwhile, alterations in gene expression and DNA methylation occur in the non-transformed cells of the tumor microenvironment as well [36,46], implying that tumor-stromal communication may play a role in this progression step. Two models of the _in situ_-to-invasive carcinoma transition have been proposed, focusing on the “seed” or the “soil”, respectively [23]. The “escape” model hypothesizes that genetic changes and clonal selection in combination will give rise to a subpopulation of tumor epithelial cells with an ability to invade out of the duct and into the surrounding tissue. The “release” model proposes that the abnormal microenvironment such as phenotypic changes of myoepithelial cells, infiltration of leukocytes, and accumulation of myofibroblasts, will lead to the disruption of the basement membrane (BM) and let the tumor epithelial cells freely spread in the stroma. Because changes in the tumor epithelial cells are likely to induce microenvironmental changes, the combination of the two models (changes in both the epithelial and stromal cell compartments) is what really explains progression to invasion. Multiple recent publications support the “release” hypothesis. Capillary blood vessels may invade and breach the BM in DCIS, creating an escape route for the cancer cells [65]. Focal breakdown of myoepithelial cell layer and BM at sites of white blood cell infiltration have also been observed in DCIS [66]. Emphasizing the necessity of changes in both in “seed” and “soil” for progression, epithelial cell clusters overlying the disrupted myoepithelial layers were different from adjacent cells within the same duct with respect of ER (estrogen receptor) status, frequency or pattern of LOH and/or MSI, and expression of tumor progression related genes, normal stem cell and proliferation markers, and showed invasion into the stroma and blood vessels-like structures [67–69]. Since tumor-stromal interactions are bi-directional, identification of the initiating events requires further study.
Nevertheless, myoepithelial cells are essential regulators of DCIS-to-invasive carcinoma transition because they produce the BM and form a physical barrier around tumor epithelial cells. Normal myoepithelial cells have been recognized as “natural tumor suppressors” and function as gatekeepers of tumor progression [21–23]. DCIS-associated myoepithelial cells demonstrate altered gene expression or DNA methylation profiles including loss of differentiation markers and augmented levels of pro-angiogenic and invasive genes [36,46]. Tumor myoepithelial cells were also unable to influence the polarity of breast epithelial cells in 3-dimensional collagen cultures due to their lack of production of BM constituent laminin 1 [70]. Myoepithelial cells of high grade DCIS exhibit strong expression of PAI-1 (plasminogen activator inhibitor type-1), which may resolve the interaction between uPAR (urokinase plasminogen activator receptor) present on the myoepithelium and vitronectin in the BM, and result in the detachment of the myoepithelial cells from the BM followed by tumor cell infiltration [71]. More importantly, tumor-associated myoepithelial cells express higher levels of several BM-degrading enzymes including MMPs, compared to their normal counterparts [36]. Therefore, contrary to the role of normal myoepithelial cells in BM synthesis and maintenance, progressive changes in the DCIS myoepithelial cells may lead to gradual degradation of BM. One the other hand, the basement membrane induces and maintains myoepithelial cell differentiation [72], therefore forming a positive feedback loop. Similarly, high levels of MMPs in myofibroblasts and fibroblasts can also contribute to ECM remodeling and indirectly regulate myoepithelial cell differentiation. Thus, molecular and cellular components of the microenvironment may shift the balance between BM synthesis and degradation, and control the _in situ_-to-invasive carcinoma transition (Fig. 2).
Figure 2. Events involved in the in situ to invasive breast carcinoma transition.
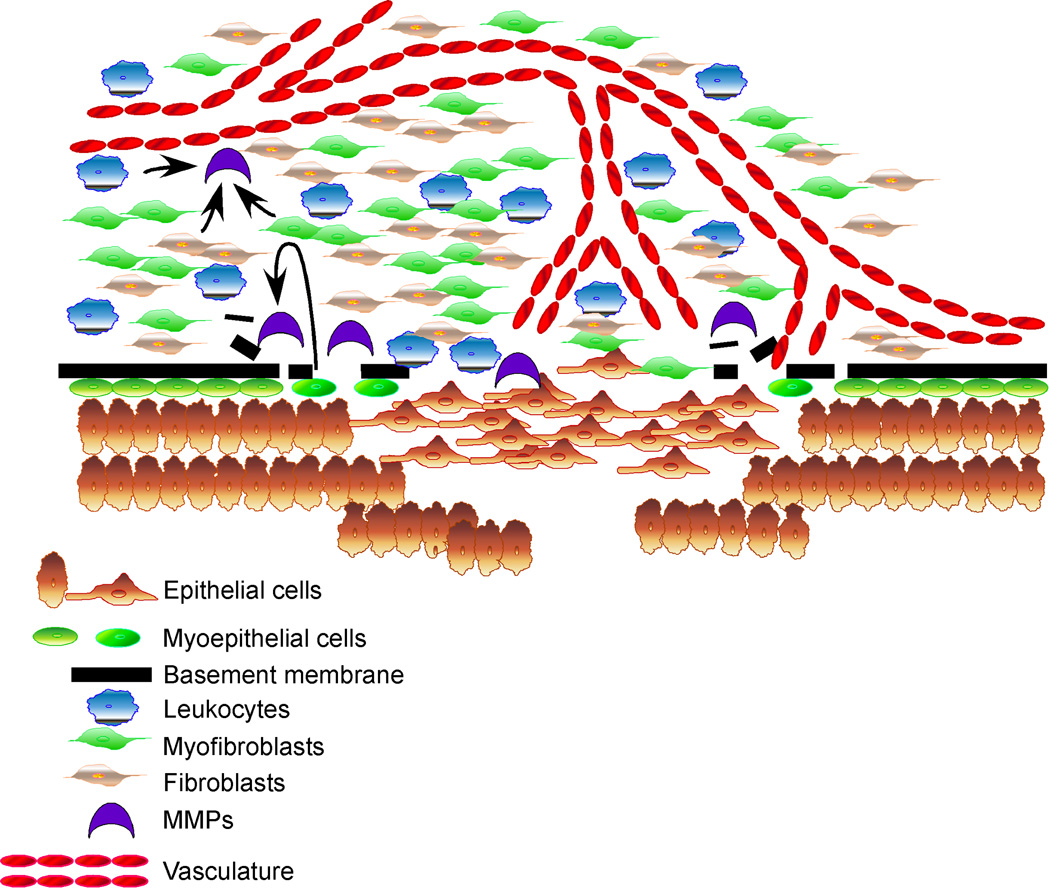
Progression to invasion is marked by the disappearance of myoepithelial layer, disruption of the basement membrane, accompanied by infiltration of leukocytes, accumulation of myofibroblasts, and intrusion of capillary vessels. High levels of MMPs produced by DCIS associated myoepithelial cells, fibroblasts, myofibroblasts, and leukocytes all contribute to the degradation of the basement membrane and tumor progression.
Conclusions
Tumor initiation and progression are determined by the molecular and phenotypic alterations arising in the tumor epithelial cells as well as in their microenvironment. Thus, combined targeting of both the “seed” and the “soil” may be a more effective approach for cancer prevention and treatment.
Acknowledgements
Studies in this laboratory are supported by Novartis, NIH (CA89393, CA94074, and CA116235), DOD (W81XWH-07-1-029), ACS (RSG-05-154-01-MGO), and Avon Foundation grants to KP, and Susan G. Komen Foundation fellowship (PDF042234) to MH.
Disclosure Statement
K.P. receives research support from and is a consultant to Novartis Pharmaceuticals, Inc. K.P. also receives research support from Biogen Idec, Inc. and is a consultant to and stock shareholder of Aveo Pharmaceuticals, Inc. K.P. and M.H. are also co-inventors of a patent application on DNA methylation changes occurring in the tumor microenvironment.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98–101. [PubMed] [Google Scholar]
- 2.Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radisky D, Hagios C, Bissell MJ. Tumors are unique organs defined by abnormal signaling and context. Semin Cancer Biol. 2001;11:87–95. doi: 10.1006/scbi.2000.0360. [DOI] [PubMed] [Google Scholar]
- 4.Tlsty TD, Hein PW. Know thy neighbor: stromal cells can contribute oncogenic signals. Curr Opin Genet Dev. 2001;11:54–59. doi: 10.1016/s0959-437x(00)00156-8. [DOI] [PubMed] [Google Scholar]
- 5.Weinberg R, Mihich E. Eighteenth annual pezcoller symposium: tumor microenvironment and heterotypic interactions. Cancer Res. 2006;66:11550–11553. doi: 10.1158/0008-5472.CAN-06-3149. [DOI] [PubMed] [Google Scholar]
- 6.Howlett AR, Bissell MJ. The influence of tissue microenvironment (stroma and extracellular matrix) on the development and function of mammary epithelium. Epithelial Cell Biol. 1993;2:79–89. [PubMed] [Google Scholar]
- 7.Elenbaas B, Weinberg RA. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res. 2001;264:169–184. doi: 10.1006/excr.2000.5133. [DOI] [PubMed] [Google Scholar]
- 8.Ronnov-Jessen L, Petersen OW, Bissell MJ. Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol Rev. 1996;76:69–125. doi: 10.1152/physrev.1996.76.1.69. [DOI] [PubMed] [Google Scholar]
- 9.van den Hooff A. Stromal involvement in malignant growth. Adv Cancer Res. 1988;50:159–196. doi: 10.1016/s0065-230x(08)60437-6. [DOI] [PubMed] [Google Scholar]
- 10.Willis RA. Fourth Edition. London: Butterworths and Company; 1967. Pathology of Tumours. [Google Scholar]
- 11.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 12.Schor SL, Schor AM, Rushton G. Fibroblasts from cancer patients display a mixture of both foetal and adult-like phenotypic characteristics. J Cell Sci. 1988;90(Pt 3):401–407. doi: 10.1242/jcs.90.3.401. [DOI] [PubMed] [Google Scholar]
- 13.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 14.van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 15.Weigelt B, Glas AM, Wessels LF, Witteveen AT, Peterse JL, van't Veer LJ. Gene expression profiles of primary breast tumors maintained in distant metastases. Proc Natl Acad Sci U S A. 2003;100:15901–15905. doi: 10.1073/pnas.2634067100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chin K, de Solorzano CO, Knowles D, Jones A, Chou W, Rodriguez EG, Kuo WL, Ljung BM, Chew K, Myambo K, et al. In situ analyses of genome instability in breast cancer. Nat Genet. 2004;36:984–988. doi: 10.1038/ng1409. [DOI] [PubMed] [Google Scholar]
- 17.Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, Payette T, Pistone M, Stecker K, Zhang BM, et al. Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci U S A. 2003;100:5974–5979. doi: 10.1073/pnas.0931261100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Porter D, Lahti-Domenici J, Keshaviah A, Bae YK, Argani P, Marks J, Richardson A, Cooper A, Strausberg R, Riggins GJ, et al. Molecular markers in ductal carcinoma in situ of the breast. Mol Cancer Res. 2003;1:362–375. [PubMed] [Google Scholar]
- 19.Yao J, Weremowicz S, Feng B, Gentleman RC, Marks JR, Gelman R, Brennan C, Polyak K. Combined cDNA array comparative genomic hybridization and serial analysis of gene expression analysis of breast tumor progression. Cancer Res. 2006;66:4065–4078. doi: 10.1158/0008-5472.CAN-05-4083. [DOI] [PubMed] [Google Scholar]
- 20.Dolberg DS, Bissell MJ. Inability of Rous sarcoma virus to cause sarcomas in the avian embryo. Nature. 1984;309:552–556. doi: 10.1038/309552a0. [DOI] [PubMed] [Google Scholar]
- 21.Barsky SH, Karlin NJ. Myoepithelial cells: autocrine and paracrine suppressors of breast cancer progression. J Mammary Gland Biol Neoplasia. 2005;10:249–260. doi: 10.1007/s10911-005-9585-5. [DOI] [PubMed] [Google Scholar]
- 22.Gudjonsson T, Adriance MC, Sternlicht MD, Petersen OW, Bissell MJ. Myoepithelial cells: their origin and function in breast morphogenesis and neoplasia. J Mammary Gland Biol Neoplasia. 2005;10:261–272. doi: 10.1007/s10911-005-9586-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Polyak K, Hu M. Do myoepithelial cells hold the key for breast tumor progression? J Mammary Gland Biol Neoplasia. 2005;10:231–247. doi: 10.1007/s10911-005-9584-6. [DOI] [PubMed] [Google Scholar]
- 24.Sadlonova A, Novak Z, Johnson MR, Bowe DB, Gault SR, Page GP, Thottassery JV, Welch DR, Frost AR. Breast fibroblasts modulate epithelial cell proliferation in three-dimensional in vitro co-culture. Breast Cancer Res. 2005;7:R46–R59. doi: 10.1186/bcr949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••25.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186.. Using mice transplanted with trackable bone marrow cells, the authors demonstrated that primary tumors mobilize haematopoietic progenitors from the bone-marrow and these cells form clusters at future metastatic sites prior to the arrival of tumor cells. This is the first study to investigate the role of haematopoietic progenitors in the homing and growth of metastatic tumor cells.
- 26.Kaplan RN, Rafii S, Lyden D. Preparing the "soil": the premetastatic niche. Cancer Res. 2006;66:11089–11093. doi: 10.1158/0008-5472.CAN-06-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the 'pre-metastatic niche': within bone and beyond. Cancer Metastasis Rev. 2006;25:521–529. doi: 10.1007/s10555-006-9036-9. [DOI] [PubMed] [Google Scholar]
- 28.Kaplan RN, Psaila B, Lyden D. Niche-to-niche migration of bone-marrow-derived cells. Trends Mol Med. 2007;13:72–81. doi: 10.1016/j.molmed.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 29.Psaila B, Kaplan RN, Port ER, Lyden D. Priming the 'soil' for breast cancer metastasis: the pre-metastatic niche. Breast Dis. 2006;26:65–74. doi: 10.3233/bd-2007-26106. [DOI] [PubMed] [Google Scholar]
- 30.Tan TT, Coussens LM. Humoral immunity, inflammation and cancer. Curr Opin Immunol. 2007;19:209–216. doi: 10.1016/j.coi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 31.Maffini MV, Soto AM, Calabro JM, Ucci AA, Sonnenschein C. The stroma as a crucial target in rat mammary gland carcinogenesis. J Cell Sci. 2004;117:1495–1502. doi: 10.1242/jcs.01000. [DOI] [PubMed] [Google Scholar]
- 32.Medina D, Kittrell F. Stroma is not a major target in DMBA-mediated tumorigenesis of mouse mammary preneoplasia. J Cell Sci. 2005;118:123–127. doi: 10.1242/jcs.01597. [DOI] [PubMed] [Google Scholar]
- 33.Ono M, Izumi H, Yoshida S, Goto D, Jimi S, Kawahara N, Shono T, Ushiro S, Ryuto M, Kohno K, et al. Angiogenesis as a new target for cancer treatment. Cancer Chemother Pharmacol. 1996;38 Suppl:S78–S82. doi: 10.1007/s002800051044. [DOI] [PubMed] [Google Scholar]
- 34.Luo Y, Zhou H, Krueger J, Kaplan C, Lee SH, Dolman C, Markowitz D, Wu W, Liu C, Reisfeld RA, et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J Clin Invest. 2006;116:2132–2141. doi: 10.1172/JCI27648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loeffler M, Kruger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. 2006;116:1955–1962. doi: 10.1172/JCI26532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, Porter D, Hu M, Chin L, Richardson A, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6:17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 37.Montel V, Mose ES, Tarin D. Tumor-stromal interactions reciprocally modulate gene expression patterns during carcinogenesis and metastasis. Int J Cancer. 2006;119:251–263. doi: 10.1002/ijc.21757. [DOI] [PubMed] [Google Scholar]
- 38.Jessani N, Humphrey M, McDonald WH, Niessen S, Masuda K, Gangadharan B, Yates JR, 3rd, Mueller BM, Cravatt BF. Carcinoma and stromal enzyme activity profiles associated with breast tumor growth in vivo. Proc Natl Acad Sci U S A. 2004;101:13756–13761. doi: 10.1073/pnas.0404727101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fukino K, Shen L, Matsumoto S, Morrison CD, Mutter GL, Eng C. Combined total genome loss of heterozygosity scan of breast cancer stroma and epithelium reveals multiplicity of stromal targets. Cancer Res. 2004;64:7231–7236. doi: 10.1158/0008-5472.CAN-04-2866. [DOI] [PubMed] [Google Scholar]
- 40.Fukino K, Shen L, Patocs A, Mutter GL, Eng C. Genomic instability within tumor stroma and clinicopathological characteristics of sporadic primary invasive breast carcinoma. Jama. 2007;297:2103–2111. doi: 10.1001/jama.297.19.2103. [DOI] [PubMed] [Google Scholar]
- 41.Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet. 2002;32:355–357. doi: 10.1038/ng1013. [DOI] [PubMed] [Google Scholar]
- 42.Kurose K, Hoshaw-Woodard S, Adeyinka A, Lemeshow S, Watson PH, Eng C. Genetic model of multi-step breast carcinogenesis involving the epithelium and stroma: clues to tumour-microenvironment interactions. Hum Mol Genet. 2001;10:1907–1913. doi: 10.1093/hmg/10.18.1907. [DOI] [PubMed] [Google Scholar]
- 43.Moinfar F, Man YG, Arnould L, Bratthauer GL, Ratschek M, Tavassoli FA. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res. 2000;60:2562–2566. [PubMed] [Google Scholar]
- 44.Weber F, Fukino K, Sawada T, Williams N, Sweet K, Brena RM, Plass C, Caldes T, Mutter GL, Villalona-Calero MA, et al. Variability in organ-specific EGFR mutational spectra in tumour epithelium and stroma may be the biological basis for differential responses to tyrosine kinase inhibitors. Br J Cancer. 2005;92:1922–1926. doi: 10.1038/sj.bjc.6602557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weber F, Shen L, Fukino K, Patocs A, Mutter GL, Caldes T, Eng C. Total-genome analysis of BRCA1/2-related invasive carcinomas of the breast identifies tumor stroma as potential landscaper for neoplastic initiation. Am J Hum Genet. 2006;78:961–972. doi: 10.1086/504090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••46.Hu M, Yao J, Cai L, Bachman KE, van den Brule F, Velculescu V, Polyak K. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005;37:899–905. doi: 10.1038/ng1596.. Using a novel genome-wide DNA methylation profiling method (MSDK), the authors demonstrated that DNA methylation changes occur in all major cell types (epithelial and myoepithelial cells, and fibroblasts) composing the tumor during breast tumor progression. This is the first study describing epigenetic changes in tumor stromal cells of any tumor type.
- 47.Fiegl H, Millinger S, Goebel G, Muller-Holzner E, Marth C, Laird PW, Widschwendter M. Breast cancer DNA methylation profiles in cancer cells and tumor stroma: association with HER-2/neu status in primary breast cancer. Cancer Res. 2006;66:29–33. doi: 10.1158/0008-5472.CAN-05-2508. [DOI] [PubMed] [Google Scholar]
- 48.Hanson JA, Gillespie JW, Grover A, Tangrea MA, Chuaqui RF, Emmert-Buck MR, Tangrea JA, Libutti SK, Linehan WM, Woodson KG. Gene promoter methylation in prostate tumor-associated stromal cells. J Natl Cancer Inst. 2006;98:255–261. doi: 10.1093/jnci/djj051. [DOI] [PubMed] [Google Scholar]
- 49.Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, Alison MR, Wright NA. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004;64:8492–8495. doi: 10.1158/0008-5472.CAN-04-1708. [DOI] [PubMed] [Google Scholar]
- 50.Creighton CJ, Bromberg-White JL, Misek DE, Monsma DJ, Brichory F, Kuick R, Giordano TJ, Gao W, Omenn GS, Webb CP, et al. Analysis of tumor-host interactions by gene expression profiling of lung adenocarcinoma xenografts identifies genes involved in tumor formation. Mol Cancer Res. 2005;3:119–129. doi: 10.1158/1541-7786.MCR-04-0189. [DOI] [PubMed] [Google Scholar]
- 51.Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–245. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zutter MM, Santoro SA, Staatz WD, Tsung YL. Re-expression of the alpha 2 beta 1 integrin abrogates the malignant phenotype of breast carcinoma cells. Proc Natl Acad Sci U S A. 1995;92:7411–7415. doi: 10.1073/pnas.92.16.7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Camps JL, Chang SM, Hsu TC, Freeman MR, Hong SJ, Zhau HE, von Eschenbach AC, Chung LW. Fibroblast-mediated acceleration of human epithelial tumor growth in vivo. Proc Natl Acad Sci U S A. 1990;87:75–79. doi: 10.1073/pnas.87.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Noel A, De Pauw-Gillet MC, Purnell G, Nusgens B, Lapiere CM, Foidart JM. Enhancement of tumorigenicity of human breast adenocarcinoma cells in nude mice by matrigel and fibroblasts. Br J Cancer. 1993;68:909–915. doi: 10.1038/bjc.1993.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- ••56.Cheng N, Bhowmick NA, Chytil A, Gorksa AE, Brown KA, Muraoka R, Arteaga CL, Neilson EG, Hayward SW, Moses HL. Loss of TGF-beta type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-alpha-, MSP- and HGF-mediated signaling networks. Oncogene. 2005;24:5053–5068. doi: 10.1038/sj.onc.1208685.. By specifically deleting TGF-beta type II receptor in mouse mammary fibroblasts, the authors demonstrated that blocking TGFβ signaling in fibroblasts promotes the growth and invasion of mammary carcinoma cells via upregulation of TGFα, MSP, and HGF signaling. This study suggested that a dynamic interaction exists between tumor epithelial and surrounding stromal cells, and implicated the potential complications associated with the therapeutic inhibition of certain pathways in all cell types.
- 57.Cheng N, Chytil A, Shyr Y, Joly A, Moses HL. Enhanced hepatocyte growth factor signaling by type II transforming growth factor-beta receptor knockout fibroblasts promotes mammary tumorigenesis. Cancer Res. 2007;67:4869–4877. doi: 10.1158/0008-5472.CAN-06-3381. [DOI] [PubMed] [Google Scholar]
- 58.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 59.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 60.Bogenrieder T, Herlyn M. Axis of evil: molecular mechanisms of cancer metastasis. Oncogene. 2003;22:6524–6536. doi: 10.1038/sj.onc.1206757. [DOI] [PubMed] [Google Scholar]
- 61.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- •62.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799.. The authors used in vivo selection in mice to isolate variants of MDA-MB-231 breast cancer cell line with increased metastatic growth in the lungs. Gene expression profiling of these cells identified a set of genes associated with lung metastasis, many of which encode for secreted or cell surface proteins. This study demonstrated the importance of autocrine/paracrine factors in metastasis.
- 63.Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8:341–352. doi: 10.1038/nrg2101. [DOI] [PubMed] [Google Scholar]
- •64.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188.. In this study, the authors demonstrated that co-injection of bone marrow-derived mesenchymal stem cells increases the metastatic potential of human breast carcinoma cells via upregulation of CCL5/CCR5 signaling using a subcutaneous xenograft system. The metastatic potential of the cancer cells was strictly dependent on interaction with mesenchymal stem cells, thus, the microenvironment has a reversible influence on the phenotype of cancer cells.
- 65.Cocker R, Oktay MH, Sunkara JL, Koss LG. Mechanisms of progression of ductal carcinoma in situ of the breast to invasive cancer. A hypothesis. Med Hypotheses. 2007;69:57–63. doi: 10.1016/j.mehy.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 66.Yousefi M, Mattu R, Gao C, Man YG. Mammary ducts with and without focal myoepithelial cell layer disruptions show a different frequency of white blood cell infiltration and growth pattern: implications for tumor progression and invasion. Appl Immunohistochem Mol Morphol. 2005;13:30–37. doi: 10.1097/00129039-200503000-00006. [DOI] [PubMed] [Google Scholar]
- 67.Man YG, Tai L, Barner R, Vang R, Saenger JS, Shekitka KM, Bratthauer GL, Wheeler DT, Liang CY, Vinh TN, et al. Cell clusters overlying focally disrupted mammary myoepithelial cell layers and adjacent cells within the same duct display different immunohistochemical and genetic features: implications for tumor progression and invasion. Breast Cancer Res. 2003;5:R231–R241. doi: 10.1186/bcr653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Man YG, Zhang Y, Shen T, Zeng X, Tauler J, Mulshine JL, Strauss BL. cDNA expression profiling reveals elevated gene expression in cell clusters overlying focally disrupted myoepithelial cell layers: implications for breast tumor invasion. Breast Cancer Res Treat. 2005;89:199–208. doi: 10.1007/s10549-004-2049-6. [DOI] [PubMed] [Google Scholar]
- 69.Man YG, Shen T, Weisz J, Berg PE, Schwartz AM, Mulshine JL, Sang QX, Nieburgs HE. A subset of in situ breast tumor cell clusters lacks expression of proliferation and progression related markers but shows signs of stromal and vascular invasion. Cancer Detect Prev. 2005;29:323–331. doi: 10.1016/j.cdp.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 70.Gudjonsson T, Ronnov-Jessen L, Villadsen R, Rank F, Bissell MJ, Petersen OW. Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. J Cell Sci. 2002;115:39–50. doi: 10.1242/jcs.115.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hildenbrand R, Arens N. Protein and mRNA expression of uPAR and PAI-1 in myoepithelial cells of early breast cancer lesions and normal breast tissue. Br J Cancer. 2004;91:564–571. doi: 10.1038/sj.bjc.6601990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Streuli CH, Bailey N, Bissell MJ. Control of mammary epithelial differentiation: basement membrane induces tissue-specific gene expression in the absence of cell-cell interaction and morphological polarity. J Cell Biol. 1991;115:1383–1395. doi: 10.1083/jcb.115.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]