Impaired Replication Capacity of Acute/Early Viruses in Persons Who Become HIV Controllers (original) (raw)
Abstract
Human immunodeficiency virus type 1 (HIV-1) controllers maintain viremia at <2,000 RNA copies/ml without antiretroviral therapy. Viruses from controllers with chronic infection were shown to exhibit impaired replication capacities, in part associated with escape mutations from cytotoxic-T-lymphocyte (CTL) responses. In contrast, little is known about viruses during acute/early infection in individuals who subsequently become HIV controllers. Here, we examine the viral replication capacities, HLA types, and virus sequences from 18 HIV-1 controllers identified during primary infection. g ag-protease chimeric viruses constructed using the earliest postinfection samples displayed significantly lower replication capacities than isolates from persons who failed to control viremia (P = 0.0003). Protective HLA class I alleles were not enriched in these early HIV controllers, but viral sequencing revealed a significantly higher prevalence of drug resistance mutations associated with impaired viral fitness in controllers than in noncontrollers (6/15 [40.0%] versus 10/80 [12.5%], P = 0.018). Moreover, of two HLA-B57-positive (B57+) controllers identified, both harbored, at the earliest time point tested, signature escape mutations within Gag that likewise impair viral replication capacity. Only five controllers did not express “protective” alleles or harbor viruses with drug resistance mutations; intriguingly, two of them displayed typical B57 signature mutations (T242N), suggesting the acquisition of attenuated viruses from B57+ donors. These data indicate that acute/early stage viruses from persons who become controllers have evidence of reduced replication capacity during the initial stages of infection which is likely associated with transmitted or acquired CTL escape mutations or transmitted drug resistance mutations. These data suggest that viral dynamics during acute infection have a major impact on HIV disease outcome.
Human immunodeficiency virus type I (HIV-1)-infected individuals who control viremia spontaneously without antiviral therapy have been termed HIV controllers (3, 18, 21, 48, 52). Unraveling the mechanisms associated with this phenotype should provide important insights regarding HIV pathogenesis and could contribute to vaccine development.
Host and viral genetics, as well as host innate and adaptive immune responses, influence the rate of disease progression in HIV-1 infection (reviewed in reference 18). Several studies have reported the correlation between in vitro HIV replication capacity and level of plasma virus loads or disease progression in individuals with chronic infection (6, 13, 35, 45, 50, 55). Studies of HIV-1 elite controllers (EC), who control viremia to below the limit of detection in commercial assays, have revealed the presence of replication-competent viruses in these individuals (7), although these viruses appear to be less fit based on studies of envelope (35) and Gag-protease (45). This fitness defect in the chronic phase of infection is due at least in part to fitness-impairing mutations induced by cytotoxic-T-lymphocyte (CTL) responses restricted by “protective” HLA class I alleles (46).
In contrast, little is known about viruses obtained from the acute/early phase of infection in persons who subsequently become HIV-1 controllers, largely due to the difficulty in enrolling such people during the acute/early phase of infection. The characterization of acute/early-phase viruses in individuals who subsequently achieve low set-point virus loads is of paramount importance to our understanding of the mechanisms of HIV-1 control.
In the present study, we analyzed acute/early-phase plasma HIV RNA sequences from 18 untreated individuals who were diagnosed during the acute/early phase and subsequently became controllers (<2,000 RNA copies/ml). We compared these to sequences from a group of HIV-1 noncontrollers enrolled similarly during acute/early infection. We also generated chimeric viruses carrying patient-derived gag-protease sequences from acute/early-phase infection and compared the viral replication capacities of the chimeric viruses from controllers and from noncontrollers.
We observed that the chimeric viruses derived from controllers have significantly reduced replicative capacities compared to those from noncontrollers. Moreover, we observed that at least 80% of these individuals who go on to become controllers featured transmission of attenuated drug-resistant viruses, transmission of HLA-B57-restricted CTL escape variants to HLA-mismatched recipients, selection of attenuated CTL escape variants in HLA-B57-positive (B57+) recipients, or combinations of these factors. Taken together, these results indicate that the initial viral dynamics have a major influence on the subsequent course of disease.
MATERIALS AND METHODS
Study subjects and blood sample collection.
All study participants were diagnosed during acute/early infection and remained untreated for a minimum of 1 year. Acute/early infection was defined according to published criteria (Acute Infection, Early Disease Research Program [AIEDRP] sponsored by NIAID) (28, 33). HIV controllers after primary infection (C-PI) were defined as HIV-1-infected subjects with acute/early infection who subsequently controlled viremia to less than 2,000 RNA copies/ml without antiviral treatment on at least 3 determinations over at least a 12-month period. Plasma and peripheral blood mononuclear cells (PBMC) from 18 C-PI were collected from multiple sites in the United States and Australia. Samples for 17 of these were obtained through the AIEDRP network. An additional subject (C-PI_13) was recruited at Massachusetts General Hospital. For individuals recruited through the AIEDRP network, the estimated date of infection (EDI) was calculated according to the AIEDRP criteria (37). For comparison, a group of 80 noncontrollers diagnosed during acute/early infection (noncontrollers after primary infection [NC-PI]) who failed to subsequently control viremia were drawn from AIEDRP and a previously described multicenter cohort with sites in the United States, Australia, and Germany (11). The participants from outside the AIEDRP network were subcategorized according to AIEDRP criteria as well. Written informed consent was obtained from all participants. The current study was approved by the appropriate institutional review boards, including that of Massachusetts General Hospital.
HLA typing.
Human leukocyte antigen (HLA) class I typing of C-PI was determined using an in-house sequence-based method involving locus-specific nested PCR on extracted DNA from plasma or PBMC, followed by DNA sequence analysis and allele interpretation (12). HLA typing of NC-PI was performed as previously described (11).
Viral RNA isolation, reverse transcription-PCR, and sequencing.
One to 3 ml of plasma from C-PI (or 0.5 ml for NC-PI) was centrifuged at 14,000 rpm (20,800 × g) for 2 h at 4°C, and viral RNA was extracted using a Qiagen viral RNA blood mini kit (Qiagen, Inc.). Reverse transcription-PCR and sequencing were performed as described previously (44). Briefly, nested reverse transcription-PCR was attempted for all HIV coding sequences from C-PI but only for gag and pol from NC-PI, such that comparisons between the groups were limited to these sequences. Bidirectional population (“bulk”) DNA sequencing was performed on an ABI 3730xl automated DNA sequencer. Data were analyzed using Sequencher software (Gene Codes). If the secondary peak was observed in both the 5′ and 3′ direction readings and the height of the secondary peak exceeded 25% of the dominant peak height in at least one of the two sequences, the nucleotides were interpreted to exist in mixtures at that position. Multiple alignment was performed using ClustalW. Contamination and/or sample mix-ups were ruled out by constructing a maximum-likelihood phylogenic tree (DNAml incorporated in BioEdit). Viral subtypes were determined using entire gag sequences with REGA HIV-1 Subtyping Tool version 2.0, available through the Stanford HIV Drug Resistance Database (http://dbpartners.stanford.edu/RegaSubtyping/). Likewise, drug resistance mutations were examined using the HIVdb Program Sequence Analysis tool (http://hivdb.stanford.edu/pages/algs/sierra_sequence.html).
Generation of chimeric viruses.
Generation of gag-protease chimeric NL4-3 viruses, viral titration, and the replication capacity assay were performed as described previously (45). Briefly, gag-protease reverse transcription-PCR amplicons were cotransfected with linearized Δ_gag-protease_ pNL4-3 vector into a long terminal repeat (LTR)-driven green fluorescent protein (GFP) reporter T cell line of CEM origin (GXR cells) (10), and chimeric viruses were obtained by homologous recombination. The patient origins of the resulting chimeric virus stocks were verified by comparing their sequences to the original plasma HIV RNA sequences (not shown). Titration was performed by infecting GXR cells with the chimeric viruses and measuring GFP expression by flow cytometry. The titer data were used to adjust input viral volumes in the subsequent replication capacity assay to obtain a multiplicity of infection (MOI) of 0.002 on day 2. GFP expression was monitored daily for a week. Replication capacity was expressed as the slope of the natural log of the percentage of GFP-expressing cells between days 3 and 6. Replication capacity assays were performed in duplicate, examining all of the viruses at the same time, and the average of the relative value to that of wild-type NL4-3 virus for each subject was used for comparisons.
Statistical analysis.
Statistical analyses of continuous variables were performed using the Mann-Whitney U test. Comparisons of categorical value were performed using Fisher's exact test. A P value of <0.05 was considered statistically significant.
Nucleotide sequence accession numbers.
Plasma viral sequences obtained in this study were submitted to GenBank under accession nmbers GU367395 to GU367447 and GU367449 to GU367592.
RESULTS
Kinetics of plasma viremia during primary infection in persons who become HIV controllers.
Little is known about the levels and kinetics of viremia during acute/early infection among individuals who subsequently become HIV-1 controllers. To address this, we identified 18 subjects diagnosed at the time of primary infection who maintained viral loads of <2,000 RNA copies/ml for at least a year in the absence of therapy (HIV controllers after primary infection [C-PI]). For each of these subjects, longitudinal samples were available for analysis of the kinetics of plasma viral load following untreated primary infection.
As shown in Fig. 1 A and B, these 18 subjects achieved HIV control within 200 days after the estimated date of infection (EDI); they were then followed for a median of 1,089 (range, 744 to 2,079) days. Six of the 18 C-PI subsequently achieved elite control (<50 RNA copies/ml for at least 1 year) during follow-up (Fig. 1A), and 12 of the C-PI controlled viremia at between 50 and 2,000 RNA copies/ml (Fig. 1B). For those who achieved elite control, the time to viral loads of less than 50 RNA copies/ml varied widely. One subject was diagnosed with early infection at 130 days post estimated date of infection (EDI), when the viral load was already less than 50 RNA copies/ml, and another achieved persistently undetectable viral loads at day 170, whereas another subject (C-PI_18) achieved elite control relatively slowly (604 days post-EDI; data not shown). One subject (C-PI_12) initially achieved a stable viremia of less than 2,000 RNA copies/ml until 710 days, at which point there was a blip in viremia and then a decline to below the limits of detection (<50 RNA copies/ml) (Fig. 1C). During the average-3-year follow-up, only 3 of the 18 subjects lost control, as evidenced by an increase in viral load to over 2,000 RNA copies/ml (Fig. 1D, E, and F), including one person initially meeting the criteria for elite control (C-PI_04) (Fig. 1D).
FIG. 1.

Kinetics of plasma virus loads during acute/early phase of infection in individuals who subsequently achieve viremia control (<2,000 RNA copies/ml). (A) Data for six controllers after primary infection (C-PI) who achieved elite control (EC; <50 RNA copies/ml) during the follow-up period. Each line represents the plasma virus loads of an individual patient; the dashed line indicates 2,000 RNA copies/ml. The blue line shows data for C-PI_18, who achieved <50 RNA copies/ml after 600 days post-EDI and, therefore, displayed >50 RNA copies/ml during the period shown in the figure. (B) Data for twelve C-PI who remained viremic during the follow-up period. Each line represents the plasma virus loads of an individual patient; the dashed line indicates 2,000 RNA copies/ml. VC, viremic controllers. (C) Data for C-PI_12, who achieved elite control after 700 days post-EDI after experiencing a blip in viremia. (D) Data for C-PI_04, who achieved elite control after 470 days post-EDI and also experienced virologic escape after 1,500 days post-EDI. (E and F) Data for C-PI_02 and 03, who experienced virologic escape. ART, antiretroviral therapy.
While the mean viral loads during acute infection have been reported to be in excess of 5 × 106 RNA copies/ml in other studies (31), the virus loads during acute phase in the HIV controllers studied here never exceeded 1 × 106 RNA copies/ml (Fig. 1A and B). Although it is important to note that the magnitude and timing of acute-phase peak viremia were likely not identified in most cases (as is also the case with other published studies), these observations nevertheless suggest that persons who are able to control HIV spontaneously following primary infection may have lower levels of viremia during acute infection. These data indicate that viremia control can be initiated during the acute/early phase of infection and that when early control is achieved, it is largely maintained over the first few years of infection in persons who initially achieve this equilibrium. Moreover, they show that elite controller status may not be achieved until two or more years after primary infection.
HLA class I allele distribution in subjects who subsequently become HIV controllers.
Among HIV controllers in the chronic phase of infection, there is an overrepresentation of “protective” HLA class I alleles, in particular, HLA-B57 and -B27 (4, 43, 48). To determine the potential role of host genetics in viral control in this cohort, we first performed high-resolution HLA typing. The frequency of individuals expressing HLA-B57 was the same in those who became HIV controllers (C-PI) as in those with primary infection who were defined as noncontrollers (NC-PI) (2/18 versus 5/80, P = 0.6) (Fig. 2 A) but was lower than in an updated cohort of chronic HIV controllers (C-chronic [reference 48 and data not shown]) (224/719, P = 0.074). Likewise, the frequency of individuals expressing this and other so-called “protective” HLA class I alleles (defined as HLA-B13, -B27, -B51, and -B57/B*1516/B*1517/B*5801; see references 23, 25, 29, 32, and 47) among the C-PI did not differ from the frequency of such individuals among the NC-PI in the present study (5/18 versus 24/80, P = 1.0) (Fig. 2B) but, again, was significantly lower than in the chronic HIV controller cohort (5/18 versus 417/719, P = 0.014) (Fig. 2B).
FIG. 2.

Proportions of the individuals expressing protective HLA class I alleles. (A) Proportions of individuals expressing HLA-B57 among controllers after primary infection (C-PI), noncontrollers after primary infection (NC-PI), and chronic HIV controllers (C-chronic). (B) Proportions of individuals expressing protective HLA class I alleles (B13, B27, B51, and B57/B*1516/B*1517/B*5801) among C-PI, NC-PI, and C-chronic.
Taken together, these data suggest that persons who achieve viral control following acute/early infection differ in host genetics from those HIV controllers classified using chronic-phase clinical and phenotypic definitions in that they are not enriched for the usual protective alleles associated with chronic HIV control (23, 25, 29, 32, 47).
Chimeric NL4-3 viruses carrying g ag-protease derived from individuals who go on to become HIV controllers display reduced replication capacities.
Our finding that protective HLA alleles were not enriched in this group suggested that factors other than host genetics might influence early control of viremia and led us to examine viral factors that might contribute to enhanced control. We recently reported that chimeric NL4-3 viruses carrying g ag-protease derived from chronic elite controllers display reduced replication capacities compared to the replication capacities of those derived from chronic progressors (45). To determine the relative fitness of acute/early viruses in these subjects, we constructed chimeric gag-protease viruses from the earliest viral isolates from both C-PI and NC-PI subjects. In this analysis, gag-protease genes were amplified from plasma HIV RNA and chimeric viruses were generated for 15 subtype B-infected C-PI (C-PI_13 was infected with a unique recombinant form with a clade A backbone and, therefore, was excluded from the following analyses). As a control, we randomly chose 45 of the 80 NC-PI (median baseline virus load, 186,000 RNA copies/ml [interquartile range, 6,160 to 750,100]). As in previous studies (9, 45, 46, 53), replication capacity assays were performed and the slope of the natural log of viral spread in culture between days 3 and 6 was calculated. All viruses were assayed in the same experiment run in duplicate, and the results are presented as the values relative to the results for the wild-type NL4-3 virus.
As shown in Fig. 3 A, chimeric viruses carrying gag-protease derived from individuals who go on to become controllers displayed significantly reduced replication capacities compared to those from individuals who became noncontrollers (C-PI versus NC-PI, 0.84 versus 0.98, P = 0.0003). Since many of the NC-PI were diagnosed during the acute phase of infection (AIEDRP group 1), whereas most of the C-PI were diagnosed during early infection (AIEDRP group 3), there was a concern that the observed difference might be a reflection of differences between acute and early infection. Therefore, we repeated the analysis, stratifying the 45 NC-PI into AIEDRP group 1 (acute, n = 25) or group 3 (early, n = 20), but found no statistically significant difference between them (0.95 versus 0.99, P = 0.23), whereas chimeric viruses carrying gag-protease from the C-PI displayed significantly reduced replication capacities compared to the replication capacities of viruses from acute and early NC-PI (P = 0.005 and 0.0003, respectively) (Fig. 3B). These data indicate that defects in viral fitness are strongly associated with the ability to control viremia following primary infection.
FIG. 3.
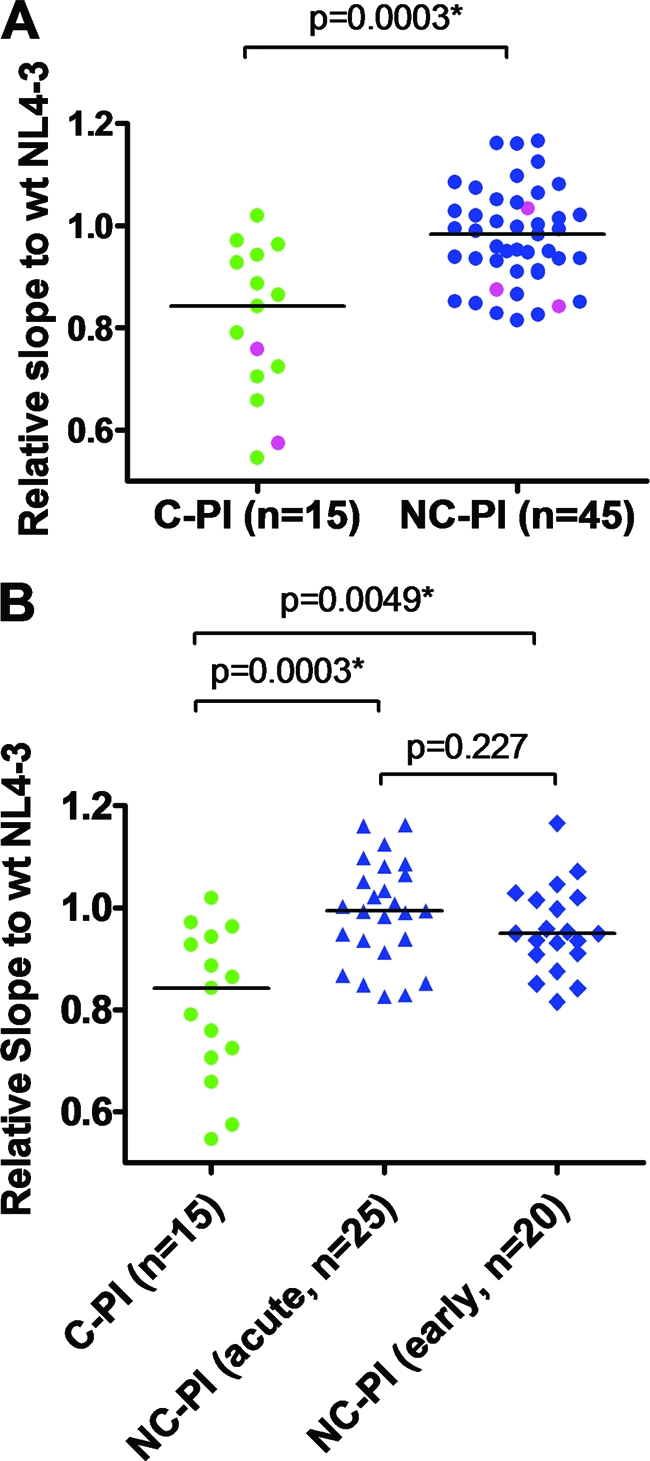
Replication capacities of chimeric NL4-3 viruses carrying gag-protease derived from the acute phase of infection in controllers and noncontrollers. Replication capacities were compared between clade B-infected C-PI (n = 15) and NC-PI (n = 45). Average results of duplicate experiments for each virus normalized to the replication capacity of wild-type (wt) NL4-3 virus were plotted (expressed as the slope of the natural log of viral spread between days 3 and 6; see Materials and Methods). (A) Data for all of the chimeric viruses derived from individuals in acute/early phase of infection. The pink dots indicate viruses carrying protease inhibitor (PI) resistance mutations. (B) Data are the same as in the analysis shown in panel A, but the data for viruses derived from the NC-PI are stratified according to phase of infection (acute or early). Horizontal lines show medians of the results.
CTL escape mutations are present in viral sequences obtained from the acute/early phase in the persons who subsequently become HIV controllers.
We recently reported that attenuation of the replication capacity of chimeric viruses carrying gag-protease derived from chronic elite controllers is associated with specific HLA class I alleles, indicating virus attenuation due to immune selection pressure (45). Since we observed the viruses in the C-PI to be attenuated, we next examined plasma viral sequences for evidence of CTL-driven escape mutations from the earliest available time point (median, 86 days, and range, 33 to 203 days post-EDI).
As shown in Tables 1 and 2, both C-PI expressing HLA-B57 (C-PI_01 and 11) harbored the B57-associated Gag T242N escape mutation within the TW10 (TSTLQEQIGW, Gag amino acids 240 to 249) epitope known to affect viral replication capacity (9, 22, 36, 40). One of the gag compensatory mutations, I223V, that is known to partially restore replication capacity impaired by the T242N mutation (9) was observed in C-PI_11, but none of these compensatory mutations was observed in C-PI_01 (Table 2). The relative replication capacities of chimeric NL4-3 viruses carrying gag-protease derived from these individuals were substantially reduced (0.864 and 0.724, respectively). Only one of the 45 NC-PI whose chimeric virus was tested for replication capacity expressed HLA-B57. The autologous viral sequence obtained at 59 days post-EDI exhibited the fitness-impairing T242N mutation but also the H219Q compensatory mutation, and the relative replication capacity of the chimeric virus derived from this subject was slightly reduced (0.852). Although this subject was categorized as an NC-PI, the plasma virus load was only 3,680 RNA copies/ml on day 59 post-EDI, indicating that viremia had been relatively well controlled in this subject. Taken together, the replication capacities of chimeric viruses carrying gag-protease obtained during acute infection from HLA-B57-positive subjects appeared moderately reduced, which might be attributable to the typical fitness-impairing T242N escape mutation.
TABLE 1.
HLA class I alleles, viral antiretroviral drug resistance mutations, and B57 footprint profiles in early controllers
| C-PI | HLA class I allelesc | HLA-B57 footprintd | Viral drug resistance mutation(s) to: | ||
|---|---|---|---|---|---|
| PI | NRTI | NNRTI | |||
| 01 | A25/A68/B18/B57/Cw06/Cw12 | T242N (I147L) | None | None | None |
| 02 | A02/A31/B08/B44/Cw05/Cw07 | (I147L) | None | None | None |
| 03 | A01/A68/B08/B44/Cw05/Cw07 | None | M46L, I54FL, I84V, N88D, L90 M | M41L, D67N, T69D, M184V, L210W, T215Y | A98G, K103N, Y318F |
| 04 | A26/A31/B07/B18/Cw07/Cw07 | None | D30N, M46I, I84V, N88D, L90 M | M41L, E44D, D67N, K70R, L74I, V118I, L210Y, T215Y, K219Q | None |
| 05 | A01/A32/B35/B44/Cw02/Cw05 | A146P (N252G) | None | None | None |
| 06 | A01/A01/B*1517/B49/Cw07/Cw12 | G248A A146N (I147 M) | None | D67N, T215S, K219E, G333E | None |
| 07 | A02/A02/B07/B*5801/Cw07/Cw07 | (G248E) | None | M41L | None |
| 08 | A03/A68/B14/B44/Cw07/Cw08 | None | None | None | None |
| 10 | A23/A31/B39/B52/Cw07/Cw12 | T242N A146P (I147L) | None | None | None |
| 11 | A01/A02/B44/B57/Cw05/Cw06 | T242N G248A (I147L) | None | None | None |
| 12 | A01/A31/B37/B51/Cw01/Cw06 | (I147L) G248A (M250I) | None | None | None |
| 13a | A03/A31/B35/B35/Cw04/Cw04 | N/D | None | None | None |
| 14 | A02/A32/B35/B44/Cw04/Cw05 | None | None | M184V | G190A |
| 15 | A11/A31/B27/B35/Cw01/Cw01 | (I147L) | None | None | None |
| 16 | A02/A29/B44/B49/Cw07/Cw16 | NA | NAb | NA | NA |
| 17 | A02/A02/B44/B49/Cw07/Cw16 | NA | NA | NA | NA |
| 18 | A02/A24/B*1501/B41/Cw03/Cw17 | None | None | None | K103N P225H |
| 20 | A02/A03/B*1501/B56/Cw01/Cw03 | T242N G248A (I147L) | None | None | None |
TABLE 2.
Plasma viral Gag protein sequences in 15 clade B-infected early controllersa
| C-PI | HLA-B57 analogue | Mutation(s) in indicated part of Gag sequencea | |
|---|---|---|---|
| 133 PIVQNLQGQMVHQAISPRTLNAWVK 157 | 213 DRLHPVHAGPIAPGQMREPRGSDIAGTTSTLQEQIGWMTNNPPI 256 | ||
| 01 | B57 | ..............L.......... | .............................N.............. |
| 02 | None | ..............L.......... | ..T...............................V....H.... |
| 03 | None | ......................... | ............................................ |
| 04 | None | ..I...................... | ..........A...............S............S.... |
| 05 | None | .....I.......P........... | ......Q...V............................G.... |
| 06 | B*1517 | .............NM.......... | ......Q...V....I...................A...H.... |
| 07 | B*5801 | ......................... | ..V................................E...S..A. |
| 08 | None | ......................... | ..M....................................S.... |
| 10 | None | .............PL.......... | .............................N.............. |
| 11 | B57 | ..............L.......... | ..........V..................N.....A...S.... |
| 12 | None | ..............L.......... | ..........V........................A.I...... |
| 14 | None | .....M................... | ............................................ |
| 15 | None | ..............L.......... | .......................................S.... |
| 18 | None | .....M................... | ..M....................................H.... |
| 20 | None | ..............L.......... | ..T..........................N.....A........ |
Evidence of transmitted CTL escape mutants from HLA-B57+ donors.
Recently, it was reported that individuals who acquired HIV from B57+ donors have a better clinical course at least during the first year of infection (15), suggesting that the acquisition of attenuated viruses may benefit the recipient even in the absence of the restricting protective allele. Therefore, we investigated whether some C-PI might have acquired viruses from donors expressing protective HLA alleles. Examining Gag protein sequences in the C-PI, we observed the signature B57 escape mutation T242N that compromises replication capacity (9, 40) in two of the 5 C-PI who expressed no other protective alleles (C-PI_10 and 20) (Tables 1 and 2), and there was a trend of a higher frequency of the T242N mutation in the C-PI than in the NC-PI (2/5 versus 4/47). The viruses in C-PI_10 also exhibited the A146P/I147L signature escape mutations within and flanking the B57 ISW9 epitope (Gag amino acids 147 to 155) (20), adding further evidence that the viruses were transmitted from a B57+ donor. The A146P/I147L mutations in conjunction with the T242N mutation have been shown to further reduce viral replication capacity (8). The relative replication capacity of the chimeric NL4-3 virus derived from C-PI_10 was extremely reduced (0.546), but the replication capacity of the virus derived from C-PI_20 was only minimally reduced (0.928). For C-PI_10, follow-up samples were available at 20 months after the EDI, but despite persistent low-level virus production in vivo, no reversions of B57-associated mutations (A146P/I147L/T242N) were observed (data not shown). These data suggest that transmission of viruses attenuated by CTL escape mutations in the HLA-B57+ donors may account for some of the C-PI and indicate that transmitted CTL escape mutations may impair the replication capacity and pathogenicity of HIV following transmission.
High frequency of drug resistance mutations in plasma viral sequences in individuals who go on to become HIV controllers.
Viral fitness can be impaired by defects in other HIV genes. In addition to gag sequences, we obtained plasma viral sequences for other HIV genes from the C-PI (16 pol, 14 vif, vpr, and vpu, 14 partial envelope, 11 nef, and 14 rev and tat sequences); however, similar to earlier studies of chronic elite controllers (44), virus sequencing in this cohort revealed no gross viral genetic defects (data not shown). Since selection pressures by antiretroviral drugs are known to impair viral replication capacity as well, we looked for the presence of “major” resistance mutations (as defined by the Stanford HIV Drug Resistance Database) in autologous viral sequences obtained during the acute phase of infection from the controllers and compared the frequency of these mutations to their frequency in sequences from noncontrollers.
Analysis of protease and reverse transcriptase (RT) sequences revealed a significantly higher prevalence of antiretroviral resistance mutations in C-PI (6/15 [40.0%]) than in NC-PI (10/80 [12.5%]) (P = 0.018) (Fig. 4 A), some of which have been demonstrated to reduce viral replication capacity (M184V, L210W, T215Y, etc.) (26, 27, 39, 59). Among individuals who went on to become controllers, we observed two cases of protease inhibitor (PI) resistance mutations, 4 cases of nucleoside analogue reverse transcriptase inhibitor (NRTI) resistance mutations, and 3 cases of non-nucleoside analogue reverse transcriptase inhibitor (NNRTI) resistance mutations (Table 1). In addition, three of these cases exhibited multiclass drug resistance, which was also significantly more frequent than in NC-PI (3/15 versus 1/80, P = 0.012) (Fig. 4B and Table 1). Notably, excluding individuals expressing protective HLA alleles, the frequency of individuals infected with drug-resistant strains reached 60% among C-PI versus 8.5% in NC-PI (6/10 versus 4/47, P = 0.0035) (Fig. 4C), and the frequency of multiclass drug resistance strains reached 30% (Fig. 4D). Taking into consideration that resistance mutations can negatively impact viral replicative capacity (17, 39, 56, 58), these results suggest that the transmission of drug-resistant variants contributes to viremia control during the early phase of infection in these individuals. It is important to note that in the present study, we analyzed replication capacity only for gag-protease chimeric viruses and, thus, that resistance mutations within RT were not evaluated. Among the total 60 viruses tested for replication capacity, five viruses (2 from C-PI and 3 from NC-PI) carried PI resistance mutations (Fig. 3A). Except for one virus derived from an NC-PI, all displayed moderately to severely impaired replicative capacities: the relative replication capacities of viruses derived from the two C-PI were 0.760 (C-PI_03) and 0.575 (C-PI_04), and viruses from two of the NC-PI carrying PI resistance mutations were moderately impaired (0.875 and 0.842).
FIG. 4.

Proportions of individuals diagnosed during acute/early phase and carrying viral strains with major drug resistance mutations (see Materials and Methods) are shown. (A) Data for individuals infected with strains with major PI, NRTI, and NNRTI resistance mutations. (B) Data for individuals infected with multiclass drug-resistant strains. (C) Data for individuals without protective HLA class I alleles infected with strains with major PI, NRTI, and NNRTI resistance mutations. (D) Data for individuals without protective HLA class I alleles infected with multiclass drug-resistant strains.
Follow-up pol sequences were available 6 months later from two C-PI, both of whom became viremic controllers (50 to 2,000 RNA copies/ml). No change in resistance profile was observed in subject C-PI_06, whereas in subject C-PI_03, four of the 9 initially documented RT resistance mutations (M184V, L210F, and T215Y for NRTI and Y318F for NNRTI) (Table 1) reverted to the wild type (data not shown). However, plasma viral loads remained low in both individuals.
In addition, we were able to obtain the plasma pol sequence from subject C-PI_04, who became an elite controller despite lacking protective HLA alleles but experienced virologic escape after 4 years of infection (Fig. 1D). At that time, 2 of the NRTI resistance mutations (K70R and L74I) had reverted to the wild type, and the T215Y mutation partially reverted to T215C/S when the plasma viral load rose to 1,300 RNA copies/ml around 5 years post-EDI. Although the L74I and T215Y mutations are known to affect viral replicative fitness only moderately (39), the K70R mutation in conjunction with the M41L mutation strongly attenuates virus (30), suggesting that these reversions may have contributed to the virologic escape in this case.
DISCUSSION
Here, we demonstrate that viruses from persons who spontaneously control HIV following primary infection show impaired replicative capacity in the earliest stages of infection, indicating that characteristics of the infecting virus have a significant impact on viral set point and virus-host dynamics. By constructing chimeric viruses expressing the gag-protease region of patient isolates and inserting them into a laboratory strain backbone, we show that viruses derived from persons who are able to achieve viral loads of less than 2,000 RNA copies/ml are attenuated compared to contemporaneous viruses derived from persons who go on to develop progressive infection. Of subjects confirmed to be infected with clade B virus, 12/15 (80%) were recipients of potentially attenuated drug-resistant strains or viruses selected in B57+ donors or expressed protective HLA alleles that selected for less fit viruses (summarized in Table 1). Remarkably, two cases had more than one factor: they expressed B57 analogues and were infected with NRTI-resistant strains. Together, these data indicate that viral dynamics in the earliest stages of HIV infection have a major impact on the course of disease.
In these persons who achieved viral control following primary infection, there was no enrichment of HLA alleles associated with long-term control found in chronic-controller cohorts. Instead, we saw enrichment for other factors that can attenuate virus replication, including transmission of drug-resistant strains and strains containing attenuating CTL escape mutations. The surprising finding that HLA-B57 was not enriched among controllers after primary infection may be related to the fact that persons expressing this allele are less symptomatic during acute infection and, thus, may have been less likely to be recognized and recruited in the acute phase of infection (1). Another important issue raised by this observation is the durability of control that is achieved early after acute infection. Indeed, one C-PI who achieved elite control status but did progress later lacked protective HLA alleles. However, the majority of persons without protective alleles were able to persistently control viremia, in one case (C-PI_03) even after reversion of some of the drug resistance mutations, suggesting that early attenuation of virus may have sustained consequences. Additional follow-up will be required to answer this question.
The subjects in this study were largely selected from subjects enrolled in the Acute Infection, Early Disease Research Program, based on their ability to achieve controller status following documented primary infection. The criteria consisted of at least 1 year of viral loads of less than 2,000 RNA copies/ml, used to define HIV controllers because this is a level at which disease progression and transmission are much less likely (49). Of the 18 individuals who went on to become HIV controllers, 6 eventually met the criteria for elite control, with viral loads persisting at below 50 RNA copies/ml. Since one of the C-PI who achieved elite control (C-PI_04) and two C-PI who became viremic controllers (C-PI_02 and C-PI_03) later progressed, early control does not necessarily predict long-term control. Of note, none of them expressed protective HLA alleles, suggesting an important role of host genetics in eventual outcome.
Although we did not show any direct impact of individual mutations observed here on viral replicative capacity, there are several studies that demonstrate a fitness cost of PI resistance mutations in particular (41, 42, 60) and of CTL escape mutations within the Gag protein (9, 40, 53). Indeed, the viruses from two of the B57+ C-PI were carrying the T242N escape mutation. In addition to these, we found putative B57 footprint mutations in the individuals expressing HLA-B*1517 (C-PI_06) and HLA-B*5801 (C-PI_07) that are known to cross-present B57 TW10 epitopes (23, 36). The autologous viral sequence for C-PI_06 did not have any of the typical signature B57 footprint mutations A146P/I147L/T242N but contained the G248A mutation that is associated with HLA-B57. Uncommon substitutions at the sites associated with HLA-B57 (A146N/I147M) were also observed (Tables 1 and 2) (20, 36). Likewise, none of the typical signature B57 mutations A146P/I147L/T242N were seen in C-PI_07, but the autologous viral sequence displayed a rare mutation (G248E) that has been reported to be selected in B57+ subjects during primary infection (5). Although we do not have direct experimental evidence of a viral fitness cost due to these mutations, substitutions at these sites have been previously shown to impair viral replication capacity (8, 46). Indeed, the relative replication capacities of the chimeric NL4-3 viruses carrying gag-protease derived from these persons were reduced (0.887 and 0.658, respectively). Taken together, viruses in these 2 C-PI expressing HLA-B57 analogues likely harbored allele-specific mutations which might have contributed to compromised viral replication capacity.
We observed the typical HLA-B57 footprint mutation (T242N) in plasma viral sequences in two of the C-PI expressing none of the protective HLA alleles, supporting previous reports of better early clinical outcome of recipients who acquired viruses from B57-positive donors (15). By analyzing viral sequences in the rest of the C-PI, we found other mutations within and flanking the TW10 epitope that had likely been selected in B57+ donors: autologous virus from C-PI_12 expressing the protective allele HLA-B51 had rare G248A/M250I mutations that were previously shown to be observed in B57+ chronic elite controllers and to impair viral replication capacity in vitro (46), and the replication capacity of the gag-protease chimeric NL4-3 virus derived from this person was reduced (0.843). Another non-B57 analogue subject (C-PI_05) harbored the B57 signature mutation A146P and a rare N252G mutation in the flanking region of the TW10 epitope that had been observed in a B57 elite controller and demonstrated to affect viral replicative capacity (46). However, the replication capacity of the chimeric NL4-3 virus for this person was not appreciably reduced (0.943). These data suggest that there were additional individuals who potentially acquired viruses from B57-positive donors and went on to become controllers.
In the current study, we did not examine the function of the reverse transcriptase or the impact of individual resistance mutations on RT function, but there are a number of publications that also support the theory presented here (19, 26, 27, 30, 38, 39, 57, 59). For instance, the M184V mutation observed in C-PI_03 and 14 is selected by lamivudine and is known to strongly reduce viral replicative fitness (39, 59); the T215Y mutation observed in C-PI_03 and 04 has a similar effect, albeit moderately (26, 39), and in addition, the L210W mutation that was observed in C-PI_03 and remained at the later time point has been shown to have a strong negative impact on viral replication capacity (27). Moreover, there was a report that the number of NRTI resistance mutations inversely correlates with viral replicative capacity (51), further supporting the idea that viruses from the acute phase of infection carrying multiple resistance mutations in some of the individuals who go on to become controllers are attenuated.
While the difference in replication capacities of chimeric viruses derived from the acute/early phase of infection in controllers and noncontrollers was significant, chimeric viruses derived from some controllers displayed only minimal reductions in replication capacity, suggesting that a better early clinical course cannot be explained solely by attenuation of Gag-Protease function. Impairment of HIV replication by mutations in other genes, like nef deletions (16, 34), or by other mechanisms may also be important. Therefore, it will be warranted to investigate the function of other HIV proteins, including RT and envelope, in future studies.
The hypothesis presented here, that viral attenuation affects the course of disease following acute infection, has been proposed previously (2, 35, 50) and is supported by other studies showing that the transmission of escape mutations selected by protective alleles in the donor provides a clinical advantage to the recipient (15, 24). Moreover, a recent study of children born to B57+ mothers revealed reduced viral loads in the B57+ children despite the transmission of B57 CTL escape mutants (54). In addition, there is a report of an inverse relationship between plasma viral load and the number of genotypic resistance mutations during primary infection (14). These data strongly suggest that the transmission of less fit viruses due to attenuating antiretroviral resistance mutations and CTL escape mutations could result in a favorable clinical course in the recipients, at least during the early phase of infection. It should be noted that our findings might imply that the spread of drug-resistant variants will increase the proportion of individuals who can control viremia in the early phase of infection; however, further investigations are necessary in order to clarify this point.
In conclusion, viruses obtained during the acute/early phase of infection from persons who subsequently become controllers were attenuated. This observation can be partly explained by the transmission of drug-resistant strains and/or CTL escape variants from B57+ donors or by de novo CTL escape mutations in B57+ recipients. During an average of 3 years of follow-up, the plasma virus load remained stable in the majority of subjects, even in one in whom apparent fitness-enhancing reversions were observed. These data suggest that viral attenuation in acute infection may have a long-term impact on control, but further longitudinal studies are warranted to reveal the role of viral fitness in long-term control of HIV-1 infection.
Acknowledgments
This work was supported by grants AI028568 and AI030914 (B.D.W.), AI55356 (E.C.), and AI041534 and AI047033 (M.M.) from the NIAID/NIH and by the Howard Hughes Medical Institute, the Harvard University Center for AIDS Research (HU CFAR), the Mark and Lisa Schwartz Foundation, the International AIDS Vaccine Initiative (IAVI), and the Bill and Melinda Gates Foundation. Z.L.B. is supported by a New Investigator Award from the Canadian Institutes for Health Research (CIHR). NCHECR is funded by the Australian Government Department of Health and Ageing and is affiliated with the Faculty of Medicine, the University of New South Wales. A.D.K. is supported by an NHMRC Practitioner Fellowship.
Samples and data from Australia were collected through the Primary Infection network consisting of R. Finlayson, Taylor Square Clinic, Darlinghurst, NSW; M. Bloch, Holdsworth House, Darlinghurst, NSW; R. Macfarland, East Sydney Doctors, Darlinghurst, NSW; C. Workman AIDS Initiative, Darlinghurst, NSW; N. Roth, Prahan Market Clinic, Prahan, Victoria; and David Cooper, St. Vincent's Hospital, Darlinghurst, NSW.
We thank Mary Carrington and Yuko Yuki for HLA class I typing of a subset of the noncontrollers.
The ideas and opinions expressed in the manuscript are solely the responsibility of the authors and are not necessarily shared by the NIH or other funding sources, Massachusetts General Hospital, or its affiliates.
The authors declare no conflicts of interest related to this study.
Footnotes
▿
Published ahead of print on 26 May 2010.
REFERENCES
- 1.Altfeld, M., M. M. Addo, E. S. Rosenberg, F. M. Hecht, P. K. Lee, M. Vogel, X. G. Yu, R. Draenert, M. N. Johnston, D. Strick, T. M. Allen, M. E. Feeney, J. O. Kahn, R. P. Sekaly, J. A. Levy, J. K. Rockstroh, P. J. Goulder, and B. D. Walker. 2003. Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS 17**:**2581-2591. [DOI] [PubMed] [Google Scholar]
- 2.Arien, K. K., G. Vanham, and E. J. Arts. 2007. Is HIV-1 evolving to a less virulent form in humans? Nat. Rev. Microbiol. 5**:**141-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailey, J. R., K. G. Lassen, H. C. Yang, T. C. Quinn, S. C. Ray, J. N. Blankson, and R. F. Siliciano. 2006. Neutralizing antibodies do not mediate suppression of human immunodeficiency virus type 1 in elite suppressors or selection of plasma virus variants in patients on highly active antiretroviral therapy. J. Virol. 80**:**4758-4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey, J. R., T. M. Williams, R. F. Siliciano, and J. N. Blankson. 2006. Maintenance of viral suppression in HIV-1-infected HLA-B*57+ elite suppressors despite CTL escape mutations. J. Exp. Med. 203**:**1357-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailey, J. R., H. Zhang, B. W. Wegweiser, H. C. Yang, L. Herrera, A. Ahonkhai, T. M. Williams, R. F. Siliciano, and J. N. Blankson. 2007. Evolution of HIV-1 in an HLA-B*57-positive patient during virologic escape. J. Infect. Dis. 196**:**50-55. [DOI] [PubMed] [Google Scholar]
- 6.Blaak, H., M. Brouwer, L. J. Ran, F. de Wolf, and H. Schuitemaker. 1998. In vitro replication kinetics of human immunodeficiency virus type 1 (HIV-1) variants in relation to virus load in long-term survivors of HIV-1 infection. J. Infect. Dis. 177**:**600-610. [DOI] [PubMed] [Google Scholar]
- 7.Blankson, J. N., J. R. Bailey, S. Thayil, H. C. Yang, K. Lassen, J. Lai, S. K. Gandhi, J. D. Siliciano, T. M. Williams, and R. F. Siliciano. 2007. Isolation and characterization of replication-competent human immunodeficiency virus type 1 from a subset of elite suppressors. J. Virol. 81**:**2508-2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boutwell, C. L., C. F. Rowley, and M. Essex. 2009. Reduced viral replication capacity of human immunodeficiency virus type 1 subtype C caused by cytotoxic-T-lymphocyte escape mutations in HLA-B57 epitopes of capsid protein. J. Virol. 83**:**2460-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brockman, M. A., A. Schneidewind, M. Lahaie, A. Schmidt, T. Miura, I. Desouza, F. Ryvkin, C. A. Derdeyn, S. Allen, E. Hunter, J. Mulenga, P. A. Goepfert, B. D. Walker, and T. M. Allen. 2007. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J. Virol. 81**:**12608-12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brockman, M. A., G. O. Tanzi, B. D. Walker, and T. M. Allen. 2006. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J. Virol. Methods 131**:**134-142. [DOI] [PubMed] [Google Scholar]
- 11.Brumme, Z. L., C. J. Brumme, J. Carlson, H. Streeck, M. John, Q. Eichbaum, B. L. Block, B. Baker, C. Kadie, M. Markowitz, H. Jessen, A. D. Kelleher, E. Rosenberg, J. Kaldor, Y. Yuki, M. Carrington, T. M. Allen, S. Mallal, M. Altfeld, D. Heckerman, and B. D. Walker. 2008. Marked epitope- and allele-specific differences in rates of mutation in human immunodeficiency type 1 (HIV-1) Gag, Pol, and Nef cytotoxic T-lymphocyte epitopes in acute/early HIV-1 infection. J. Virol. 82**:**9216-9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brumme, Z. L., C. J. Brumme, D. Heckerman, B. T. Korber, M. Daniels, J. Carlson, C. Kadie, T. Bhattacharya, C. Chui, J. Szinger, T. Mo, R. S. Hogg, J. S. Montaner, N. Frahm, C. Brander, B. D. Walker, and P. R. Harrigan. 2007. Evidence of differential HLA class I-mediated viral evolution in functional and accessory/regulatory genes of HIV-1. PLoS Pathog. 3**:**e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell, T. B., K. Schneider, T. Wrin, C. J. Petropoulos, and E. Connick. 2003. Relationship between in vitro human immunodeficiency virus type 1 replication rate and virus load in plasma. J. Virol. 77**:**12105-12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chin, B. S., J. Choi, J. G. Nam, M. K. Kee, S. D. Suh, J. Y. Choi, C. Chu, and S. S. Kim. 2006. Inverse relationship between viral load and genotypic resistance mutations in Korean patients with primary HIV type 1 infections. AIDS Res. Hum. Retroviruses 22**:**1142-1147. [DOI] [PubMed] [Google Scholar]
- 15.Chopera, D. R., Z. Woodman, K. Mlisana, M. Mlotshwa, D. P. Martin, C. Seoighe, F. Treurnicht, D. A. de Rosa, W. Hide, S. A. Karim, C. M. Gray, and C. Williamson. 2008. Transmission of HIV-1 CTL escape variants provides HLA-mismatched recipients with a survival advantage. PLoS Pathog. 4**:**e1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deacon, N. J., A. Tsykin, A. Solomon, K. Smith, M. Ludford-Menting, D. J. Hooker, D. A. McPhee, A. L. Greenway, A. Ellett, C. Chatfield, V. A. Lawson, S. Crowe, A. Maerz, S. Sonza, J. Learmont, J. S. Sullivan, A. Cunningham, D. Dwyer, D. Dowton, and J. Mills. 1995. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 270**:**988-991. [DOI] [PubMed] [Google Scholar]
- 17.Deeks, S. G., R. Hoh, T. B. Neilands, T. Liegler, F. Aweeka, C. J. Petropoulos, R. M. Grant, and J. N. Martin. 2005. Interruption of treatment with individual therapeutic drug classes in adults with multidrug-resistant HIV-1 infection. J. Infect. Dis. 192**:**1537-1544. [DOI] [PubMed] [Google Scholar]
- 18.Deeks, S. G., and B. D. Walker. 2007. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 27**:**406-416. [DOI] [PubMed] [Google Scholar]
- 19.Deval, J., K. L. White, M. D. Miller, N. T. Parkin, J. Courcambeck, P. Halfon, B. Selmi, J. Boretto, and B. Canard. 2004. Mechanistic basis for reduced viral and enzymatic fitness of HIV-1 reverse transcriptase containing both K65R and M184V mutations. J. Biol. Chem. 279**:**509-516. [DOI] [PubMed] [Google Scholar]
- 20.Draenert, R., S. Le Gall, K. J. Pfafferott, A. J. Leslie, P. Chetty, C. Brander, E. C. Holmes, S. C. Chang, M. E. Feeney, M. M. Addo, L. Ruiz, D. Ramduth, P. Jeena, M. Altfeld, S. Thomas, Y. Tang, C. L. Verrill, C. Dixon, J. G. Prado, P. Kiepiela, J. Martinez-Picado, B. D. Walker, and P. J. Goulder. 2004. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J. Exp. Med. 199**:**905-915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emu, B., E. Sinclair, H. Hatano, A. Ferre, B. Shacklett, J. N. Martin, J. M. McCune, and S. G. Deeks. 2008. HLA class I-restricted T-cell responses may contribute to the control of human immunodeficiency virus infection, but such responses are not always necessary for long-term virus control. J. Virol. 82**:**5398-5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feeney, M. E., Y. Tang, K. Pfafferott, K. A. Roosevelt, R. Draenert, A. Trocha, X. G. Yu, C. Verrill, T. Allen, C. Moore, S. Mallal, S. Burchett, K. McIntosh, S. I. Pelton, M. A. St. John, R. Hazra, P. Klenerman, M. Altfeld, B. D. Walker, and P. J. Goulder. 2005. HIV-1 viral escape in infancy followed by emergence of a variant-specific CTL response. J. Immunol. 174**:**7524-7530. [DOI] [PubMed] [Google Scholar]
- 23.Frahm, N., S. Adams, P. Kiepiela, C. H. Linde, H. S. Hewitt, M. Lichterfeld, K. Sango, N. V. Brown, E. Pae, A. G. Wurcel, M. Altfeld, M. E. Feeney, T. M. Allen, T. Roach, M. A. St. John, E. S. Daar, E. Rosenberg, B. Korber, F. Marincola, B. D. Walker, P. J. Goulder, and C. Brander. 2005. HLA-B63 presents HLA-B57/B58-restricted cytotoxic T-lymphocyte epitopes and is associated with low human immunodeficiency virus load. J. Virol. 79**:**10218-10225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goepfert, P. A., W. Lumm, P. Farmer, P. Matthews, A. Prendergast, J. M. Carlson, C. A. Derdeyn, J. Tang, R. A. Kaslow, A. Bansal, K. Yusim, D. Heckerman, J. Mulenga, S. Allen, P. J. Goulder, and E. Hunter. 2008. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J. Exp. Med. 205**:**1009-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goulder, P. J., R. E. Phillips, R. A. Colbert, S. McAdam, G. Ogg, M. A. Nowak, P. Giangrande, G. Luzzi, B. Morgan, A. Edwards, A. J. McMichael, and S. Rowland-Jones. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 3**:**212-217. [DOI] [PubMed] [Google Scholar]
- 26.Harrigan, P. R., S. Bloor, and B. A. Larder. 1998. Relative replicative fitness of zidovudine-resistant human immunodeficiency virus type 1 isolates in vitro. J. Virol. 72**:**3773-3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harrigan, P. R., I. Kinghorn, S. Bloor, S. D. Kemp, I. Najera, A. Kohli, and B. A. Larder. 1996. Significance of amino acid variation at human immunodeficiency virus type 1 reverse transcriptase residue 210 for zidovudine susceptibility. J. Virol. 70**:**5930-5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hecht, F. M., L. Wang, A. Collier, S. Little, M. Markowitz, J. Margolick, J. M. Kilby, E. Daar, B. Conway, and S. Holte. 2006. A multicenter observational study of the potential benefits of initiating combination antiretroviral therapy during acute HIV infection. J. Infect. Dis. 194**:**725-733. [DOI] [PubMed] [Google Scholar]
- 29.Honeyborne, I., A. Prendergast, F. Pereyra, A. Leslie, H. Crawford, R. Payne, S. Reddy, K. Bishop, E. Moodley, K. Nair, M. van der Stok, N. McCarthy, C. M. Rousseau, M. Addo, J. I. Mullins, C. Brander, P. Kiepiela, B. D. Walker, and P. J. Goulder. 2007. Control of human immunodeficiency virus type 1 is associated with HLA-B*13 and targeting of multiple gag-specific CD8+ T-cell epitopes. J. Virol. 81**:**3667-3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeeninga, R. E., W. Keulen, C. Boucher, R. W. Sanders, and B. Berkhout. 2001. Evolution of AZT resistance in HIV-1: the 41-70 intermediate that is not observed in vivo has a replication defect. Virology 283**:**294-305. [DOI] [PubMed] [Google Scholar]
- 31.Kassutto, S., K. Maghsoudi, M. N. Johnston, G. K. Robbins, N. C. Burgett, P. E. Sax, D. Cohen, E. Pae, B. Davis, K. Zachary, N. Basgoz, E. M. D'Agata, V. DeGruttola, B. D. Walker, and E. S. Rosenberg. 2006. Longitudinal analysis of clinical markers following antiretroviral therapy initiated during acute or early HIV type 1 infection. Clin. Infect. Dis. 42**:**1024-1031. [DOI] [PubMed] [Google Scholar]
- 32.Kiepiela, P., A. J. Leslie, I. Honeyborne, D. Ramduth, C. Thobakgale, S. Chetty, P. Rathnavalu, C. Moore, K. J. Pfafferott, L. Hilton, P. Zimbwa, S. Moore, T. Allen, C. Brander, M. M. Addo, M. Altfeld, I. James, S. Mallal, M. Bunce, L. D. Barber, J. Szinger, C. Day, P. Klenerman, J. Mullins, B. Korber, H. M. Coovadia, B. D. Walker, and P. J. Goulder. 2004. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 432**:**769-775. [DOI] [PubMed] [Google Scholar]
- 33.Kilby, J. M., H. Y. Lee, J. D. Hazelwood, A. Bansal, R. P. Bucy, M. S. Saag, G. M. Shaw, E. P. Acosta, V. A. Johnson, A. S. Perelson, and P. A. Goepfert. 2008. Treatment response in acute/early infection versus advanced AIDS: equivalent first and second phases of HIV RNA decline. AIDS 22**:**957-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirchhoff, F., T. C. Greenough, D. B. Brettler, J. L. Sullivan, and R. C. Desrosiers. 1995. Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N. Engl. J. Med. 332**:**228-232. [DOI] [PubMed] [Google Scholar]
- 35.Lassen, K. G., M. A. Lobritz, J. R. Bailey, S. Johnston, S. Nguyen, B. Lee, T. Chou, R. F. Siliciano, M. Markowitz, and E. J. Arts. 2009. Elite suppressor-derived HIV-1 envelope glycoproteins exhibit reduced entry efficiency and kinetics. PLoS Pathog. 5**:**e1000377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leslie, A. J., K. J. Pfafferott, P. Chetty, R. Draenert, M. M. Addo, M. Feeney, Y. Tang, E. C. Holmes, T. Allen, J. G. Prado, M. Altfeld, C. Brander, C. Dixon, D. Ramduth, P. Jeena, S. A. Thomas, A. St. John, T. A. Roach, B. Kupfer, G. Luzzi, A. Edwards, G. Taylor, H. Lyall, G. Tudor-Williams, V. Novelli, J. Martinez-Picado, P. Kiepiela, B. D. Walker, and P. J. Goulder. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 10**:**282-289. [DOI] [PubMed] [Google Scholar]
- 37.Little, S. J., S. D. Frost, J. K. Wong, D. M. Smith, S. L. Pond, C. C. Ignacio, N. T. Parkin, C. J. Petropoulos, and D. D. Richman. 2008. Persistence of transmitted drug resistance among subjects with primary human immunodeficiency virus infection. J. Virol. 82**:**5510-5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maeda, Y., D. J. Venzon, and H. Mitsuya. 1998. Altered drug sensitivity, fitness, and evolution of human immunodeficiency virus type 1 with pol gene mutations conferring multi-dideoxynucleoside resistance. J. Infect. Dis. 177**:**1207-1213. [DOI] [PubMed] [Google Scholar]
- 39.Martinez-Picado, J., and M. A. Martinez. 2008. HIV-1 reverse transcriptase inhibitor resistance mutations and fitness: a view from the clinic and ex vivo. Virus Res. 134**:**104-123. [DOI] [PubMed] [Google Scholar]
- 40.Martinez-Picado, J., J. G. Prado, E. E. Fry, K. Pfafferott, A. Leslie, S. Chetty, C. Thobakgale, I. Honeyborne, H. Crawford, P. Matthews, T. Pillay, C. Rousseau, J. I. Mullins, C. Brander, B. D. Walker, D. I. Stuart, P. Kiepiela, and P. Goulder. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J. Virol. 80**:**3617-3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinez-Picado, J., A. V. Savara, L. Shi, L. Sutton, and R. T. D'Aquila. 2000. Fitness of human immunodeficiency virus type 1 protease inhibitor-selected single mutants. Virology 275**:**318-322. [DOI] [PubMed] [Google Scholar]
- 42.Martinez-Picado, J., A. V. Savara, L. Sutton, and R. T. D'Aquila. 1999. Replicative fitness of protease inhibitor-resistant mutants of human immunodeficiency virus type 1. J. Virol. 73**:**3744-3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Migueles, S. A., A. C. Laborico, H. Imamichi, W. L. Shupert, C. Royce, M. McLaughlin, L. Ehler, J. Metcalf, S. Liu, C. W. Hallahan, and M. Connors. 2003. The differential ability of HLA B*5701+ long-term nonprogressors and progressors to restrict human immunodeficiency virus replication is not caused by loss of recognition of autologous viral gag sequences. J. Virol. 77**:**6889-6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miura, T., M. A. Brockman, C. J. Brumme, Z. L. Brumme, J. M. Carlson, F. Pereyra, A. Trocha, M. M. Addo, B. L. Block, A. C. Rothchild, B. M. Baker, T. Flynn, A. Schneidewind, B. Li, Y. E. Wang, D. Heckerman, T. M. Allen, and B. D. Walker. 2008. Genetic characterization of human immunodeficiency virus type 1 in elite controllers: lack of gross genetic defects or common amino acid changes. J. Virol. 82**:**8422-8430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miura, T., M. A. Brockman, Z. L. Brumme, C. J. Brumme, F. Pereyra, A. Trocha, B. L. Block, A. Schneidewind, T. M. Allen, D. Heckerman, and B. D. Walker. 2009. HLA-associated alterations in replication capacity of chimeric NL4-3 viruses carrying gag-protease from elite controllers of human immunodeficiency virus type 1. J. Virol. 83**:**140-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miura, T., M. A. Brockman, A. Schneidewind, M. Lobritz, F. Pereyra, A. Rathod, B. L. Block, Z. L. Brumme, C. J. Brumme, B. Baker, A. C. Rothchild, B. Li, A. Trocha, E. Cutrell, N. Frahm, C. Brander, I. Toth, E. J. Arts, T. M. Allen, and B. D. Walker. 2009. HLA-B57/B*5801 human immunodeficiency virus type 1 elite controllers select for rare gag variants associated with reduced viral replication capacity and strong cytotoxic T-lymphocyte recognition. J. Virol. 83**:**2743-2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O'Brien, S. J., X. Gao, and M. Carrington. 2001. HLA and AIDS: a cautionary tale. Trends Mol. Med. 7**:**379-381. [DOI] [PubMed] [Google Scholar]
- 48.Pereyra, F., M. M. Addo, D. E. Kaufmann, Y. Liu, T. Miura, A. Rathod, B. Baker, A. Trocha, R. Rosenberg, E. Mackey, P. Ueda, Z. Lu, D. Cohen, T. Wrin, C. J. Petropoulos, E. S. Rosenberg, and B. D. Walker. 2008. Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J. Infect. Dis. 197**:**563-571. [DOI] [PubMed] [Google Scholar]
- 49.Quinn, T. C., M. J. Wawer, N. Sewankambo, D. Serwadda, C. Li, F. Wabwire-Mangen, M. O. Meehan, T. Lutalo, and R. H. Gray. 2000. Viral load and heterosexual transmission of human immunodeficiency virus type 1. Rakai Project Study Group. N. Engl. J. Med. 342**:**921-929. [DOI] [PubMed] [Google Scholar]
- 50.Quinones-Mateu, M. E., S. C. Ball, A. J. Marozsan, V. S. Torre, J. L. Albright, G. Vanham, G. van Der Groen, R. L. Colebunders, and E. J. Arts. 2000. A dual infection/competition assay shows a correlation between ex vivo human immunodeficiency virus type 1 fitness and disease progression. J. Virol. 74**:**9222-9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ross, L., N. Parkin, and R. Lanier. 2008. The number of HIV major NRTI mutations correlates directly with other antiretroviral-associated mutations and indirectly with replicative capacity and reduced drug susceptibility. AIDS Res. Hum. Retroviruses 24**:**617-620. [DOI] [PubMed] [Google Scholar]
- 52.Saez-Cirion, A., C. Lacabaratz, O. Lambotte, P. Versmisse, A. Urrutia, F. Boufassa, F. Barre-Sinoussi, J. F. Delfraissy, M. Sinet, G. Pancino, and A. Venet. 2007. HIV controllers exhibit potent CD8 T cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc. Natl. Acad. Sci. U. S. A. 104**:**6776-6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneidewind, A., M. A. Brockman, R. Yang, R. I. Adam, B. Li, S. Le Gall, C. R. Rinaldo, S. L. Craggs, R. L. Allgaier, K. A. Power, T. Kuntzen, C.-S. Tung, M. X. Labute, S. M. Mueller, T. Harrer, A. J. McMichael, P. J. R. Goulder, C. Aiken, C. Brander, A. D. Kelleher, and T. M. Allen. 2007. Escape from the dominant HLA-B27 restricted CTL response in Gag is associated with a dramatic reduction in HIV-1 replication. J. Virol. 81**:**12382-12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schneidewind, A., Y. Tang, M. A. Brockman, E. G. Ryland, J. Dunkley-Thompson, J. C. Steel-Duncan, M. A. St. John, J. A. Conrad, S. A. Kalams, F. Noel, T. M. Allen, C. D. Christie, and M. E. Feeney. 2009. Maternal transmission of human immunodeficiency virus escape mutations subverts HLA-B57 immunodominance but facilitates viral control in the haploidentical infant. J. Virol. 83**:**8616-8627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Troyer, R. M., K. R. Collins, A. Abraha, E. Fraundorf, D. M. Moore, R. W. Krizan, Z. Toossi, R. L. Colebunders, M. A. Jensen, J. I. Mullins, G. Vanham, and E. J. Arts. 2005. Changes in human immunodeficiency virus type 1 fitness and genetic diversity during disease progression. J. Virol. 79**:**9006-9018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Villena, C., J. G. Prado, M. C. Puertas, M. A. Martinez, B. Clotet, L. Ruiz, N. T. Parkin, L. Menendez-Arias, and J. Martinez-Picado. 2007. Relative fitness and replication capacity of a multinucleoside analogue-resistant clinical human immunodeficiency virus type 1 isolate with a deletion of codon 69 in the reverse transcriptase coding region. J. Virol. 81**:**4713-4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weber, J., B. Chakraborty, J. Weberova, M. D. Miller, and M. E. Quinones-Mateu. 2005. Diminished replicative fitness of primary human immunodeficiency virus type 1 isolates harboring the K65R mutation. J. Clin. Microbiol. 43**:**1395-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.White, K. L., N. A. Margot, T. Wrin, C. J. Petropoulos, M. D. Miller, and L. K. Naeger. 2002. Molecular mechanisms of resistance to human immunodeficiency virus type 1 with reverse transcriptase mutations K65R and K65R+M184V and their effects on enzyme function and viral replication capacity. Antimicrob. Agents Chemother. 46**:**3437-3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoshimura, K., R. Feldman, E. Kodama, M. F. Kavlick, Y. L. Qiu, J. Zemlicka, and H. Mitsuya. 1999. In vitro induction of human immunodeficiency virus type 1 variants resistant to phosphoralaninate prodrugs of Z-methylenecyclopropane nucleoside analogues. Antimicrob. Agents Chemother. 43**:**2479-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zimmer, J. M., F. Roman, C. Lambert, A. Jonckheer, A. Vazquez, J. M. Plesseria, J. Y. Servais, K. Covens, J. Weber, K. Van Laethem, J. C. Schmit, A. M. Vandamme, M. E. Quinones-Mateu, and M. De Maeyer. 2008. Impact on replicative fitness of the G48E substitution in the protease of HIV-1: an in vitro and in silico evaluation. J. Acquir. Immune Defic. Syndr. 48**:**255-262. [DOI] [PubMed] [Google Scholar]