Protein S-nitrosylation in health and disease: a current perspective (original) (raw)
. Author manuscript; available in PMC: 2011 Jun 2.
Summary
Protein _S_-nitrosylation conveys a large part of the ubiquitous influence of nitric oxide on cellular signal transduction, and accumulating evidence indicates important roles for _S_-nitrosylation both in normal physiology and in a broad spectrum of human diseases. Here we review recent findings that implicate _S_-nitrosylation in cardiovascular, pulmonary, musculoskeletal and neurological (dys)function, as well as in cancer. The emerging picture shows that, in many cases, pathophysiology correlates with hypo- or hyper-_S_-nitrosylation of specific protein targets, rather than a general cellular insult due to loss of or enhanced nitric oxide synthase activity. In addition, it is increasingly evident that dysregulated _S_-nitrosylation can result not only from alterations in the expression, compartmentalization and/or activity of nitric oxide synthases but can also reflect a contribution from denitrosylases, including prominently the _S_-nitrosoglutathione (GSNO)-metabolizing enzyme, GSNO reductase. Finally, because exogenous mediators of protein _S_-nitrosylation or denitrosylation can substantially affect the development or progression of disease, potential therapeutic agents that modulate _S_-nitrosylation could well have broad clinical utility.
Keywords: nitric oxide, _S_-nitrosylation, _S_-nitrosoglutathione, cysteine, GSNOR, heart failure, asthma, nitrosative stress
Introduction
In mammalian cells, the L-Arg-dependent nitric oxide (NO) synthases – neuronal NOS (nNOS, NOS1), inducible NOS (iNOS, NOS2) and endothelial NOS (eNOS, NOS3) – are the major sources of endogenous NO, and the stimulus-coupled activation or induction of NO synthases has been shown to mediate or modulate a broad range of cellular signaling pathways. The physiological influence of NO is exerted predominantly through the posttranslational modification and functional regulation of proteins. It was first established that nitrosylation of heme iron within soluble guanylate cyclase activates the enzyme to generate cyclic GMP, and thereby subserves NO-based vasoactivity. However, hemes do not generally elicit cellular signaling involving posttranslational modification of proteins, and thus an explanation for most NO-based bioactivity was not apparent. Subsequently, a large body of experimental evidence has demonstrated that _S_-nitrosylation of Cys residues within a broad functional spectrum of proteins conveys a large part of the ubiquitous influence of NO on cellular function [1].
Expression of iNOS is induced in many mammalian cell types by a variety of stressors or injury, and the cytotoxic action of NO generated in particular by phagocytic cells raised the possibility that NO generated at relatively high and sustained levels by iNOS could compromise cellular function through a generalized nitrosative stress. However, the emerging recognition that NO is involved in a multiplicity of cellular signal transduction pathways, through protein _S_-nitrosylation, pointed to the possibility that dysregulated _S_-nitrosylation could contribute to pathophysiologies characteristic of a wide range of disease states [2]. The relatively recent development of improved methods for the analysis of protein _S_-nitrosylation [3,4] has allowed the identification of numerous _S_-nitrosylated proteins (SNO-proteins) whose levels of _S_-nitrosylation can be altered in disease, and the emerging picture shows that hypo- or hyper-_S_-nitrosylation of these specific protein targets (which result in alterations in protein function) are directly implicated in the etiology and symptomatology of an increasing number of human diseases, including prominently disorders of the cardiovascular, musculoskeletal and nervous systems (Table 1). In a number of cases, the specific Cys residues that are the loci of (patho)physiological regulation by _S_-nitrosylation have been identified.
Table 1.
_S_-nitrosylated peptides and proteins that have been implicated in mammalian (patho)physiology.

The molecular mechanisms underlying the (dys)regulation of _S_-nitrosylation, and possible approaches to therapeutically altering SNO-protein levels, have also begun to receive increasing attention. Although NOS expression and activity are obvious governors of _S_-nitrosylation, the co-localization of NOS enzymes with target proteins, including direct interactions, appears in many cases to be an important determinant of _S_-nitrosylation under physiological conditions, and accordingly, aberrant NOS localization appears to be involved in at least several diseases (Fig. 1). Such deficits can have a genetic basis. For example, in a variant of long QT syndrome, a mutation in an nNOS-scaffolding protein results in disinhibition of nNOS and aberrant _S_-nitrosylation of a cardiac ion channel [5]. In addition to localization, it has been established that the transfer of NO groups between proteins and glutathione governs a cellular equilibrium between small-molecular-weight and protein _S_-nitrosothiols (SNOs) (Fig. 2). In mouse models, genetic ablation of _S_-nitrosoglutathione reductase (GSNOR), the enzyme that is principally responsible for GSNO metabolism, results in enhanced levels of SNO-proteins and significantly attenuates experimental asthma and heart-failure [6,7], while increasing the severity of endotoxic shock [8]. Finally, the therapeutic potential of agents that affect _S_-nitrosylation is being explored with promising results (Table 2). These agents have the potential to restore deficient SNO-proteins to physiological levels or to otherwise influence cellular signaling pathways that are mediated or modulated by _S_-nitrosylation. For example, SNO-repleting agents have been shown to be highly efficacious in the setting of inflammation, including experimental models of lung injury, stroke and multiple sclerosis in which _S_-nitrosylation appears to play a major role in expression of the innate immune response [9–12]. This review focuses on the numerous proteins and signaling pathways that are regulated by _S_-nitrosylation, in the context of diseases in which aberrant _S_-nitrosylation has recently been implicated.
Fig. 1. NOS-dependent mechanisms of _S_-nitrosylation.
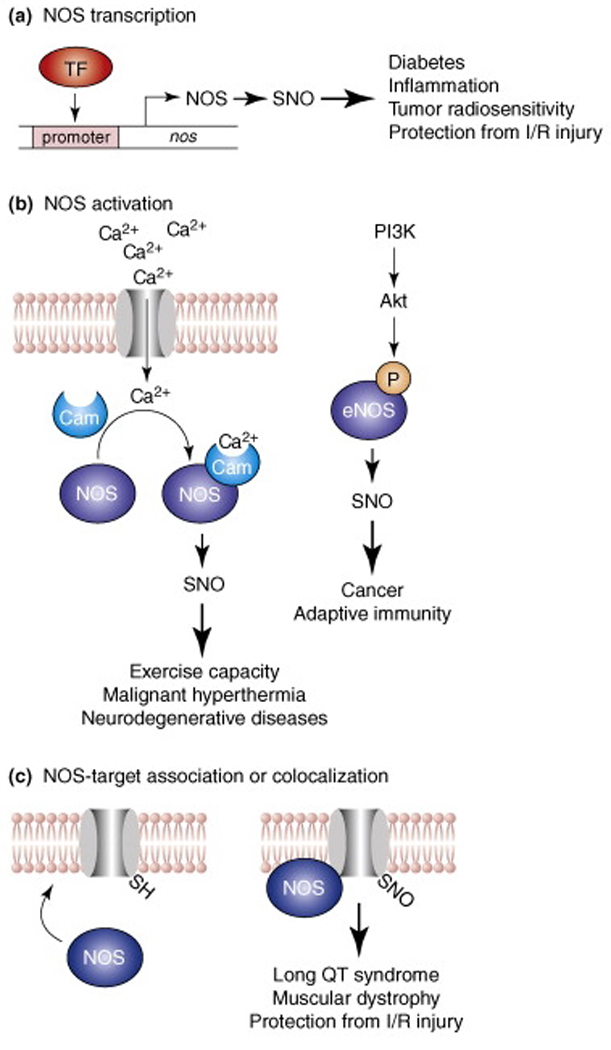
Three principal mechanisms for regulation of NO synthase (NOS)-dependent protein _S_-nitrosylation and the (patho)physiology associated with the induction or disruption of these mechanisms are shown. (a) Binding of a transcription factor (TF) to NOS promoter induces expression of the gene encoding NOS. (b) (Left) influx into the cytosol of extracellular or internal store-derived Ca2+ promotes Ca2+-calmodulin (CaM) binding and activation of eNOS and nNOS. Alternatively (right), phosphoinositide-3 kinase (PI3K) activates protein kinase B (Akt), which phosphorylates and activates eNOS. (c) Subcellular compartmentation (co-localization) of NOS and its substrates, which may involve a direct interaction (as in the illustrated case of a membrane-intercalated ion channel) is an important determinant of the target specificity of _S_-nitrosylation, and dysregulated co-localization can result in hyper- or hypo-_S_-nitrosylation.
Fig. 2. Regulation of SNO homeostasis by _S_-nitrosoglutathione reductase (GSNOR).
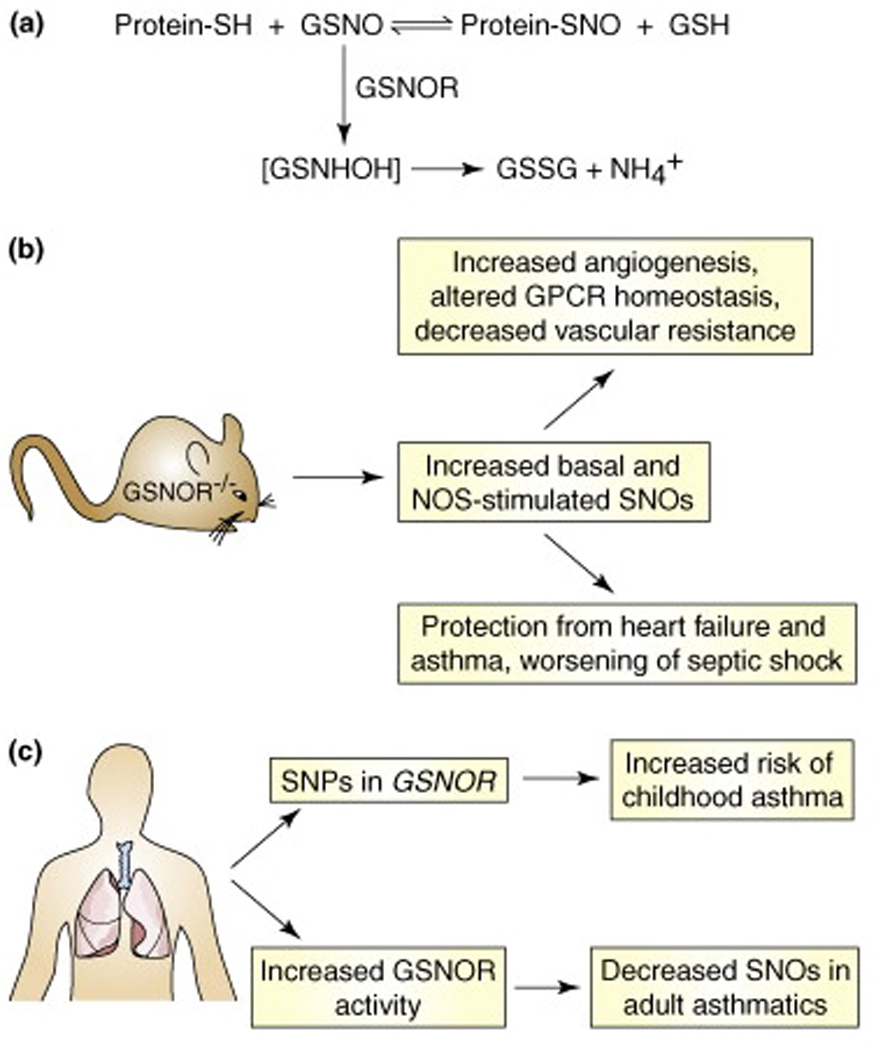
(a) In cells, the low-mass _S_-nitrosothiol, _S_-nitrosoglutathione (GSNO), is in equilibrium with a subset of protein _S_-nitrosothiols. GSNO is metabolized by the enzyme GSNOR, and cells and tissues lacking GSNOR exhibit increased levels of SNO-proteins. (b) Analyses in GSNOR-knockout mice (GSNOR−/−) have revealed numerous roles for protein _S_-nitrosylation. In particular, knockout animals exhibit low systemic vascular resistance and increased cardiac output under basal conditions, and are protected from mocardial infarction and allergen-induced airway hyperreactivity. (c) Mutations in GSNOR, as well as increased airway GSNOR expression and activity, are associated with human asthma.
Table 2.
Amelioration of disease/tissue injury in humans and in animal models by treatments that affect _S_-nitrosylation.

Modulation of SNO production: NO synthases and organic nitrates
In tissues and extracellular fluids, SNO levels are likely to reflect NOS activity and, accordingly, can be modulated through altered expression or activity of enzymes that control the availability of endogenous NOS substrates (e.g. l-Arg) or endogenous NOS inhibitors (e.g. asymmetric dimethylarginine, ADMA). Additionally, a number of enzymes in the l-Arg/NO pathway are also targets of regulatory _S_-nitrosylation [1], including arginase, which catabolizes l-Arg. Arginase 1 (Arg1; cytosolic arginase) is activated through enhanced multimerization resulting from _S_-nitrosylation at cysteine 303 (C303) [13]. Arg1 _S_-nitrosylation and activation coincide with increased iNOS expression in the aortic endothelium of aging rats [13] and may contribute to age-related endothelial dysfunction and vascular stiffness [14]. Levels of the bronchodilator GSNO are low in asthmatic airways [2], and a causal link between SNO depletion and arginase levels has been suggested [15]: Arg1 and Arg2 are upregulated in human asthma and in experimental asthma models [16,17], and single nucleotide polymorphisms (SNPs) in genes encoding Arg1 and Arg2 are associated with increased risk of childhood asthma [18]. Finally, pharmacological inhibition of arginase leads to increased SNO levels in bronchial epithelia of allergen-challenged mice, and although it can also increase inflammation [15], arginase inhibition correlates with protection from allergen-induced airway hyperresponsivity (AHR) in several asthma models [16,19]. These data suggest that altering NO homeostasis and SNO levels might ameliorate the asthmatic phenotype.
NO or low-mass SNOs can also be produced pharmacologically from organic nitrovasodilators, chief among which is nitroglycerin (glyceryl trinitrate; GTN). The vasodilatory properties of GTN arise from its metabolism by mitochondrial aldehyde dehydrogenase (mtALDH) and the concerted production of NO bioactivity by mitochondria [20]. However, prolonged use of GTN results in tachyphylaxis to the drug itself (mechanism-based tolerance) as well as diminished responsiveness to other nitrovasodilators (cross-tolerance). _S_-nitrosylation is increasingly implicated in these effects [21], with soluble guanylate cyclase (sGC) serving as a major locus of effect (Box 1). Both exposure to low-mass SNOs and eNOS activation result in _S_-nitrosylation of sGC, and the modification coincides with a reduction in NO-stimulated sGC activity [22]. Two cysteines residues in the sGC alpha/beta heterodimer are the targets of _S_-nitrosylation, C243 of the alpha-chain (αC243) and C122 of the beta-chain (βC122). Of these, βC122 is in proximity to the heme-binding site, although mutation of either residue partially prevents SNO-induced sGC densitization [22]. These effects of NO/SNO are essentially recapitulated in cellular and animal models of nitrate tolerance, and, in addition, partial sGC denitrosylation and restoration of sGC activity are observed after treatment with N-acetylcysteine (NAC) [23]. Thus, denitrosylation of SNO-sGC might underlie some of the clinical benefit of NAC therapy for nitrate tolerance.
Box 1. Protein _S_-nitrosylation can be regulated independently of changes in NOS expression, activity or colocalization with substrates.
- Anion Exchange Protein 1 (AE1/band 3). Decreased transnitrosylative transfer of NO from SNO-Hb to the erythrocyte membrane protein band 3 in sickle cell anemia, resulting in decreased NO-based vasoactivity of red blood cells [49].
- Guanylyl cyclase. Nitroglycerin-mediated NO production by mitochondrial aldehyde dehydrogenase (mtALDH) and _S_-nitrosylation of both mtALDH and guanylate cyclase in tolerance to nitrovasodilators [23].
- Microbial proteins. Transition metal-dependent protein _S_-nitrosylation in yeast, implicated in nitrosative stress that subserves innate immunity [139].
- Hemoglobin. Superoxide dismutase (SOD)-catalyzed _S_-nitrosylation of hemoglobin within RBCs, proposed as a mechanism to regulate hypoxic vasodilation and SNO homeostasis [140]; the O2-catalyzed (allosterically-coupled) auto-_S_-nitrosylation of Hb wherein NO transfers from oxidized heme to thiol, proposed as an integral aspect of the human respiratory cycle [48,141].
- NMDA receptor. Copper-induc_e_d _S_-nitrosylation of neuronal NMDA receptors, suggested as a mechanism for copper-dependent protection against NMDA receptor-mediated excitotoxicity [142]
- Mitochondrial proteins and GAPDH. Expression of mutant SOD results in decreased S-nitrosylation of mitochondrial proteins and GAPDH in an experimental model of amyotrophic lateral sclerosis [152].
Therapy with the NO donor isosorbide dinitrate (in combination with hydralazine) markedly reduces mortality in African-Americans with heart failure [24]. Morbidity and mortality in heart failure patients results from either arrhythmia or ventricular dysfunction that is due at least in part to downregulation of β-adrenergic receptors (β-AR) and dysregulation of calcium homeostasis. It is therefore of interest that NO bioactivity has been strongly associated with both arrhythmia and pump failure [25–27], and recent studies support the possibility that _S_-nitrosylation of cardiac proteins ameliorates heart failure by reversing calcium leakage [28] and normalizing β-AR expression [27].
GSNO reductase and its regulation by NO/SNO – implications for asthma
A major mechanism for protein _S_-nitrosylation in vivo is the reversible transnitrosylation of protein thiols by GSNO, the predominant low-mass SNO (Figure 2) [2]. GSNO is the preferred physiological substrate for glutathione-dependent formaldehyde dehydrogenase (class III alcohol dehydrogenase, ADH III, which in methylotropic bacteria may also metabolize formaldehyde), whereasalcohols are apparently not physiological substrates of ADH III. Thus the enzyme has been renamed GSNO reductase (GSNOR). GSNOR metabolizes GSNO to glutathione disulfide and a glutathione N-hydroxysulfenamide intermediate, which is ultimately reduced to ammonia (Figure 2). Analyses in GSNOR-knockout mice have helped elucidate the physiological roles of both GSNO and protein _S_-nitrosylation. GSNOR-knockout mice have markedly increased levels of SNO-proteins, which demonstrates a role for GSNO/GSNOR in SNO-protein homeostasis. These mice exhibit increased mortality in endotoxic shock, and SNO-protein levels and mortality are attenuated by iNOS inhibitors. In contrast GSNOR-knockout mice are protected from experimental myocardial infarction, due at least in part to _S_-nitrosylation-mediated stabilization of hypoxia-inducible factor HIF-1α and increased angiogenesis ([7]; see below).
There is also accumulating evidence from both human studies and experimental models that GSNOR plays a significant role in the etiology and symptomatology of asthma. It is well established that airway SNOs are depleted in asthma [2], and it has recently been reported that this decrease is associated with increased GSNOR levels and activity in airway lining fluid (obtained by bronchoalveolar lavage) of adult asthmatics [29]. In addition, SNPs in GSNOR are associated with a decreased (1 or 2 copies of minor allele of SNP rs1154404) or increased (homozygotes of minor allele of SNP rs28730619) risk of childhood asthma; haplotypes with the major allele of rs1154404 and minor allele of rs28730619 have an increased risk for childhood asthma [30]. Further, one SNP (rs1154400) is also associated with decreased responsiveness to {Greek beta}-agonist therapy in African-American children with asthma [153]. The effects of these SNPs on GSNOR protein expression, localization and/or activity have not yet been characterized. However, GSNOR activity in the lungs of 36 human asthmatics correlated directly with airway hyperreactivity [29]. In an experimental asthma model, GSNOR-knockout mice exhibit lower basal airway tone and less AHR to methacholine challenge following sensitization with ovalbumin (OVA) [6]. This protection is associated with an increase in iNOS-derived SNO-proteins in the lungs of GSNOR-knockout mice following OVA challenge [6], and is reversed by NO synthase inhibitors. Lungs from GSNOR-knockout mice also have increased levels of β-adrenergic receptors (β-ARs) and their airways are resistant to β-agonist-induced desensitization in vitro, suggesting that GSNO promotes airway relaxation through the regulation of β-AR signaling (Box 2) [6].
Box 2. Regulation of G protein-coupled receptor signaling—pulmonary and cardiovascular consequences.
Several lines of evidence point to a role for _S_-nitrosylation in the regulation of GPCR desensitization (loss of G-protein signaling) and downregulation that characterize heart failure. Diminished responsiveness (tachyphylaxis) to β-agonists, as assessed by vasodilation or cardiac contractility, is potentiated in vivo by NOS inhibitors and can be reversed by administration of low-mass SNOs [27,143]. GSNOR-knockout mice are protected from tachyphylaxis to β-agonists [6], and hearts and lungs from these mice display increased basal β-AR expression [27]. In addition, GSNO inhibits isoproterenol-induced downregulation of cardiac β-ARs in a mouse model of ‘heart failure’ [27]. In vitro, exogenous SNOs or eNOS overexpression inhibit desensitization and internalization of the β2-AR, in concert with inhibition of β-AR phosphorylation and β-arrestin translocation to the plasma membrane [27]. These effects are consistent with inhibition of the G protein-coupled receptor kinase, GRK2, by _S_-nitrosylation of a crucial Cys (C340) in the catalytic domain, mutation of which abolishes GPCR-mediated _S_-nitrosylation of GRK2 as well as the eNOS-dependent regulation of β2-AR internalization [27]. These data establish a possible mechanism for the tonic inhibition of cardiac and vascular β-AR desensitization by endogenous NO, and identify GRK2 as a regulatory locus. Inasmuch as both asthma and heart failure are characterized by attenuated β-AR signaling that results from increased receptor desensitization and downregulation, these disease states might reflect dysregulated GRK2 _S_-nitrosylation, one potential underlying mechanism being diminished NOS activity (which could result in part from inhibition of eNOS by GRK2 itself [144]). In addition, _S_-nitrosylation of dynamin and β-arrestin2 (βarr2) regulates GPCR internalization [56,145]. _S_-nitrosylation of C607 within dynamin promotes its mutimerization and activation, and _S_-nitrosylation of C410 within βarr2 promotes its interaction with clathrin/AP-2. Notably, SNO-βarr2 levels, and the affiliations of βarr2 with clathrin/AP-2, are enhanced in GSNOR-knockout mice [145].
NO also appears to modulate GSNOR expression. Inhibition of constitutive NOS activity increases GSNOR mRNA in mouse lungs [31], whereas overexpression of vascular endothelial growth factor (VEGF) results in large, NO-dependent increases in GSNOR transcript levels that appear to be mediated through induction of eNOS and iNOS [31]. The increase in GSNOR expression (although not verified at the protein level) suggests a mechanism for VEGF-induced AHR. GSNOR transcription is stimulated by specificity factor 1 (Sp1) and repressed by Sp3/Sp4 [32]. By analogy to the GSNO-dependent regulation of cystic fibrosis trans-membrane conductance regulator (CFTR) transcription by Sp3/Sp1 [33], low levels of NO/SNO (arising from constitutive NOS activity) might repress GSNOR transcription through augmented Sp3 binding, whereas high levels of NO/SNO (e.g. induced by VEGF) might activate GSNOR transcription by simultaneously inhibiting Sp3 binding and potentiating Sp1 binding [33]. Collectively, these studies demonstrate a role for GSNO in SNO-protein homeostasis, suggest that (dys)regulation of GSNOR expression may be important in the etiology of airway disease and strongly support a role for protein _S_-nitrosylation in NOS-dependent (patho)physiology.
Ras _S_-nitrosylation – adaptive immunity and tumor maintenance
Many GTPases within the Ras superfamily contain redox-sensitive Cys residues that are susceptible to _S_-nitrosylation [34]. In the cases of H-, K- and N-Ras, NO promotes the conversion by guanine nucleotide exchange of inactive, GDP-bound Ras to its active, GTP-bound form. Ras activation is coincident with _S_-nitrosylation of C118, which resides within the nucleotide-binding domain; mutation of C118 abolishes NO-induced Ras activation [34]. Although _S_-nitrosylation of Ras is closely coupled to NOS activity, it has been proposed that higher oxides of NO (e.g. NO2•) affect guanine nucleotide exchange by C118 thiyl radical-catalyzed oxidation and, finally, release of the guanine cofactor [35,36]. In this context, _S_-nitrosylation might prevent or promote further nucleotide turnover (depending on whether Cys radical formation is inhibited or enhanced) and/or serve as a marker of NO-based activation.
In T-lymphocytes, eNOS undergoes rapid and sustained activation by both phosphoinositide 3-kinase/protein kinase B (PI3K–Akt)- and Ca2+-dependent pathways upon binding of the T-cell receptor (TCR) to antigen-presenting cells (APCs) (Figure 4) [37]. In T-cells, eNOS is localized to the Golgi complex, and its activation (at the immunological synapse between the T-cell and APC) drives N-Ras-dependent ERK activation [37]. Importantly, only Golgi-localized N-Ras, and not plasma membrane-associated K-Ras, is activated and _S_-nitrosylated by eNOS, demonstrating a requirement for compartmentalization of eNOS with its target [38,39]. Mutation of C118 abrogates N-Ras _S_-nitrosylation and activation as well as TCR-dependent apoptosis [38], suggesting a role for _S_-nitrosylation in the negative selection of highly reactive T-cells in the thymus.
Ras _S_-nitrosylation is also implicated in the initiation of tumorigenesis and maintenance of established tumors. In tumor cells, oncogenic (mutant) K-Ras activates eNOS by PI3K–Akt-dependent phosphorylation of eNOS at S1117, which results in _S_-nitrosylation of wild-type N-and H-Ras [40]. S1117-phosphorylated eNOS is elevated in tumors isolated from patients with pancreatic cancer, and overexpression of either S1117A eNOS, or C118S N- or H-Ras inhibits pancreatic tumor growth [40]. Although C118 of Ras appears to be a principal locus of action of tumorigenic nitric oxide, NO might play a multifaceted role in cancer [41], and _S_-nitrosylation of numerous proteins could influence the cancer phenotype (Box 3; [42]).
Box 3. Targets of _S_-nitrosylation in cancer.
_S_-nitrosylation of a number of proteins can be linked directly or indirectly to tumor proliferation (see [42] for review):
- Pro-angiogenic targets include HIF-1α, dynamin, Ras, COX-2 (see text) and PTEN [146].
- Anti-apoptotic targets include NF-κB p65 (see text), caspases [107], p53 [147] and Bcl-2 [148].
- Additional proliferative targets include methionine adenosyl transferase [149].
- DNA repair enzymes suppressed by _S_-nitrosylation include 8-oxoguanine glycosylase (Ogg1) and _O_6-alkylguanine-DNA alkyltransferase (AGT) [150,151].
_S_-nitrosylation has also been implicated in the effects of several anti-cancer therapeutics:
- BCNU (see text) and auranofin [107] are likely to inhibit cellular denitrosylation and induce general nitrosative stress.
- TRAIL appears to induce iNOS and activate SNO-GAPDH-dependent carcinoma cell death (see text).
Aberrant hemoglobin _S_-nitrosylation and the human respiratory cycle – sickle cell anemia, banked blood and pulmonary arterial hypertension
Accumulating evidence supports a role for red blood cell (RBC) SNOs, which originate from _S_-nitroso-(βCys93)hemoglobin (SNO-Hb), in mediating oxygen tension (PO2)-dependent vasodilatory activity [43] within the human respiratory cycle. In the resultant 3-gas (NO, O2, CO2) model of the respiratory cycle, RBCs liberate NO-based bioactivity to enable efficient O2 delivery (which is primarily a function of blood flow). This activity of RBCs appears to require the transfer of SNO from Hb to RBC low-mass and/or membrane protein thiols (e.g. erythrocyte band 3) and finally to circulating or vascular thiols [44–46]. Ex vivo arterial vasodilation by human RBCs, triggered by hypoxia (e.g. 1% O2), does not require NOS activity or intact endothelium, but depends critically upon RBC-SNO levels [47]. Moreover, this vasodilatory activity is actuated in a graded manner across a PO2 range of 60-7 mm Hg, representative of physiological O2 gradients in the microcirculation [48].
The loss of SNO-Hb, or defective transfer of SNO from Hb to acceptor thiols, might underlie numerous pathological states [2], and accumulating data suggest that the restoration of SNO levels (‘SNO repletion’) can ameliorate these conditions. Sickle cell Hb (HbS; Glu6Val Hb) is impaired in both its ability to form SNO-HbS and to transfer the SNO to the RBC membrane, resulting in impaired hypoxic vasodilation, and the magnitudes of these deficits correlate with clinical severity in patients with sickle cell anemia [49]. Hypoxic vasoactivity (and membrane SNO content) of sickle RBCs can be at least partially restored by treatment with NO [49]. In addition, storage of blood (under conditions employed in blood banking) leads to a time-dependent loss of SNO-Hb and impairment of hypoxic vasodilation by RBCs, both of which are restored upon SNO repletion [50,51]. These data provide a rationale for the well-established inability of stored blood to fully correct anemia-associated deficits in tissue oxygen delivery, and they suggest that SNO repletion might be effective in reducing morbidity and mortality associated with transfusion. Finally, RBCs exposed to prolonged hypoxia are defective in SNO-Hb formation, and in the lung, exhibit enhanced hypoxic pulmonary vasoconstrictive activity and are deficient in increasing blood oxygenation; these deficiencies are reversed by SNO repletion [52]. Consistent with these findings, hypoxemic patients with pulmonary aterial hypertension (PAH) have markedly reduced SNO-Hb levels, and hypoxic vasodilation by their RBCs is impaired both ex vivo and in vivo [52]. Treatment of PAH patients with the _S_-nitrosylating gas, ethyl nitrite (ENO), normalizes SNO-Hb levels and RBC vasodilation, and improves oxygenation and lowers pulmonary arterial pressures [52]. In addition, ENO has shown clinical benefits in the syndrome of persistent hypertension of the newborn [53].
Control of hypoxic signaling – tumor radiotherapy, protection from myocardial ischemia and development of pulmonary arterial hypertension
HIF-1, a master transcriptional regulator, is activated at low oxygen tension through the stabilization of its alpha subunit (HIF-1α). Under normoxia, hydroxylation of HIF-1α by an O2-dependent prolyl hydroxylase promotes its binding with and ubiquitylation by the E3 ligase complex containing pVHL and subsequent proteasomal degradation. Accumulating evidence suggests that, under normoxia, NO/SNO stablizes HIF-1α (thus mimicking hypoxia) [1].
_S_-nitrosylation-dependent HIF-1α stabilization is implicated in the resistance of solid tumors to ionizing radiation [54]. Irradiation of tumors results in increased HIF-1 activity and VEGF levels, coincident with macrophage- and iNOS-dependent stabilization of HIF-1α through its oxygen-dependent degradation (ODD) domain. _S_-nitrosylation appears to prevent HIF-1α degradation by inhibiting the binding of the Von Hippel-Lindau disease tumor suppressor pVHL to the hydroxylated ODD, and the effects of exogenous SNO or iNOS (on both the ODD–pVHL interaction and HIF-1α stability) require _S_-nitrosylation of C533 in the ODD domain [54]. In addition, NOS inhibitors act synergistically with radiotherapy to reduce the rate of tumor growth in mouse models of breast cancer and melanoma, and tumor growth is substantially slower in radiation-treated iNOS-knockout versus wild-type mice [54]. These data are consistent with a role for tumor vascularization, mediated by NO, in tumor survival [41]. Although NOS inhibitors may have therapeutic potential in cancers in which paracrine (macrophage-derived) or autocrine (eNOS-derived; see discussion of Ras _S_-nitrosylation) NO plays a role, it should be noted that NO may also sensitize tumors to chemo-, radio- or immuno-therapies [55].
HIF-1α stabilization, resulting from increased endogenous GSNO, also appears to underlie amelioration of myocardial infarction. GSNOR-knockout mice have reduced myocardial infarct size and preserved ventricular systolic and diastolic function and maintain tissue oxygenation following left coronary artery ligation [7]. These effects are associated with increases in myocardial capillary density, increased levels and activity of HIF-1α and elevated HIF-1α _S_-nitrosylation in GSNOR-knockout mice [7]. These data identify a role for endogenous SNO in angiogenesis and myocardial protection and, more generally, suggest novel therapeutic approaches to modulate angiogenesis and preserve cardiac function. It should be noted that these data do not rule out the possibility that additional, pro-angiogenic proteins and/or pathways might be (dys)regulated by _S_-nitrosylation. For example, _S_-nitrosylation of dynamin stimulates its GTPase activity [56,57] and potentiates endothelial cell survival signaling [57], and _S_-nitrosylation of JNK3 phosphatase MKP7, which promotes endothelial proliferation [58], might also promote (S)NO-dependent angiogenesis.
HIF-1α stabilization by _S_-nitrosothiols is also implicated in the development of PAH. Mice treated with NAC or _S_-nitroso-N-acetylcysteine (SNOAC) for several weeks develop PAH, mimicking the effects of chronic hypoxia [46]. eNOS-knockout mice are protected from the increases in relative right ventricular weight and systolic pressure induced by NAC (but not SNOAC), implicating SNOAC in the effects of NAC [46]. The bioactivity of NAC appears to originate from SNO-Hb, as SNOAC is formed from NAC in blood coincident with declines in SNO-Hb, and SNO-Hb levels are lower and SNOAC formation from blood is impaired in eNOS-knockout mice [46]. Declines in SNO-Hb and increases in SNOAC are facilitated by increasing hypoxia, a consequence of SNO-Hb instability upon deoxygenation. NAC/SNOAC treatment stabilizes whole-lung HIF1 DNA-binding activity; C162 of pVHL, a residue known to be essential for binding of the adaptor protein elongin C, is implicated in the effects of SNOAC [46]. These data suggest that PAH may be caused either by deficiency in SNO-Hb and/or by excess in low-mass _S_-nitrosothiols: deficiency of SNO-Hb might impair hypoxia-mediated release of NO bioactivity to counter pulmonary vasoconstriction, whereas excess SNO simulates sustained hypoxia, which ultimately increases pulmonary pressure (secondary to vascular remodeling). PAH might also be a potential side effect of chronic NAC therapy.
(Dys)regulated _S_-nitrosylation in the heart through differential NOS expression and localization – contractility, ischemia and long QT
The importance of NOS subcellular compartmentalization is well-exemplified in the heart, where eNOS and nNOS are differentially localized and exhibit opposite effects on myocardial contractility [25,59,60]. eNOS, associated primarily with sarcolemmal caveolae, attenuates β-AR-dependent myocardial contractility [61]; inhibition of the L-type Ca2+ channel (LTCC), at least in part by _S_-nitrosylation [26], appears to underlie this effect. By contrast, nNOS is primarily localized to the sarcoplasmic reticulum (SR) and activates the cardiac ryanodine receptor/Ca2+ channel (RyR2) by _S_-nitrosylation [62], resulting in release of Ca2+ to the cytosol and enhanced catecholamine-stimulated contractility. Accordingly, nNOS- but not eNOS-knockout mice exhibit hypo-_S_-nitrosylation of RyR2 and diastolic Ca2+ leakage with arrhythmia characteristic of sudden cardiac death (SCD) [28]. Sex-related differences in NOS expression/localization also appear to modulate Ca2+ handling. Hearts [63] and myocytes [26,63] from female mice show less isoproterenol (ISO)-induced SR Ca2+ loading compared with males, evidently owing to the translocation of nNOS to the sarcolemma and _S_-nitrosylation of the LTCC [26]. The redistribution of nNOS by ISO, as well as the upregulation of NOS isoforms (due at least in part, to 17β-estradiol-dependent gene expression [64,65]), appears to underlie the protection of female hearts from ischemia/reperfusion (I/R) injury [26]. However, the redistribution of nNOS to the plasma membrane is also observed in the hearts of humans with idiopathic dilated cardiomyopathy [66,67] and of rodents with experimental myocardial infarction [66,68]. Thus, aberrant RyR2 and LTCC _S_-nitrosylation might also explain some of the characteristic dysfunction of the failing heart.
Dysregulated _S_-nitrosylation that results from altered NOS-target associations has also been linked to long QT syndrome (LQTS), an inherited disease that is characterized by a prolonged QT interval, which can result in fainting and sudden cardiac death from arrhythmia. A mutation in α1-syntrophin (A390V SNTA1), identified in a patient with LQTS, results in release of the plasma membrane calcium-ATPase 4b (a negative regulator of nNOS activity) from a complex containing SNTA1, nNOS and the cardiac sodium channel SCN5A [5]. Heterologous expression of A390V SNTA1 results in NOS-activity-dependent increases in sodium channel gating and in both peak and late sodium currents (_I_na). These effects coincide with hyper-_S_-nitrosylation of SCN5A, which is suggested as a mechanism for regulation of _I_na. A linkage has also been found between SNPs in NOS1AP (CAPON; a positive regulator of nNOS activity) and both altered QT interval [69,70] and risk of SCD [70], although it is unknown whether aberrant NOS activity or ion channel _S_-nitrosylation underlies these phenotypes. Endogenously derived NO has been shown to influence QT interval through _S_-nitrosylation of the slowly activating delayed-rectifier K+ channel (IKs) [71,72]. Gender differences in QT duration and susceptibility to ventricular arrhythmia have been linked to IKs currents [73]. Thus, sex-related differences in cardiac (dys)function may reflect differences in NOS activity/localization and _S_-nitrosylation of cardiac ion channels/transporters.
_S_-nitrosylation in disorders of skeletal muscle
The skeletal muscle ryanodine receptor (RyR) isoform (RyR1) is activated by _S_-nitrosylation of C3635, which reduces the inhibitory effect of Ca2+–CaM on the channel [74]. A number of mutations in RyR1 (e.g. Y522S) are associated with malignant hyperthermia and related diseases that are characterized by involuntary muscle contractures, tissue lysis (rhabdomyolysis) and sudden death in response to elevated environmental temperatures [75,76]. Heterozygous RyR1Y522S/wt mutant mice, which have a malignant hyperthemia phenotype, have elevated ratios of oxidized-to-reduced glutathione, and RyR1 is hyper-_S_-nitrosylated and -_S_-glutathionylated in these mice, in association with temperature-dependent increases in cytosolic Ca2+ levels [75]. Prolonged Ca2+ leakage ultimately results in inadequate force generation and mitochondrial damage. NOS inhibitors and ascorbate treatment, which reverse NO- but not GSH-based modifications of the channels, ameliorate the classic temperature-sensitivity of the channel. Furthermore, NAC treatment in vitro and prolonged administration in vivo reverse NO-based modifications of the channel and ameliorate the phenotype [75]. These data suggest a cycle in which the mutation in RyR1 causes Ca2+ leakage, leading to increased NOS activity and further _S_-nitrosylation and activation of the channel (i.e. a SNO-induced feed-forward mechanism). Thus, _S_-nitrosylation is primarily responsible for the increased temperature sensitivity of the RyR characteristic of malignant hyperthermia; oxidative stress appears to be a secondary consequence of increased NOS activity.
Hyper-_S_-nitrosylation of RyR1, and consequent channel leakage, is also observed in mice (and humans) subjected to repeated exercise [77] as well as in dystrophin-deficient (mdx) mice [78] that serve as a model of Duchenne muscular dystrophy (DMD), a genetic disorder characterized by progressive neuromuscular degeneration and eventual paralysis and death. Exercise-induced RyR1 _S_-nitrosylation appears to result from an increase in eNOS expression [77], whereas _mdx_-associated hypernitrosylation is mediated by an RyR-affiliated iNOS that is upregulated by inflammatory cytokines. In addition to altering RyR1-CaM interactions, hyper-_S_-nitrosylation of RyR1 correlates with the loss of binding between RyR1 and the peptidyl-prolyl cis-trans isomerase calstabin1 (FKBP12/FKBP1A; a protein that stabilizes the closed channel conformation), which contributes at least in part to channel leakage and decreased exercise capacity [77]. A novel small molecule (S107), which stabilizes the interaction of FKBP12 and RyR1, largely rescues these effects [77]. Human DMD results from mutations in the gene encoding dystrophin, which forms a complex with nNOS and syntrophin, and it has long been known that nNOS is mostly absent from skeletal muscle in DMD patients [79]. However, in mdx mice, iNOS is upregulated (and co-immunoprecipitates with RyR1), coincident with RyR1 hyper-_S_-nitrosylation [78]. These effects also lead to destabilization of the FKBP12–RyR1 complex, and treatment of mdx mice with S107 for two weeks restores RyR1-bound FKBP12, decreases Ca2+ leakage, ameliorates muscle damage and improves muscle function [78]. It will be of interest to determine whether S107 can alter other pathologies associated with RyR1 hyper-_S_-nitrosylation, including malignant hyperthermia.
Although S107 apparently acts to reverse the deleterious effects of iNOS induction in the mdx mouse, overexpression of eNOS also appears to be ameliorative. Expression of constitutively active eNOS restores muscle differentiation and mimics the effects of histone deactylase (HDAC) inhibitors, which appear to induce myogenesis through transcriptional activation of follistatin [80]. HDAC2 is upregulated in mdx skeletal muscle, and _S_-nitrosylation of HDAC2 by eNOS inhibits its deacetylase activity [81]. This contrasts with the report that _S_-nitrosylation of HDAC2 (at C262 and C274) by nNOS in neurons facilitates its release from chromatin without altering deacetylase activity [82]. Whether these mechanistic differences reflect NO source, cell type or additional parameters (and whether nNOS is a physiological regulator of HDAC2 in skeletal muscle) has not been determined, and these factors may impact the approach to therapy; in particular, whereas all isoforms of NOS might hyper-_S_-nitrosylate RyR, which is likely to have deleterious consequences, the underlying cause of hypernitrosylation, and the extent to which the nitrosative stress is localized to the sarcoplasmic reticulum, or is more generalized, will vary in different muscle disorders. It remains possible that appropriately targeted NO therapies, perhaps in conjunction with treatments that ameliorate RyR hypernitrosylation, will have beneficial effects in one or more disorders.
iNOS-induced _S_-nitrosylation in diabetes
Accumulating evidence links insulin resistance in type 2 diabetes to NO production and protein _S_-nitrosylation [83]. iNOS expression is increased in mouse models of diabetes, and iNOS-knockout mice fed a high-fat diet develop obesity but have improved glucose tolerance and normal insulin sensitivity (in vivo) and insulin-stimulated glucose uptake (ex vivo) compared with diet-induced obese wild-type mice [84]. iNOS-dependent _S_-nitrosylation of protein kinase B (PKB/Akt), the insulin receptor β-subunit (IRβ) and insulin receptor substrate 1 (IRS-1) are all markedly increased in diabetic mouse models and might underlie the effects of iNOS on insulin signaling [85,86]. NO/SNO inhibits insulin-stimulated Akt activity, and mutation of C224 within Akt abolishes both Akt _S_-nitrosylation and its inhibition by low-mass SNOs [85]. iNOS activity or exogenous GSNO inhibit the tyrosine kinase activity of IRβ [86]; a specific target of regulatory _S_-nitrosylation has not been identified, although the enzyme possesses at least one potential regulatory cysteine (C860) [87]. Finally, _S_-nitrosylation of IRS-1 might promote its ubiquitylation-dependent degradation [86,88]. _S_-nitrosylation of each of these substrates is also increased in skeletal muscle of wild-type mice treated with lipopolysaccharide (LPS) to stimulate iNOS induction but not in iNOS-knockout mice, and LPS-stimulated insulin resistance is also abrogated in the iNOS knockout [89]. These data support a link between protein _S_-nitrosylation and abnormal toll-like receptor (TLR) signaling in type 2 diabetes [90,91], and suggest that blocking iNOS induction might provide a mechanism for restoring insulin sensitivity. It has been proposed that the beneficial effects of thiazoledinediones, peroxisome proliferator-activated receptor-γ agonists that are used clinically for the treatment of diabetes mellitus, might be attributable to their inhibition of cytokine-induced iNOS expression [84]. Indeed, in diet-induced obese rats, the thiazoledinedione rosiglitazone largely abrogates _S_-nitrosylation of IRβ, IRS-1 and Akt, concomitant with a reduction in iNOS expression, amelioration of insulin resistance and restoration of insulin signaling [86]. Similar effects could underlie the improvement in insulin signaling by acute physical exercise, which reduces iNOS expression as well as IRS-1 and Akt _S_-nitrosylation [92].
Nitrosative stress in neurodegenerative diseases
Dysregulated protein _S_-nitrosylation (which can result from over-activation of NMDA receptors) appears to be prevalent in neurodegenerative disorders characterized, in particular, by the accumulation of misfolded proteins [2,93]. Hyper-_S_-nitrosylation of parkin, protein disulfide isomerase (PDI), peroxiredoxin 2 (Pdx2), X-linked inhibitor of apoptosis (XIAP) and dynamin-related protein 1 (Drp1) is observed in brains from patients with neurological disorders (including Alzheimer’s, sporadic Parkinson’s and diffuse Lewy body diseases; Table 1), and _S_-nitrosylation of these proteins is implicated in stress-induced neuronal death [94–99]. _S_-nitrosylation initially appears to increase the E3 ligase activity of parkin and promote its auto-ubiquitylation [95], and ultimately results in its inhibition [94,95]. Zn-binding Cys residues in the RING domain of parkin have been identified as targets of _S_-nitrosylation [95]. NO inhibits both the ubiquitin-dependent degradation of parkin substrates (including synphilin-1) and the protection by parkin from cell death induced by protease inhibitors in α-synuclein and synphilin-1-expressing cells [94,95]. PDI is _S_-nitrosylated at one or both of the Cys thiols in each of its dithiol (Cys-Gly-His-Cys) active sites, resulting in inhibition of disulfide isomerase activity and accumulation of misfolded synphilin-1 in the endoplasmic reticulum [96]. Cumulative _S_-nitrosylation of parkin and PDI might underlie the accumulation of misfolded and ubiquitylated proteins that ultimately leads to cell death. _S_-nitrosylation of Cys residues within the baculoviral IAP-repeat (BIR) motifs of XIAP is implicated in inhibition of the antiapoptotic function of the enzyme (which requires a direct interaction with caspases), but in contrast to parkin, low-mass SNOs do not compromise the E3 ligase activity of XIAP [98]. Pdx2, a 2-Cys peroxiredoxin, is inactivated by _S_-nitrosylation of both its catalytic and resolving cysteine residues (C51 and C172, respectively), sensitizing dopaminergic neurons to H2O2-dependent cell death [97]. Increased nitrosative stress, and Pdx2 _S_-nitrosylation, might contribute to the loss of dopaminergic neurons in Parkinson’s disease [97]. It is notable that compounds that inhibit mitochondrial complex I (rotenone) or induce a Parkinson-like phenotype (MPTP/MPP+) also induce _S_-nitrosylation of each of these four proteins in vitro or in vivo [95–98]. Although a specific mechanism has not been elucidated, these data support the idea that, in neurons, nitrosative and oxidative stress might be linked. Finally, it has recently been shown that _S_-nitrosylation of Drp1 is enhanced in the brains of Alzheimer’s (but not of Parkinson’s) patients [99]. Similar to the effects of _S_-nitrosylation on dynamin (Box 1), _S_-nitrosylation of C644 within Drp1 promotes its multimerization and thereby mitochondrial fission, which causes neuronal damage [99]. Furthermore, exposure of nNOS-expressing cells to β-amyloid protein (a crucial effector of neurotoxicity in Alzheimer’s disease) results in Drp1 _S_-nitrosylation, suggesting a role for _S_-nitrosylation in β-amyloid-associated neurotoxicity.
SNO-GAPDH and cell death – therapeutic implications in neurodegeneration and cancer
Nuclear accumulation of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a hallmark of stress-induced cell death [100]. Stimulation of nNOS in neurons or of iNOS in macrophages (by NMDA and cytokines, respectively) triggers GAPDH _S_-nitrosylation at its active site, C145, which promotes the association of GAPDH with the E3 ubiquitin ligase Siah1, resulting in nuclear translocation of the SNO-GAPDH–Siah1 complex [101]. Within the nucleus, SNO-GAPDH stabilizes Siah1, which facilitates the ubiquitylation and degradation of nuclear proteins [101], and independently SNO-GAPDH is acetylated by and consequently activates the acetyltransferase p300/CREB binding protein, leading to the acetylation/activation of downstream targets, including the tumor supressor p53 [102]; both of these mechanisms promote cell death.
Accumulating evidence suggests that this pathway might be therapeutically relevant. Deprenyl and CGP-3466, drugs that protect neurons from stress-induced apoptosis [103] and slow the progression of Parkinson’s disease [104], bind to GAPDH [105] and inhibit both GAPDH _S_-nitrosylation and the GAPDH–Siah1 interaction in MPTP-treated mice [103]. Deprenyl also induces the expression of thioredoxin [106], a denitrosylase for GAPDH [107]. Conversely, BCNU (carmustine), a chemotherapeutic agent often used to treat brain and other cancers, increases SNO-GAPDH levels [108], providing a mechanism for sensitization of tumor cells to GAPDH-dependent death. _S_-nitrosylation of GAPDH and its nuclear accumulation, as well as NOS-dependent apoptosis, are also observed in thyroid carcinoma cells treated with TNF-related apoptosis-inducing ligand (TRAIL) [109], a promising cancer therapeutic [110]; iNOS is most likely the source of NO [109,111]. It has been suggested that this mechanism contributes to the tumor-selective effects of TRAIL [110].
Regulation of prostaglandin synthesis – preconditioning in ischemia-reperfusion injury
Prostaglandin synthesis by cyclooxygenase (COX) is coupled to iNOS activation in cells and tissues, and _S_-nitrosylation is implicated in this effect. iNOS binds directly to and activates COX-2 by _S_-nitrosylation of a single cysteine (C526) proximal to the substrate (arachidonic acid; AA) binding site [112], and COX-2 _S_-nitrosylation and prostaglandin E2 formation are attenuated by blocking iNOS–COX-2 interaction [112]. Similarly, binding and _S_-nitrosylation of COX-2 by nNOS appears to mediate NMDA receptor-induced neurotoxicity [113]. Upstream of COX-2, the formation of AA by cytosolic phospholipase A2α (cPLA2α) is similarly regulated. In particular, iNOS activates cPLA2α by _S_-nitrosylation of C152 [114], located within the N-terminal regulatory domain [115]. However, rather than interacting directly with iNOS, cPLA2α forms a ternary complex with COX-2 and iNOS, and induction of COX-2 is crucially required for cPLA2α _S_-nitrosylation and activation by iNOS [114].
These findings suggest that blocking the _S_-nitrosylation of COX-2 and/or cPLA2α might prove ameliorative in settings of inflammation, cancer and neurotoxicity. By contrast, the activation of COX-2 by _S_-nitrosylation is implicated in the cardioprotective effects of 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors (statins) [116,117], and in the protective effects of ischemia- or drug-induced preconditioning [118,119] in ischemia-reperfusion (I/R) injury [120,121]. Although the molecular targets of prostanoids have not been elucidated fully [118], the protective effect of the statin atorvastatin (ATV) in reducing myocardial infarct size (when administered before ischemia or upon reperfusion) appears to involve both the up-regulation and PI3K/Akt-dependent activation of eNOS [117], the NF-κB-dependent induction of iNOS and COX-2, and finally COX-2 _S_-nitrosylation and activation [116,117]. Consistent with these findings, the ATV-dependent reduction in infarct size after I/R is lost in both eNOS-knockout and iNOS-knockout mice (in which COX-2 upregulation and/or _S_-nitrosylation are abrogated) [117], and, in wild-type mice, ATV-dependent increases in myocardial 6-keto-PGF1α are blocked by an iNOS inhibitor without concomitant changes in COX-2 levels [116].
Anti-inflammatory activities of endogenous and exogenous _S_-nitrosothiols
Protein _S_-nitrosylation by iNOS-derived NO occurs downstream of TLR activation and can be proinflammatory; targets of TLR-dependent _S_-nitrosylation include surfactant protein D, which is increasingly implicated in lung inflammation [122,123]. However, numerous additional studies point to a role for _S_-nitrosylation in the feedback inhibition of TLR-mediated signaling. NOS and/or low-mass SNOs inhibit the expression both of iNOS [124–126] and of multiple cytokines (interleukin-1β (IL-1β) [127], IL-12 p40 subunit [128], IL-6, IL-8 and MIP-2 [129]), and induce the expression of the anti-inflammatory mediators IRAK-M [130] and SOCS-1 [131]. Possible mechanisms include: 1) inhibition of poly(ADP-ribose) polymerase isoform 1 (PARP-1) binding to the iNOS promoter via PARP-1 _S_-nitrosylation [(the site(s) of _S_-nitrosylation are unknown) [125]; 2) attenuation of NF-κB p50-p65 activity, including its binding to iNOS promoter DNA, by _S_-nitrosylation of a conserved Cys residue (C38 of p65) at the NF-κB–DNA interface [120,124]; 3) _S_-nitrosylation (by eNOS) at C216 of the adaptor protein MyD88 (which couples TLR to NF-κB activation), resulting in the inhibition of its LPS-induced membrane association and binding to the sorting adaptor TIRAP, an interaction crucial for the recruitment of MyD88 to TLRs [129]; 4) _S_-nitrosylation at C179 of the inhibitory κB kinase (which phosphorylates and promotes the degradation of the NF-κB inhibitory protein, IκB), resulting in its inhibition and the attenuation of NF-κB activation through IκB [121]. In addition to providing molecular bases for the anti-inflammatory actions of low-mass SNOs, these findings also suggest multiple potential targets for SNO-based therapy.
Anti-inflammatory and ameliorative roles for GSNO have been demonstrated in numerous diseases and disease models. For example, in a model of multiple sclerosis (experimental autoimmune encephalomyelitis; EAE), oral GSNO greatly improves clinical scores, attenuates infiltration of immune cells into the CNS, protects against severe demyelination and causes downregulation of proinflammatory cytokines and iNOS [12]. The protective role of GSNO in EAE might reflect disruption of leukocyte trafficking, inasmuch as GSNO pretreatment inhibits monocyte adhesion to TNFα-activated endothelial cells in vitro. This effect appears to be mediated through downregulation of endothelial cell adhesion molecules and _S_-nitrosylation of the p65 subunit of NF-κB [12]. Similarly, GSNO protects against experimental autoimmune uveitis, a model of human uveitis, a leading cause of blindness, with concomitant inhibition of proinflammatory cytokine expression [132]. Finally, GSNO is implicated as the barrier-inducing factor produced by enteric glia to regulate intestinal barrier function and inflammation [133]. Under conditions of glial cell ablation, GSNO protects against intestinal permeability, intestinal inflammation, TNF-α expression and mortality, and GSNO also inhibits human intestinal mucosal barrier permeability and restores colonic permeability in colon biopsies from patients with Crohn’s disease [133]. Whether GSNO deficiency, due to a disruption in the enteric glial network, plays a role in Crohn’s disease pathology is as yet undetermined.
The gaseous _S_-nitrosylating agent ethyl nitrite (ENO, which can be delivered by inhalation) might be more efficacious in some diseases than either GSNO, which requires processing (to _S_-nitrosocysteine or _S_-nitroso-cysteinylglycine) for cellular uptake, or inhaled NO (iNO), which has lower target specificity and a propensity to produce toxic byproducts. Antiinflammatory effects of ENO are observed in models of bronchopulmonary dysplasia (BPD) and acute lung injury/acute respiratory distress syndrome (ALI/ARDS). Whereas experimental BPD is induced in newborn rats exposed to hyperoxia, simultaneous exposure to hyperoxia and ENO protects rat pups from BPD, reduces cytokine expression and inhibits bronchiolar epithelial NF-κB activation [9]. Consistent with its higher specificity for thiols, the effects of ENO are superior to those of iNO, which has not been proven to reduce the incidence of BPD [134] and shows a very narrow window of efficacy. ENO also offers protection against lung inflammation and injury in mice exposed to aerosolized LPS – a model of ALI/ARDS [10]. Interestingly, LPS alone induces protein denitrosylation in the lung, although the effect on inflammation is unknown [10]. ENO increases protein SNOs in airway lining fluid and in the lung, increases NF-κB p65 _S_-nitrosylation and inhibits LPS-induced NF-κB DNA-binding [10]. These findings point to the potential for ENO as a preventative measure in ALI/ARDS, in contrast to iNO, which has failed to show a reduction in mortality or other outcome benefit [135,136], and which might even be associated with increased morbidity [136]. Thus, although NO production by one or more forms of NOS has been implicated in disease etiology (in particular as a proinflammatory agent), the onset of disease and/or the severity of symptoms can be attenuated by _S_-nitrosylating agents.
Future directions
The broad purview of protein _S_-nitrosylation in normal and disturbed cell function presents, in principle, novel therapeutic opportunities in a wide range of human diseases. These opportunities remain largely untapped. In addition, the improvement and dissemination of new methodologies for the analysis of protein _S_-nitrosylation – which have enabled many of the recent discoveries detailed here and which have been reviewed recently [137,138] – will allow the role of aberrant _S_-nitrosylation to be elucidated in additional, related and dissimilar disease states. The available data already indicate that, (i) in the case of skeletal muscle, a collection of disorders share in common dysregulated _S_-nitrosylation of RyR1 [75,77,78]; (ii) in the case of RBCs, seemingly disparate disorders share in common dysregulated _S_-nitrosylation of hemoglobin, which can influence disease symptomatology and which may be viewed as a new type of hemoglobinopathy; and (iii) in the case of Parkinson’s disease, dysregulated _S_-nitrosylation of multiple disparate proteins might contribute to a common phenotype. However, it should be noted that, in many cases, it remains to be determined whether alterations in _S_-nitrosylation are causal in the development of disease. In addition, a number of disease-modifying drugs, including statins, deprenyl and BCNU, can exert ameliorative effects at least in part through modulation of _S_-nitrosylation.
Further progress may be expected from the rapid and continuing accumulation of knowledge in several salient areas. It is increasingly apparent that protein _S_-nitrosylation is spatially and temporally regulated not only by the direct interaction or compartmentation of SNO targets with NO synthases, but also by the enzymes that remove SNO from glutathione (e.g. GSNOR) and proteins (e.g. thioredoxin [107]). These discoveries suggest novel mechanisms for the dysregulation of protein _S_-nitrosylation in disease and add to an expanding list of potential therapeutic targets. Finally, the related elucidation of a genetic basis in some cases of dysregulated _S_-nitrosylation, although still in its infancy, points to the possibility that molecular medicine will offer new avenues for treatment through the targeted repair (or pharmacological rescue) of the genetic defects that seem likely to underlie a significant number of pathophysiologies in which dysregulated _S_-nitrosylation plays a role.
Acknowledgements
This work was supported by grants U19-ES012496, HL075443 and HL059130 from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hess DT, et al. Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 2.Foster MW, et al. S-nitrosylation in health and disease. Trends Mol. Med. 2003;9:160–168. doi: 10.1016/s1471-4914(03)00028-5. [DOI] [PubMed] [Google Scholar]
- 3.Jaffrey SR, et al. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 4.Gow AJ, et al. Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. J. Biol. Chem. 2002;277:9637–9640. doi: 10.1074/jbc.C100746200. [DOI] [PubMed] [Google Scholar]
- 5.Ueda K, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc. Natl. Acad. Sci. U. S. A. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Que LG, et al. Protection from experimental asthma by an endogenous bronchodilator. Science. 2005;308:1618–1621. doi: 10.1126/science.1108228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lima B, et al. Endogenous S-nitrosothiols protect against myocardial injury. Proc. Natl. Acad. Sci. U. S. A. 2009;106:6297–6302. doi: 10.1073/pnas.0901043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu L, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 9.Auten RL, et al. Inhaled ethyl nitrite prevents hyperoxia-impaired postnatal alveolar development in newborn rats. Am. J. Respir. Crit. Care Med. 2007;176:291–299. doi: 10.1164/rccm.200605-662OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marshall HE, et al. Protection from LPS-induced lung injury by augmentation of airway S-nitrosothiols. Am. J. Respir. Crit. Care Med. 2009;180:11–18. doi: 10.1164/rccm.200807-1186OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 11.Khan M, et al. Cerebrovascular protection by various nitric oxide donors in rats after experimental stroke. Nitric Oxide. 2006;15:114–124. doi: 10.1016/j.niox.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 12.Prasad R, et al. GSNO attenuates EAE disease by S-nitrosylation-mediated modulation of endothelial-monocyte interactions. Glia. 2007;55:65–77. doi: 10.1002/glia.20436. [DOI] [PubMed] [Google Scholar]
- 13.Santhanam L, et al. Inducible NO synthase dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ. Res. 2007;101:692–702. doi: 10.1161/CIRCRESAHA.107.157727. [DOI] [PubMed] [Google Scholar]
- 14.Santhanam L, et al. Arginase and vascular aging. J. Appl. Physiol. 2008;105:1632–1642. doi: 10.1152/japplphysiol.90627.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ckless K, et al. Inhibition of arginase activity enhances inflammation in mice with allergic airway disease, in association with increases in protein S-nitrosylation and tyrosine nitration. J. Immunol. 2008;181:4255–4264. doi: 10.4049/jimmunol.181.6.4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.North ML, et al. Functionally important role for arginase 1 in the airways hyperresponsiveness of asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;296:L911–L920. doi: 10.1152/ajplung.00025.2009. [DOI] [PubMed] [Google Scholar]
- 17.Maarsingh H, et al. Arginine homeostasis in allergic asthma. Eur. J. Pharmacol. 2008;585:375–384. doi: 10.1016/j.ejphar.2008.02.096. [DOI] [PubMed] [Google Scholar]
- 18.Salam MT, et al. Roles of arginase variants, atopy, and ozone in childhood asthma. J. Allergy Clin. Immunol. 2009;123:596–602. doi: 10.1016/j.jaci.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maarsingh H, et al. Arginase inhibition protects against allergen-induced airway obstruction, hyperresponsiveness, and inflammation. Am. J. Respir. Crit. Care Med. 2008;178:565–573. doi: 10.1164/rccm.200710-1588OC. [DOI] [PubMed] [Google Scholar]
- 20.Chen Z, Stamler JS. Bioactivation of nitroglycerin by the mitochondrial aldehyde dehydrogenase. Trends Cardiovasc. Med. 2006;16:259–265. doi: 10.1016/j.tcm.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Stamler JS. Nitroglycerin-mediated S-nitrosylation of proteins: a field comes full cycle. Circ. Res. 2008;103:557–559. doi: 10.1161/CIRCRESAHA.108.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sayed N, et al. Desensitization of soluble guanylyl cyclase, the NO receptor, by S-nitrosylation. Proc. Natl. Acad. Sci. U. S. A. 2007;104:12312–12317. doi: 10.1073/pnas.0703944104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sayed N, et al. Nitroglycerin-Induced S-nitrosylation and desensitization of soluble guanylyl cyclase contribute to nitrate tolerance. Circ. Res. 2008;103:606–614. doi: 10.1161/CIRCRESAHA.108.175133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor AL, et al. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N. Engl. J. Med. 2004;351:2049–2057. doi: 10.1056/NEJMoa042934. [DOI] [PubMed] [Google Scholar]
- 25.Hare JM, Stamler JS. NO/redox disequilibrium in the failing heart and cardiovascular system. J. Clin. Invest. 2005;115:509–517. doi: 10.1172/JCI200524459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun J, et al. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel α1 subunit and reduced ischemia/reperfusion injury. Circ. Res. 2006;98:403–411. doi: 10.1161/01.RES.0000202707.79018.0a. [DOI] [PubMed] [Google Scholar]
- 27.Whalen EJ, et al. Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129:511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez DR, et al. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A. 2007;104:20612–20617. doi: 10.1073/pnas.0706796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Que L, et al. S-Nitrosoglutathione reductase: an important regulator in asthma. Am. J. Respir. Crit. Care Med. 2009;180:226–231. doi: 10.1164/rccm.200901-0158OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu H, et al. Genetic variation in S-nitrosoglutathione reductase (GSNOR) and childhood asthma. J. Allergy Clin. Immunol. 2007;120:322–328. doi: 10.1016/j.jaci.2007.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhandari V, et al. Essential role of nitric oxide in VEGF-induced, asthma-like angiogenic, inflammatory, mucus, and physiologic responses in the lung. Proc. Natl. Acad. Sci. U. S. A. 2006;103:11021–11026. doi: 10.1073/pnas.0601057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwon HS, et al. Sp3 and Sp4 can repress transcription by competing with Sp1 for the core cis-elements on the human ADH5/FDH minimal promoter. J. Biol. Chem. 1999;274:20–28. doi: 10.1074/jbc.274.1.20. [DOI] [PubMed] [Google Scholar]
- 33.Zaman K, et al. Concentration-dependent effects of endogenous S-nitrosoglutathione on gene regulation by specificity proteins Sp3 and Sp1. Biochem. J. 2004;380:67–74. doi: 10.1042/BJ20031687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raines KW, et al. Nitric oxide cell signaling: S-nitrosation of Ras superfamily GTPases. Cardiovasc. Res. 2007;75:229–239. doi: 10.1016/j.cardiores.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 35.Heo J, et al. Mechanism of free radical nitric oxide-mediated Ras guanine nucleotide dissociation. J. Mol. Biol. 2005;346:1423–1440. doi: 10.1016/j.jmb.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 36.Heo J, Campbell SL. Ras regulation by reactive oxygen and nitrogen species. Biochemistry. 2006;45:2200–2210. doi: 10.1021/bi051872m. [DOI] [PubMed] [Google Scholar]
- 37.Ibiza S, et al. Endothelial nitric oxide synthase regulates T cell receptor signaling at the immunological synapse. Immunity. 2006;24:753–765. doi: 10.1016/j.immuni.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 38.Ibiza S, et al. Endothelial nitric oxide synthase regulates N-Ras activation on the Golgi complex of antigen-stimulated T cells. Proc. Natl. Acad. Sci. U. S. A. 2008;105:10507–10512. doi: 10.1073/pnas.0711062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iwakiri Y, et al. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc. Natl. Acad. Sci. U. S. A. 2006;103:19777–19782. doi: 10.1073/pnas.0605907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim KH, et al. Tumour maintenance is mediated by eNOS. Nature. 2008;452:646–649. doi: 10.1038/nature06778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fukumura D, et al. The role of nitric oxide in tumour progression. Nat. Rev. Cancer. 2006;6:521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 42.Floyd RA, et al. Nitric oxide and cancer development. J. Toxicol. Pathol. 2007;20:77–92. [Google Scholar]
- 43.Singel DJ, Stamler JS. Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annu. Rev. Physiol. 2005;67:99–145. doi: 10.1146/annurev.physiol.67.060603.090918. [DOI] [PubMed] [Google Scholar]
- 44.Pawloski JR, et al. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409:622–626. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- 45.Lipton AJ, et al. S-Nitrosothiols signal the ventilatory response to hypoxia. Nature. 2001;413:171–174. doi: 10.1038/35093117. [DOI] [PubMed] [Google Scholar]
- 46.Palmer LA, et al. S-Nitrosothiols signal hypoxia-mimetic vascular pathology. J. Clin. Invest. 2007;117:2592–2601. doi: 10.1172/JCI29444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diesen DL, et al. Hypoxic vasodilation by red blood cells: evidence for an S-nitrosothiol-based signal. Circ. Res. 2008;103:545–553. doi: 10.1161/CIRCRESAHA.108.176867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McMahon TJ, et al. Nitric oxide in the human respiratory cycle. Nat. Med. 2002;8:711–717. doi: 10.1038/nm718. [DOI] [PubMed] [Google Scholar]
- 49.Pawloski JR, et al. Impaired vasodilation by red blood cells in sickle cell disease. Proc. Natl. Acad. Sci. U. S. A. 2005;102:2531–2536. doi: 10.1073/pnas.0409876102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reynolds JD, et al. S-Nitrosohemoglobin deficiency: a mechanism for loss of physiological activity in banked blood. Proc. Natl. Acad. Sci. U. S. A. 2007;104:17058–17062. doi: 10.1073/pnas.0707958104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bennett-Guerrero E, et al. Evolution of adverse changes in stored RBCs. Proc. Natl. Acad. Sci. U. S. A. 2007;104:17063–17068. doi: 10.1073/pnas.0708160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McMahon TJ, et al. A nitric oxide processing defect of red blood cells created by hypoxia: deficiency of S-nitrosohemoglobin in pulmonary hypertension. Proc. Natl. Acad. Sci. U. S. A. 2005;102:14801–14806. doi: 10.1073/pnas.0506957102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moya MP, et al. Inhaled ethyl nitrite gas for persistent pulmonary hypertension of the newborn. Lancet. 2002;360:141–143. doi: 10.1016/S0140-6736(02)09385-6. [DOI] [PubMed] [Google Scholar]
- 54.Li F, et al. Regulation of HIF-1α stability through S-nitrosylation. Mol. Cell. 2007;26:63–74. doi: 10.1016/j.molcel.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yasuda H. Solid tumor physiology and hypoxia-induced chemo/radio-resistance: novel strategy for cancer therapy: nitric oxide donor as a therapeutic enhancer. Nitric Oxide. 2008;19:205–216. doi: 10.1016/j.niox.2008.04.026. [DOI] [PubMed] [Google Scholar]
- 56.Wang G, et al. Nitric oxide regulates endocytosis by S-nitrosylation of dynamin. Proc. Natl. Acad. Sci. U. S. A. 2006;103:1295–1300. doi: 10.1073/pnas.0508354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang-Decker N, et al. Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin-2. J. Cell Sci. 2007;120:492–501. doi: 10.1242/jcs.03361. [DOI] [PubMed] [Google Scholar]
- 58.Pi X, et al. SDF-1α stimulates JNK3 activity via eNOS-dependent nitrosylation of MKP7 to enhance endothelial migration. Proc. Natl. Acad. Sci. U. S. A. 2009;106:5675–5680. doi: 10.1073/pnas.0809568106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Massion PB, et al. Regulation of the mammalian heart function by nitric oxide. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2005;142:144–150. doi: 10.1016/j.cbpb.2005.05.048. [DOI] [PubMed] [Google Scholar]
- 60.Barouch LA, et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–339. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- 61.Massion PB, Balligand JL. Modulation of cardiac contraction, relaxation and rate by the endothelial nitric oxide synthase (eNOS): lessons from genetically modified mice. J. Physiol. 2003;546:63–75. doi: 10.1113/jphysiol.2002.025973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu L, et al. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 63.Cross HR, et al. Ca2+ loading and adrenergic stimulation reveal male/female differences in susceptibility to ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H481–H489. doi: 10.1152/ajpheart.00790.2001. [DOI] [PubMed] [Google Scholar]
- 64.Chambliss KL, Shaul PW. Estrogen modulation of endothelial nitric oxide synthase. Endocr. Rev. 2002;23:665–686. doi: 10.1210/er.2001-0045. [DOI] [PubMed] [Google Scholar]
- 65.Siow RC, et al. Cardiovascular targets for estrogens and phytoestrogens: transcriptional regulation of nitric oxide synthase and antioxidant defense genes. Free Radic. Biol. Med. 2007;42:909–925. doi: 10.1016/j.freeradbiomed.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 66.Bendall JK, et al. Role of myocardial neuronal nitric oxide synthase-derived nitric oxide in β-adrenergic hyporesponsiveness after myocardial infarction-induced heart failure in rat. Circulation. 2004;110:2368–2375. doi: 10.1161/01.CIR.0000145160.04084.AC. [DOI] [PubMed] [Google Scholar]
- 67.Damy T, et al. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet. 2004;363:1365–1367. doi: 10.1016/S0140-6736(04)16048-0. [DOI] [PubMed] [Google Scholar]
- 68.Damy T, et al. Up-regulation of cardiac nitric oxide synthase 1-derived nitric oxide after myocardial infarction in senescent rats. FASEB J. 2003;17:1934–1936. doi: 10.1096/fj.02-1208fje. [DOI] [PubMed] [Google Scholar]
- 69.Chang KC, et al. CAPON modulates cardiac repolarization via neuronal nitric oxide synthase signaling in the heart. Proc. Natl. Acad. Sci. U. S. A. 2008;105:4477–4482. doi: 10.1073/pnas.0709118105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kao WH, et al. Genetic variations in nitric oxide synthase 1 adaptor protein are associated with sudden cardiac death in US white community-based populations. Circulation. 2009;119:940–951. doi: 10.1161/CIRCULATIONAHA.108.791723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bai CX, et al. Role of nitric oxide in Ca2+ sensitivity of the slowly activating delayed rectifier K+ current in cardiac myocytes. Circ. Res. 2005;96:64–72. doi: 10.1161/01.RES.0000151846.19788.E0. [DOI] [PubMed] [Google Scholar]
- 72.Asada K, et al. Redox- and calmodulin-dependent S-nitrosylation of the KCNQ1 channel. J. Biol. Chem. 2009;284:6014–6020. doi: 10.1074/jbc.M807158200. [DOI] [PubMed] [Google Scholar]
- 73.Bai CX, et al. Nontranscriptional regulation of cardiac repolarization currents by testosterone. Circulation. 2005;112:1701–1710. doi: 10.1161/CIRCULATIONAHA.104.523217. [DOI] [PubMed] [Google Scholar]
- 74.Sun J, et al. Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. Proc. Natl. Acad. Sci. U. S. A. 2001;98:11158–11162. doi: 10.1073/pnas.201289098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Durham WJ, et al. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133:53–65. doi: 10.1016/j.cell.2008.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stamler JS, et al. A SNO storm in skeletal muscle. Cell. 2008;133:33–35. doi: 10.1016/j.cell.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 77.Bellinger AM, et al. Remodeling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. Proc. Natl. Acad. Sci. U. S. A. 2008;105:2198–2202. doi: 10.1073/pnas.0711074105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bellinger AM, et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009;15:325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chang WJ, et al. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc. Natl. Acad. Sci. U. S. A. 1996;93:9142–9147. doi: 10.1073/pnas.93.17.9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Minetti GC, et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat. Med. 2006;12:1147–1150. doi: 10.1038/nm1479. [DOI] [PubMed] [Google Scholar]
- 81.Colussi C, et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. U. S. A. 2008;105:19183–19187. doi: 10.1073/pnas.0805514105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nott A, et al. S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature. 2008;455:411–415. doi: 10.1038/nature07238. [DOI] [PubMed] [Google Scholar]
- 83.Kaneki M, et al. Nitrosative stress and pathogenesis of insulin resistance. Antioxid. Redox Signal. 2007;9:319–329. doi: 10.1089/ars.2006.1464. [DOI] [PubMed] [Google Scholar]
- 84.Perreault M, Marette A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat. Med. 2001;7:1138–1143. doi: 10.1038/nm1001-1138. [DOI] [PubMed] [Google Scholar]
- 85.Yasukawa T, et al. S-Nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J. Biol. Chem. 2005;280:7511–7518. doi: 10.1074/jbc.M411871200. [DOI] [PubMed] [Google Scholar]
- 86.Carvalho-Filho MA, et al. S-Nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance. Diabetes. 2005;54:959–967. doi: 10.2337/diabetes.54.4.959. [DOI] [PubMed] [Google Scholar]
- 87.Maggi D, et al. Cys860 in the extracellular domain of insulin receptor β-subunit is critical for internalization and signal transduction. Endocrinology. 1998;139:496–504. doi: 10.1210/endo.139.2.5744. [DOI] [PubMed] [Google Scholar]
- 88.Sugita H, et al. Inducible nitric-oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells. J. Biol. Chem. 2005;280:14203–14211. doi: 10.1074/jbc.M411226200. [DOI] [PubMed] [Google Scholar]
- 89.Carvalho-Filho MA, et al. Targeted disruption of iNOS prevents LPS-induced S-nitrosation of IRβ/IRS-1 and Akt and insulin resistance in muscle of mice. Am. J. Physiol. Endocrinol. Metab. 2006;291:E476–E482. doi: 10.1152/ajpendo.00422.2005. [DOI] [PubMed] [Google Scholar]
- 90.Kim F, et al. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ. Res. 2007;100:1589–1596. doi: 10.1161/CIRCRESAHA.106.142851. [DOI] [PubMed] [Google Scholar]
- 91.Reyna SM, et al. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes. 2008;57:2595–2602. doi: 10.2337/db08-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pauli JR, et al. Acute physical exercise reverses S-nitrosation of the insulin receptor, insulin receptor substrate 1 and protein kinase B/Akt in diet-induced obese Wistar rats. J. Physiol. 2008;586:659–671. doi: 10.1113/jphysiol.2007.142414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nakamura T, Lipton SA. Emerging roles of S-nitrosylation in protein misfolding and neurodegenerative diseases. Antioxid. Redox Signal. 2008;10:87–101. doi: 10.1089/ars.2007.1858. [DOI] [PubMed] [Google Scholar]
- 94.Chung KK, et al. S-Nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 95.Yao D, et al. Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. U. S. A. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Uehara T, et al. S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 97.Fang J, et al. S-Nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson's disease. Proc. Natl. Acad. Sci. U. S. A. 2007;104:18742–18747. doi: 10.1073/pnas.0705904104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tsang AH, et al. S-Nitrosylation of XIAP compromises neuronal survival in Parkinson's disease. Proc. Natl. Acad. Sci. U. S. A. 2009;106:4900–4905. doi: 10.1073/pnas.0810595106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cho DH, et al. S-Nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sawa A, et al. Glyceraldehyde-3-phosphate dehydrogenase: nuclear translocation participates in neuronal and nonneuronal cell death. Proc. Natl. Acad. Sci. U. S. A. 1997;94:11669–11674. doi: 10.1073/pnas.94.21.11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hara MR, et al. S-Nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 102.Sen N, et al. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat. Cell Biol. 2008;10:866–873. doi: 10.1038/ncb1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hara MR, et al. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc. Natl. Acad. Sci. U. S. A. 2006;103:3887–3889. doi: 10.1073/pnas.0511321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.LeWitt PA, Taylor DC. Protection against Parkinson's disease progression: clinical experience. Neurotherapeutics. 2008;5:210–225. doi: 10.1016/j.nurt.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kragten E, et al. Glyceraldehyde-3-phosphate dehydrogenase, the putative target of the antiapoptotic compounds CGP 3466 and R-(-)-deprenyl. J. Biol. Chem. 1998;273:5821–5828. doi: 10.1074/jbc.273.10.5821. [DOI] [PubMed] [Google Scholar]
- 106.Andoh T, et al. Role of the redox protein thioredoxin in cytoprotective mechanism evoked by (-)-deprenyl. Mol. Pharmacol. 2005;68:1408–1414. doi: 10.1124/mol.105.012302. [DOI] [PubMed] [Google Scholar]
- 107.Benhar M, et al. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Forrester MT, et al. Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. J. Biol. Chem. 2007;282:13977–13983. doi: 10.1074/jbc.M609684200. [DOI] [PubMed] [Google Scholar]
- 109.Du ZX, et al. Involvement of glyceraldehyde-3-phosphate dehydrogenase in tumor necrosis factor-related apoptosis-inducing ligand-mediated death of thyroid cancer cells. Endocrinology. 2007;148:4352–4361. doi: 10.1210/en.2006-1511. [DOI] [PubMed] [Google Scholar]
- 110.Johnstone RW, et al. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer. 2008;8:782–798. doi: 10.1038/nrc2465. [DOI] [PubMed] [Google Scholar]
- 111.Cantarella G, et al. Trail interacts redundantly with nitric oxide in rat astrocytes: potential contribution to neurodegenerative processes. J. Neuroimmunol. 2007;182:41–47. doi: 10.1016/j.jneuroim.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 112.Kim SF, et al. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310:1966–1970. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 113.Tian J, et al. S-Nitrosylation/activation of COX-2 mediates NMDA neurotoxicity. Proc. Natl. Acad. Sci. U. S. A. 2008;105:10537–10540. doi: 10.1073/pnas.0804852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xu L, et al. Activation of cytosolic phospholipase A2α through nitric oxide-induced S-nitrosylation. Involvement of inducible nitric-oxide synthase and cyclooxygenase-2. J. Biol. Chem. 2008;283:3077–3087. doi: 10.1074/jbc.M705709200. [DOI] [PubMed] [Google Scholar]
- 115.Nalefski EA, et al. Delineation of two functionally distinct domains of cytosolic phospholipase A2, a regulatory Ca2+-dependent lipid-binding domain and a Ca2+-independent catalytic domain. J. Biol. Chem. 1994;269:18239–18249. [PubMed] [Google Scholar]
- 116.Atar S, et al. Atorvastatin-induced cardioprotection is mediated by increasing inducible nitric oxide synthase and consequent S-nitrosylation of cyclooxygenase-2. Am. J. Physiol. Heart Circ. Physiol. 2006;290:H1960–H1968. doi: 10.1152/ajpheart.01137.2005. [DOI] [PubMed] [Google Scholar]
- 117.Ye Y, et al. The role of eNOS, iNOS, and NF-κB in upregulation and activation of cyclooxygenase-2 and infarct size reduction by atorvastatin. Am. J. Physiol. Heart Circ. Physiol. 2008;295:H343–H351. doi: 10.1152/ajpheart.01350.2007. [DOI] [PubMed] [Google Scholar]
- 118.Xuan YT, et al. Mechanism of cyclooxygenase-2 upregulation in late preconditioning. J. Mol. Cell. Cardiol. 2003;35:525–537. doi: 10.1016/s0022-2828(03)00076-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jiang X, et al. COX-2 mediates morphine-induced delayed cardioprotection via an iNOS-dependent mechanism. Life Sci. 2006;78:2543–2549. doi: 10.1016/j.lfs.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 120.Marshall HE, Stamler JS. Inhibition of NF-κB by S-nitrosylation. Biochemistry. 2001;40:1688–1693. doi: 10.1021/bi002239y. [DOI] [PubMed] [Google Scholar]
- 121.Reynaert NL, et al. Nitric oxide represses inhibitory κaB kinase through S-nitrosylation. Proc. Natl. Acad. Sci. U. S. A. 2004;101:8945–8950. doi: 10.1073/pnas.0400588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Guo CJ, et al. S-Nitrosylation of surfactant protein-D controls inflammatory function. PLoS Biol. 2008;6:e266. doi: 10.1371/journal.pbio.0060266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Atochina-Vasserman EN, et al. Immune reconstitution during Pneumocystis lung infection: disruption of surfactant component expression and function by S-nitrosylation. J. Immunol. 2009;182:2277–2287. doi: 10.4049/jimmunol.0802775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kelleher ZT, et al. NOS2 regulation of NF-κB by S-nitrosylation of p65. J. Biol. Chem. 2007;282:30667–30672. doi: 10.1074/jbc.M705929200. [DOI] [PubMed] [Google Scholar]
- 125.Yu Z, et al. Nitric oxide-dependent negative feedback of PARP-1 trans-activation of the inducible nitric-oxide synthase gene. J. Biol. Chem. 2006;281:9101–9109. doi: 10.1074/jbc.M511049200. [DOI] [PubMed] [Google Scholar]
- 126.Khan M, et al. S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J. Cereb. Blood Flow Metab. 2005;25:177–192. doi: 10.1038/sj.jcbfm.9600012. [DOI] [PubMed] [Google Scholar]
- 127.Schroeder RA, et al. Endotoxin-mediated nitric oxide synthesis inhibits IL-1β gene transcription in ANA-1 murine macrophages. Am. J. Physiol. 1999;277:C523–C530. doi: 10.1152/ajpcell.1999.277.3.C523. [DOI] [PubMed] [Google Scholar]
- 128.Xiong H, et al. Inhibition of interleukin-12 p40 transcription and NF-κB activation by nitric oxide in murine macrophages and dendritic cells. J. Biol. Chem. 2004;279:10776–10783. doi: 10.1074/jbc.M313416200. [DOI] [PubMed] [Google Scholar]
- 129.Into T, et al. Regulation of MyD88-dependent signaling events by S nitrosylation retards toll-like receptor signal transduction and initiation of acute-phase immune responses. Mol. Cell. Biol. 2008;28:1338–1347. doi: 10.1128/MCB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.del Fresno C, et al. Nitric oxide activates the expression of IRAK-M via the release of TNF-α in human monocytes. Nitric Oxide. 2004;10:213–220. doi: 10.1016/j.niox.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 131.Gonzalez-Leon MC, et al. Nitric oxide induces SOCS-1 expression in human monocytes in a TNF-α-dependent manner. J. Endotoxin Res. 2006;12:296–306. doi: 10.1179/096805106X118843. [DOI] [PubMed] [Google Scholar]
- 132.Haq E, et al. S-Nitrosoglutathione prevents interphotoreceptor retinoid-binding protein (IRBP161–180)-induced experimental autoimmune uveitis. J. Ocul. Pharmacol. Ther. 2007;23:221–231. doi: 10.1089/jop.2007.0023. [DOI] [PubMed] [Google Scholar]
- 133.Savidge TC, et al. Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology. 2007;132:1344–1358. doi: 10.1053/j.gastro.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 134.Kinsella JP, et al. Early inhaled nitric oxide therapy in premature newborns with respiratory failure. N. Engl. J. Med. 2006;355:354–364. doi: 10.1056/NEJMoa060442. [DOI] [PubMed] [Google Scholar]
- 135.Taylor RW, et al. Low-dose inhaled nitric oxide in patients with acute lung injury: a randomized controlled trial. J. Am. Med. Assoc. 2004;291:1603–1609. doi: 10.1001/jama.291.13.1603. [DOI] [PubMed] [Google Scholar]
- 136.Adhikari NK, et al. Effect of nitric oxide on oxygenation and mortality in acute lung injury: systematic review and meta-analysis. Br. Med. J. 2007;334:779. doi: 10.1136/bmj.39139.716794.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Forrester MT, et al. Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic. Biol. Med. 2009;46:119–126. doi: 10.1016/j.freeradbiomed.2008.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Forrester MT, et al. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat. Biotechnol. 2009;27:557–559. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Foster MW, et al. A genetic analysis of nitrosative stress. Biochemistry. 2009;48:792–799. doi: 10.1021/bi801813n. [DOI] [PubMed] [Google Scholar]
- 140.Romeo AA, et al. Superoxide dismutase targets NO from GSNO to Cysβ93 of oxyhemoglobin in concentrated but not dilute solutions of the protein. J. Am. Chem. Soc. 2003;125:14370–14378. doi: 10.1021/ja0289752. [DOI] [PubMed] [Google Scholar]
- 141.Gow AJ, Stamler JS. Reactions between nitric oxide and haemoglobin under physiological conditions. Nature. 1998;391:169–173. doi: 10.1038/34402. [DOI] [PubMed] [Google Scholar]
- 142.Schlief ML, et al. Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc. Natl. Acad. Sci. U. S. A. 2006;103:14919–14924. doi: 10.1073/pnas.0605390103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Whalen EJ, et al. βAdrenoceptor dysfunction after inhibition of NO synthesis. Hypertension. 2000;36:376–382. doi: 10.1161/01.hyp.36.3.376. [DOI] [PubMed] [Google Scholar]
- 144.Liu S, et al. A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat. Med. 2005;11:952–958. doi: 10.1038/nm1289. [DOI] [PubMed] [Google Scholar]
- 145.Ozawa K, et al. S-Nitrosylation of β-arrestin regulates β-adrenergic receptor trafficking. Mol. Cell. 2008;31:395–405. doi: 10.1016/j.molcel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yu CX, et al. Redox regulation of PTEN by S-nitrosothiols. Mol. Pharmacol. 2005;68:847–854. doi: 10.1124/mol.104.010504. [DOI] [PubMed] [Google Scholar]
- 147.Calmels S, et al. Nitric oxide induces conformational and functional modifications of wild-type p53 tumor suppressor protein. Cancer Res. 1997;57:3365–3369. [PubMed] [Google Scholar]
- 148.Chanvorachote P, et al. Nitric oxide regulates cell sensitivity to cisplatin-induced apoptosis through S-nitrosylation and inhibition of Bcl-2 ubiquitination. Cancer Res. 2006;66:6353–6360. doi: 10.1158/0008-5472.CAN-05-4533. [DOI] [PubMed] [Google Scholar]
- 149.Ruiz F, et al. Nitric oxide inactivates rat hepatic methionine adenosyltransferase in vivo by S-nitrosylation. Hepatology. 1998;28:1051–1057. doi: 10.1002/hep.510280420. [DOI] [PubMed] [Google Scholar]
- 150.Jaiswal M, et al. Human Ogg1, a protein involved in the repair of 8-oxoguanine, is inhibited by nitric oxide. Cancer Res. 2001;61:6388–6393. [PubMed] [Google Scholar]
- 151.Liu L, et al. Inactivation and degradation of O6-alkylguanine-DNA alkyltransferase after reaction with nitric oxide. Cancer Res. 2002;62:3037–3043. [PubMed] [Google Scholar]
- 152.Schonhoff CM, et al. S-nitrosothiol depletion in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U. S. A. 2006;103:2404–2409. doi: 10.1073/pnas.0507243103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Moore PE, et al. Genetic variations of GSNOR and ADBR2 influence response to albuterol in African-American children with severe asthma. Pediatr. Pulmonol. 2009;44:649–654. doi: 10.1002/ppul.21033. [DOI] [PubMed] [Google Scholar]