IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia (original) (raw)
. Author manuscript; available in PMC: 2010 Jul 12.
Published in final edited form as: Nat Med. 2008 Feb 10;14(3):275–281. doi: 10.1038/nm1710
Abstract
Emerging evidence supports the concept that T helper type 17 (TH17) cells, in addition to mediating autoimmunity, have key roles in mucosal immunity against extracellular pathogens. Interleukin-22 (IL-22) and IL-17A are both effector cytokines produced by the TH17 lineage, and both were crucial for maintaining local control of the Gram-negative pulmonary pathogen, Klebsiella pneumoniae. Although both cytokines regulated CXC chemokines and granulocyte colony–stimulating factor production in the lung, only IL-22 increased lung epithelial cell proliferation and increased transepithelial resistance to injury. These data support the concept that the TH17 cell lineage and its effector molecules have evolved to effect host defense against extracellular pathogens at mucosal sites.
IL-17 is a proinflammatory cytokine implicated in many autoimmune diseases1–3. Recently, it has been shown that IL-17–producing T cells belong to a unique subset of T cells, TH17 cells, that develop under the control of transforming growth factor-β and IL-6 in mice and IL-23 and IL-1β in humans via the transcription factor RORγt (refs. 4–10). IL-17A and IL-17F are important molecules in neutrophil recruitment and granulopoiesis2. IL-23 has also been shown to be crucial as an upstream regulator of IL-17 production in vivo11,12. Mice with a homozygous deletion of the gene encoding IL-17 receptor A (Il17ra), which can bind both IL-17A and IL-17F (ref. 13), are susceptible to lung infection with the Gram-negative bacteria K. pneumoniae. These mice do not mount a normal mucosal granulocyte colony–stimulating factor (G-CSF) response, which leads to defective granulopoiesis in response to the infection14. Antibodies to IL-17A have also been shown to impair intra-abdominal abscess formation in response to Bacteroides fragilis15 and host defense against Bordetella pertussis16 and Mycoplasma pulmonis17.
IL-22 is an IL-10 family cytokine member18 that was originally thought to be predominantly expressed in TH1 cells and has been shown to induce the antimicrobial peptides human β-defensin 2 and β-defensin 3; with IL-17A, IL-22 augments expression of the calcium-binding proteins S100A7, S100A8 and S100A9 in skin keratinocytes19,20. It has recently been shown that TH17 cells grown in vitro also produce IL-22, and in vitro production was optimized when IL-23 was present20.
On the basis of this evidence, we hypothesized that IL-22 may be a crucial effector molecule in host defense against Gram-negative bacterial infection in the lung. Here we show that human bronchial epithelial (HBE) cells express IL-22R and that IL-22 and IL-17 induce host defense genes, as well as increase the clonogenic potential of HBE cells. Using an experimental model of Gram-negative pneumonia, we show that IL-22 is produced in a time-dependent fashion, similarly to IL-17A and IL-17F (ref. 12), and is regulated by IL-23. Neutralization of IL-22 resulted in a marked bacterial dissemination from the lung that was exacerbated by the absence of IL-17A. IL-22 also increased the expression of host defense genes in mouse lung epithelium and, among these, lipocalin-2 was required for IL-22–augmented epithelial antimicrobial activity in vitro. These data demonstrate that IL-22 has a crucial role in mucosal host defense, and they support the concept that the TH17 cell lineage and the effector molecules produced by these cells evolved to effect host defense against extracellular pathogens at mucosal sites.
Results
The production of IL-17A and IL-17F in infected tissue is regulated by IL-23 (ref. 12); however, IL-23 has been shown to be only marginally involved in primary infections with pathogens that require TH1 immunity, such as Mycobacterium tuberculosis21. Similar to mice deficient in IL-23 (_Il23a_−/−), mice deficient in IL-17RA, which is required for IL-17A and IL-17F signaling13, are not more susceptible to H37Rv M. tuberculosis infection than wild-type mice (Supplementary Fig. 1 online). Moreover, IL-17RA signaling is not required for host defense against another TH1-dependent pathogen, Listeria monocytogenes (Supplementary Fig. 2 online), and these mice have normal interferon-γ responses upon antigen recall to the pathogen (Supplementary Fig. 2). This is consistent with normal TH1 immunity and host resistance against L. monocytogenes in _Il22_−/− mice22. These data suggest that TH17 cells may be important for host defense against extracellular pathogens because they produce IL-17A and IL-22.
Expression of IL-22R and activity of IL-22 on HBE cells
IL-22R is expressed on polarized, ciliated, primary HBE cells (Fig. 1a), and its expression at the mRNA level is not altered after 24-h stimulation with IL-17A, IL-22 or both cytokines (Supplementary Fig. 3a online). To examine gene expression, we stimulated HBE cells with 10 ng/ml IL-17A, 20 ng/ml IL-22 or a combination of both for 24 h and harvested total RNA for microarray analysis. The combination of IL-22 with IL-17A markedly induced several host defense genes, including those encoding human β-defensin 2 (DEFB4), psoriasin (S100A7) and calgranulin C (S100A12), as well as IL19, CSF_3, IL1F9, DUOX2, CXCL1, CXCL5, CXCL_9 and CCL3 (Fig. 1b). We confirmed CSF3 induction both at the mRNA level by real-time PCR (Supplementary Fig. 3b) and at the protein level (Fig. 1c). Moreover, we observed a significant increase in IL-17A–induced IL-6 production in HBE cells by IL-22 (Fig. 1d).
Figure 1.

Primary HBE cells express IL-22R, and stimulation with IL-22 and IL-17A leads to the upregulation of host defense genes and increases clonogenic frequency. (a) Primary HBE cells were stained with a goat antibody to IL-22R. Fifteen percent of HBE cells were positive for IL-22R (red); controls with secondary antibody only are shown in purple. (b) Heat map of genes expressed in HBE cells after 24 h stimulation with media, 20 ng/ml IL-22, 10 ng/ml IL-17A or a combination of 20 ng/ml IL-22 and 10 ng/ml of IL-17A (n = 3 per condition). (c) G-CSF abundance in basolateral media of HBE cells after stimulation with IL-17A, IL-22 or both (n = 5–6 from three individual donors per condition). (d) IL6 abundance in basolateral media of HBE cells after stimulation with IL-17A, IL-22 or both (n = 5–6 from three individual donors per condition). (e) Primary HBE cells were seeded at specific densities into a 96-well plate containing media, 200 ng/ml of IL-22 or 100 ng/ml of IL-17A. IL-22 increased the clonogenicity of cells more than IL-17A or media alone did (n = 3–4 from three individual donors per condition). (f) We developed 10-μm wounds in primary HBE cells before stimulating them with media alone or 20 ng/ml of IL-22 (n = 5–6 from three individual donors per condition). For c–f, *P < 0.05 compared with media control and error bars represent means ± s.e.m.
As IL-22 has been implicated in wound repair in the skin5, we examined whether IL-22 increases the clonogenic frequency of HBE cells and whether IL-22 affects epithelial barrier function. Treatment with IL-22 at a concentration of 200 ng/ml (but not 20 ng/ml, data not shown) significantly increased the clonogenic potential of HBE cells compared to treatment with media alone, or compared to treatment with 100 ng/ml of IL-17A (Fig. 1e). We next evaluated whether IL-22 affected the maintenance of transepithelial resistance in injured epithelium. We created a 10-μM wound in well-differentiated, polarized HBE cells and found that the addition of IL-22 at a dose as low as 20 ng/ml significantly enhanced recovery of epithelial resistance (Fig. 1f). The recovery of resistance occurred as early as 6 h after the injury, whereas control cells recovered over a 36-h time period. IL-17A at doses of 10 or 100 ng/ml had no activity in this assay (data not shown).
Requirement of IL-22 in host defense against K. pneumoniae
To examine whether IL-22 is important for mucosal host defense in the lung, we used a well-established model of K. pneumoniae pulmonary infection. Similar to the previously observed time course for IL-17A and IL-17F production (ref. 12), IL-22 was detectable at the message level as early as 6 h (Fig. 2a) and at the protein level in lung homogenate as early as 16 h in mice infected with K. pneumoniae (Fig. 2b), and its expression was significantly elevated (P < 0.05) compared to that in uninfected mice. Additionally, it has been reported that TH17 cells express CCR4 and CCR6 (ref. 23), and thus we assessed the expression of the genes encoding the ligands for these receptors, Ccl17, Ccl20 and Ccl22. Both Ccl17 and Ccl20 expression was significantly increased (P < 0.05) at 4 h in lung tissue of mice infected with K. pneumoniae (Supplementary Fig. 4 online), and Ccl20 expression continued to increase at 16 h. Ccl22 expression, however, showed a steady decline in this model (Supplementary Fig. 4).
Figure 2.
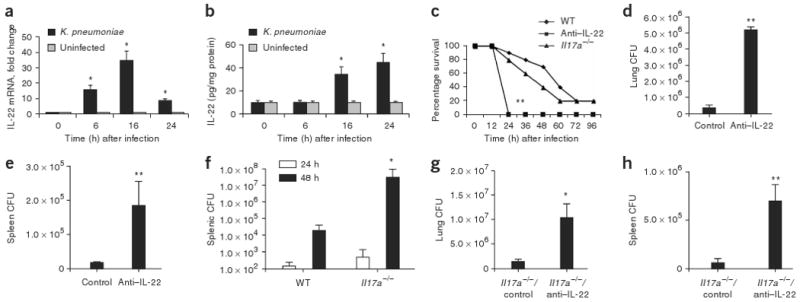
IL-22 expression is elevated in mice infected with K. pneumoniae, and neutralizing IL-22 leads to decreased bacterial clearance from lung and spleen. Mice were infected with K. pneumoniae and killed at the designated time points. (a) IL-22 mRNA was measured by quantitative real-time PCR. Error bars represent means ± s.e.m. (b) IL-22 protein abundance was measured by ELISA and normalized per mg of protein. Error bars represent means ± s.e.m. (c) C57BL/6 and _Il17a_−/− mice were infected with K. pneumoniae and evaluated for survival. Mice given antibody to IL-22 (anti–IL-22) were moribund at 24 h compared to WT or _Il17a_−/− mice (n = 8–10, **P < 0.01 by log-rank test compared to _Il17a_−/− or WT mice). (d,e) K. pneumoniae burden in the lung (d) or spleen (e) 24 h after infection in control or anti–IL-22–treated mice. (f) K. pneumoniae burden in the spleen in WT or _Il17a_−/− mice at 24 and 48 h after infection. (g) K. pneumoniae burden in the lung 24 h after infection in control or anti–IL-22–treated _Il17a_−/− mice. n = 5–6 per group, *P < 0.05 compared with controls for panels a,b,f,g and **P < 0.01 compared with controls for d,e. All values are mean ± s.e.m. (h) K. pneumoniae burden in the spleen 24 h after infection in control or anti–IL-22–treated _Il17a_−/− mice (n = 5–6 per group and **P < 0.01 compared with controls).
We then investigated the potential cellular sources of IL-22 in our model system. _Rag2_−/−_Il2rg_−/− mice (deficient in natural killer, T and B cells) were infected with K. pneumoniae, and their lungs were harvested at 24 h after infection (Supplementary Fig. 5a online). As expected, _Rag2_−/−_Il2rg_−/− mice produced no detectable IL-22 compared to their infected, wild-type (WT) counterparts. To further confirm that T cells produce IL-22 in response to K. pneumoniae, we isolated CD90+ T cells from lungs harvested from infected mice and uninfected controls and then assessed IL-22 precursor frequency by enzyme-linked immunosorbent spot (ELISPOT). There was a significant increase in IL-22–producing CD90+ cells in _K. pneumoniae_–infected mice compared to uninfected controls (Supplementary Fig. 5b). There were no spots from CD90− cells (data not shown).
We next determined the survival of WT and _Il17a_−/− mice treated with control antibody or antibody to IL-22 and challenged with 1 × 104 colony-forming units (CFU) of K. pneumoniae (Fig. 2c). Compared to WT mice or _Il17a_−/− mice given control antibody, WT mice treated with antibody to IL-22 were moribund 24 h after infection and had to be killed. Mice treated with antibody to IL-22 had significantly (P < 0.05) greater numbers of CFU in the lung compared to control mice (Fig. 2d) and also showed increased dissemination to the spleen (Fig. 2e). Administration of antibodies to IL-22 did not affect induction of IL-17A in this model (Supplementary Fig. 6 online). Consistent with a more crucial role for IL-22 than for IL-17A in mucosal host defense, increased dissemination to the spleen was not detectable in _Il17a_−/− mice until 48 h after infection (Fig. 2f). Moreover, administration of IL-22–specific antibody to _Il17a_−/− mice before infection with K. pneumoniae resulted in significantly greater bacterial growth in the lung (Fig. 2g) and significantly more bacterial dissemination to the spleen (Fig. 2h) at 24 h after infection. To exclude the possibility of IL-17F compensation in _Il17a_−/− mice, we also assessed IL-17F levels in WT and _Il17a_−/− mice, and, at 18 h after infection, IL-17F was similarly induced in both strains of mice (data not shown).
Similarly to _Il17ra_−/− (ref. 14) and _Il23a_−/− (ref. 12) mice, _Il17a_−/− mice had significantly lower amounts of G-CSF and CXCL1 in the bronchoalveolar lavage (BAL) fluid at 24 h compared to WT mice (Fig. 3a,b). However, compared to WT mice, mice treated with antibody to IL-22 showed no difference in BAL G-CSF and CXCL1 protein concentrations (Fig. 3a,b). Despite the lack of a function for IL-22 in regulation of G-CSF and CXCL1 in WT mice, _Il17a_−/− mice given antibody to IL-22 had lower Csf3 abundance compared to _Il17a_−/− mice given control antibody, but this difference was not statistically significant. Moreover, CXCL1 expression was partially dependent on both IL-17A and IL-22, as it was lower in _Il17a_−/− mice treated with antibody to IL-22 (Fig. 3b). In contrast to G-CSF, IL-22 was crucial for IL-6 production in the lung, as both antibody to IL-22–treated WT and _Il17a_−/− mice had statistically significant reductions in BAL IL-6 abundance 24 h after infection (Fig. 3c). A similar pattern was also observed with CCL3 (Fig. 3d), where mice treated with antibody to IL-22 had significantly lower CCL3 in the BAL fluid at 24 h after infection compared to control mice. In the absence of IL-17A, antibody neutralization of IL-22 resulted in a further reduction in IL-6 and CCL3 abundance, suggesting a requirement for both IL-17A and IL-22 in this response.
Figure 3.

IL-22 and IL-17A regulation of pulmonary cytokines and chemokines in K. pneumoniae infection. WT or _Il17a_−/− mice were infected with K. pneumoniae and treated with 50 μg anti–IL-22 or control antibody intratracheally. At 24 h after antibody treatment, mice were killed and BAL was assayed for G-CSF (a), CXCL1 (b), IL6 (c) or CCL3 (d) by Luminex (n = 5–6 per group and *P < 0.05 or **P < 0.01 compared with controls. Error bars represent means ± s.e.m.).
IL-22 and its role in chemokine expression require IL-23
IL-23 is a key factor for the survival of TH17 cells, and thus we investigated whether IL-23 is required for IL-22 elaboration in vivo. The induction of IL-22 mRNA requires IL-23, as IL-22 expression was markedly attenuated in _Il23a_−/− mice (Fig. 4a). We next examined whether administration of recombinant IL-22, IL-17A or both intratracheally 12 h after infection with 1 × 103 CFU of K. pneumoniae could augment mucosal host defense. Even at this lower bacterial inoculum, as previously reported, _Il23a_−/− mice treated with vehicle had higher organism burdens in the lung compared to WT mice (Fig. 4b). Treatment with recombinant IL-22, IL-17A or both significantly reduced recoverable CFU in the lung at 24 h (Fig. 4b). Furthermore, there was significantly less bacterial dissemination to the spleen in mice given recombinant IL-22, IL-17A or both cytokines (Fig. 4c).
Figure 4.

IL-22 production in vivo requires IL-23. (a) WT and _Il23a_−/− mice were infected with 1 × 103 CFU K. pneumoniae and killed at the designated time points after infection. IL-22 mRNA was measured by quantitative real-time PCR and graphed as fold change over time (n = 5–6 per group. *P < 0.05 compared with _Il23a_−/−-veh. Error bars represent means ± s.e.m.). WT and _Il23a_−/− mice were infected with K. pneumoniae, and 12 h after infection, _Il23a_−/− mice were treated with 1 μg of vehicle (PBS), IL-22, IL-17A or both cytokines. CFU were plated from lung homogenate (b) or spleen (c) at the 24-h time point (n = 5–6 per group. *P < 0.05 compared with all other groups. Error bars represent means ± s.e.m.).
As IL-22R is expressed in epithelial cells, we tested whether this is the site of action of recombinant IL-22 in the lung using in situ hybridization for Cxcl1 and the closely related Cxcl2 and Cxcl9 (Fig. 5), as these genes have been shown to be regulated by IL-17A in vivo in pulmonary infection14,24. WT mice showed Cxcl9 transcripts in both airway cells and alveolar epithelial cells (Fig. 5). _Il23a_−/− mice showed less staining in lung tissue (Fig. 5); however, administration of IL-22, IL-17A or the combination of IL-22 with IL-17A augmented hybridization signals for Cxcl1, Cxcl2 and Cxcl9 expression (Fig. 5). Moreover, IL-22 treatment resulted in increased transcripts for Cxcl1, Cxcl2 and Cxcl9 in bronchiole epithelium (Fig. 5, black arrows), whereas administration of IL-17A or the combination of IL-22 and IL-17A increased expression in bronchiole epithelium as well as in distal lung epithelium (Fig. 5, gray arrows).
Figure 5.

_Il23a_−/− mice rescued with IL-22, IL-17A and both cytokines have augmented Cxcl1, Cxcl2 and Cxcl9 expression in airways and alveolar epithelium. WT and _Il23a_−/− mice were infected with K. pneumoniae, and 12 h after infection, _Il23a_−/− mice were rescued with 1 μg of vehicle (PBS), IL-22, IL-17A or both cytokines. A representative in situ hybridization for Cxcl1, Cxcl2 and Cxcl9 is depicted. _Il23a_−/− mice had reduced staining for Cxcl1, Cxcl2 and Cxcl9 in lung tissue compared with WT mice. After administration of IL-22, IL-17A or combination of IL-22 and IL-17A, _Il23a_−/− mice had significantly increased expression of all three chemokines both in distal airway (black arrows) and alveolar epithelium (gray arrows).
IL-22 regulates antimicrobial activity of lung epithelium
To investigate the gene expression and antimicrobial activity of IL-22 in mouse lung epithelium, we studied polarized primary mouse tracheal epithelial cells (MTECs). Cells were stimulated with 20 ng/ml IL-22 and 10 ng/ml IL-17, and Csf3 protein abundance was significantly (P < 0.05) increased by the combination of IL-22 and IL-17 (Fig. 6a). This was also the case with Cxcl1 (Fig. 6b). To analyze gene expression in these cells, we performed microarray analysis. There was upregulation of several host defense genes, including Lcn2 (encoding lipocalin-2), Cxcl1, Cxcl5, Pigr (encoding the polymeric immunoglobulin receptor) and Cxcl9 (Fig. 6c). Lipocalin-2 is a protein known to have a function in innate immune response through its ability to sequester iron from bacterial organisms such as Escherichia coli25,26.
Figure 6.

IL-22 augments the antimicrobial activity of MTEC cells in vitro. IL-22 is produced abundantly from cells isolated from lymph node explants of individuals with cystic fibrosis. MTECs from WT mice were stimulated for 24 h with media (control), 20 ng/ml recombinant mouse IL-22, 10 ng/ml IL-17A or both cytokines. Csf3 (a) and Cxcl1 (b) abundance was measured in basolateral supernatants (n = 6 per condition, *P < 0.05 compared with media control. Error bars represent means ± s.e.m.). (c) Heat map of MTECs after 24 h stimulation with media, 20 ng/ml IL-22, 10 ng/ml IL-17A or both cytokines (n = 3 per condition). (d) MTECs harvested from _Lcn2_−/− mice were stimulated for 24 h with media, IL-22, IL-17A or the combination of both and subsequently infected with 1 × 104 CFU of K. pneumoniae apically for 8 h, and apical CFUs were then determined by plating several dilutions of the supernatants on tryptic soy agar (n = 3–5 per condition, *P < 0.05 compared with media control). (e) BAL samples from consenting subjects with cystic fibrosis and subjects without cystic fibrosis (control) were obtained. IL-23, IL-17A, IL-17F and IL-22 protein abundance was measured by ELISA (age range 2–18 years; n = 15 cystic fibrosis and n = 8 control; error bars represent means ± s.e.m.). T cells isolated from human explanted lymph nodes from individuals with cystic fibrosis (n = 5) and individuals without cystic fibrosis (n = 2) were unstimulated or stimulated with ConA, and IL-17A protein abundance (f) was measured by LINCOplex; IL-22 and IL-17F abundance (g) was measured by ELISA at 24–48 h.
To investigate whether IL-22 has an antimicrobial function in vitro, we applied K. pneumoniae apically to MTECs that had been pretreated with IL-22, IL-17A or both cytokines. IL-22 significantly (P < 0.05) augmented killing of K. pneumoniae (Fig. 6d). This was dependent on the expression of lipocalin-2 as IL-22–induced killing was significantly abrogated in MTEC cultures derived from _Lcn2_−/− mice compared to WT controls (Fig. 6d).
T cells from explanted cystic fibrosis lymph nodes produce IL-22
Gram-negative bacterial pneumonia is common in immunocompromised individuals such as organ transplant recipients27. In order to study IL-22 in this cohort, we obtained samples of BAL fluid from seven subjects who were lung transplant recipients with bacterial pneumonia. Although most subjects had elevated CXCL8 levels (a measure of sample integrity), none had any significant detectable levels of IL-22 or IL-17A (Supplementary Table 1 online). This was not surprising to us in light of the fact that all individuals were maintained on T cell immunosuppression to prevent rejection of their transplanted lungs. These data are in contrast to the elevated sputum IL-17A and IL-17F levels observed during Pseudomonas aeruginosa lung infection in subjects with cystic fibrosis who have normal T cell function28. On the basis of these observations, we investigated IL-22 expression during this gram-negative bacterial lung infection.
We studied banked BAL fluid samples from individuals with cystic fibrosis undergoing a clinical exacerbation with P. aeruginosa infection and from control individuals without cystic fibrosis. Compared to the controls, IL-23, IL-17A and IL-17F were significantly elevated in BAL fluid of subjects with cystic fibrosis (Fig. 6e). Of note, very little IL-22 was detected in these samples (Fig. 6e). Based on the fact that in our mouse pneumonia model, IL-22 levels were higher in lung tissue than in BAL, we sought to determine whether IL-22 responses were elevated in lung lymphoid tissue in humans. We isolated cells from explanted hilar lymph nodes from individuals with end-stage cystic fibrosis undergoing lung transplantation (n = 5), in addition to uninfected control lung lymph nodes from individuals without cystic fibrosis (n = 2). Compared to uninfected lung lymph nodes from individuals without cystic fibrosis, dissociated lymph nodes cells from _P. aeruginosa_–infected subjects with cystic fibrosis had significantly increased spontaneous production of IL-17A (Fig. 6f), IL-17F and IL-22 (Fig. 6g), and production of these cytokines were stimulated with concanavalin A (ConA), a glycoprotein that stimulates T lymphocyte proliferation (Fig. 6f,g).
Discussion
These data demonstrate that IL-22 has a more crucial role than IL-17A in mucosal host defense and in our model of experimental bacterial pneumonia. IL-22 expression in vivo is regulated by IL-23, and rescue of _Il23a_−/− mice is associated with increased lung epithelial expression of CXCL1, CXCL2 and CXCL9. Similar to the situation in _Il17ra_−/− mice, IL-17A was crucial for the induction of G-CSF, a key regulator of granulopoiesis in response to an infectious challenge14, in the lung. However, IL-22 was more important than IL-17A in regulating pulmonary IL-6 and CCL3 production. Studies in _Il17a_−/− mice show that IL-6 and CCL3 are coordinately regulated by both TH17 effectors, suggesting that the synergy observed with IL-17A and IL-22 in human bronchial epithelial cells in terms of gene expression is relevant in the in vivo setting as well. Additional activities of IL-22 not shared by IL-17A were the effect of increasing clonogenic frequencies of HBE cells, as well enhancing repair of transepithelial resistance. These activities of IL-22 may be crucial for maintaining barrier function at epithelial surfaces. In support of this, WT and _Il17a_−/− mice that received antibody to IL-22 had more rapid bacterial dissemination outside of the lung compared with mice given control antibody. In vitro MTEC studies show that a key gene regulated by IL-22 in the epithelium is lipocalin-2, which is required for epithelial host defense against K. pneumoniae in vitro. This is consistent with the fact that _Lcn2_−/− mice are susceptible to certain Gram-negative bacteria, depending upon which siderophores they express25,26.
Furthermore, we show that IL-17R signaling is dispensable for host defense to a primary challenge with M. tuberculosis or L. monocytogenes, two intracellular pathogens that require TH1 immunity. These data strongly suggest that the TH17 lineage evolved to mediate host defense at mucosal and serosal surfaces against extracellular pathogens. Additionally, neutralization of IL-17A has been shown to abrogate host defense against lung pathogens, such as K. pneumoniae and _M. pulmoni_s14,17, systemic infection with the fungus Candida albicans29, as well as in the setting of intra-abdominal infection, B. fragilis15 and E. coli30. Potential host defense mechanisms downstream from IL-22 and IL-17A, in addition to granulopoiesis, are the regulation of chemokine gradients and the mucosal production of antimicrobial proteins such as β-defensins31 and calgranulins20. It is noteworthy that IL-22 responses are significantly elevated in lung lymphoid tissue from human subjects with cystic fibrosis. This is consistent with elevated levels of antimicrobial peptides in the sputum and BAL of these individuals32 and may explain in part why bacteremia with P. aeruginosa is rare in these individuals.
Although neither IL-23 nor IL-17R signaling is required for host defense against primary infection with M. tuberculosis, vaccine-induced protection with the BCG tuberculosis vaccine requires the local recruitment of IL-17A–producing T cells24. In this study, IL-17A regulated the expression of CXCL9 and CXCL10, thereby regulating TH1 cell recruitment24. It has recently been shown that IL-17A is also required for vaccine-induced immunity against _S. pneumonia_e33 and B. pertussis16. The vaccine-induced immunity observed in S. pneumoniae infection was independent of antibody production33, suggesting that TH17 cells may regulate mucosal immunity apart from humoral immune responses. Thus, adjuvants that are capable of regulating IL-22 or IL-17A production may serve as robust adjuvants where one desires strong mucosal immunity. A caveat of this approach is the fact that TH17 cells are also important in autoimmunity1,34. Recently, it has been shown that IL-27 is a key negative regulator of TH17 immunity35,36 and may be crucial for limiting TH17-induced pathology in the setting of infection. In summary, both IL-22 and IL-17A have independent roles in maintaining host defense against K. pneumoniae, and these results raise the possibility for new therapies to use these cytokines to limit or prevent mucosal infections.
Methods
Human bronchial epithelial cell cultures
Primary HBE cells were provided by the Tissue Core Laboratory at the University of Pittsburgh or purchased from Cambrex (Lonza). We grew primary cells as polarized air-liquid interface (ALI) cultures as previously described28. We stimulated HBE cells apically and basolaterally for 24 h with media (control), recombinant human IL-22, IL-17A (both R&D Systems) or both cytokines.
Mouse tracheal epithelial cell cultures
We cultured MTECs as described by You et al.37. We stimulated cells growing at the ALI with IL-22 and IL-17A. We pretreated cells harvested from _Lcn2_−/− mice similarly and infected them with 1 × 104 CFU of K. pneumoniae apically for 8 h.
Staining for interleukin-22 receptor and flow cytometry
We stained HBE cells with polyclonal goat antibody to human IL-22R (Novus Biologicals) and Alexa Fluor 488–conjugated donkey antibody to goat IgG (Molecular Probes) and then detected stained cells with the FACSAria flow cytometer.
Limiting-dilution assay
To determine clonogenic frequency, we performed a limiting-dilution assay as previously described38. We sorted primary HBE cells with a FACSAria flow cytometer directly into media (control), IL-22 or IL-17A (R&D Systems). After 72 h, we fixed the cells and stained them with 0.5% crystal violet.
Wound-repair assay
We created 10-μm wounds in HBE cells with a pipette tip. We measured transepithelial resistance over 36 h. We then treated the cells with media, IL-22 or IL-17A.
Quantitative RT-PCR
We isolated total RNA from cultured cells and lung homogenate and used the real-time PCR detection system iCycler iQ (Bio-Rad) to detect the genes of interest. We purchased gene-specific primers and probes for IL-22, IL-22R, G-CSF, CCL17, CCL20 and CCL22, as well as CXCL1, CXCL2 and CXCL9, from Applied Biosystems. Data are expressed using the CT method and normalized to the housekeeping gene Rn18s (18s rRNA).
Microarray
Microarray was performed using the human 133A 2.0 and mouse 430 2.0 gene chip platforms (Affymetrix) by the University of Pittsburgh Genomics and Proteomics Core Laboratory. We analyzed the data with GeneSifter microarray analysis software.
Mice
We purchased 6–8-week-old male C57BL/6 and IL-23 p19 subunit–deficient mice from the National Institute of Cancer and Genentech, respectively. _Il17a_−/− mice were kindly provided by Y. Iwakura and _Lcn2_−/− mice were kindly provided by T. Mak. Common gamma (γc) _Rag2_−/− mice and their age-matched C57BL/10ScNCR controls were obtained from Taconic. Mice were housed in specific pathogen–free rooms within animal care facilities of the Children's Hospital of Pittsburgh. All mouse experiments were approved by the Children's Hospital of Pittsburgh Animal Research and Care Committee.
Infection model
We anesthetized mice using the isofluorane inhalation method (IsoFlo, Abbott Laboratories) and gave them 50 μl of K. pneumoniae strain 43816 serotype 2 via retropharyngeal instillation at an inoculum of 1 × 104 CFU/ml. We dissected the spleen and whole lung from the mice and homogenized them in PBS for CFU plating. For studies involving IL-22 neutralization, we gave WT or _Il17a_−/− mice either control antibody (goat IgG) or 50 μg of goat antibody to mouse IL-22 (R&D Systems) intratracheally immediately before infection with K. pneumoniae. For IL-22 and IL-17A rescue experiments, we gave _Il23a_−/− mice vehicle (PBS) or 1 μg of mouse recombinant IL-22, IL-17A or both cytokines 12 h after K. pneumoniae infection. We then killed the mice at 24 h after K. pneumoniae infection.
Interleukin-22, interleukin-17A and interleukin-17F protein determination
We used murine and human IL-22 Quantikine kits and a human IL-17F Duoset (R&D Systems) to quantify those cytokines. We measured IL-17A protein levels by a 22-plex mouse Luminex assay (Linco/Millipore).
Chemokine cDNA cloning and in situ hybridization
We obtained mouse cDNAs encompassing the open reading frames of CXCL1, CXCL2 and CXCL9 by RT-PCR through a two-step approach as described39, using primers based on mouse cDNA sequences available in GenBank. Template RNAs were from B16/M05 mouse adenocarcinoma cells (kindly provided by H. Okada) or mouse spleen cells. We performed stringent in situ hybridization (21-day exposures) as described40.
Interleukin-22 enzyme-linked immunosorbent spot
We infected C57BL/6 WT mice with K. pneumoniae and harvested whole lungs. We collected and plated CD90+ cells. We performed the ELISPOT assay with monoclonal antibody to mouse IL-22 and biotinylated antibody to mouse IL-22 (R&D Systems).
Human bronchoalveolar lavage samples
BAL samples (1–9) were provided by S.H. Informed consent was obtained from all subjects, and these studies were approved by the University of Pittsburgh Institutional Review Board.
Human lymph node cultures
Individuals with cystic fibrosis undergoing lung transplant at the University of Pittsburgh Medical Center and Children's Hospital of Pittsburgh consented to donate their explanted lung tissue for study. These studies were approved by the University of Pittsburgh Institutional Review Board. We dissected lung explants to extract hilar lymph nodes. We processed lymph nodes for single cell suspension and stimulated cells with Con A (Sigma) as positive controls.
Statistical analysis
All data are presented as the means ± s.e.m. We determined statistical significance (*P < 0.05 or **P < 0.01) by _t_-test or ANOVA. We performed survival analysis using the log-rank test.
Accession codes
Gene Expression Omnibus microarray accession code, GSE10240.
Supplementary Material
Supplementary Data
Acknowledgments
The authors would like to acknowledge support from the following Public Health Service grants: 5R01HL079142 (J.K.K.), P50HL084932, and P30DK072506 (J.K.K. and J.M.P.). We would like to thank Y. Iwakura (Center for Experimental Medicine, The Institute of Medical Science, The University of Tokyo) for the _Il17a_−/− mice, and T. Mak (University Health Network, Toronto) for the _Lcn2_−/− mice.
Footnotes
Author Contributions: S.J.A. performed all the experiments depicted in Figures 1, 2b,d,e,g,h, 3, 4, 6a–d and g and wrote the manuscript. Y.R.C. performed the experiment depicted in Figure 6f. M.F. and M.Z. performed experiments depicted in Figure 2a,c,f. D.J.A. provided MTECs and assisted in experiments depicted in Figure 6a–c. D.A.P. assisted in experiments depicted in Figures 2g,h and 3. P.J.D. assisted in experiments depicted in Figure 4. F.M. performed experiments depicted in Figure 6e. T.A.R. and K.G. performed in situ hybridization as shown in Figure 5. J.E. performed the experiments with L. monocytogenes (Supplementary Fig. 2). C.A.M. performed the experiments with M. tuberculosis (Supplementary Fig. 1). S.H. provided BAL samples and helpful discussions in experiments depicted in Supplementary Table 1. J.L.K., J.M.P. and M.M.M. supplied HBE cells and helped performed experiments for Figure 1. Y.I. provided _Il17A_−/− mice, as well as useful discussion in experimental design. J.K.K. was responsible for experimental design, microarray analysis and final manuscript preparation.
Note: Supplementary information is available on the Nature Medicine website.
References
- 1.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 2.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 3.Lubberts E. The role of IL-17 and family members in the pathogenesis of arthritis. Curr Opin Investig Drugs. 2003;4:572–577. [PubMed] [Google Scholar]
- 4.Harrington LE, et al. Interleukin 17–producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 5.Mangan PR, et al. Transforming growth factor-β induces development of the TH17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 6.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 7.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17–producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Ivanov II, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 9.Wilson NJ, et al. Development, cytokine profile and function of human interleukin 17–producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 10.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17–producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 11.Langrish CL, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Happel KI, et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med. 2005;202:761–769. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toy D, et al. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol. 2006;177:36–39. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- 14.Ye P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony–stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–528. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chung DR, et al. CD4+ T cells mediate abscess formation in intra-abdominal sepsis by an IL-17–dependent mechanism. J Immunol. 2003;170:1958–1963. doi: 10.4049/jimmunol.170.4.1958. [DOI] [PubMed] [Google Scholar]
- 16.Higgins SC, Jarnicki AG, Lavelle EC, Mills KH. TLR4 mediates vaccine-induced protective cellular immunity to Bordetella pertussis: role of IL-17–producing T cells. J Immunol. 2006;177:7980–7989. doi: 10.4049/jimmunol.177.11.7980. [DOI] [PubMed] [Google Scholar]
- 17.Wu Q, et al. IL-23–dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect. 2007;9:78–86. doi: 10.1016/j.micinf.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dumoutier L, Louahed J, Renauld JC. Cloning and characterization of IL-10–related T cell–derived inducible factor (IL-TIF), a novel cytokine structurally related to IL-10 and inducible by IL-9. J Immunol. 2000;164:1814–1819. doi: 10.4049/jimmunol.164.4.1814. [DOI] [PubMed] [Google Scholar]
- 19.Wolk K, et al. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 20.Liang SC, et al. Interleukin (IL)-22 and IL-17 are coexpressed by TH17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khader SA, et al. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-γ responses if IL-12p70 is available. J Immunol. 2005;175:788–795. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- 22.Zenewicz LA, et al. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity. 2007;27:647–659. doi: 10.1016/j.immuni.2007.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Acosta-Rodriguez EV, et al. Surface phenotype and antigenic specificity of human interleukin 17–producing T helper memory cells. Nat Immunol. 2007;8:639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 24.Khader SA, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 25.Berger T, et al. Lipocalin 2–deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2006;103:1834–1839. doi: 10.1073/pnas.0510847103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flo TH, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 27.Chakinala MM, Trulock EP. Pneumonia in the solid organ transplant patient. Clin Chest Med. 2005;26:113–121. doi: 10.1016/j.ccm.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 28.McAllister F, et al. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-α and granulocyte colony–stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. J Immunol. 2005;175:404–412. doi: 10.4049/jimmunol.175.1.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti–Candida albicans host defense in mice. J Infect Dis. 2004;190:624–631. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 30.Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. Resident Vδ1+ γδ T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J Immunol. 2007;178:4466–4472. doi: 10.4049/jimmunol.178.7.4466. [DOI] [PubMed] [Google Scholar]
- 31.Kao CY, et al. IL-17 markedly up-regulates β-defensin-2 expression in human airway epithelium via JAK and NF-κB signaling pathways. J Immunol. 2004;173:3482–3491. doi: 10.4049/jimmunol.173.5.3482. [DOI] [PubMed] [Google Scholar]
- 32.Singh PK, et al. Production of β-defensins by human airway epithelia. Proc Natl Acad Sci USA. 1998;95:14961–14966. doi: 10.1073/pnas.95.25.14961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malley R, et al. Antibody-independent, interleukin-17A–mediated, cross-serotype immunity to pneumococci in mice immunized intranasally with the cell wall polysaccharide. Infect Immun. 2006;74:2187–2195. doi: 10.1128/IAI.74.4.2187-2195.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langrish CL, et al. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev. 2004;202:96–105. doi: 10.1111/j.0105-2896.2004.00214.x. [DOI] [PubMed] [Google Scholar]
- 35.Stumhofer JS, et al. Interleukin 27 negatively regulates the development of inter-leukin 17–producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 36.Batten M, et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17–producing T cells. Nat Immunol. 2006;7:929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 37.You Y, Richer EJ, Huang T, Brody SL. Growth and differentiation of mouse tracheal epithelial cells: selection of a proliferative population. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1315–L1321. doi: 10.1152/ajplung.00169.2002. [DOI] [PubMed] [Google Scholar]
- 38.Taswell C. Limiting dilution assays for the determination of immunocompetent cell frequencies. I. Data analysis. J Immunol. 1981;126:1614–1619. [PubMed] [Google Scholar]
- 39.Franks TJ, et al. Lung pathology of severe acute respiratory syndrome (SARS): a study of 8 autopsy cases from Singapore. Hum Pathol. 2003;34:743–748. doi: 10.1016/S0046-8177(03)00367-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fallert BA, Reinhart TA. Improved detection of simian immunodeficiency virus RNA by in situ hybridization in fixed tissue sections: combined effects of temperatures for tissue fixation and probe hybridization. J Virol Methods. 2002;99:23–32. doi: 10.1016/s0166-0934(01)00378-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data