The Growing Scope of Applications of Genome-scale Metabolic Reconstructions: the case of E. coli (original) (raw)
. Author manuscript; available in PMC: 2011 Jun 6.
Published in final edited form as: Nat Biotechnol. 2008 Jun;26(6):659–667. doi: 10.1038/nbt1401
Abstract
The number and scope of methods developed to interrogate and use metabolic network reconstructions has significantly expanded since the first review of the use of constraint-based analysis in Nature Biotechnology some 14 years ago. In particular, the Escherichia coli metabolic network reconstruction has reached the genome-scale and has been broadly adapted. Specifically, it has been used to address a broad spectrum of basic and practical applications, falling into five main categories: 1) metabolic engineering, 2) model-directed discovery, 3) interpretations of phenotypic screens, 4) analysis of network properties, and 5) studies of evolutionary processes. With these accomplishments in hand, the field is expected to move forward and seek to further, i) broaden the scope and content of network reconstructions, ii) develop new and novel in silico analysis tools, and iii) expand in adaptation to uses of proximal and distal causation in biology. Taken together, these efforts will solidify a mechanistic genotype-phenotype relationship for microbial metabolism.
The availability of reconstructed metabolic networks for microorganisms has increased rapidly in recent years, and a growing number of research groups are reconstructing metabolic networks for organisms of interest1. A network reconstruction represents a highly curated set of primary biological information for a particular organism and thus can be considered a biochemically, genetically and genomically structured (BiGG) data base1, 2. A curated BiGG data base (de facto a knowledge base) can be converted into a mathematical format (i.e., an in silico model), and used to computationally assess phenotypic properties using a variety of computational methods2, 3. Genome-scale reconstructions are thus, a key step in quantifying the genotype-phenotype relationship and can be used to ‘bring genomes to life’4. The purpose of this review is to summarize and classify applications utilizing the E. coli reconstruction to answer a broad spectrum of biological questions. These studies provide both an up to date review of the applications of constraint-based analysis and a guide to similar applications for the growing number of organisms for which genome-scale reconstructions are becoming available.
The Key Steps in the Formulation of Genome-scale Metabolic Network Models
The four key steps in the formulation and use of genome-scale models are illustrated in Fig. 1. Foundational to the process is the generation of global, or genome-scale, omics data. Omics data, along with legacy information (i.e., the ‘bibliome’) and small-scale detailed experiments, can be used to define the interactions amongst the biological components that are used to reconstruct organism-specific networks1. Network reconstruction is also an iterative, on-going process that continually integrates data in a formal fashion as it becomes available5. As a result, a current and well curated genome-scale network reconstruction is a common denominator for those studying systems biology of an organism. An in depth review on the bottom-up reconstruction process can be found in2 and will not be described here.
Figure 1. Formulation and use of GEMs as a four-step process.
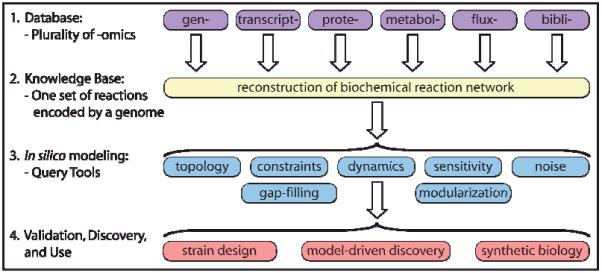
Formulation and use of GEMs as a four-step process. Step 1, the process is based on a variety of high-throughput data sets (i.e., omics data) and a comprehensive assessment of the literature (i.e., bibliomic data). Step 2, all of the data types are used to reconstruct the list of biochemical transformations that make up a network as well as their genetic basis1. In principal, the network is unique. Step 3, the data contained in the reconstruction can be formally represented (i.e., in the form of matrices and logical statements) that can be mathematically characterized by a variety of methods. Step 4, the computational model enables a broad spectrum of applications, as reviewed in this article. Figure adapted from2
The arrow from step 2 to step 3 in Fig. 1 involves a somewhat subtle, but critical, transition. With the definition of systems boundaries and other details, a network reconstruction can be converted into a mathematical format that can be computationally interrogated and subsequently used for experimental design2. Thus, a network reconstruction is converted into a Genome-scale Model (GEM)3. This arrow represents a bridge between the realms of high-throughput data/bioinformatics on one hand and systems science on the other. A network reconstruction (or BiGG knowledge base) is accessible to all and significant strides have been made to make computation with GEMs more readily accessible and free of use6-11. This availability of both genome-scale reconstruction and GEMs has unleashed creativity in research groups around the world and resulted in the series of studies reviewed below.
The E. coli Metabolic Reconstruction
The 18-year history of reconstruction of the E. coli metabolic network (summarized in Fig. 2), has culminated in a network containing a total number of 1,260 ORF metabolic functions12-19. This reconstruction represents 48% of the total experimentally annotated ORF functions in the E. coli genome (Table 1). It should be noted that the function of 92% of the 1,260 gene products have been experimentally verified. Reconstruction of the E. coli network has thus, approached an exhaustion of known metabolic gene functions and it is now being used in a prospective fashion to discover new metabolic capabilities (see below). The reconstruction of the E. coli metabolic network represents the best-developed genome-scale network to date and it has proven to be a platform for a variety of computational analyses. Three successive E. coli GEMs17-19 have been used as the basis for over 60 detailed studies reviewed below.
Figure 2. The ongoing reconstruction of the E. coli metabolic network.

History of the E. coli metabolic reconstruction. Shown are six milestone efforts contributing to the reconstruction of the E. coli metabolic network. For each of the six reconstructions 12-19, the number of included reactions (blue diamonds), genes (green triangles) and metabolites (purple squares) are displayed. Also listed are noteworthy properties that each successive reconstruction provided over previous efforts. For example, Varma & Palsson13, 14 included amino acid and nucleotide biosynthesis pathways in addition to the content that Majewski & Domach12 characterized. The start of the genomic era92 (1997) marked a significant increase in included reconstruction components for each successive iteration. The reaction, gene and metabolite values for pre-genomic era reconstructions were estimated from the content outlined in each publication and in some cases, encoding genes for reactions were unclear.
Table 1.
Properties of the most current E. coli metabolic reconstruction19
| Included Genes | 1260 | (28%) d |
|---|---|---|
| Experimentally Based Function | 1161 | (92%) |
| Computationally Predicted Function | 99 | (8%) |
| Unique Functional Proteins | 1148 | |
| Multigene Complexes | 167 | |
| Genes Involved in Complexes | 415 | |
| Instances of Isozymes a | 346 | |
| Reactions | 2077 | |
| Metabolic Reactions | 1387 | |
| Unique Metabolic Reactions b | 1339 | |
| Cytoplasmic | 1187 | |
| Periplasmic | 192 | |
| Extracellular | 8 | |
| Transport Reactions | 690 | |
| Cytoplasm to Periplasm | 390 | |
| Periplasm to Extracellular | 298 | |
| Cytoplasm to Extracellular | 2 | |
| Gene - Protein - Reaction associations | ||
| Gene Associated (Metabolic / Transport) | 1294 / 625 | |
| Spontaneous / Diffusion Reactions c | 16 / 9 | |
| Total (Gene Associated and No Association Needed) | 1310 / 634 | (94%) |
| No Gene Association (Metabolic / Transport) | 77 / 56 | (6%) |
| Exchange reactions | 304 | |
| Metabolites | ||
| Unique Metabolites b | 1039 | |
| Cytoplasmic | 951 | |
| Periplasm | 418 | |
| Extracellular | 299 |
Ask not what you can do for a reconstruction, but what a reconstruction can do for you
A growing number of research groups utilize the E. coli GEM for predicting, interpreting and understanding E. coli phenotypic states and function, in addition, the reconstruction itself has been used as a context for the interpretation of large amounts of experimental data. Applications of the E. coli GEM range from pragmatic to theoretical studies, and can be classified into five general categories (Fig. 3): 1) metabolic engineering20-30; 2) biological discovery31-37; 3) assessment of phenotypic behavior19, 38-63; 4) biological network analysis64-79; and 5) studies of bacterial evolution80-82. The in silico methods used to probe the E. coli GEM in each study are summarized in Fig. 4. It should be noted that these methods perform an assessment of the solution spaces associated with the mathematical representation of a reconstruction2; these methods are categorized as unbiased and biased methods3. The latter category relies on an observer bias that is stated through an objective function (that is now beginning to be experimentally examined83) and is utilized in most of the studies reviewed here use the general application of flux balance analysis (FBA)84-86. Each category of application is now detailed, with emphasis on the first three that have the greatest practical utility.
Figure 3. Applications of the Genome-Scale Model (GEM) of E. coli.

Uses of the E. coli reconstructions divided into five categories. (A) A drawing of a predicted effect from a loss of function mutation in a simple system is shown. Metabolic engineering studies have investigated in silico strain design using E. coli metabolic reconstructions to overproduce desired products20-30. (B) Recent studies utilizing the reconstruction in a prospective manner have aimed to use the current biochemical and genetic information included in the metabolic network along with additional data types to drive biological discovery, such as predicting genes encoding for orphan reactions32, 33, 35-37. (C) Utilizing the reconstruction in phenotypic studies, computational analyses have examined gene19, 46, 51, 53, 63, metabolite44, 60 and reaction39, 47, 48, 58 essentiality along with considering thermodynamics19, 40, 47, 49, 52, 54, 55, 57, 61 to make better predictions about the physiological state (i.e., the active pathways) of the cell for a given environmental condition. (D) The E. coli reconstructions have been used to analyze and interpret the intrinsic properties of biological networks. One example being finding coupled reaction activities66 (as shown in the drawing) across different growth conditions. (E) Using the network reconstruction, evolutionary studies have examined the cellular network in the context of adaptive evolution events81, horizontal gene transfer80, 81 and minimal metabolic network evolution (as shown in the drawing)82.
Figure 4. Summary of the in silico methods utilized in published E. coli GEM studies.

This heatmap characterizes the incorporation of different computational methods into studies utilizing genome-scale models of E. coli. A dark box indicates that a particular method (one method per row) was utilized in a corresponding study (one citation per column); the frequency of usage of a particular method is given on the right. Studies were grouped into one of five general categories and studies examining phenotypic behavior were further divided into three subgroups. Studies that contributed new experimental growth data are also marked along the bottom offset row.
Applications of GEMs to metabolic engineering of E. coli
Through the application of computational methods that incorporate linear, mixed integer linear, and non-linear programming, it has been demonstrated that model-directed strain design can lead to increased metabolite production20-30. In these studies, the E. coli GEM is principally used to analyze the metabolite production potential of E. coli and identify metabolic interventions needed to enable the production of the product of interest. Thus, E. coli strains have been systematically designed through in silico analysis to overproduce target metabolites such as lycopene23, 24, lactic acid25, ethanol26, succinic acid27, 28, L-valine29, L-threonine30, additional amino acids21, as well as diverse products from hydrogen to vanillin22. Select exemplary metabolic engineering applications will be described in more detail.
To increase the production of an already high producing strain, a systematic computational search was developed24 to explore the E. coli metabolic network and report gene deletions that diverted metabolic flux towards the desired product. This process resulted a knock-out strain, that when constructed, showed a two-fold increase in the production of lycopene over the parental strain. In this analysis, the computational algorithm MOMA41 and the _i_JE66018 E. coli GEM were utilized to sequentially examine additive genetic deletions that would improve lycopene production while maintaining cell viability. Strain designs were constructed through genetic manipulations using the predicted modifications and it was found that this computational approach yielded the twofold increase in production rate over a previously engineered overproducing strain and an 8.5 fold increase over wild-type production harboring only a lycopene biosynthesis plasmid24. Strain performance was evaluated by monitoring lycopene production through enzymatic assays and mutant growth rates. In addition, the strain designs identified computationally were compared to mixed combinatorial transposon mutagenesis and it was found that the maximum production observed could be designed solely using the systematic GEM aided computational method23, 24. Furthermore, a deleterious effect was observed when targets identified in individual computational designs were combined in an attempt to achieve an overall more desirable phenotype. Thus, the overall systematic effects from individual designs were not additive and needed to be interpreted in the context of the entire network.
Two studies producing the amino acids L-valine29 and L-threonine30 have demonstrated the broad usage of GEM aided computation for strain design. In the first study, GEM aided modeling was employed in three different areas to increase the production of L-threonine to industrial titers30. In one instance, in silico modeling was used to identify the optimal activity of a key enzymatic reaction towards maximum L-threonine production using a parametric sensitivity analysis that compared reaction activity to L-threonine production rate. The optimal activity prediction was subsequently used to tune the overexpression of the gene which encodes for this enzymatic reaction through comparison to base-line activity and the result was a production increase. This method proved to be vital to the success of this strain, as a previous transcription profiling guided attempt at overexpression resulted in an undesirable surplus of activity and was detrimental to L-threonine production. For the same strain, a GEM aided flux analysis in conjunction with mRNA expression data levels also guided the elimination of negative regulation on a gene which encoded for a reaction that channeled flux towards the final product. The third use of the GEM for the design of this strain occurred when an unwanted byproduct was observed in the culture medium and computation was utilized to divert the flux from this byproduct to L-threonine30 through overexpression of another key gene encoded activity. The second analysis applied the systematic computational search algorithm previously described24 to the updated E. coli GEM MBEL9797 (similar to the _i_JR904 GEM17) to improve L-valine production. The in silico analysis of beneficial knock-outs to divert flux towards the desired product once again resulted in a significant increase in the production of the desired metabolite over an existing overproducing strain; more than a two-fold increase in this case29. Furthermore, in this same study, a number of additional metabolic engineering approaches to increase overproduction were performed (i.e., relieving feedback inhibition and regulation through attenuation, removing competing pathways, up-regulation of primary biosynthetic pathways, and overexpression of exporting machinery). When compared to each of the other individual strain modifications, the in silico GEM aided interventions resulted in the greatest increase in L-valine production29. Taken together, these two studies demonstrate the broad applications for which GEMs can be utilized to design strains not only in a de novo fashion, but to make further improvements on strains through integrating and interpreting experimental data.
Several other strain designs utilizing E. coli GEMs have been reported. In a combined computational and experimental study, the bi-level optimization algorithm OptKnock20 and _i_JR90417 were utilized to overproduce lactate in E. coli25. The algorithm OptKnock optimizes two objective functions, biomass formation and product secretion, to produce strains that will couple the excretion of a desirable product to the growth rate. Using adaptive evolution with growth rate selection pressure, the lactate producing strains designed using OptKnock were found to possess this growth-coupling property. Growth rate, uptake and secretion rate profiles were the measures by which this property was examined and thus this study demonstrated the utility of adaptive evolution as a design tool87. Additional noteworthy examples of GEM aided design are two studies which demonstrated27, 28 that GEM modeling using _i_JR90417 was beneficial to screen genes that were deemed to be important for succinate production. Combinatorial knock-outs that were predicted to be overproducers in silico were experimentally verified to display the same overproducing phenotype in vivo. Furthermore, this method had an advantage over using comparative genomics for strain design, which was also performed in one of the studies27.
Taken together, a growing number of metabolic engineering studies demonstrate the use of GEMs to generate strain designs that are often non-intuitive and non-obvious. An excellent example of a non-intuitive strain improvement outlined in this section was when modeling was used to not only study the effect of a gene removal, but to tune the expression of a gene to an optimally predicted level, that when expressed too highly, was detrimental to product formation. Genome-scale reconstructions thus allow the examination and simulation of metabolism as an integrated network, circumventing the possible shortcomings of methods that rely on manual assessment of a limited number of interactions and fail to detect non-intuitive causal interactions. With the growing availability of organism and strain specific GEMs, applications for designing microbial strains for industrial production are expected to continue to grow. This growth expectation is in part based on the on-going reconstruction of additional cellular processes, such as transcriptional regulation and protein production. Computations based on genome-scale models are also beginning to influence other areas of industrial microbiology such as generation of renewable energy88-90 and bioremediation89.
Directing Discovery: GEM-driven discovery in E. coli
GEMs can provide a guide to biological discovery. This capability is based on comparison of computed and actual experimental outcomes. Given the fact that BiGG knowledge bases are incomplete and that they contain gaps91, they provide a context for systematic discovery of missing information. The comparison between computation and experiments are summarized in Fig. 5 highlighting how agreements and disagreements are analyzed.
Figure 5. Comparison of computation and experimental data: identification of agreements and disagreements.

The comparison of GEM computation and organism-specific experimental measurements identifies agreements and disagreements. The phenotypic outcomes are tabulated for genetic perturbations examined in a given environment (e.g., growth or no growth). A ‘+’ indicates that a given phenotype is not affected by the perturbation, and ‘−’ indicates it does. Each outcome of comparison has a different implication; 1: consistency check - a perturbation has no affect on the property being measured and modeling predicts the same; 4: validation - the perturbation affects the experimental outcome and modeling with the GEM predicts this outcome; 2: identification of missing content - when GEM modeling fails to predict the positive confirmation of the property being measured, this outcome indicates that there is missing content in the GEM and can lead to the identification of specific areas for biological discovery; 3: identification of errors, inconsistencies or missing context-specific information – a positive prediction for the measured property and an opposite experimental observation indicates a possible error in the current organism-specific knowledge or that additional context-specific information is lacking from the GEM or modeling method (e.g., transcriptional regulation).
The current area of most significant interest is to direct discovery efforts towards characterizing unknown ORFs in the E. coli genome. Ten years after the first release of the complete genome-sequence92, many unknown ORFs still exist in the E. coli genome (see Supplementary Table 1), with many of these likely to encode metabolic functions. ORF discovery utilizing GEMs also has significant potential to impact not only how new and less studied genomes are annotated, but to fill out the missing pieces in E. coli metabolism.
To address this challenge, algorithms have been developed to determine the probable gene candidates that fill knowledge gaps in the E. coli and other network reconstructions. These algorithms utilize global network topology and genomic correlations, such as genome context and protein fusion events32, as well as local network topology and/or phylogenetic profiles32, 33. Similar tools has been developed which utilize mRNA coexpression93 and which can evaluate more general metabolic pathway databases94. In addition to these network topology-based methods, an optimization based procedure has also been developed to fill network gaps and evaluate reaction reversibility along with adding additional transport and intracellular reactions from databases of known metabolic reactions36. These studies produce specific targets for drill-down experiments needed for confirmation of these computationally generated hypotheses.
Two recent studies have integrated a combined computational and experimental approach to aid the ORF discovery process in E. coli through utilizing the GEM and high-throughput phenotype data35, 37. The first study utilized an iterative process35 in which, 1) differences in modeling predictions and high-throughput growth phenotype data were identified, 2) potential missing reactions that remedy these disagreements were algorithmically determined, 3) bioinformatics was utilized to identify likely encoding ORFs, and 4) resulting targeted ORFs were cloned and experimentally characterized. Application of this process led to the functional characterization of eight ORFs that are involved in transport, regulatory and metabolic functions in E. coli35. The discovery process was aided by a high-throughput growth phenotyping analysis and the genome-wide single-gene mutant collection95, along with other characterization analyses such as targeted expression profiling. The second GEM-based analysis which resulted in ORF discovery utilized network topology to examine orphan reactions in the E. coli network (i.e., reactions known to exist in E. coli that have not been linked to an encoding gene) identified by the previously mentioned network topology-based gap-filling algorithms32, 33, 93. The basic premise behind these algorithms is the utilization of an orphan reaction’s network neighbors as constraints to assign metabolic function. With the resulting tentative ORF assignment, biochemical characterization studies utilizing genetic mutants95, analysis of growth under different substrate conditions, and expression data were all utilized to characterize and assign function to an orphan ORF that is responsible for a metabolic conversion that has been known for 25 years37.
Further studies in this category of biological discovery applications (not focused on ORF identification) have utilized GEMs of E. coli to identify potential bottleneck reactions in the metabolic network34 and as of yet uncharacterized transcription factor target interactions in E. coli31. The aforementioned study targeting the elucidation of regulatory and metabolic interactions in E. coli developed an iterative procedure focused on reconciling computational and experimental discrepancies stemming from high-throughput growth phenotype and gene expression data where selected expression changes were validated using RT-PCR31. With the advancement of high-throughput technologies to test the hypotheses generated from computational studies, these and similar algorithmic approaches are likely to continue to aid in the quest to achieve full functional annotation of the E. coli genome and its context-specific uses.
Phenotypic Functions: GEM aided assessment
The area where the E. coli GEMs has been most extensively utilized is for the examination and quantitative interpretation of metabolic physiology for wild-type, genetically perturbed and adaptively evolved strains of E. coli19, 38-63. These efforts have implications in both the quantitative and qualitative understanding of physiological states of the cell. Furthermore, these efforts have examined E. coli physiology for a vast number of given genetic and environmental conditions and incorporation of the developed methods will have an impact on future design of biological systems and modeling approaches. A large subset of these studies of phenotypic behavior aim to utilize thermodynamic laws and information to refine phenotype predictions of GEMs and to incorporate metabolomic and fluxomic data into modeling19, 40, 47, 49, 52, 54, 55, 57, 61.
A set of distinct computational methods using GEMs have been developed to determine the physiological state of E. coli after genetic perturbations41, 45, 50. These studies have utilized 13C flux measurements and growth rate phenotype data to evaluate the predictability of the developed algorithms when compared to experimental observations. Whereas comparisons to flux data from wild-type and E. coli mutants reveals that the computational algorithm MOMA41 provides better predictions for transient growth rates (early post perturbation state), the algorithm ROOM45 (and basic FBA) was found to be more successful in predicting final steady-state growth rates and overall lethality45. These algorithms have been utilized, in addition to basic FBA, for genome-wide essentiality screens, as now outlined.
A range of computational studies have sought to understand phenotypes through determining the essential genes19, 46, 51, 53, 63, metabolites44, 60 and reactions39, 47, 48, 58 in the E. coli metabolic network. A common benchmark for examining GEM predictive ability is to determine the agreement with growth phenotype data from knock-out collections of E. coli. Such studies will be further enabled by the recent availability of a comprehensive single-gene knock-out library for E. coli95 (for example19, 53). Implications for examining network essentiality in E. coli include determining network essentiality in similar organisms39, 48, 53, 58, deciphering network makeup and enzyme dispensability (i.e., measures of robustness)46, 58, 60, aiding in metabolic network annotation, validation and refinement44, and even rescuing knock-out strains through additional gene deletions63, to name a few. The predictive capability of the E. coli GEM, as demonstrated by these studies, has been instrumental in the adaptation of its use. One particular study examining knock-out phenotypes has demonstrated that the E. coli GEM was able to predict the outcomes of adaptively evolved strains to a high degree (78%) when knock-out E. coli strains were grown in a number of different substrate environments by examining growth rates at the beginning and end of adaptive evolution43. This study represents a demonstration of a GEM’s ability to look at adaptive behavior (or ‘distal’ causation96), in addition to immediate behavior (or ‘proximal’ causation96). Predictive capability is expected to improve through examining growth behavior across a greater number of environments (additional phenotyping screens will be necessary) and with an increase of integration of additional cellular processes. Genetic perturbations have played a key role in the study of the genotype-phenotype relationship in biology and GEMs can be used to mechanistically interpret the results and predict the outcomes of such perturbations.
Incorporating thermodynamic information into E. coli GEMs has shown promise in narrowing predictions of allowable physiological states in a given environment19, 40, 47, 49, 52, 54, 55, 57, 61 and in identifying reactions likely to be subject to active allosteric or genetic regulation49, 54. This field is progressing rapidly and should prove to increase the predictive capabilities of genome-scale modeling through the addition of governing thermodynamic physiochemical constrains. One particular analysis incorporating compound formation and reaction energies for the content of the GEM based on _i_JR90417 identified reactions that are likely to be effectively irreversible for any realistic metabolite concentration54. The hypothesis was advanced that these reactions are candidates for cellular regulation in their respective pathways since enzyme regulation will likely be the dominant mechanism for control of flux through these reactions54.
The addition of thermodynamics enables the analysis of metabolomic data in the context of a reconstruction. A study utilizing high-throughput metabolomic data and GEMs proposed likely regulatory interactions by deciphering the metabolite concentrations in the context of overall network functionality49. Not only did the metabolomic data benefit computations by constraining the system using physiological measurements, but the computational predictions were also able to validate quantitative metabolomic data sets for consistency through providing a functional context to relate metabolite concentrations. This application is one example of how metabolomic data will directly influence modeling and metabolite concentration data is likely to greatly influence future metabolic modeling due to its intimate connection with GEM content. Similar work incorporating other quantitative values with FBA, such as metabolite concentrations57 and flux ratios at branch points in metabolism56 is also appearing.
Applying a different physiochemical constraint, molecular crowding, a framework has also been developed to incorporate spatial constraints into FBA59. The functional states predicted with this method (i.e., FBA with molecular crowding, FBAwMC) and the E. coli GEM were validated against generated growth, substrate, and production rate data along with gene expression profiles and enzyme activity measures to demonstrate predictive accuracy, including substrate preferentiality, when examining growth in complex substrate environments59, 62. Overall, these studies which incorporate reaction thermodynamics and additional cellular constraints should further narrow the range of allowable functional network states that can made based on stoichiometry alone and thus improve the utility of GEMs.
In addition to analyses on the genomic scale, a number of studies modeling the metabolism of E. coli on a smaller-scale have been performed. These analyses typically utilize models containing approximately 100 reactions or less and most often, focus on incorporating non-linear analysis to understand quantitative experimental data (e.g., isotopomer modeling). With the advancement of computational power and developed platforms, the networks that can be analyzed will grow in size97. Given that the results produced from analyses such as isotopomer modeling have been shown to be highly dependent on the content of a reduced model, the logical starting point for building such models is the E. coli GEM97. A number of noteworthy studies have been conducted with reduced models, but not detailed here as they are outside the scope of this review.
Systems Biology: Analysis of network properties
E. coli is generally viewed as having the most complete characterization of any model organism98, 99. Due to the incorporation of thousands of metabolic interactions with relatively high reliability (e.g., 92% of the genes included in the latest reconstruction of E. coli19 have experimentally determined annotated functions99, Table 1), validated genome-scale reconstructions of E. coli have become popular resources for the analysis of various network properties64-79. The methods designed to analyze the underlying network structure of E. coli metabolism, some characterizing its interplay with regulation, have been developed to determine a number of physiological features. These features include the most probable active pathways and utilized metabolites under all possible growth conditions67, 69, 73, 75, the existence of alternate optimal solutions and their physiological significance65, conserved intracellular pools of metabolites68, coupled reaction activities66 and their relationship to gene co-expression77, metabolite coupling71, metabolite utilization72, the organization of metabolic networks64, 76, strategies for E. coli to incorporate metabolic redundancy78, and the dominant functional states of the network across various environments70, 74, 79. These findings are both driven by biased approaches utilizing FBA and biomass objective function optimization and by unbiased approaches such as graph-based analyses (see Fig. 4). One noteworthy study utilizing the GEM outlined network examined thousands of different potential growth conditions and observed a ‘high-flux backbone’ in E. coli that both carried high levels of flux across the different environmental conditions and was composed of a relatively small set of enzymatic reactions67. This result can be of practical importance for synthetic biology efforts aimed towards manipulating flux within biological systems. Furthermore, this finding was hypothesized to be a universal feature of metabolic activity in all cells and was consistent with flux measurements from 13C labeling experiments67.
The studies in this category have a common systems biology theme; namely the development and subsequent demonstration of methods that identify sets of reactions or metabolites with correlated or coordinated functions and systematic relationships. The systems biology that these methods enable and demonstrate has potential implications for, i) antimicrobial drug-target discovery68, 69, ii) aiding the development of additional metabolic reconstructions66, 68, iii) guiding genetic manipulations66, iv) improving metabolic engineering applications67, 68, and v) increasing the general understanding of biological network behavior65, 74, 77 and resilience78. The role that the E. coli GEM has taken is a comprehensive and curated set of up to date metabolic knowledge; thus providing a scaffold for these large-scale computations.
Bacterial evolution: GEM aided studies of distal causation
The GEMs of E. coli have been used to examine the process of bacterial evolution80-82. Specifically, the network reconstructions have been used to interpret adaptive evolution events81, horizontal gene transfer80, 81 and evolution to minimal metabolic networks82. These studies, which utilize the E. coli reconstruction as an organism-specific genetic and metabolic content database, and the corresponding GEM, have been able to provide insight into evolutionary events through combining known physiological data (e.g., in various environmental conditions) with hypotheses and in silico computation. Examining the evolution of minimal metabolic networks through simulation demonstrated that it was possible to predict the gene content of close relatives of E. coli by examining the necessity of genes and reactions in the overall context of the system functionality for a specific lifestyle82. Similarly, by re-examining network functionality in a number of different environments and through the utilization of comparative genomics, it was shown that recent evolutionary events (i.e., horizontal gene transfer) likely resulted from a response to a change in environment81. Furthermore, computational analysis led to the additional conclusion that these horizontal gene transfer events are more likely if the host organism contains an enzyme that catalyzes a coupled metabolic flux related to the transferred enzyme’s function80, 81. Taken together, these studies demonstrate the importance of having high-quality curated reconstructions to enable studies on an organism’s response to environmental changes and for understanding the fundamental forces driving bacterial evolution.
Closing
The myriad of studies described in this review highlights the rapid development and use of genome-scale reconstruction and derived computational models to address a growing spectrum of basic research and applied problems. The experience with genome-scale reconstructions has demonstrated that they are a common denominator in the systems analysis of metabolic functions. With the recognition of its basic paradigms and a growing spectrum of practical uses enabled, there are several exciting challenges that this field now faces. Accordingly, further development is necessary, and three major areas where it will be influential are now discussed; i) network reconstructions and the reconstruction process, ii) computational BiGG query tools (i.e., modeling), and iii) application to proximal and distal causation in biology.
The scope of reconstructions is bound to grow, representing more and more BiGG knowledge in the structured format of a GEM91. Growth in scope in the near-term will on one front, involve the transcriptional and translational machinery of bacterial cells100-102. Such an extension will enable a range of studies including the direct inclusion of proteomic data, fine graining of growth requirements and the explicit consideration of secreted protein products. Another expansion in scope in the near-term is the reconstruction of the genome-scale transcriptional regulatory network (TRN). Such reconstruction at the genome-scale is now enabled by new experimental technologies, such as ChIP-chip103. Experimental interrogation of the currently available TRN suggests that we know about one-fourth to one-third of its content31, indicating that there is much to be discovered. Once reconstructed, the TRN will allow computational predictions of the context-specific uses of the E. coli genome and the responses of two-component signaling systems. Taken together, these near-term expansions in content will encompass the activity of apparently 2000 ORFs in the E. coli genome.
Mid-term expansions in scope will include the growth cycle, shock responses and additional cellular functions. Such a reconstruction should eventually be a comprehensive representation of the chemical reactions and transactions enabled by _E. coli_’s gene products. Longer-term reconstruction may begin to address the 3-dimensional organization of the bacterial cell. In particular, high-resolution ChIP-chip data on the DNA binding protein could enable the estimation of the topological arrangement of the genome, and potentially elucidate the structure of the cell wall and other cellular structures that will allow us a full 3-dimensional reconstruction of E. coli.
We now know how to represent BiGG data in either a stoichiometric format or in the form of causal relationships104 and how to use them to perform several lines of computational inquiries. Computational query tools of GEMs will continue to be developed. New advances will likely include modularization methods, use of fluxomic data and eventually kinetics. As the scope and content of the reconstruction grows, the need to modularize its content becomes more pressing. Fine or course grained views of cellular processes are needed for different applications. For instance, as previously mentioned, current computational limitations force the reduction in a network for the analysis of isotopomer data, and a rational way to carry out such reduction is needed. Given the systemic nature of fluxomic data and its phenotypic relevance, there is a pressing need to increase the size of the networks that can be analyzed for experimental measurement and estimation of flux states. Finally, although detailed kinetic models of microbial functions may currently be mostly of academic interest, we will most likely be able to construct them in the mid-term based on advances with metabolomic and fluxomic data, in addition to the developments that are occurring with the incorporation of thermodynamic information. Such large-scale kinetic models are likely to differ from those resulting from traditional approaches for construction of kinetic models as, they come with different challenges.
As this review shows, the scope of applications of genome-scale reconstructions and GEMs is growing. Going forward, we wish to comment on three categories of applications: growth in coverage (i.e., gap-filling), engineering (i.e., synthetic biology), and the development of fundamental understanding. Growth in coverage will come through discovery of missing network components. For instance, the latest metabolic reconstruction, _i_AF1260, contains 14% blocked reactions19. This disconnected content means that we have knowledge gaps that have arisen due to characterization of individual gene products outside the context of a given physiological function (i.e., outside a defined pathway). Metabolomic profiling is one measure that will provide us with the missing upstream or downstream routes to such dead ends in the network. Also, an expansion of scope in modeling will allow for further investigation of network content, such as tRNA charging reactions that are currently in this blocked reaction set19. Furthermore, growing metabolomic data suggests that we are discovering the existence of several new metabolites. Pathways that include these metabolites need to be discovered. Methods exist to compute missing pathways between molecules105 that can be applied to such data. Such pathways, in turn, will lead to experimental programs to discover novel gene functions and to validate or refute the existence of such pathways. Similarly, we expect that a number of the components of TRNs are missing, such as new sRNA molecules (see Supplementary Table 1). Clearly, well QC/QA’ed reconstructions will help in guiding us to comprehensive genome-scale representation of all major cellular processes in bacteria at the BiGG data level of resolution that, in turn, enables GEMs of growing coverage and resolution. The scope of this effort has been described as being; “… 10 times more ambitious and 100 times more important for mankind [compared with Human Genome Project]…” Hans Westerhoff106.
Predictive models allow for design. In fact, in engineering, there is ‘nothing more useful that a good theory.’ As this review demonstrates, genomics and high-throughput technologies have enabled the construction of predictive computational models. The scope of such predictions is limited at the moment, but with the growing scope and coverage of genome-scale reconstructions and advancements in the development of computational tools, this scope will broaden. Not only will GEMs influence design in synthetic biology, but their influence in discovery of cellular content will provide a more complete picture of the environment (i.e., the parts list in the cell) in which future synthetically engineered constructs and circuits will be placed. The impact of GEMs on synthetic biology is thus likely to be notable; ranging from the provision of the cellular-context of a small-scale gene circuit design to engineering of the entire genome-scale network towards fundamentally new and useful (i.e., production) phenotypes.
Finally, we can speculate about the deep scientific impact that comprehensive predictive GEMs will have on our understanding of the living process. A comprehensive view of cellular functions will allow us to study the fundamental properties of both the underlying energy and information flows in living organisms. Such a view is likely to deeply affect our understanding of both distal and proximal causation in biology.
Supplementary Material
table
Acknowledgements
We would like to thank Andrew Joyce, Jennifer Reed, Daniel Segre, Nathan Price, Markus Herrgard and Christian Barrett for their invaluable insight. AF is supported by National Institutes of Health R01 GM057089 grant.
References
- 1.Reed JL, Famili I, Thiele I, Palsson BO. Towards multidimensional genome annotation. Nat Rev Genet. 2006;7:130–141. doi: 10.1038/nrg1769. [DOI] [PubMed] [Google Scholar]
- 2.Palsson BO. Systems biology: properties of reconstructed networks. Cambridge University Press; New York: 2006. [Google Scholar]
- 3.Price ND, Reed JL, Palsson BO. Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat Rev Microbiol. 2004;2:886–897. doi: 10.1038/nrmicro1023. [DOI] [PubMed] [Google Scholar]
- 4.Frazier ME, Johnson GM, Thomassen DG, Oliver CE, Patrinos A. Realizing the potential of the Genome Revolution: The Genomes to life Program. Science. 2003;300:290–293. doi: 10.1126/science.1084566. [DOI] [PubMed] [Google Scholar]
- 5.Reed JL, Palsson BO. Thirteen Years of Building Constraint-Based In Silico Models of Escherichia coli. J Bacteriol. 2003;185:2692–2699. doi: 10.1128/JB.185.9.2692-2699.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Becker SA, et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox. Nat. Protocols. 2007;2:727–738. doi: 10.1038/nprot.2007.99. [DOI] [PubMed] [Google Scholar]
- 7.Lee SY, et al. Systems-level analysis of genome-scale in silico metabolic models using MetaFluxNet. Biotechnol. Bioproc. Eng. 2005;10:425–431. [Google Scholar]
- 8.Klamt S, Saez-Rodriguez J, Gilles ED. Structural and functional analysis of cellular networks with CellNetAnalyzer. BMC systems biology. 2007;1:2. doi: 10.1186/1752-0509-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raman K, Chandra N. 11th SBML Forum. 2006.
- 10.Luo RY, Liao S, Zeng SQ, Li YX, Luo QM. FluxExplorer: A general platform for modeling and analyses of metabolic networks based on stoichiometry. Chinese Science Bulletin. 2006;51:689–696. [Google Scholar]
- 11.Hucka M, et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics. 2003;19:524–531. doi: 10.1093/bioinformatics/btg015. [DOI] [PubMed] [Google Scholar]
- 12.Majewski RA, Domach MM. Simple constrained optimization view of acetate overflow in E. coli. Biotechnol Bioeng. 1990;35:732–738. doi: 10.1002/bit.260350711. [DOI] [PubMed] [Google Scholar]
- 13.Varma A, Boesch BW, Palsson BO. Stoichiometric interpretation of Escherichia coli glucose catabolism under various oxygenation rates. Appl Environ Microbiol. 1993;59:2465–2473. doi: 10.1128/aem.59.8.2465-2473.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varma A, Boesch BW, Palsson BO. Biochemical production capabilities of Escherichia coli. Biotechnol Bioeng. 1993;42:59–73. doi: 10.1002/bit.260420109. [DOI] [PubMed] [Google Scholar]
- 15.Pramanik J, Keasling JD. Stoichiometric model of Escherichia coli metabolism: Incorporation of growth-rate dependent biomass composition and mechanistic energy requirements. Biotechnol Bioeng. 1997;56:398–421. doi: 10.1002/(SICI)1097-0290(19971120)56:4<398::AID-BIT6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 16.Pramanik J, Keasling JD. Effect of Escherichia coli biomass composition on central metabolic fluxes predicted by a stoichiometric model. Biotechnol Bioeng. 1998;60:230–238. doi: 10.1002/(sici)1097-0290(19981020)60:2<230::aid-bit10>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 17.Reed JL, Vo TD, Schilling CH, Palsson BO. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR) Genome Biology. 2003;4:R54.51–R54.12. doi: 10.1186/gb-2003-4-9-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edwards JS, Palsson BO. The Escherichia coli MG1655 in silico metabolic genotype: Its definition, characteristics, and capabilities. Proc Natl Acad Sci U S A. 2000;97:5528–5533. doi: 10.1073/pnas.97.10.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feist AM, et al. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol. 2007;3 doi: 10.1038/msb4100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burgard AP, Pharkya P, Maranas CD. Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng. 2003;84:647–657. doi: 10.1002/bit.10803. [DOI] [PubMed] [Google Scholar]
- 21.Pharkya P, Burgard AP, Maranas CD. Exploring the overproduction of amino acids using the bilevel optimization framework OptKnock. Biotechnol Bioeng. 2003;84:887–899. doi: 10.1002/bit.10857. [DOI] [PubMed] [Google Scholar]
- 22.Pharkya P, Burgard AP, Maranas CD. OptStrain: a computational framework for redesign of microbial production systems. Genome Res. 2004;14:2367–2376. doi: 10.1101/gr.2872004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alper H, Jin YS, Moxley JF, Stephanopoulos G. Identifying gene targets for the metabolic engineering of lycopene biosynthesis in Escherichia coli. Metab Eng. 2005;7:155–164. doi: 10.1016/j.ymben.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 24.Alper H, Miyaoku K, Stephanopoulos G. Construction of lycopene-overproducing E. coli strains by combining systematic and combinatorial gene knockout targets. Nat Biotechnol. 2005;23:612–616. doi: 10.1038/nbt1083. [DOI] [PubMed] [Google Scholar]
- 25.Fong SS, et al. In silico design and adaptive evolution of Escherichia coli for production of lactic acid. Biotechnol Bioeng. 2005;91:643–648. doi: 10.1002/bit.20542. [DOI] [PubMed] [Google Scholar]
- 26.Pharkya P, Maranas CD. An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab Eng. 2006;8:1–13. doi: 10.1016/j.ymben.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Lee SJ, et al. Metabolic engineering of Escherichia coli for enhanced production of succinic acid, based on genome comparison and in silico gene knockout simulation. Appl Environ Microbiol. 2005;71:7880–7887. doi: 10.1128/AEM.71.12.7880-7887.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Q, Chen X, Yang Y, Zhao X. Genome-scale in silico aided metabolic analysis and flux comparisons of Escherichia coli to improve succinate production. Appl Microbiol Biotechnol. 2006;V73:887–894. doi: 10.1007/s00253-006-0535-y. [DOI] [PubMed] [Google Scholar]
- 29.Park JH, Lee KH, Kim TY, Lee SY. Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proc Natl Acad Sci U S A. 2007;104:7797–7802. doi: 10.1073/pnas.0702609104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee KH, Park JH, Kim TY, Kim HU, Lee SY. Systems metabolic engineering of Escherichia coli for L-threonine production. Mol Syst Biol. 2007;3:149. doi: 10.1038/msb4100196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BO. Integrating high-throughput and computational data elucidates bacterial networks. Nature. 2004;429:92–96. doi: 10.1038/nature02456. [DOI] [PubMed] [Google Scholar]
- 32.Chen L, Vitkup D. Predicting genes for orphan metabolic activities using phylogenetic profiles. Genome Biol. 2006;7:R17. doi: 10.1186/gb-2006-7-2-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kharchenko P, Chen L, Freund Y, Vitkup D, Church GM. Identifying metabolic enzymes with multiple types of association evidence. BMC Bioinformatics. 2006;7 doi: 10.1186/1471-2105-7-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herrgard MJ, Fong SS, Palsson BO. Identification of genome-scale metabolic network models using experimentally measured flux profiles. PLoS Comput Biol. 2006;2:e72. doi: 10.1371/journal.pcbi.0020072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reed JL, et al. Systems approach to refining genome annotation. Proc Natl Acad Sci U S A. 2006;103:17480–17484. doi: 10.1073/pnas.0603364103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar V. Satish, Dasika MS, Maranas CD. Optimization based automated curation of metabolic reconstructions. BMC Bioinformatics. 2007;8:212. doi: 10.1186/1471-2105-8-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuhrer T, Chen L, Sauer U, Vitkup D. Computational prediction and experimental verification of the gene encoding the NAD+/NADP+-dependent succinate semialdehyde dehydrogenase in Escherichia coli. J Bacteriol. 2007 doi: 10.1128/JB.01027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edwards JS, Ibarra RU, Palsson BO. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat Biotechnol. 2001;19:125–130. doi: 10.1038/84379. [DOI] [PubMed] [Google Scholar]
- 39.Burgard AP, Vaidyaraman S, Maranas CD. Minimal reaction sets for Escherichia coli metabolism under different growth requirements and uptake environments. Biotechnol Prog. 2001;17:791–797. doi: 10.1021/bp0100880. [DOI] [PubMed] [Google Scholar]
- 40.Beard DA, Liang SD, Qian H. Energy balance for analysis of complex metabolic networks. Biophys J. 2002;83:79–86. doi: 10.1016/S0006-3495(02)75150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Segre D, Vitkup D, Church GM. Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci U S A. 2002;99:15112–15117. doi: 10.1073/pnas.232349399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ibarra RU, Edwards JS, Palsson BO. Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature. 2002;420:186–189. doi: 10.1038/nature01149. [DOI] [PubMed] [Google Scholar]
- 43.Fong SS, Palsson BO. Metabolic gene-deletion strains of Escherichia coli evolve to computationally predicted growth phenotypes. Nat Genet. 2004;36:1056–1058. doi: 10.1038/ng1432. [DOI] [PubMed] [Google Scholar]
- 44.Imielinski M, Belta C, Halasz A, Rubin H. Investigating metabolite essentiality through genome-scale analysis of Escherichia coli production capabilities. Bioinformatics. 2005;21:2008–2016. doi: 10.1093/bioinformatics/bti245. [DOI] [PubMed] [Google Scholar]
- 45.Shlomi T, Berkman O, Ruppin E. Regulatory on/off minimization of metabolic flux changes after genetic perturbations. Proc Natl Acad Sci U S A. 2005;102:7695–7700. doi: 10.1073/pnas.0406346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghim CM, Goh KI, Kahng B. Lethality and synthetic lethality in the genome-wide metabolic network of Escherichia coli. J theor Biol. 2005;237:401–411. doi: 10.1016/j.jtbi.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 47.Henry CS, Jankowski MD, Broadbelt LJ, Hatzimanikatis V. Genome-scale thermodynamic analysis of Escherichia coli metabolism. Biophys J. 2006;90:1453–1461. doi: 10.1529/biophysj.105.071720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Samal A, et al. Low degree metabolites explain essential reactions and enhance modularity in biological networks. BMC Bioinformatics. 2006;7:118. doi: 10.1186/1471-2105-7-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kümmel A, Panke S, Heinemann M. Putative regulatory sites unraveled by network-embedded thermodynamic analysis of metabolome data. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100074. 2006.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wunderlich Z, Mirny LA. Using the topology of metabolic networks to predict viability of mutant strains. Biophys J. 2006;91:2304–2311. doi: 10.1529/biophysj.105.080572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerdes S, et al. Essential genes on metabolic maps. Curr Opin Biotechnol. 2006;17:448–456. doi: 10.1016/j.copbio.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 52.Kümmel A, Panke S, Heinemann M. Systematic assignment of thermodynamic constraints in metabolic network models. BMC Bioinformatics. 2006;7 doi: 10.1186/1471-2105-7-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Joyce AR, et al. Experimental and Computational Assessment of Conditionally Essential Genes in Escherichia coli. J Bacteriol. 2006;188:8259–8271. doi: 10.1128/JB.00740-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henry CS, Broadbelt LJ, Hatzimanikatis V. Thermodynamics-Based Metabolic Flux Analysis. Biophys. J. 2007;92:1792–1805. doi: 10.1529/biophysj.106.093138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ederer M, Gilles ED. Thermodynamically feasible kinetic models of reaction networks. Biophysical Journal. 2007;92:1846–1857. doi: 10.1529/biophysj.106.094094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choi HS, Kim TY, Lee DY, Lee SY. Incorporating metabolic flux ratios into constraint-based flux analysis by using artificial metabolites and converging ratio determinants. J Biotechnol. 2007;129:696–705. doi: 10.1016/j.jbiotec.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 57.Hoppe A, Hoffmann S, Holzhutter HG. Including metabolite concentrations into flux balance analysis: thermodynamic realizability as a constraint on flux distributions in metabolic networks. BMC systems biology. 2007;1 doi: 10.1186/1752-0509-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guimera R, Sales-Pardo M, Amaral LA. A network-based method for target selection in metabolic networks. Bioinformatics. 2007;23:1616–1622. doi: 10.1093/bioinformatics/btm150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beg QK, et al. Intracellular crowding defines the mode and sequence of substrate uptake by Escherichia coli and constrains its metabolic activity. Proc Natl Acad Sci U S A. 2007;104:12663–12668. doi: 10.1073/pnas.0609845104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim PJ, et al. Metabolite essentiality elucidates robustness of Escherichia coli metabolism. Proc Natl Acad Sci U S A. 2007;104:13638–13642. doi: 10.1073/pnas.0703262104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Warren PB, Jones JL. Duality, thermodynamics, and the linear programming problem in constraint-based models of metabolism. Physical review letters. 2007;99:108101. doi: 10.1103/PhysRevLett.99.108101. [DOI] [PubMed] [Google Scholar]
- 62.Vazquez A, et al. Impact of the solvent capacity constraint on E. coli metabolism. BMC systems biology. 2008;2:7. doi: 10.1186/1752-0509-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Motter AE, Gulbahce N, Almaas E, Barabasi AL. Predicting synthetic rescues in metabolic networks. Mol Syst Biol. 2008;4:168. doi: 10.1038/msb.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gagneur J, Jackson DB, Casari G. Hierarchical analysis of dependency in metabolic networks. Bioinformatics. 2003;19:1027–1034. doi: 10.1093/bioinformatics/btg115. [DOI] [PubMed] [Google Scholar]
- 65.Mahadevan R, Schilling CH. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab Eng. 2003;5:264–276. doi: 10.1016/j.ymben.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 66.Burgard AP, Nikolaev EV, Schilling CH, Maranas CD. Flux Coupling Analysis of Genome-Scale Metabolic Network Reconstructions. Genome. Res. 2004;14:301–312. doi: 10.1101/gr.1926504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Almaas E, Kovacs B, Vicsek T, Oltvai ZN, Barabasi AL. Global organization of metabolic fluxes in the bacterium Escherichia coli. Nature. 2004;427:839–843. doi: 10.1038/nature02289. [DOI] [PubMed] [Google Scholar]
- 68.Nikolaev EV, Burgard AP, Maranas CD. Elucidation and structural analysis of conserved pools for genome-scale metabolic reconstructions. Biophys J. 2005;88:37–49. doi: 10.1529/biophysj.104.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Almaas E, Oltvai ZN, Barabasi AL. The Activity Reaction Core and Plasticity of Metabolic Networks. PLoS Comput Biol. 2005;1:e68. doi: 10.1371/journal.pcbi.0010068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barrett CL, Herring CD, Reed JL, Palsson BO. The global transcriptional regulatory network for metabolism in Escherichia coli attains few dominant functional states. Proc Natl Acad Sci U S A. 2005;102:19103–19108. doi: 10.1073/pnas.0505231102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Becker SA, Price ND, Palsson BO. Metabolite coupling in genome-scale metabolic networks. BMC Bioinformatics. 2006;7 doi: 10.1186/1471-2105-7-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Imielinski M, Belta C, Rubin H, Halasz A. Systematic Analysis of Conservation Relations in Escherichia coli Genome-Scale Metabolic Network Reveals Novel Growth Media. Biophys J. 2006;90:2659–2672. doi: 10.1529/biophysj.105.069278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beasley JE, Planes FJ. Recovering metabolic pathways via optimization. Bioinformatics. 2007;23:92–98. doi: 10.1093/bioinformatics/btl554. [DOI] [PubMed] [Google Scholar]
- 74.Shlomi T, Eisenberg Y, Sharan R, Ruppin E. A genome-scale computational study of the interplay between transcriptional regulation and metabolism. Mol Syst Biol. 2007;3:101. doi: 10.1038/msb4100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Almaas E. Optimal flux patterns in cellular metabolic networks. Chaos (Woodbury, N.Y. 2007;17:026107. doi: 10.1063/1.2737828. [DOI] [PubMed] [Google Scholar]
- 76.Sales-Pardo M, Guimera R, Moreira AA, Amaral LA. Extracting the hierarchical organization of complex systems. Proc Natl Acad Sci U S A. 2007;104:15224–15229. doi: 10.1073/pnas.0703740104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Notebaart RA, Teusink B, Siezen RJ, Papp B. Co-regulation of metabolic genes is better explained by flux coupling than by network distance. PLoS Comput Biol. 2008;4:e26. doi: 10.1371/journal.pcbi.0040026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mahadevan R, Lovley DR. The degree of redundancy in metabolic genes is linked to mode of metabolism. Biophys J. 2008;94:1216–1220. doi: 10.1529/biophysj.107.118414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Samal A, Jain S. The regulatory network of E. coli metabolism as a Boolean dynamical system exhibits both homeostasis and flexibility of response. BMC systems biology. 2008;2:21. doi: 10.1186/1752-0509-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pal C, Papp B, Lercher MJ. Horizontal gene transfer depends on gene content of the host. Bioinformatics. 2005;21(Suppl 2):ii222–ii223. doi: 10.1093/bioinformatics/bti1136. [DOI] [PubMed] [Google Scholar]
- 81.Pal C, Papp B, Lercher MJ. Adaptive evolution of bacterial metabolic networks by horizontal gene transfer. Nat Genet. 2005;37:1372–1375. doi: 10.1038/ng1686. [DOI] [PubMed] [Google Scholar]
- 82.Pal C, et al. Chance and necessity in the evolution of minimal metabolic networks. Nature. 2006;440:667–670. doi: 10.1038/nature04568. [DOI] [PubMed] [Google Scholar]
- 83.Schuetz R, Kuepfer L, Sauer U. Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol Syst Biol. 2007;3 doi: 10.1038/msb4100162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Varma A, Palsson BO. Metabolic Flux Balancing: Basic concepts, Scientific and Practical Use. Nat Biotechnol. 1994;12:994–998. [Google Scholar]
- 85.Edwards JS, Ramakrishna R, Schilling CH, Palsson BO. In: Metabolic Engineering. Lee SY, Papoutsakis ET, editors. Marcel Deker; 1999. [Google Scholar]
- 86.Kauffman KJ, Prakash P, Edwards JS. Advances in flux balance analysis. Curr Opin Biotechnol. 2003;14:491–496. doi: 10.1016/j.copbio.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 87.Fraser-Liggett CM. Insights on biology and evolution from microbial genome sequencing. Genome Res. 2005;15:1603–1610. doi: 10.1101/gr.3724205. [DOI] [PubMed] [Google Scholar]
- 88.Bro C, Regenberg B, Forster J, Nielsen J. In silico aided metabolic engineering of Saccharomyces cerevisiae for improved bioethanol production. Metab Eng. 2006;8:102–111. doi: 10.1016/j.ymben.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 89.Mahadevan R, et al. Characterization of Metabolism in the Fe(III)-Reducing Organism Geobacter sulfurreducens by Constraint-Based Modeling. Appl. Environ. Microbiol. 2006;72:1558–1568. doi: 10.1128/AEM.72.2.1558-1568.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Feist AM, Scholten JCM, Palsson BO, Brockman FJ, Ideker T. Modeling methanogenesis with a genome-scale metabolic reconstruction of Methanosarcina barkeri. Mol Syst Biol. 2006;2:1–14. doi: 10.1038/msb4100046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Breitling R, Vitkup D, Barrett MP. New surveyor tools for charting microbial metabolic maps. Nat Rev Microbiol. 2008;6:156–161. doi: 10.1038/nrmicro1797. [DOI] [PubMed] [Google Scholar]
- 92.Blattner FR, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 93.Kharchenko P, Vitkup D, Church GM. Filling gaps in a metabolic network using expression information. Bioinformatics. 2004;20(Suppl 1):I178–I185. doi: 10.1093/bioinformatics/bth930. [DOI] [PubMed] [Google Scholar]
- 94.Green ML, Karp PD. A Bayesian method for identifying missing enzymes in predicted metabolic pathway databases. BMC Bioinformatics. 2004;5:76. doi: 10.1186/1471-2105-5-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100050. 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mayr E. This is biology : the science of the living world. Belknap Press of Harvard University Press; Cambridge, Mass: 1997. [Google Scholar]
- 97.Suthers PF, et al. Metabolic flux elucidation for large-scale models using 13C labeled isotopes. Metab Eng. 2007;9:387–405. doi: 10.1016/j.ymben.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Janssen P, Goldovsky L, Kunin V, Darzentas N, Ouzounis CA. Genome coverage, literally speaking. The challenge of annotating 200 genomes with 4 million publications. EMBO Rep. 2005;6:397–399. doi: 10.1038/sj.embor.7400412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Riley M, et al. Escherichia coli K-12: a cooperatively developed annotation snapshot-2005. Nucleic Acids Res. 2006;34:1–9. doi: 10.1093/nar/gkj405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Allen TE, Palsson BO. Sequenced-Based Analysis of Metabolic Demands for Protein Synthesis in Prokaryotes. J theor Biol. 2003;220:1–18. doi: 10.1006/jtbi.2003.3087. [DOI] [PubMed] [Google Scholar]
- 101.Mehra A, Hatzimanikatis V. An algorithmic framework for genome-wide modeling and analysis of translation networks. Biophys J. 2006;90:1136–1146. doi: 10.1529/biophysj.105.062521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Thomas R, Paredes CJ, Mehrotra S, Hatzimanikatis V, Papoutsakis ET. A model-based optimization framework for the inference of regulatory interactions using time-course DNA microarray expression data. BMC Bioinformatics. 2007;8:228. doi: 10.1186/1471-2105-8-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee TI, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 104.Gianchandani EP, Papin JA, Price ND, Joyce AR, Palsson BO. Matrix Formalism to Describe Functional States of Transcriptional Regulatory Systems. PLoS Comput Biol. 2006;2:e101. doi: 10.1371/journal.pcbi.0020101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li C, et al. Computational discovery of biochemical routes to specialty chemicals. Chemical Engineering Science. 2004;59:5051–5060. [Google Scholar]
- 106.Holden C. Alliance launched to model E. coli. Science. 2002;297:1459–1460. doi: 10.1126/science.297.5586.1459a. [DOI] [PubMed] [Google Scholar]
- 107.Edwards JS, Ramakrishna R, Palsson BO. Characterizing the metabolic phenotype: a phenotype phase plane analysis. Biotechnol Bioeng. 2002;77:27–36. doi: 10.1002/bit.10047. [DOI] [PubMed] [Google Scholar]
- 108.Schuster S, Hilgetag C. On elementary flux modes in biochemical reaction systems at steady state. Journal of Biological Systems. 1994;2:165–182. [Google Scholar]
- 109.Schilling CH, Letscher D, Palsson BO. Theory for the systemic definition of metabolic pathways and their use in interpreting metabolic function from a pathway-oriented perspective. J theor Biol. 2000;203:229–248. doi: 10.1006/jtbi.2000.1073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
table