Mechanosensitivity of Nav1.5, a voltage-sensitive sodium channel (original) (raw)
Abstract
The voltage-sensitive sodium channel Nav1.5 (encoded by SCN5A) is expressed in electromechanical organs and is mechanosensitive. This study aimed to determine the mechanosensitive transitions of Nav1.5 at the molecular level. Nav1.5 was expressed in HEK 293 cells and mechanosensitivity was studied in cell-attached patches. Patch pressure up to −50 mmHg produced increases in current and large hyperpolarizing shifts of voltage dependence with graded shifts of half-activation and half-inactivation voltages (Δ_V_1/2) by ∼0.7 mV mmHg−1. Voltage dependence shifts affected channel kinetics by a single constant. This suggested that stretch accelerated only one of the activation transitions. Stretch accelerated voltage sensor movement, but not rate constants for gate opening and fast inactivation. Stretch also appeared to stabilize the inactivated states, since recovery from inactivation was slowed with stretch. Unitary conductance and maximum open probability were unaffected by stretch, but peak current was increased due to an increased number of active channels. Stretch effects were partially reversible, but recovery following a single stretch cycle required minutes. These data suggest that mechanical activation of Nav1.5 results in dose-dependent voltage dependence shifts of activation and inactivation due to mechanical modulation of the voltage sensors.
Introduction
Voltage-sensitive ion channels may also be mechanosensitive
Voltage-sensitive ion channels are nano-machines that serve as biological transistors with well-defined primary functions (Hille, 2001). Many voltage-sensitive ion channels are expressed in contractile organs, which prompted consideration of mechanical modulation of ion channel function. The mechanosensitivity of voltage-sensitive sodium-selective ion channels (Nav channels) was first noted in mechanically manipulated Loligo nerve fibres, where an increase in hydrostatic pressure depolarized the membrane potential and decreased the amplitude of the action potential (Baker et al. 1962). Since the advent of molecular cloning, the list of eukaryotic voltage-sensitive ion channels that are also mechanosensitive has grown. These include representatives from the families of calcium (Farrugia et al. 1999; Calabrese et al. 2002; Kraichely et al. 2009), potassium (Gu et al. 2001; Laitko & Morris, 2004) and sodium (Tabarean et al. 1999; Ou et al. 2003; Morris & Juranka, 2007; Wang et al. 2009) channels.
The mechanosensitivity of voltage-sensitive channels is not surprising, as the conformational rearrangements required for both sensing the voltage (Schmidt & Mackinnon, 2008) and for gating involve physical changes in the shape of the channel (Zhao et al. 2004). The conformational states undergone by the channel during the gating process have various sizes and shapes, and any of these transitions could be mechanosensitive (Gu et al. 2001; Calabrese et al. 2002; Strege et al. 2003_a_; Laitko & Morris, 2004; Laitko et al. 2006; Morris & Juranka, 2007; Kraichely et al. 2009). The mechanosensitivity can also be influenced by the channel environment. Most notably, crystallographic data suggest that the voltage sensors lie at the periphery of the channel and interact with the boundary lipids (Jiang et al. 2003; Milescu et al. 2009). Therefore, an emerging concept is that the function of the voltage sensors is modulated by the surrounding lipids. In turn, channel gating also impacts the structural and mechanical states of the bilayer (Schmidt & Mackinnon, 2008; Beyder & Sachs, 2009; Kraichely et al. 2009; Milescu et al. 2009).
Voltage-sensitive sodium selective ion channels are mechanosensitive
Mechano-regulation of Nav channels in electromechanical tissues is of particular interest. Given the steep voltage sensitivity of these channels, even small changes in channel kinetics can affect physiology. Two laboratories have demonstrated the mechanosensitivity of a rat muscle Na+ channel, Nav1.4 (SkM1) (Shcherbatko et al. 1999; Tabarean et al. 1999). Both studies showed irreversible changes in steady-state and transient kinetic parameters as a result of patch suction in cell-attached patches of Xenopus oocytes expressing Nav1.4 (Tabarean et al. 1999). Suction of 30 mmHg induced a large (−20 mV) shift in the voltage dependence of steady-state activation and inactivation. Moreover, the channels in the patch underwent a permanent conversion from predominately ‘slow’ to ‘fast’ kinetics of activation and inactivation. As these effects were irreversible, the authors hypothesized an involvement of the cytoskeleton. They showed that patch excision and microtubule disruption accelerated the shift to the ‘fast’ kinetics mode. Shcherbatko et al. (1999) extended their study to the peripheral nerve sodium channel Nav1.7 (PN1) with similar results. Recently, Nav1.6, localized at the axonal nodes of Ranvier, was found to have a similar irreversible hyperpolarizing shift in voltage dependence in response to mechanical stimulation (Wang et al. 2009).
Nav1.5 is a voltage-sensitive sodium channel found in two human electromechanical systems, the heart (Gellens et al. 1992) and the gut (Lyford & Farrugia, 2003). Mutations in Nav1.5 result in disease. In the heart, Nav1.5 mutations are known to cause long QT 3 (LQT3), Brugada syndrome, cardiac conduction disease and dilated cardiomyopathy (Bennett et al. 1995; Ackerman, 1998). In the gut, mutations in Nav1.5 are associated with an increased incidence of abdominal pain syndromes (Locke et al. 2006). It also appears that mechanical sensitivity of Nav1.5 may play a role in pathophysiology of irritable bowel syndrome. Recently, a mutant Nav1.5 (G298S) from a patient with irritable bowel syndrome was found to be less responsive to shear stress than wild-type Nav1.5 (Saito et al. 2009).
In both the heart and gut, there is continual oscillating movement, making the effect of mechanical stress particularly relevant. We and others have demonstrated mechanosensitivity of Nav1.5 on a macroscopic level (Strege et al. 2003_b_; Morris & Juranka, 2007). For Nav1.5 channels expressed in HEK cells, shear stress increased peak current by 27% and accelerated activation (Strege et al. 2003_a_). Later, in Xenopus oocyte cell-attached patches, Morris & Juranka (2007) showed that suction produced a reversible 40% acceleration of Nav1.5 activation and inactivation. This suggested a small negative shift in midpoints of ∼2–4 mV. A subsequent study found a more significant shift of 10 mV for −30 mmHg (Banderali et al. 2010). The authors also examined stretch effects on two LQT3 Nav1.5 mutants (R1623Q and R1626P). As these mutant channels have previously been shown to have uncoupled activation and inactivation, it was surprising that stretch did not have a differential effect on the activation and inactivation of the channels. These studies have established stretch sensitivity of Nav1.5. However, the precise effects of stretch on the specific gating transitions remain unclear.
A kinetic model of Nav channels is complex
To understand the effects of stretch on rate constants requires that mechanical effects be considered with respect to the effects of stretch on discrete gating transitions. Previous studies on whole cells and cell-attached patches yielded large macroscopic currents (Shcherbatko et al. 1999; Tabarean et al. 1999; Morris & Juranka, 2007). These demonstrated mechanical modulation of Nav and specifically Nav1.5, but these studies lacked single channel data and detailed analysis of the distinct transitions in the Nav gating scheme. Our aim was to characterize the mechanosensitivity of Nav channels by determining the stretch-dependent transitions. A practical difficulty is that Nav channels have complex kinetic properties, and while multiple models have been proposed, a consensus kinetic model does not exist (Armstrong & Bezanilla, 1977; Bezanilla & Armstrong, 1977; Armstrong, 2006). In order to better understand which gating transitions are mechanosensitive we examined mechanosensitivity using a highly simplified five state model. This model simply outlines the widely accepted gating transitions of the Nav channels (Fig. 1). At rest (hyperpolarized), the voltage-sensitive Na+ channel is closed (state C) and the voltage sensors are in the rest position. A depolarization activates the channels via a series of voltage-dependent steps, but will be denoted here by a single step C→CA with q representing voltage dependence. From the activated state, the channel can proceed to an open and conducting state via a _voltage_-independent transition (CA→O). Inactivation may occur from both the open (O) and the activated (CA) states. When a large depolarization drives all channels to activation, opening of the cytoplasmic ion channel gate makes the inactivation site accessible and allows the inactivation particle to quickly close the channel (O→IO). At potentials negative with respect to channel opening, channels may enter the inactivated state directly from the activated (CA) state without opening, also called closed state inactivation (CA→IA). Recovery from the inactivated states (IA and IO) is followed by the voltage sensors returning to their deactivated position (CA→C). In this study, we examined the effects of mechanical perturbations on the specific transitions in this gating scheme.
Figure 1. Simplified Nav gating model.
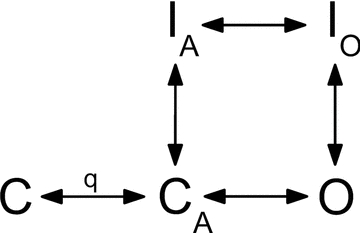
In the resting state all channels are in state C. With depolarization, channels proceed through the voltage-dependent (q) states, as the four voltage sensors move into the activated positions. From this activated state (CA), channels may either proceed to inactivation (CA→IA) or to the open state (O). The transition to the open conducting state (CA→O) is independent of voltage, representing opening of the intracellular gate. Shortly after opening, the channel is inactivated by an intracellular inactivation gate (O→IO). Recovery from the inactivated states may proceed without channel opening, so IO→IA→CA.
We used Nav1.5-transfected HEK cells and stretched cell-attached patches. A single stretch of the membrane caused a persistent increase in current and hyperpolarizing shifts in the midpoint of activation and inactivation. We show that the shifts in voltage dependence were due to an acceleration of a single voltage sensing step. Channel gate function, inactivation from open states (fast inactivation), and unitary conductance were unaffected by stretch. Stretch slowed recovery from inactivation, suggesting that the inactivated states were stabilized. These data also suggest that mechanosensitivity of Nav1.5 may play a significant role in the behaviour of the channel in mechanically active organs.
Methods
Pipette fabrication
Patch pipettes were pulled from 2.7-inch length Corning 7040 glass tubing (Corning Inc., Corning, NY, USA; 1.65 mm OD, 1.15 mm ID), using a four stage pull by a Sutter P-97 electrode puller (Sutter Instrument Co., Novato, CA, USA). The tips were intentionally pulled to be blunt (bullet-nosed). After pulling, the final tip geometry was achieved by fire polishing. In general, fire polishing was done at higher than normal temperature with the tip farther from the glass coated filament than customary for construction of low noise pipettes. These procedures produced a rounding of the tip without much narrowing of the internal diameter. The final tip diameter could be controlled by adjusting the duration of fire polishing. Several iterative experiments with different tip geometries allowed the desired recordings in about half of the patches. In addition, the selected tip geometry produced patches that were consistently sensitive to patch pressure.
Cell culture
Human embryonic kidney cells 293 (HEK) (American Type Culture Collection, Manassas, VA, USA) were grown in T25 flasks in minimum essential medium, 10% horse serum, non-essential amino acids, sodium pyruvate and penicillin–streptomycin (Invitrogen, Carlsbad, CA, USA). Cells were grown to about 75% confluency. One day prior to the experiments, flasks were transfected with SCN5A encoding the hH1C3 variant (the most prevalent variant in the gut; Strege and others 2003_b_), and with green fluorescent protein (GFP) pEGFP-C1 (Clontech, Palo Alto, CA, USA), using Lipofectamine 2000 reagent and OPTI-MEM I reduced serum medium (Invitrogen) (Saito and others 2009). On the morning of the experiments, cells were trypsinized and resuspended in Ringer solution, and plated onto the recording chamber 15 min prior to the initiation of experiments.
Recording solutions
The bath solution contained (in mm): 149.2 Na+, 159 Cl−, 4.7 K+, 2.5 Ca2+, and 5.0 Hepes (and 5.5 mm glucose for the whole cell experiments). Pipette solution was made of bath solution plus 10 μm GdCl3 to minimize contamination by endogenous cation selective stretch channels (Morris et al. 2006). The bath solution was made in bulk, but gadolinium solution was prepared on the day of the experiments. Since use of gadolinium (Caldwell et al. 1998) has been shown to affect Nav channels (Elinder & Arhem, 1994), we determined the minimal amount required. The addition of 10 μm GdCl3 to the pipette solution was sufficient in blocking endogenous cation-selective stretch currents (Fig. S1 in the online Supplemental Material). For whole cell experiments, the intracellular (pipette) solution was (in mm): 135 K+, 130 CH3SO3−, 5 Na+, 5 Mg2+, 5 Hepes, and 2 EGTA in the tip with 0.6 mm amphotericin B added to the backfill (final DMSO concentration was less than 1%).
Mechanical stimuli
Transfected cells were identified by fluorescence microscopy, and cell-attached patches were formed using standard techniques. Gentle suction was applied to make a gigaseal and patch pressure was then released. Seal history is important for such experiments (Morris et al. 2006), therefore we recorded the pressures required for patch formation. Patches were typically formed at <5 mmHg. Patches with seals that were formed at =10 mmHg were discarded. Pressure was stepped to the desired level for a few seconds prior to protocol execution, and then stepped back to 0 mmHg between successive datasets.
A significant impediment to studying the mechanosensitivity of ion channels is the inability to apply a precise mechanical stimulus. Mechanical stress is difficult to apply reproducibly and hence local stress is often ill-defined. For experimental systems that aim to combine voltage- and mechano-clamp, application of pressure to a voltage-clamped membrane patch is the most reproducible stimulus available (Suchyna et al. 2009). Patch pressure was modulated with a pneumatic transducer (DPM-1, Bio-Tek, Winooski, VT, USA). Determination of the actual stimulus at the patch is complicated because the geometry is not spherical, and the patch anatomy is inhomogeneous and changing in time (Suchyna et al. 2009). Therefore, we focused on differential effects within the same patch (Opsahl & Webb, 1994). Video recordings demonstrated patch creep at suction greater than −30 mmHg (Fig. S2). We performed experiments at pressures as high as −120 mmHg, but due to patch creep most of the quantitative analysis was from 0 and −30 mmHg. To minimize drift caused by time-dependent changes in the patch properties (Morris et al. 2006), we limited the data acquisition to 30 seconds at elevated pressure.
Electrophysiology
An Axopatch 200A amplifier, Digidata 1322A, and Clampex 9.0 software (Molecular Devices, Sunnyvale, CA, USA) were used for voltage-clamp and data acquisition. Previous work showed that large transmembrane voltages (100+ mV) applied over seconds produced patch creep, which caused activation of mechanosensitive channels in Xenopus oocytes (Gil et al. 1999). To prevent this creep, we kept the holding potential at 0 mV.
Average current data collection & analysis
The fast inactivation of Nav1.5 channels is complete in only a few milliseconds, but recovery requires hundreds of milliseconds at a holding potential of −100 mV (Bezanilla & Armstrong, 1977). However, channels can be more rapidly restored by a strong hyperpolarization (Bezanilla & Armstrong, 1977). We found that full recovery from inactivation can be accomplished within a few milliseconds (τ= 0.59 ms) using short (2–5 ms) hyperpolarizing pulses to −204 mV (maximum functional range of our amplifier) (Fig. S3). By minimizing intersweep intervals, we were able to acquire sufficient single channel events over a 30 s period at elevated pressure. P/4 subtraction was used to minimize capacity transients (holding potential (HP) = 0 mV).
Average traces resembled macroscopic currents and were treated as such. We used Origin 7.0 (OriginLab Co., Northampton, MA, USA) to fit normalized peak currents (I/_I_max) that were plotted against applied voltage using a two state Boltzmann model (I/I_max=A/[1 + exp((V_1/2−_V)/d_V)]), where I is current, _I_max is maximal current, A is a scaling factor, V_1/2 is the voltage at half-maximal current, and d_V is the slope of the sigmoidal gating curve.
Single channel data collection and analysis
A sequence of 200–1000 steps maximized the number of events while keeping the protocol time to approximately 30 s. A separate protocol to P/4 (HP = 0 mV) was acquired after each sequence and was scaled and subtracted offline. Data were analysed in QuB, a freely available software package (Qin et al. 1996; Qin, 2004). Events were indexed and exported to Origin 7.0, where data were analysed statistically. The number of open channels in the patch was assumed to obey a binomial distribution of the total number (Horn, 1991). If the number of channels in the patch is n, and the open probability is _P_o, then for a number of sweeps m, the probability (k) that the maximum observed level is the total number of channels in the patch is 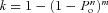 . Since activation and inactivation overlap temporally the maximum _P_o is less than 1.0 and is difficult to estimate. To estimate the maximum _P_o and to count the number of channels in the patch, we used patches with a small number of channels (n < 10), and depolarized them to (0, +5 and +10 mV), where _P_o should be maximal and acquired _m_= 600 sweeps. Under these conditions, we could count up to five channels with greater than 90% confidence (_k_ > 0.9) for _P_o > 0.3. _P_o at peak current was calculated from a single sweep using _P_o=_I_avg/_I_peak.
. Since activation and inactivation overlap temporally the maximum _P_o is less than 1.0 and is difficult to estimate. To estimate the maximum _P_o and to count the number of channels in the patch, we used patches with a small number of channels (n < 10), and depolarized them to (0, +5 and +10 mV), where _P_o should be maximal and acquired _m_= 600 sweeps. Under these conditions, we could count up to five channels with greater than 90% confidence (_k_ > 0.9) for _P_o > 0.3. _P_o at peak current was calculated from a single sweep using _P_o=_I_avg/_I_peak.
Results
Membrane stretch increases peak current and accelerates both activation and inactivation
Average currents in response to a typical voltage ladder protocol (Fig. 2_A_) for a single patch at 0 mmHg and −30 mmHg are shown in Fig. 2_B_ and C. To bring channels out of inactivation, we used a 5 ms pulse to maximum hyperpolarization allowed by the amplifier (−204 mV) (Fig. S3). This allowed satisfactory averaging of currents as small as 3 pA over the 30 s stretch steps.
Figure 2. Averaged patch currents increase in peak and accelerate with stretch.
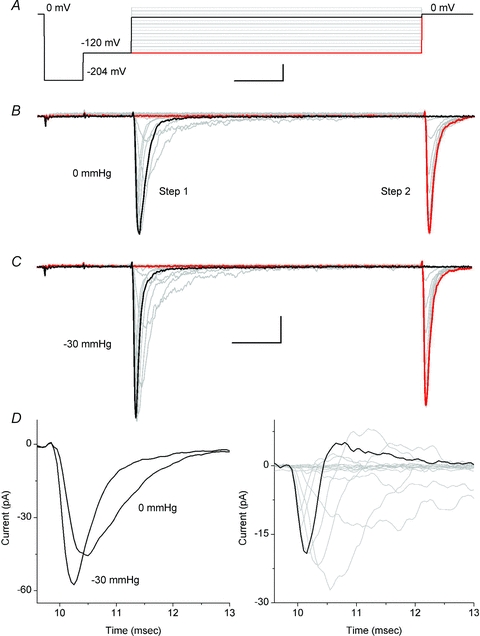
A, a typical voltage protocol. From holding potential (0 mV), each 30 ms-long ladder step (starting from −120 to 20 mV in 10 mV increments) was preceded by 5 ms hyperpolarizing pulses to −204 mV and −120 mV (scale 5 ms and 50 mV). Ten averages were obtained for each averaged current experiment. B and C, average patch current at rest (0 mmHg) (B) and average patch current when stretched by a −30 mmHg stimulus (C). Peak currents evoked at the beginning of each ladder step (Step 1) and immediately after stepping to 0 mV at the end of each ladder step (Step 2) were used to analyse voltage dependence of activation and inactivation, respectively. For Step 1 black traces show patch currents in response to a depolarization to −10 mV. For Step 2 black traces show no current at 0 mV after a ‘prepulse’ of −10 mV, and red traces show current at 0 mV after a ‘prepulse’ to −120 mV. For B and C scale is 5 ms and 10 pA. D, left panel highlights the early portion of Step 1 currents at 0 and −30 mmHg for a depolarization to −10 mV. These traces clearly demonstrate that peak currents increased with stretch and showed accelerated kinetics. The right panel shows the difference in Step 1 currents from left panel (black) and at all voltages (grey) (I(t)0 mmHg−I(t)−30 mmHg).
At 0 mmHg, the average peak current of Step 1 was 47 ± 24 pA (Fig. 2_B_). Patch pressure produced a robust increase in the peak value (Fig. 2_B_–D). At −30 mmHg, for 8 of 10 patches examined Step 1 peak currents increased 33% to 63 ± 29 pA (_n_= 10) (Fig. 2_C_). Suction increased peak currents significantly for −20, −30 and −40 mmHg by 1.32 ± 0.21, 1.33 ± 0.27 and 1.37 ± 0.34, respectively (_n_= 6, P < 0.05 compared with 0 mmHg, _P_ > 0.05 between pressures). Peak currents for −10 mmHg and −50 mmHg increased but did not reach statistical significance. At low suction (−10 mmHg) the mechanical stimulus was likely to have been too small relative to background variations while at −50 mmHg patch creep may have changed patch properties (Fig. S2). Positive pressure also caused an increase in current (data not shown), showing that membrane tension and not applied pressure was the key variable.
Stretch also had effects on the kinetics of activation and inactivation. For the traces highlighted in black (depolarization to −10 mV) in Fig. 2_B_ and C, pressure accelerated the time to reach peak current (activation) from 0.49 ms to 0.64 ms (30%) (Fig. 2_D_, left panel). Fast inactivation, which was estimated from the kinetics of current decay from peak current, was also accelerated. When inactivation was fitted to a single exponential, the time constant decreased (accelerated) with stretch from 0.81 to 0.68 ms (29%). The difference in time dependence of current with stretch demonstrated a mix of excitatory effects at small depolarizations (near resting potential) and transient excitatory mixed with later inhibitory effects of large depolarizations at large depolarizations (Fig. 2_D_, right panel).
Stretch left-shifts voltage dependence of steady state activation and inactivation
The voltage ladder protocol shown in Fig. 2_A_ was used to assess the macroscopic behaviour for voltage dependence of activation and steady-state inactivation. Voltage dependence of activation was measured as peak currents evoked at the beginning of each ladder step (Step 1 peak). Steady-state inactivation, or availability, was measured as peak currents immediately after stepping to 0 mV at the end of each ladder step (Step 2 peak). These results are qualitatively similar to the macroscopic (whole cell) data (Makielski et al. 2003). Mean current–voltage (I–V) curves for a typical experiment show that pressure induced a significant shift in the steady-state voltage dependence of activation and inactivation (Fig. 3_A_ and B). When fitted to a two state Boltzmann model, at 0 mmHg the half-point of activation (_V_1/2) was −33 ± 9 mV (R_= 1.0) with a slope (d_V) of 5.6 ± 0.3 mV (_R_= 0.91) (_n_= 6). _V_1/2 compared well to previous experiments with whole cell currents where _V_1/2 was reported as −40 ± 2 mV and d_V_= 4.9 ± 0.3 mV (Makielski et al. 2003). The wide range of _V_1/2 values in cell-attached experiments was likely due to variable resting potential and tension (Suchyna et al. 2009). To quantify voltage errors secondary to the resting potential of the cell we measured the resting potential of HEK cells transfected with Nav1.5. First, a gigaohm seal was established, and amphotericin was allowed sufficient time to perforate the patch. Then, resting voltage was recorded in current clamp mode (0 pA injected). Resting voltage of these cells was −8.1 ± 4.1 mV (_n_= 7).
Figure 3. Suction effects on voltage dependence of Nav1.5 activation and inactivation showing a marked leftward shift.
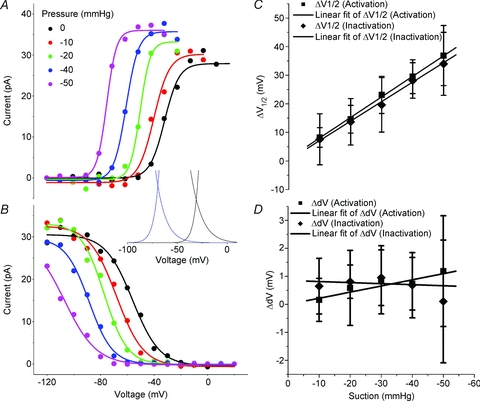
A, Two state Boltzmann model fits of voltage dependence of activation at pressures 0 mmHg to −50 mmHg. B, Two state Boltzmann model fits of steady-state voltage dependence of inactivation at pressures 0 mmHg to −50 mmHg. Inset shows window currents resulting from overlap of the voltage dependence of activation and inactivation at 0 mmHg and at −40 mmHg showing the leftward shift in the window current with suction. C, shift in half-point (Δ_V_1/2) of activation (squares) and inactivation (diamonds) (Δ_V_1/2=_V_1/2,0 mmHg−_V_1/2,testP) with response to increasing amplitude suction stimuli from −10 to −50 mmHg. Linear fits of the shift in activation and inactivation were −0.72 ± 0.18 mV mmHg−1 (_R_= 0.97) and −0.68 ± 0.28 mV mmHg−1 (R_= 0.99), respectively. D, shift in slope (Δd_V) of activation (squares) and inactivation (diamonds) (Δd_V_= d_V_1/2,0 mmHg− d_V_1/2,testP) with response to increasing amplitude suction stimuli from −10 to −50 mmHg. Linear fits of the Boltzmann slope of activation and inactivation were 0.022 ± 0.035 mV mmHg−1 (_R_= 0.30) and −0.0038 ± 0.041 mV mmHg−1 (_R_= 0.24), respectively.
To minimize the effects of patch to patch variation on our data, we analysed individual patches and compared the relative effects. From tension at rest (0 mmHg applied to patch), we applied pressure stimuli as short 30 s steps with steps back to resting tension in between stimuli. When compared to resting tension, in all 10 of 10 stretched patches, mean current–voltage (I–V) curves (Fig. 3) showed a significant shift in the steady state voltage dependence of activation and inactivation. In the voltage range tested (−10 to −50 mmHg), suction-driven shifts of _V_1/2 in Fig. 3 were linear with pressure (_R_= 0.97, _R_= 0.99, respectively) and in the hyperpolarized direction by −0.72 ± 0.18 mV mmHg−1 (_n_= 4) for activation and −0.68 ± 0.28 mV mmHg−1 for inactivation (_n_= 4) (Fig. 3C). Shifts in half-point of activation were similar when patches were stepped to −30 mmHg in a single step (−19.0 ± 8.2 mV, _n_= 6) or after a couple of increasing steps of increasing pressure (−23.1 ± 6.5 mV, n_= 4). Boltzmann slopes (d_V) showed a trend toward steeper activation but this was not statistically significant at any of the pressures tested (Δd_V_= 0.022 ± 0.035 mV mmHg−1 (_n_= 4) for activation and −0.0038 ± 0.041 mV mmHg−1 (_n_= 4) for inactivation) (Fig. 3_D_).
The small area below the intersection of the activation and inactivation curves is known as the window current (Fig. 3, inset). Over this voltage range, the activated channels would not be fully driven to inactivation, resulting in a steady-state current. The larger the window, the more significant is the steady-state current. The significance of the window current is enhanced if it is found over the resting voltage of the cell. We show above that stretch resulted in a leftward shift in voltage dependence of activation and inactivation, hence the window current (Fig. 3, inset). However, since the activation and inactivation were similarly affected and there was no significant change in the steepness of the Boltzmann fit, the size of the shifted window current was relatively conserved.
Positive pressure was applied to rule out effects secondary to patch curvature. It produced shifts in voltage dependence with independence of slope sensitivity similar to those observed with negative pressures (Fig. S4). The correlation of positive and negative pressure results implies that the effects were due to membrane tension (Fig. S4). However, negative pressure was more robust experimentally, and was thus used for the following experiments.
Effect of membrane stretch on activation and fast-inactivation kinetics
The currents in Fig. 2 qualitatively demonstrate an increased rate of activation and inactivation that was typically seen. The activation mechanism of sodium channels is a multi-step process. Facilitation of either the voltage sensor activation (Borjesson et al. 2008) or gate opening (Zhao et al. 2004) may left-shift activation _V_1/2 and thus produce apparent acceleration of current activation at all voltages. Furthermore, it is known that Nav activation is kinetically linked to inactivation, because it is fast and rate-limiting (Aldrich et al. 1983). Thus, acceleration of activation may also accelerate inactivation. In the next set of experiments, we examined (1) whether or not activation and inactivation are both stretch sensitive and (2) which of the activation processes (voltage sensors or gate opening) are affected by stretch.
The left panel of Fig. 4 shows the I–V curves for a typical experiment for a patch at rest (black) and stretched by −30 mmHg (red). The I–V curves demonstrate the negative shift of voltage dependence of channel activation and the increase in peak current with stretch. Points 1–5 (_V_m from −60 mV to −10 mV) were picked along the steep portion of the I–V curve where channels are most sensitive to voltage. The traces in the middle panel show the time course of activation and inactivation for currents at these voltages. Stretch increased the peak current and accelerated both activation and inactivation kinetics at all voltages. In the right panel we have normalized peaks to amplitude and time. Temporal scaling was accomplished simply by _t_scaled=t/SF, where the scaling factor (SF) with, SF =_t_peak,0 mmHg/_t_peak,30 mmHg. It is evident from the right panel of Fig. 4 that all traces could be well overlapped using a single linear scaling constant (SF). We performed this analysis in five patches and had good overlap in each case. As a result, it is evident that a single time scaling constant accounted for the complex kinetic changes.
Figure 4. Stretch acceleration of activation and inactivation scales linearly.
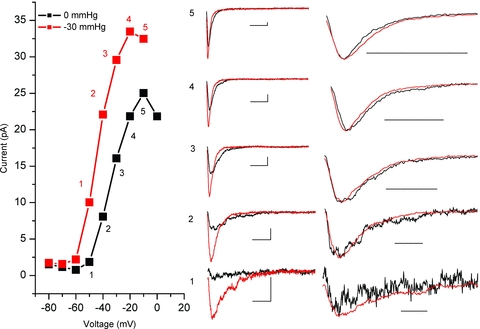
In the left panel, voltage dependent currents (I–V) at 0 mmHg (black) and −30 mmHg (red) are shown. Highlighted points 1–5 track increasing levels of activation. In the middle panel, for the voltage steps marked by 1–5, single time-dependent traces at resting tension (black) are plotted with the corresponding traces for this patch under −30 mmHg stretch (red). In the right panel, traces were normalized to peak current and temporally scaled to times of peak showing that a single time scaling constant accounts for the kinetic changes. Temporal scaling factors (SF) for this patch were 1.57, 1.89, 1.75, 1.47 and 1.71, for points 1–5, respectively.
Our data suggest that stretch accelerates activation and inactivation by a single constant. Given the kinetic link between activation and inactivation, it is most likely that stretch produces an acceleration of channel activation, and inactivation only appears faster. Furthermore, the fact that acceleration is explained by simple linear scaling suggests that either the voltage sensor activation step (C→CA) or gate opening step (CA→O) is affected, but not both.
Inactivation from closed states is enhanced by stretch
Because the kinetics are fast, it was difficult to determine which transitions were affected by stretch: was it activation (C→CA) or channel opening (CA→O)? To discriminate between these effects, we examined inactivation from closed states (CA→IC), closed state inactivation (CSI) (Fig. 1). CSI may be viewed as a ‘leak’ from the activation process. If stretch enhanced the voltage sensitive transitions (C→CA) then states CA would become more populated with stretch. In this scenario, CSI would be enhanced. On the other hand, if stretch affected the gate opening (CA→O), channels would be more likely to proceed to opening, thus decreasing the availability of channels in the CA state. Then, CSI would appear to be decreased.
Closed state inactivation is important at hyperpolarized voltages, where _P_o is low and thus channel openings are rare (Armstrong, 2006). At these voltages, the voltage sensors operate, but the channel gates do not yet open. We used a two-step voltage protocol as shown in the top panel of Fig. 5_A_. In the first part of the protocol, after a brief hyperpolarizing pulse and a step back to −140 mV for 5 ms, patches were stepped to 0 mV for 5 ms (Step 1). The same sequence then preceded a step ladder protocol from −140 to −70 mV, with each step lasting 30 ms. Step 2 was a depolarization to 0 mV to assess channel availability. In the bottom left panel of Fig. 5_A_ we show a typical set of patch currents. At 0 mmHg (black traces), Step 1 peak currents revealed the maximum available current in the patch at 0 mV (average = 74 ± 2 pA). The following step ladder did not produce currents, signifying that the channels did not activate. Yet, Step 2 peak currents for each step were smaller than the control Step 1 peak. The decrease in peak Step 2 current is proportional to closed state inactivation (CSI ∝ 1 − Step2peak/Step1peak). For this experiment, CSI0 mmHg ranged from 0.01 to 0.07 between −140 and −110 mV, meaning that less than 10% of channels inactivated from the closed state. As expected, when this patch was stretched (red traces) Peak 1 and Peak 2 were increased and kinetics accelerated. Also, stretch resulted in some of the ladder steps producing an activating current (−80 and −70 mV), consistent with the negative shift in the voltage dependence of activation. However, the relative increases in Peak 2 currents were significantly smaller than the Peak 1 controls. Under stretch, CSI−30 mmHg ranged between 0.17 and 0.37 between −140 and −110 mV. The excess CSI due to stretch (=ΔCSI = CSI0 mmHg− CSI−30 mmHg) for six patches is shown in Fig. 5_B_. Stretch resulted in an increase in CSI at all voltages but one (−110 mV). Our data show that stretch produced an apparent acceleration of inactivation from closed states. As detailed above, increased CSI with stretch suggests an increased CA availability secondary to enhanced voltage sensor operation with stretch (C→CA).
Figure 5. Closed state inactivation (CSI) increases with stretch.
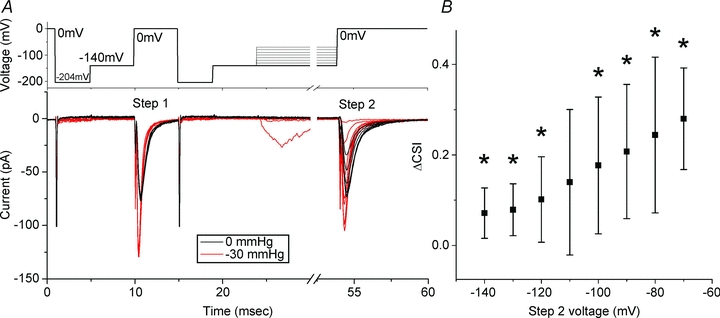
A, in the top panel a typical protocol is shown. Prior to both Step 1 and Step 2 patches were stepped through −204 and −140 mV for 4 and 5 ms, respectively. Step 1 was a depolarization to 0 mV for 5 ms, which was followed by a step ladder of 30 ms pulses from −140 to −80 mV and finally a Step 2 depolarization to 0 mV. The bottom panel shows resulting currents for a typical experiment. In black, a patch at resting tension (0 mmHg) has Step 1 and Step 2 peaks that are of nearly identical height. For non-activating Step 2 voltage pulses, the difference in Step 1 and Step 2 peak heights is proportional to closed state inactivation, so CSI ∝ 1 −_I_step2,max/_I_step1,max,0 mV. For this experiment, CSI0 mmHg was 0.07, 0.02, 0.01, 0.03, 0.18, 0.35, 0.46 for −140 to −70 mV, respectively. In red, the same patch stretched by a −30 mmHg stimulus has Step 1 peak that is disproportionately larger than Step 2 peak. For this experiment, CSI−30 mmHg values were 0.22, 0.17, 0.23, 0.37, 0.41, 0.57, for voltages −140 to −90, respectively. Steps to −80 and −70 mV activated small currents and we not include these in the analysis because of open state inactivation. Excess CSI with stretch is ΔCSI = CSI−30 mmHg− CSI0 mmHg. For this experiment, ΔCSI was 0.15, 0.16, 0.23, 0.33, 0.34, 0.39 for −140, −130, −120, −110 and −100 mV, respectively. B, average ΔCSI for 7 patches plotted against Step 2 voltage. Marked by * are voltages at which stretch resulted in more CSI than no stretch (p < 0.05 by t-test).
Recovery from open state inactivation is slower with stretch
We used another two step voltage protocol to test the mechanosensitivity of recovery from open state inactivation (_I_O) (Fig. 6_A_, inset). Patches were depolarized to 0 mV for 20 ms (Step 1) to allow fast inactivation to proceed to completion. Step 1 was followed by recovery periods of increasing length (0 to 20 ms) at holding potentials (HP) of −140 through −100 mV. Test current was then elicited by depolarization to 0 mV (Step 2). For a typical experiment at 0 mmHg (Fig. 6_A_, black traces), average Step 1 peak current was 40 ± 2 pA and at HP −120 mV, 16.5 ms was required to reach full recovery by Step 2 current (40 pA). As expected, stretch increased the peak current (44 ± 2 pA) and accelerated the kinetics of the currents evoked by Step 1 (Fig. 6_A_, red traces). Yet, while peak currents of Step 1 increased, the peak currents for Step 2 were smaller than in the non-stretch case and over 20 ms failed to attain full recovery. Recovery of Step 2 peak currents (_I_2) was an exponential function of time. Step 2 peaks were normalized to Step 1 peak and fitted to an exponential _I_2/_I_1=I_0+A(1 − e−_t /τ)) (Fig. 6_B_). The time constants of recovery from inactivation for resting tension (0 mmHg) and stretched patches (−30 mmHg) were compared as a ratio (τ0mm/τ−30mm) on a patch-by-patch basis and the ratios were then averaged (Fig. 6_C_). At all holding potentials (HPs), recovery was slower for channels that were stretched compared to unstressed channels. The results were statistically significant for all HPs, except −140 mV, where a trend to slower recovery was noted. These findings were not affected by the left-shift of the steady state inactivation curve. At voltages more hyperpolarized than −100 mV, inactivation for even the stretched channels had peaked (Fig. 3_B_), and thus we expected recovery to be unaltered. This slower recovery for stretched channels suggests that the inactivated states are more stable under stretch. The simplest interpretation is that the inactivated states are slightly larger in diameter and/or thinner.
Figure 6. Inactivation recovery is slowed with stretch.
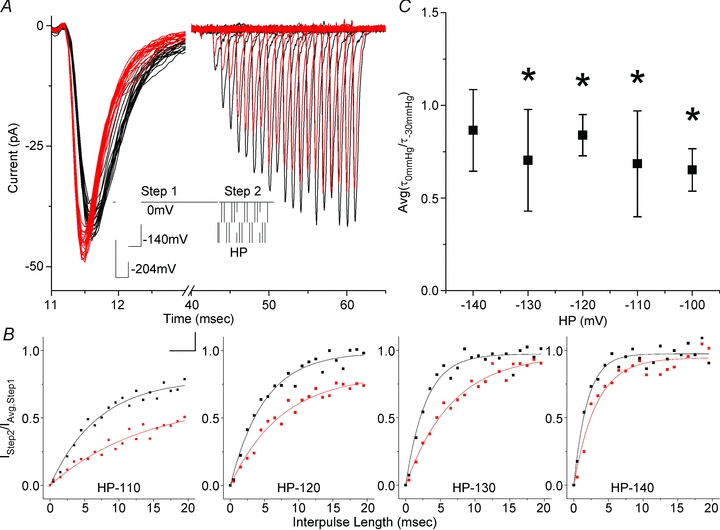
A, a two step experiment with a typical protocol is shown in the inset. All protocols had 4 ms long −204 mV and −140 mV steps preceding Step 1 to 0 mV for 20 ms to attain full inactivation. Recovery periods were variable interpulse lengths (0 to 20 ms from 0.5 ms by 1 ms increments) at HP =−140 mV, −130 mV, −120 mV, −110 mV and −100 mV before Step 2 to 0 mV to assess for recovery (scale is 10 ms and 50 mV). Currents in the figure are typical recovery at −120 mV. Stretch resulted in Step 1 current increase and acceleration of kinetics (black is 0 mmHg, red is −30 mmHg). Current inactivated fully over 20 ms Step 1 pulse (first 2 ms shown). Recovery was observed as increasing Step 2 currents. B, peak currents from Step 2 were normalized to average Step 1 peak (_I_Step2/_I_Avg,Step1) and plotted against interpulse length, showing inactivation recovery at resting pressure (0 mmHg, grey) and for a stressed patch (−30 mmHg, black). Data were fitted by an exponential described by _I_=I_0+A(1 −_e−x/τ). For this experiment, τ0 mmHg were 6.0 ± 0.60, 5.1 ± 0.43, 3.1 ± 0.31, 2.3 ± 0.34 ms and τ−30 mmHg were 16.0 ± 4.0, 7.4 ± 0.76, 6.8 ± 0.54, 3.4 ± 0.6 ms at HP −110, −120, −130, −140 mV. C, recovery from inactivation with stretch relative time constants Avg(τ0 mmHg/τ−30 mmHg) as a function of HP voltage at −140 mV (_n_= 4), −130 mV (_n_= 4), −120 mV (_n_= 5), −110 mV (_n_= 3) and −100 mV (_n_= 2).
Single channel conductance was unaltered by stretch
The total patch current is (I) =_NiP_o, where N is the number of channels, i is the single channel current and _P_o is the probability of a channel being open. Thus, an increase in total patch current (I) implies an increase in the number of available channels (N), unitary conductance (i) and/or peak open probability (_P_o). Single channel records were obtained at a range of pressures and voltages (Fig. 7_A_ and B). A typical voltage ladder protocol is shown in Fig. 7_A_. Again, we designed this protocol to have a large hyperpolarizing pulse preceding each step to bring channels out of inactivation. In this manner, we were able to collect roughly 200 traces with five steps of voltage from −50 to −10 mV over 30 seconds (overlap shown as grey shadows in Fig. 7_B_). For each voltage step, single channel events were clearly visible after the bulk of the channels in the patch inactivated (Fig. 7_B_). Traces were idealized and single channel events identified using QuB as described in Methods. Figure 7_C_ shows a typical all points amplitude histogram for traces at −30 mV for a single patch at rest (0 mmHg, grey) and at −30 mmHg (black). Multiple peaks can be identified on this histogram. The peak around 0 pA is noise, but the other peaks of decreasing height around 1.5 pA, 3.0 pA and 4.5 pA show amplitudes for one, two and three open channels, respectively. The number of events increased with applied pressure, a consistent finding for all patches (_n_= 5). Linear fits (_R_= 0.997) placed the sodium reversal potential at _E_Na=+47.9 mV. Gaussian fits showed the unitary conductance was independent of pressure (_n_= 5) (Fig. 7_B_). The single channel conductance at rest (_g_0mmHg) was 17.3 ± 0.6 pS, similar to previously published values (Hille, 2001), and not significantly different from single channel conductance with stretch (Fig. 7_D_).
Figure 7. Unitary conductance is not affected by stretch.
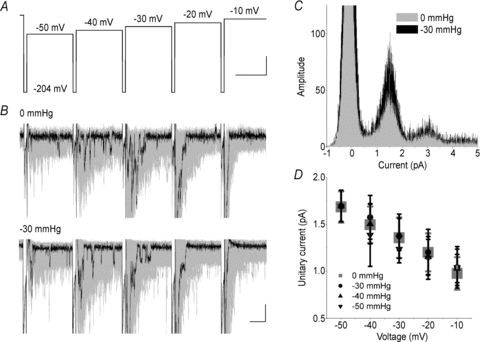
A, single channel ladder protocol was designed with a 2 ms step to −204 mV, followed by 10 mV steps from −50 mV to −10 mV. To maximize the number of single channel events, 200 ladders were acquired. B, typical currents for a patch at rest (0 mmHg) and stretched by a −30 mmHg stimulus. Black highlights a single trace. Grey shadows are overlap of all 200 traces (peaks clipped for display purposes). Segments were then cut to exclude peak activation (_t_= 0 to 6 ms) and single channel events were idealized for subsequent 26 ms. C, all points amplitude histogram showing single channel currents at 0 (grey) and −30 mmHg (black) for a typical data set at −30 mV. Amplitude histogram demonstrates qualitative overlap of peaks around 1.5, 3.0 and 4.5 pA for one, two and three channels open, respectively. Amplitude histograms were then fitted with single Gaussians. D, average Gauss peaks are plotted against voltage for pressures 0 to −50 mmHg (_n_= 5). Single channel conductances (g) were the same at all pressures, 17.3 ± 0.6, 17.7 ± 0.5, 17.3 ± 0.5, 17.2 ± 0.9, 17.0 ± 0.9, 17.3 ± 0.1 for 0 mmHg, −10 mmHg, −20 mmHg, −30 mmHg, −40 mmHg, −50 mmHg, respectively.
Stretch increased the number of active channels in the patch
With unitary conductance (i) not changing, the increases in total patch current (I) are due to an increase in the channel number (N) and/or open probability (_P_o). _P_o is difficult to estimate for voltage-sensitive Na+ channels since activation and inactivation overlap in time and _P_o does not reach unity. However, the binomial distribution predicts that for a patch with a small number of channels (<5) and _P_o > 0.25 in 600 depolarizations the likelihood of a single trace with all channels open is =80%. Data from a typical patch are presented in Fig. 8_A_. At rest, this patch had a peak current of 3.12 pA (top panel, red trace) consistent with four channels with a unitary current of _i_= 0.78 ± 0.13 pA and average current of 0.84 pA, suggesting a _P_o of 0.27. When the patch was stretched (bottom panel), peak current increased to 3.79 pA, unitary current was 0.73 ± 0.15 pA and average current rose to 1.0 pA, suggesting a _P_o of 0.26, and a channel number of five instead of four. Figure 8_B_ shows averaged data for five such patches. The top panel shows that stretched patches had more current at all voltages. The downward slope of these I–V curves suggests that maximally stimulating voltage pulses were used, and _P_o was maximal. The middle panel shows that there was no significant increase in _P_o with stretch, suggesting an increase in the number of channels (lower panel). In the bottom panel, from a starting channel number average of 5.03 ± 0.09, channel number increased by a factor of 1.1 ± 0.4, or ∼20%.
Figure 8. Open probability and channel number in a stretched patch.
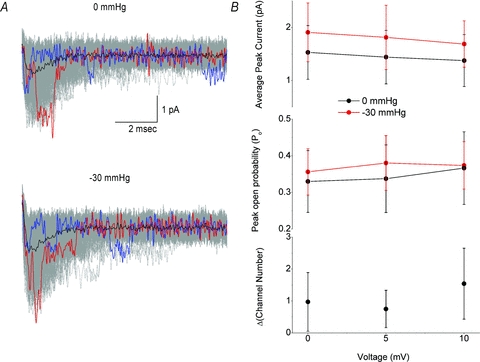
A, a cell-attached membrane patch was depolarized to 0, +5 and +10 mV. Top panel, at rest (0 mmHg), patch was depolarized to +5 mV, showing single traces (600, grey overlap), the trace with peak amplitude (red), and a trace with resolvable single channel openings (blue) and averaged current (black). Bottom panel, the same patch at −30 mmHg. B, top panel, average peak current at 0 mmHg (black) and −30 mmHg (red). Middle panel, max open probability (_P_o) at 0 mmHg (black) and −30 mmHg (red). Bottom panel, increase in active channels with stretch (ΔChannel no. = no. channels post – no. channels pre, _n_= 5). No change in _P_o was noted but a 23% increase in number of channels was seen.
Stretch-effects are partially reversible
Reversibility of stretch effects was tracked by measuring the peak current and the shift in voltage dependence of activation over time after removal of the stretch stimulus. Figure 9_A_ shows a typical patch for which a −30 mmHg produced a −26 mV shift in _V_1/2 and a 70% increase in peak current. Over the next 5 min _V_1/2 returned to near baseline values while the peak current remained elevated. For all the patches measured using this protocol, a single −30 mmHg stretch produced a Δ_V_1/2 of −17 ± 6 mV, and recovery was highly variable over the following 5 min, averaging Δ_V_1/2= 4 ± 10 mV (_n_= 5) (Fig. 9_B_). Half of the cells returned close to the original baseline, while in two other patches there was a persistent hyperpolarized shift in _V_1/2 even after 30 min. Peak currents also increased with stretch Δ_I_peak of 31% (_n_= 5). The amplitude of recovery was not uniform, and the peak current remained elevated after 5 min in all but one patch (Fig. 9_C_).
Figure 9. Stretch-induced shift in voltage dependence of activation and the increase in peak current reverse slowly.
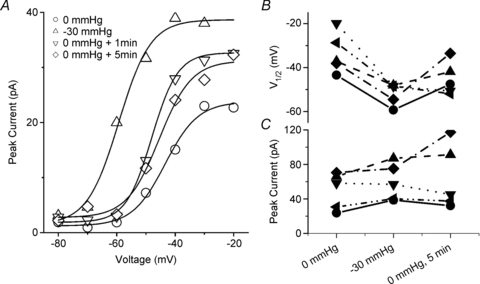
A, Typical cell attached patch was stretched once by a −30 mmHg pulse for 30 s. I–V curves were fitted by a twostate Boltzmann model with the following parameters: at 0 mmHg (_V_1/2=−43 mV, _d_V= 5.6 mV, _I_peak= 23 pA), −30 mmHg stretch (_V_1/2=−59 mV, dV = 5.4 mV, _I_peak= 39 pA), 0 mmHg + 1 min (_V_1/2=−48 mV, dV = 5.5 mV, _I_peak= 33 pA) and 0 mmHg + 5 min (_V_1/2=−45 mV, dV = 5.5 mV, _I_peak= 32 pA). B, In 5 patches, voltage at half-point of activation increased uniformly with stretch, with a variable but incomplete return toward baseline. C, Peak current also increased with stretch and in all but one patch remained elevated.
Discussion
The main findings that emerge from this study are that (1) Nav1.5 is mechanosensitive, confirming previous whole cell data (Strege et al. 2003_b_) and cell-attached patch results (Morris & Juranka, 2007), (2) stretch produces persistent dose-dependent shifts in the voltage dependence of activation and inactivation due to mechanical modulation of the voltage sensors, and (3) inactivated states are stabilized by stretch. These findings carry physiological and pathophysiological relevance for mechanically active organs where Nav1.5 is expressed.
Patch pressure results in increased tension
The pressures used in this study are routinely attained in organs where Nav1.5 is expressed. The heart and the gut generate and experience pressures in the hundreds of mmHg thousands of times a day. However, at the level of channel function, the relevant parameter is tension, not pressure. Images of the stressed patch showed that suction (≤−30 mmHg) increased the patch radius of curvature, while maintaining attachment points (Fig. S2). At more elevated pressures, patch creep and loss of stable attachment points were visible for even short stretch stimuli (30 s). Since Laplace's law dictates that Δ_T_=Δ_rP_/2, the increase in patch radius implies an increase in tension, which for a typical stimulus was on the order of 0.01 N m−1. In fact, a distinct advantage of using patches for mechano-stimulation is the well-controlled pressure application of stimulus and a robust response. However, a limitation is that there is poorly controlled resting tension in the patch, as evidenced by an increased variability of voltage sensitivity recorded at rest (_V_1/2,0mmHg± 9 mV), a several-fold increase compared to typical whole-cell measurements. This is in line with previous studies, which have also demonstrated a variable half-point of activation for sodium channels in the cell attached patches over time (Kimitsuki et al. 1990) and after excision (Fleig et al. 1994). This is most likely to be due to baseline mechanical variation of the patch.
Stretch accelerates voltage-dependent activation and stabilizes the inactivated states
Mechanical stress on a patch containing Nav1.5 channels resulted in large hyperpolarizing shifts in voltage dependence of activation and inactivation. This was a robust and striking finding evident for both positive and negative pressures applied to a patch (Fig. 3). Pressure versus Boltzmann mid-points of activation and inactivation were linear dose-response fits. The slope of Boltzmann curves did not change significantly (Fig. 3_D_). In previous papers where oocyte patches containing other Nav channels were stretched, the midpoints of voltage dependence of activation and inactivation were also left-shifted (Shcherbatko et al. 1999; Tabarean et al. 1999; Wang et al. 2009). The current data further show a quantitative dose–response relationship of voltage dependence to pressure, and thus to tension. It is unlikely that these effects were due to artifacts. For example, one may propose that voltage dependence shifts were secondary to increased access resistance for channels residing in the seal (Suchyna et al. 2009). This is unlikely for two reasons. First, the slopes of the macroscopic I–V curves produced with stretch were not significantly different from those at rest (Fig. 3). Second, increased access resistance should produce a decrease in single channel currents, whereas we observed that single channel amplitudes and Gaussian distributions were not affected with stretch (Fig. 7_C_ and D).
In the time domain, stretch accelerated both activation and fast inactivation (Fig. 2). This acceleration could be explained by the shift in the voltage half-point of activation. When time-dependent traces were amplitude normalized and temporally scaled, there was good overlap for both activation and inactivation, suggesting a single stretch constant was responsible for the stretch effects (Fig. 4). A recent related study on mechanical effects on the cardiac isoform of Nav1.5 expressed in oocytes also noted the acceleration of activation and inactivation kinetics (Morris & Juranka, 2007). Similarly, an overlap of the time-dependent traces by scaling with a single stretch factor was found. The magnitude of these stretch factors was similar to the ones reported here. Since activation and inactivation are kinetically linked in the gating of the Nav channels, scaling of the activation and inactivation kinetics by a single constant implies an increase in a single rate constant in the activation sequence of the channel (C→CA→O). The voltage-dependent steps are prime candidates due to the extensive voltage sensor–lipid interface (Milescu et al. 2009), and previous findings of mechanical sensitivity in the oocyte membranes and bilayers (Schmidt & Mackinnon, 2008). However, this is not the only possibility: for example, stabilization of the open state affects the rate-limiting voltage-independent gate opening and can similarly shift the voltage dependence of activation (Zhao et al. 2004).
The precise determination of the transitions involved in stretch sensing awaits further single channels studies. Our study provides data that implicate a single transition in the multi-step process of channel activation. Inactivation from closed states increased with pressure (Fig. 5). This fact, in combination with a linear stretch constant and similar size shifts in voltage dependence of both activation and inactivation implies that the acceleration in channel kinetics was due to stretch effects on voltage-dependent transitions and not voltage-independent activation gate opening. The data suggest that C→CA is likely to be responsible for these effects and not CA→IA. Firstly, the shift of steady-state inactivation mirrors the amount of shift in voltage dependence of activation (Figs 3_B_ and Fig. 4). Secondly, a separate stretch-sensitive transition in the Nav gating scheme would prevent simple linear scaling demonstrated in Fig. 4. If a sufficiently large depolarizing stimulus is sustained, channels proceed through the open channel sequence and inactivate by traditional open state inactivation. We found that recovery from the open inactivated state is slowed with stretch, which suggests that pressure stabilizes this inactivated state.
Increase in peak current with stretch is due to an increase in the number of active Nav1.5 channels
Single channel conductance (i) and maximum probability of channel opening (_P_o) did not change with stretch (Fig. 8) while estimates of N suggested an increase of about 20% in the number of channels. The conservation of unitary current in the face of increased peak current through the patch is also an important new finding that emerges from this study. The significance of this is that even though Nav1.5 channels can ‘sense’ mechanical stress, the pores are energetically stable and mechanically protected.
The increase in peak current appears to be a result of an increase in the number of active channels in the patch but the source of this increase is still unclear. Multiple possibilities exist. The first and most obvious is inherent to the patch method. As we have shown above, stretch results in a visible increase in membrane area and a likely alteration of pipette-membrane interface, possibly peeling some of the bilayer off the glass wall and exposing more channels. Both suction (negative pressure) and blowing (positive pressure) may peel the patch from the glass due to the resting tension in the patch (Suchyna et al. 2009). Still, we expect less peeling with suction than with blowing. Yet, blowing and suction induced shifts in peak currents that were identical. For a 30 mmHg positive pressure, peak current increase was 0.32 ± 0.10 (_n_= 3), which compares well to the 30 mmHg suction peak current increase of 0.33 ± 0.10 (_n_= 6). Thus, it is unlikely that simple peeling of the patch membrane is responsible for the increase in peak currents.
A more plausible possibility is that stretch induces an increase in membrane fluidity which allows for an enhanced diffusion of the channels from the seal into the dome of the patch (Oghalai et al. 2000). Suchyna et al. (2009) have also showed that even at fixed pressure channels can move between the seal and the patch. Mechanical stress was previously shown to change lipid bilayer fluidity and which in turn produced similar shifts in the voltage sensitivity of Nav channels (Lundbaek et al. 2004). The consequence of this in the electromechanical systems may be significant. Mechanical stress is known to be inhomogeneous for cells in vivo. Regions of cell membrane with increased fluidity may allow accumulation of channels in those locations. In fact, the asymmetry of Nav1.5 distribution in cardiac myocytes is known, and may be related to mechanical forces (Mazzone et al. 2008).
Another explanation may have to do with the possibility of awaking a subset of ‘sleepy channels’ that are already present in the patch (Matteson & Armstrong, 1982). The authors found that cooling converted some typical NaV channels into ‘sleepy’ channels that were slowly activating, but could be aroused by a very positive voltage prepulse. In our system, mechanical stretch inputs energy into the patch, so it may be similar to raising temperature and awaking some of these ‘sleepy’ channels. The physiological consequence of awaking sleepy channels is also potentially important. Increased density of channels in the areas of increased stretch would result in an inhomogeneous sodium concentration in the cell. In turn, this may affect local calcium concentration and provide feedback for excitation–contraction coupling in the areas of increased mechanical stress (Neco et al. 2010).
Stretch of tissues containing Nav1.5 channels may affect physiological function by enhancing activation and allowing a baseline conductance
Alteration of Nav1.5 function or its macromolecular complex components produces disease (Ackerman et al. 2004; Locke et al. 2006). This channel is found in the heart and gut, two systems in which electromechanical coupling is essential. We show that Nav1.5 is mechanosensitive, but in the strictest sense it is not mechano-activated; it is instead mechano-modulated (Sachs & Morris, 1998). This means that the energy input from mechanical stress is not sufficient to noticeably modulate _P_open at resting potentials. Instead, we show a large impact of mechanical stimulation on the channel function during channel activation, inactivation and recovery from inactivation. These findings are clearly important for understanding the physiology and pathophysiology of the electromechanical organs.
Contractile organs can be modelled as having a background of steady-state homogeneous stretch, and on this background is superimposed the acute stretch and simultaneous voltage activation. In this setting, we can begin to examine the potential relevance of our findings. Stretch will negatively shift the voltage dependence of activation and inactivation, producing a shift in the window current. We show that the voltage shift is substantial, amounting to tens of millivolts. For the cells that have resting potentials at the window current, this will make them hyperexcitable. The result will be an increase in the baseline current, which has clear implications for pathology, as exemplified in LQT. The Cells that rest at more hyperpolarized potentials will have a larger fraction of channels available for activation, increasing baseline chatter and also making these cells excitable. On the other hand, the cells that rest at more depolarized potentials will have fewer channels available for activation.
The assumption of uniform steady-state stretch is likely to be incorrect. First, at the level of the cell, there are time-dependent changes in stress due to the inhomogeneity of the cytoskeleton (Morone et al. 2008) and viscosity of both the cytoskeleton and the bilayer (Suchyna & Sachs, 2007). Our data agree with this notion by showing a prolonged impact of single stretch on channel operation. Second, at the level of mechanically active tissues, steady-state stretch does not hold. These tissues regularly cycle mechanical stimuli and rely on tight mechanoelectrical feedback. The asymmetry of cell surface anatomy and mechanical stress suggests that the notion of a simple shift of window current with stretch is naïve. Instead, the window current will shift in a graded fashion, producing a stretch-dependent distribution of steady state leakage over a wide voltage range. Additionally, with increased underlying activity other channel states will become increasingly important. In this situation the stability of the inactivated states is also paramount. Our results show that stretched channels will preferentially enter the inactivated states from both closed and open states and are likely to require stronger stimuli for further activation. This raises the possibility that mechanical modulation of Nav1.5 is also aimed at protection from out-of-scale mechanical stimuli. Alternatively, cells may use modification of the Nav1.5 cytoskeletal connections (Maruoka et al. 2000) and lipid domain (raft) targeting (Tamkun et al. 2007) to direct the channel localization and allow normalization of response.
In summary, our data suggest that mechanical stimulation of Nav1.5 results in dose-dependent voltage dependence shifts of activation and inactivation due to mechanical modulation of the voltage sensors. Our findings suggest that mechanical sensitivity via effects on voltage-dependent channels may play a significant role in the physiology of motile cells.
Acknowledgments
The authors would like to thank Drs Lorin Milescu, Thomas Suchyna and Catherine Morris for valuable discussions and comments on the manuscript. This work was supported by The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) R01 52766.
Glossary
Abbreviations
CSI
closed state inactivation
SF
scaling factor
Author contributions
The authors approved the final version. This work was performed in the laboratory of Gianrico Farrugia, Mayo Clinic, Rochester, MN, USA. Conception and design of the experiments: A.B., J.L.R., C.B., G.F.; collection, analysis and interpretation of data: A.B., J.L.R., C.B., P.R.S., G.F.; drafting the article or revising in critically for important intellectual content: A.B., J.L.R., F.S., G.F.
Supplemental material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Ackerman MJ. The long QT syndrome: ion channel diseases of the heart. Mayo Clin Proc. 1998;73:250–269. doi: 10.4065/73.3.250. [DOI] [PubMed] [Google Scholar]
- Ackerman MJ, Splawski I, Makielski JC, Tester DJ, Will ML, Timothy KW, Keating MT, Jones G, Chadha M, Burrow CR, Stephens JC, Xu C, Judson R, Curran ME. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm. 2004;1:600–607. doi: 10.1016/j.hrthm.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Aldrich RW, Corey DP, Stevens CF. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 1983;306:436–441. doi: 10.1038/306436a0. [DOI] [PubMed] [Google Scholar]
- Armstrong CM. Na channel inactivation from open and closed states. Proc Natl Acad Sci U S A. 2006;103:17991–17996. doi: 10.1073/pnas.0607603103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker PF, Hodgkin AL, Shaw TI. Replacement of the axoplasm of giant nerve fibres with artificial solutions. J Physiol. 1962;164:330–354. doi: 10.1113/jphysiol.1962.sp007025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banderali U, Juranka PF, Clark RB, Giles WR, Morris CE. Impaired stretch modulation in potentially lethal cardiac sodium channel mutants. Channels (Austin) 2010;4:12–21. doi: 10.4161/chan.4.1.10260. [DOI] [PubMed] [Google Scholar]
- Bennett PB, Yazawa K, Makita N, George AL., Jr Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- Beyder A, Sachs F. Electromechanical coupling in the membranes of Shaker-transfected HEK cells. Proc Natl Acad Sci U S A. 2009;106:6626–6631. doi: 10.1073/pnas.0808045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. J Gen Physiol. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borjesson SI, Hammarstrom S, Elinder F. Lipoelectric modification of ion channel voltage gating by polyunsaturated fatty acids. Biophys J. 2008;95:2242–2253. doi: 10.1529/biophysj.108.130757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese B, Tabarean IV, Juranka P, Morris CE. Mechanosensitivity of N-type calcium channel currents. Biophys J. 2002;83:2560–2574. doi: 10.1016/S0006-3495(02)75267-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell RA, Clemo HF, Baumgarten CM. Using gadolinium to identify stretch-activated channels: technical considerations. Am J Physiol Cell Physiol. 1998;275:C619–621. doi: 10.1152/ajpcell.1998.275.2.C619. [DOI] [PubMed] [Google Scholar]
- Elinder F, Arhem P. Effects of gadolinium on ion channels in the myelinated axon of Xenopus laevis: four sites of action. Biophys J. 1994;67:71–83. doi: 10.1016/S0006-3495(94)80456-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia G, Holm AN, Rich A, Sarr MG, Szurszewski JH, Rae JL. A mechanosensitive calcium channel in human intestinal smooth muscle cells. Gastroenterology. 1999;117:900–905. doi: 10.1016/s0016-5085(99)70349-5. [DOI] [PubMed] [Google Scholar]
- Fleig A, Ruben PC, Rayner MD. Kinetic mode switch of rat brain IIA Na channels in Xenopus oocytes excised macropatches. Pflugers Arch. 1994;427:399–405. doi: 10.1007/BF00374253. [DOI] [PubMed] [Google Scholar]
- Gellens ME, George AL, Jr, Chen LQ, Chahine M, Horn R, Barchi RL, Kallen RG. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci U S A. 1992;89:554–558. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil Z, Silberberg SD, Magleby KL. Voltage-induced membrane displacement in patch pipettes activates mechanosensitive channels. Proc Natl Acad Sci U S A. 1999;96:14594–14599. doi: 10.1073/pnas.96.25.14594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu CX, Juranka PF, Morris CE. Stretch-activation and stretch-inactivation of Shaker-IR, a voltage-gated K+ channel. Biophys J. 2001;80:2678–2693. doi: 10.1016/S0006-3495(01)76237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. Sunderland, MA: Sinauer Associates, Inc.; 2001. [Google Scholar]
- Horn R. Estimating the number of channels in patch recordings. Biophys J. 1991;60:433–439. doi: 10.1016/S0006-3495(91)82069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- Kimitsuki T, Mitsuiye T, Noma A. Negative shift of cardiac Na+ channel kinetics in cell-attached patch recordings. Am J Physiol Heart Circ Physiol. 1990;258:H247–254. doi: 10.1152/ajpheart.1990.258.1.H247. [DOI] [PubMed] [Google Scholar]
- Kraichely RE, Strege PR, Sarr MG, Kendrick ML, Farrugia G. Lysophosphatidyl choline modulates mechanosensitive L-type Ca2+ current in circular smooth muscle cells from human jejunum. Am J Physiol Gastrointest Liver Physiol. 2009;296:G833–839. doi: 10.1152/ajpgi.90610.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitko U, Juranka PF, Morris CE. Membrane stretch slows the concerted step prior to opening in a Kv channel. J Gen Physiol. 2006;127:687–701. doi: 10.1085/jgp.200509394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitko U, Morris CE. Membrane tension accelerates rate-limiting voltage-dependent activation and slow inactivation steps in a Shaker channel. J Gen Physiol. 2004;123:135–154. doi: 10.1085/jgp.200308965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke GR, 3rd, Ackerman MJ, Zinsmeister AR, Thapa P, Farrugia G. Gastrointestinal symptoms in families of patients with an SCN5A-encoded cardiac channelopathy: evidence of an intestinal channelopathy. Am J Gastroenterol. 2006;101:1299–1304. doi: 10.1111/j.1572-0241.2006.00507.x. [DOI] [PubMed] [Google Scholar]
- Lundbaek JA, Birn P, Hansen AJ, Sogaard R, Nielsen C, Girshman J, Bruno MJ, Tape SE, Egebjerg J, Greathouse DV, Mattice GL, Koeppe RE, 2nd, Andersen OS. Regulation of sodium channel function by bilayer elasticity: the importance of hydrophobic coupling. Effects of Micelle-forming amphiphiles and cholesterol. J Gen Physiol. 2004;123:599–621. doi: 10.1085/jgp.200308996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyford GL, Farrugia G. Ion channels in gastrointestinal smooth muscle and interstitial cells of Cajal. Curr Opin Pharmacol. 2003;3:583–587. doi: 10.1016/j.coph.2003.06.010. [DOI] [PubMed] [Google Scholar]
- Makielski JC, Ye B, Valdivia CR, Pagel MD, Pu J, Tester DJ, Ackerman MJ. A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ Res. 2003;93:821–828. doi: 10.1161/01.RES.0000096652.14509.96. [DOI] [PubMed] [Google Scholar]
- Maruoka ND, Steele DF, Billie PY, Dan P, Zhang X, Moore EDW, Fedida D. α-Actinin-2 couples to cardiac Kv1.5 channels, regulating current density and channel localization in HEK cells. FEBS Lett. 2000;473:188–194. doi: 10.1016/s0014-5793(00)01521-0. [DOI] [PubMed] [Google Scholar]
- Matteson DR, Armstrong CM. Evidence for a population of sleepy sodium channels in squid axon at low temperature. J Gen Physiol. 1982;79:739–758. doi: 10.1085/jgp.79.5.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzone A, Strege PR, Tester DJ, Bernard CE, Faulkner G, De Giorgio R, Makielski JC, Stanghellini V, Gibbons SJ, Ackerman MJ, Farrugia G. A mutation in telethonin alters Nav1.5 function. J Biol Chem. 2008;283:16537–16544. doi: 10.1074/jbc.M801744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milescu M, Bosmans F, Lee S, Alabi AA, Kim JI, Swartz KJ. Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat Struct Mol Biol. 2009;16:1080–1085. doi: 10.1038/nsmb.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morone N, Nakada C, Umemura Y, Usukura J, Kusumi A. Three-dimensional molecular architecture of the plasma-membrane-associated cytoskeleton as reconstructed by freeze-etch electron tomography. Methods Cell Biol. 2008;88:207–236. doi: 10.1016/S0091-679X(08)00412-3. [DOI] [PubMed] [Google Scholar]
- Morris CE, Juranka PF. Nav channel mechanosensitivity: activation and inactivation accelerate reversibly with stretch. Biophys J. 2007;93:822–833. doi: 10.1529/biophysj.106.101246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CE, Juranka PF, Lin W, Morris TJ, Laitko U. Studying the mechanosensitivity of voltage-gated channels using oocyte patches. Methods Mol Biol. 2006;322:315–329. doi: 10.1007/978-1-59745-000-3_22. [DOI] [PubMed] [Google Scholar]
- Neco P, Rose B, Huynh N, Zhang R, Bridge JH, Philipson KD, Goldhaber JI. Sodium-calcium exchange is essential for effective triggering of calcium release in mouse heart. Biophys J. 2010;99:755–764. doi: 10.1016/j.bpj.2010.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oghalai JS, Zhao HB, Kutz JW, Brownell WE. Voltage- and tension-dependent lipid mobility in the outer hair cell plasma membrane. Science. 2000;287:658–661. doi: 10.1126/science.287.5453.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opsahl LR, Webb WW. Lipid-glass adhesion in giga-sealed patch-clamped membranes. Biophys J. 1994;66:75–79. doi: 10.1016/S0006-3495(94)80752-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Y, Strege P, Miller SM, Makielski J, Ackerman M, Gibbons SJ, Farrugia G. Syntrophin γ2 regulates SCN5A gating by a PDZ domain-mediated interaction. J Biol Chem. 2003;278:1915–1923. doi: 10.1074/jbc.M209938200. [DOI] [PubMed] [Google Scholar]
- Qin F. Restoration of single-channel currents using the segmental k-means method based on hidden Markov modeling. Biophys J. 2004;86:1488–1501. doi: 10.1016/S0006-3495(04)74217-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F, Auerbach A, Sachs F. Estimating single-channel kinetic parameters from idealized patch-clamp data containing missed events. Biophys J. 1996;70:264–280. doi: 10.1016/S0006-3495(96)79568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs F, Morris CE. Mechanosensitive ion channels in nonspecialized cells. Rev Physiol Biochem Pharmacol. 1998;132:1–77. doi: 10.1007/BFb0004985. [DOI] [PubMed] [Google Scholar]
- Saito YA, Strege PR, Tester DJ, Locke GR, 3rd, Talley NJ, Bernard CE, Rae JL, Makielski JC, Ackerman MJ, Farrugia G. Sodium channel mutation in irritable bowel syndrome: evidence for an ion channelopathy. Am J Physiol Gastrointest Liver Physiol. 2009;296:G211–218. doi: 10.1152/ajpgi.90571.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Mackinnon R. Voltage-dependent K+ channel gating and voltage sensor toxin sensitivity depend on the mechanical state of the lipid membrane. Proc Natl Acad Sci U S A. 2008;105:19276–19281. doi: 10.1073/pnas.0810187105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbatko A, Ono F, Mandel G, Brehm P. Voltage-dependent sodium channel function is regulated through membrane mechanics. Biophys J. 1999;77:1945–1959. doi: 10.1016/S0006-3495(99)77036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strege PR, Holm AN, Rich A, Miller SM, Ou Y, Sarr MG, Farrugia G. Cytoskeletal modulation of sodium current in human jejunal circular smooth muscle cells. Am J Physiol Cell Physiol. 2003a;284:C60–66. doi: 10.1152/ajpcell.00532.2001. [DOI] [PubMed] [Google Scholar]
- Strege PR, Ou Y, Sha L, Rich A, Gibbons SJ, Szurszewski JH, Sarr MG, Farrugia G. Sodium current in human intestinal interstitial cells of Cajal. Am J Physiol Gastrointest Liver Physiol. 2003b;285:G1111–1121. doi: 10.1152/ajpgi.00152.2003. [DOI] [PubMed] [Google Scholar]
- Suchyna TM, Markin VS, Sachs F. Biophysics and structure of the patch and the gigaseal. Biophys J. 2009;97:738–747. doi: 10.1016/j.bpj.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchyna TM, Sachs F. Mechanosensitive channel properties and membrane mechanics in mouse dystrophic myotubes. J Physiol. 2007;581:369–387. doi: 10.1113/jphysiol.2006.125021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabarean IV, Juranka P, Morris CE. Membrane stretch affects gating modes of a skeletal muscle sodium channel. Biophys J. 1999;77:758–774. doi: 10.1016/S0006-3495(99)76930-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamkun MM, O'Connell KM, Rolig AS. A cytoskeletal-based perimeter fence selectively corrals a sub-population of cell surface Kv2.1 channels. J Cell Sci. 2007;120:2413–2423. doi: 10.1242/jcs.007351. [DOI] [PubMed] [Google Scholar]
- Wang JA, Lin W, Morris T, Banderali U, Juranka PF, Morris CE. Membrane trauma and Na+-leak from Nav1.6 channels. Am J Physiol Cell Physiol. 2009;297:C823–834. doi: 10.1152/ajpcell.00505.2008. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Yarov-Yarovoy V, Scheuer T, Catterall WA. A gating hinge in Na+ channels: a molecular switch for electrical signaling. Neuron. 2004;41:859–865. doi: 10.1016/s0896-6273(04)00116-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.