Single-Cell Mass Cytometry of Differential Immune and Drug Responses Across a Human Hematopoietic Continuum (original) (raw)
. Author manuscript; available in PMC: 2012 Feb 7.
Published in final edited form as: Science. 2011 May 6;332(6030):687–696. doi: 10.1126/science.1198704
Abstract
Flow cytometry is an essential tool for dissecting the functional complexity of hematopoiesis. We used single-cell “mass cytometry” to examine healthy human bone marrow, measuring 34 parameters simultaneously in single cells (binding of 31 antibodies, viability, DNA content, and relative cell size). The signaling behavior of cell subsets spanning a defined hematopoietic hierarchy was monitored with 18 simultaneous markers of functional signaling states perturbed by a set of ex vivo stimuli and inhibitors. The data set allowed for an algorithmically driven assembly of related cell types defined by surface antigen expression, providing a superimposable map of cell signaling responses in combination with drug inhibition. Visualized in this manner, the analysis revealed previously unappreciated instances of both precise signaling responses that were bounded within conventionally defined cell subsets and more continuous phosphorylation responses that crossed cell population boundaries in unexpected manners yet tracked closely with cellular phenotype. Collectively, such single-cell analyses provide system-wide views of immune signaling in healthy human hematopoiesis, against which drug action and disease can be compared for mechanistic studies and pharmacologic intervention.
Fluorescence-based flow cytometry has been fundamental to the discovery and definition of major and minor cell subsets of the immune system. Although the outline of hematopoiesis is generally understood (1), a comprehensive framework of its system-wide properties remains to be determined (2). Technological developments in flow cytometry and cell sorting [the introduction of new fluorophores, such as quantum dots (3)] have paralleled appreciation of the compartmentalization of function in the hematopoietic system and contributed to diverse fields, including immunology, stem cells (4, 5), HIV (6), cancer (7), transcription (8, 9), intracellular signaling (10, 11), apoptosis, cell cycle (12), and development of cytometry-based clinical diagnostics (13, 14). However, use of flow cytometry remains practically confined to the measurement of 6 to 10 simultaneous parameters (15). Analysis at the 11- to 15-parameter range is possible but limited by compensation needed to correct for spectral overlap that can create a source of confounding variability (16).
We used transition element isotopes not normally found in biological systems as chelated antibody tags in atomic mass spectrometric analysis of single cells to create a detailed response profile of the healthy primary human hematopoietic system with 34 simultaneously measured cellular parameters. This allowed us to take full advantage of the measurement resolution of mass spectrometry and apply it to single-cell analysis. Because the method is largely unhampered by interference from spectral overlap, it allows for the detection of considerably more simultaneous parameters than does traditional flow cytometry (17, 18). Combined with its quantitative nature, atomic mass spectrometry measurement creates a platform with which to conduct multiplexed measurement of single-cell biological parameters that can exhibit vastly different dynamic ranges during signaling or over time (such as signaling changes indicated by shifts in protein phosphorylation).
We simultaneously measured 34 parameters in each single cell in human bone marrow (BM) samples to provide an in-depth analysis of normal human hematopoietic and immunological signaling overlaid onto a detailed template of cell phenotype. Cell subset–specific signaling phenotypes of drug action in the face of clinically meaningful physiologic stimuli were localized to pathway and cell-specific boundaries, with examples in B cell signaling shown. These provide a system-wide view of signaling behaviors, expanding our view of drug action while allowing us to limit the functions that certain drugs might have on complex tissues. Given that this technology can reasonably be expected to allow for as many as 100 parameters per cell (18, 19), it affords an opportunity to increase our understanding of cell type–specific signaling responses in complex, distributed organs such as the immune system.
Performance assessment of mass cytometry
The workflow for mass cytometry is comparable with that of fluorescence flow cytometry (Fig. 1A). Antibodies coupled to distinct, stable, transition element isotopes were used to bind target epitopes on and within cells. Cells, with bound antibody-isotope conjugates, were sprayed as single-cell droplets into an inductively coupled argon plasma (created by passing argon gas through an induction coil with a high radio-frequency electric current) at approximately 5500 K. This vaporizes each cell and induces ionization of its atomic constituents. The resulting elemental ions were then sampled by a time-of-flight (TOF) mass spectrometer and quantified. The signal for each transition element isotope reporter was integrated as each cell’s constituent ions reached the detector. Currently, TOF sampling resolution enables the measurement of up to 1000 cells per second. We compared mass cytometry with conventional nine-parameter fluorescence flow cytometry in analysis of cytokine signaling through responses in human peripheral blood mononuclear cells (PBMCs) from two healthy donors (Fig. 1, B to E, and fig. S1). Seven surface antigens (CD3, CD4, CD8, CD45RA, CD56, CD20, and CD33) and two intracellular phosphoprotein epitopes [phos-phorylated signal transducer and activator of transcription 3 and 5 (pSTAT3 and pSTAT5)] were measured by means of fluorescence cytometry on two human PBMC samples treated with interleukin-2 (IL-2), IL-6, IL-10, granulocyte-monocyte colony stimulating factor (GM-CSF), or interferon-α (IFNα) to measure cytokine-mediated signaling responses in specific cell subsets. In traditional flow cytometry, forward scatter (FSC) and side scatter (SSC) measurements of laser light are used to detect the presence of a cell and to “trigger” the electronics in order to collate information as a cell “event” (the window of time during which a cell is measured). Because FSC and SSC are not currently implemented on the CyTOF platform, alternative parameters providing analogous utility were included to assist with the discrimination of single-cell events: (i) an antibody to the surface epitope CD45 (expressed on most cells measured in this study), (ii) a metal-encoded DNA intercalator to identify nucleated cells (20), and (iii) a derived parameter (“cell length”) indicating the duration of each cell’s measurement window (18).
Fig. 1.
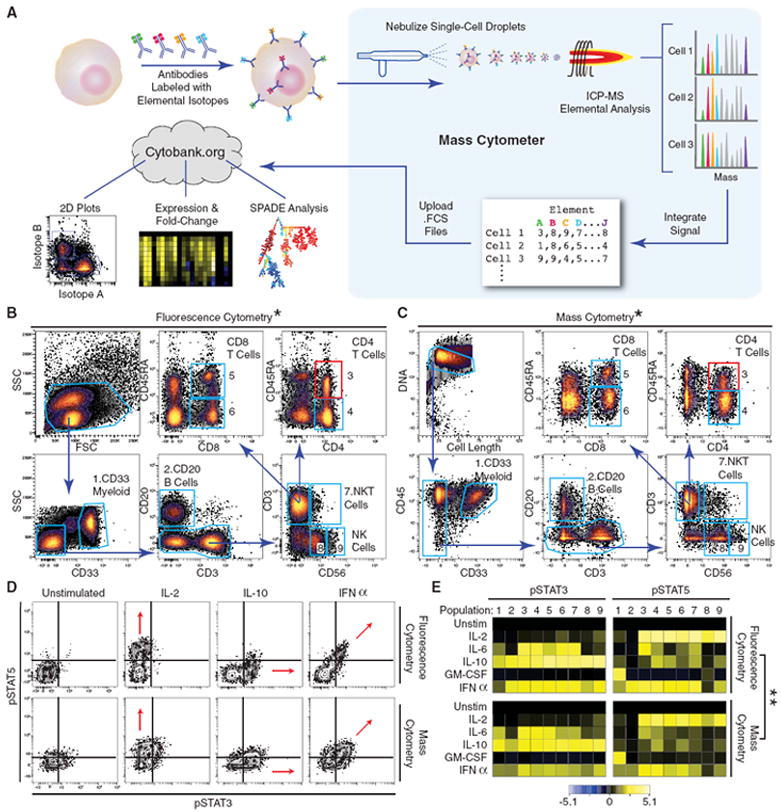
Mass cytometry profiling of immune cell response patterns. (A) Work-flow summary of mass cytometry analysis. Cells are stained with epitope-specific antibodies conjugated to transition element isotope reporters, each with a different mass. Cells are nebulized into single-cell droplets, and an elemental mass spectrum is acquired for each. The integrated elemental reporter signals for each cell can then be analyzed by using traditional flow cytometry methods as well as more advanced approaches such as heat maps of induced phosphorylation and tree plots. (B and C) Representative antibody surface-staining results and cell population definitions (“gating”) for (B) fluorescence and (C) mass cytometry analysis of fixed PBMCs from the same donor. Replicate analysis of a second donor is provided in (21) (Fig. S1A and S1B). *Pearson correlation between frequencies measured by fluorescence or mass cytometry, including both donors (r = 0.99, P < 0.000001, two-tailed t test) (table S1 and fig. S1C). (D) Induction of STAT3 and 5 phosphorylation by various ex vivo stimuli in naive CD4 +CD45RA+ T cells [(B) and (C), red boxes] as measured by (top) fluorescence and (bottom) mass cytometry. Red arrows indicate the expected shift along the STAT phosphorylation axes. (E) Heatmap summary of induced STAT phosphorylation in immune populations from the PBMC donor defined in (B) and (C) [column headers refer to blue polygons in (B) and (C)]. Responses to the indicated stimuli in each row were measured by (top) fluorescence and (bottom) mass cytometry. Color scale indicates the difference in log2 mean intensity of the stimulated condition compared with the unstimulated control. Signaling responses of a second donor are provided in (21) (fig. S1D). **Pearson correlation between signaling induction measured by fluorescence or mass cytometry, including both donors [pSTAT3: _r_ = 0.92; _P_ < 0.000001, two-tailed _t_ test (fig. S1E); pSTAT5: r = 0.89, P < 0.000001, two-tailed t test] (figs. S1E and S1F).
Fluorescence (Fig. 1B and fig. S1A) and mass (Fig. 1C and fig. S1B) cytometry analysis provided comparable results when analyzed via traditional dot plots (fig. S2). Pertinent qualities, such as reduced CD45RA expression on CD4+ T cells relative to that on CD8+ cells, were reproduced between platforms (Fig. 1, B and C). Despite use of different metrics for identifying cell events, both platforms yielded quantitatively similar frequencies (P < 0.000001) for 12 manually gated cell populations in the parallel analysis of two separate door samples (fig. S1C and table S1). Patterns of specific induction of STAT protein phosphorylation within the CD4+CD45RA+ T cell population demonstrated that both platforms could equivalently detect pSTAT3, pSTAT5, and dual pSTAT3-pSTAT5 responses to IL-10, IL-2, and IFNα, respectively (Fig. 1D). One qualitative difference between the two platforms was the mathematical correction required to address spectral overlap in the fluorescence data (termed “compensation”), a procedure not required with the atomic mass spectrometer. A second major distinction is the absence of cell-dependent background signal in the mass cytometry data. Thus, although laser-based flow cytometry detects signals from cellular autofluorescence, nonactivated cells had mass cytometric phosphoprotein intensities near zero, indicating very little background antibody binding. This manifests in atomic mass spectrometry as a narrow grouping of cell events at the low end of the dot plot axes. Qualitatively and quantitatively (P < 0.000001) similar patterns were revealed by means of fluorescence-based flow cytometry or mass cytometry in terms of magnitude of the pSTAT3 and pSTAT5 responses in cell populations across two healthy peripheral blood samples (Fig. 1E and fig. S1, D to F). An overview of the antibody quality control with testing on cell lines, human PBMCs, and bone marrow is shown in fig. S11. Taken together, mass cytometry and traditional fluorescence based approaches can produce results with equivalent informational value.
Organization and analysis of high-dimensional single-cell data
Taking advantage of the increased dimensionality of mass cytometry, we prepared a set of reagents to capture a system-wide view of immune cell types from a replicate analysis of bone marrow mononuclear cells from two healthy human donors. Thirty-one distinct transition element isotopes were used to label two antibody-staining panels for the study of healthy human bone marrow mononuclear cells. [Data are publicly available at Cytobank (www.cytobank.org/nolanlab). An “immunophenotyping” panel was designed that monitored 13 “core” surface markers and 18 subset-specific cell-surface markers to allow identification of human hematologic cell types. A “functional” panel contained the 13 core surface markers and also 18 intracellular epitopes that reflect intracellular signaling states, such as phosphorylation status of kinase substrates (21). These complementary panels allowed simultaneous biochemical analysis of intracellular signaling in rare and diverse cell subsets that were identified through in silico merging of the data. Intracellular signaling responses were determined by treating cells ex vivo with modulators such as cytokines, small molecules, or combinations thereof. Perturbation analysis has proven useful in causality determinations for signaling at the single-cell level (11, 22-25) and was applied here to enable cell subset–specific response profiles. An additional three parameters—a DNA intercalator, cell length, and a cell viability dye (21)—were included in the analysis panels, creating a total of 34 parameters in each. With an overlap of 13 core surface antibodies between the two analysis panels and the three shared additional cell features, a combined total of 52 unique single-cell parameters were measured. The resulting single-cell data set of bone marrow cells captured a snapshot of the cell types present and their corresponding regulatory signaling responses throughout development from early human hematopoietic progenitors to lineage-committed cells.
A central dogma of immunology is that cells at different stages of maturation can be characterized by the expression of unique sets of proteins on the cell surface. Such “cluster of differentiation” (CD) markers are routinely used for flow cytometric identification of cell populations. Although it is convenient to think of cells in different stages of development as having distinct, regimented profiles, hematopoiesis frequently manifests as a continuum of CD marker expression connecting the cellular lineage stages (26). Although cells might pause at recognized stages of development to which we ascribe certain phenotypes, cells also pass through transient intermediate states that connect parent populations to their progeny. As they proceed from one stage of development to the next, CD marker sets rise and fall in accordance with programmed differentiation and environmental contexts. A conventional display of the relationships between the 31 cell surface markers measured here on human bone marrow would require greater than 450 biaxial dot plots (fig. S3), making a comprehensive interpretation of the underlying cellular progression unwieldy, if not impossible.
We hypothesized that the inherent similarity of cell stages and continuity of the transitions between cell differentiation states could be used to organize high-dimensional data into ordered, continuous clusters of similar cell phenotypes that, when projected on a two-dimensional (2D) plane, would convey the relatedness of these cells in a higher dimensional space. We leveraged progressive changes in CD marker expression to organize bone marrow cells in an unsupervised manner, creating a tree-like scaffold for visualization of high-dimensional intracellular signaling behaviors in various cell types present during hematopoietic development in the bone marrow (27, 28). To accomplish this, we used SPADE (spanning-tree progression analysis of density-normalized events), a density normalization, agglomerative clustering, and minimum-spanning tree algorithm to distill multidimensional single-cell data down to interconnected clusters of rare, transitional, and abundant cell populations, which were organized and displayed as a 2D tree plot (Fig. 2A). Such a tree plot from healthy bone marrow represented the clustered expression of the cell-surface antigens that were used to build the tree in 13-dimensional space on the basis of the core surface markers conserved between our two 34-parameter analysis panels (CD3, -4, -8, -11b, -19, -20, -33, -34, -38, -45, -45RA, -90, and -123) (Fig. 2B). Each node of the plot encompasses a cluster of cells that were phenotypically similar in the 13-dimensional space defined by the core surface markers. The approach uses a minimum-spanning tree algorithm, in which each node of cells is connected to its “most related” node of cells as a means to convey the relationships between the cell clusters. The number of nodes and ultimately their boundaries is driven by a user-definable value (21). Each node describes an _n_-dimensional boundary encompassing a population of phenotypically similar cells. When connected via the minimum spanning tree, this provides a convenient approach to map complex _n_-dimensional relationships into a representative 2D structure.
Fig. 2.
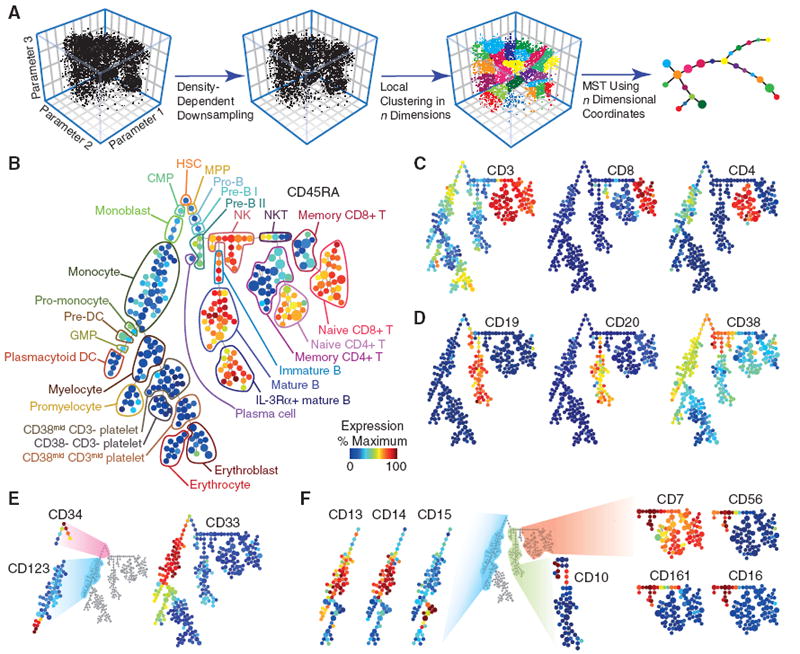
SPADE links related immune cell types in a multidimensional continuum of marker expression. (A) Summary of SPADE analysis. Single-cell data are sampled in a density-dependent fashion so as to reduce the total cell count while maintaining representation of all cell phenotypes. Neighboring cells are then grouped by unsupervised hierarchical clustering. Resulting nodes (defined as those cells within a boundary of an _n_-dimensional hull) are then linked by a minimum-spanning tree, which is flattened for 2D display.(B) Immunophenotypic progression in healthy human bone marrow. A tree plot was constructed by using 13 cell-surface antigens in healthy human bone marrow. 18 additional intracellular parameters were acquired concurrently but excluded from tree construction. The size of each circle in the tree indicates relative frequency of cells that fall within the 13-dimensional confines of the node boundaries. Node color is scaled to the median intensity of marker expression of the cells within each node, expressed as a percentage of the maximum value in the data set (CD45RA is shown). Putative cell populations were annotated manually (table S2) and are represented by colored lines encircling sets of nodes that have CD marker expression emblematic of the indicated subset designations. (C) Overlaid expression patterns of CD3, CD8, and CD4. Three markers, along with CD45RA (B), were used in clustering that helped define T cell lineages. Color scale is as described in (B). (D) Overlaid expression patterns of CD19, CD20, and CD38. Three markers were used in clustering that helped define B cell lineages. Color scale is as described in (B). (E) Overlaid expression patterns of CD34, CD123, and CD33. Three markers were used in clustering that helped define myeloid and progenitor cell lineages. Color scale is as described in (B). (F) Overlaid expression of complementary surface markers from a staining panel with 18 additional surface markers (fig. S4) by using the 13 core surface markers as landmarks (21). Overlaid expression patterns are shown for eight complementary surface markers that helped to further define the myeloid (CD13, CD14, and CD15), B cell (CD10), and NK/T cell (CD7, CD56, CD161, and CD16) portions of the SPADE representation. These markers were not used for tree construction. Color scale is as described in (B).
As such, related nodes could be mapped into traditionally described immunological cell populations as determined by the localized expression patterns of 13 directly measured surface markers (Fig. 2, B to E, and fig. S4A). A summary of evidence supporting these annotations and boundaries can be found in table S2. For instance, T cell populations were annotated on the far right branch of the tree plot based on the high expression of CD3 (Fig. 2C, bright red). The T cell markers CD4 and CD8 were expressed in mutually exclusive clusters but overlapped with CD3 expression. Density normalization enabled the display of rare cell types, such as CD34+ progenitor cells, in the same space as the more abundant differentiated cell types (Fig. 2E). The unsupervised organization of phenotypically related cell types into adjacent branches, such as CD4 and CD8 Tcells (Fig. 2C), mature and immature B cells (Fig. 2D), and different clusters of myeloid cells (Fig. 2E) collectively illustrates that the algorithmic ordering of surface marker similarity can objectively organize cell types into physiologically relevant compartments.
Although they were not used in the tree-building step, the 18 additional surface markers from the “immunophenotype”-staining panel were used to confirm and refine cell subset annotations (Fig. 2F and fig. S4B). These markers were overlaid in an unsupervised fashion onto the existing tree by assigning each cell from the immunophenotyping experiment to whichever node contained analogous cells from the functional data set according to the expression of the shared 13 core surface markers in the registration space. The accuracy of this automated overlaying approach is supported by the agreement of multiple natural killer (NK), monocyte, and B cell markers that localized to the appropriate cell populations (Fig. 2F), even though they were not used to direct the tree’s original organization. Although the tree structure derived from bone marrow data recapitulates many features of hematopoietic organization and relatedness, it is interpreted here as a map of the phenotypic relationships between diverse cell types and is not meant to imply a developmental hierarchy. Indeed, even measuring a large number of cells in a single tissue will fail to capture some developmental transitions, including (i) rapid activation (release of cytoplasmically sequestered receptors) (29); (ii) uneven surface marker partitioning during asymmetric cell division (30); and (iii) organ-specific development outside the assayed organ (maturation of T cells in the thymus). In this bone marrow data set, several well-defined cell types (such as T, NK, B, and monocyte) provide landmarks for the organization of the tree and give context to the nodes encompassing transitional and less-understood cell types. Ultimately, this approach enabled visualization of 34-dimensional bone marrow data in an intuitive graphical format. Although the algorithm over-segregated some cell types into redundant contiguous clusters, this approach has several advantages that complemented the complexity of this data set: (i) increased resolution captured unexpected and transitional cell types that escape standard classification strategies; (ii) Unsupervised analysis helped overcome the bias of subjective gating; and (iii) _n_-dimensional algorithms leveraged the multi-parameter mass cytometry data to define cell types on the basis of previously unappreciated, subtle differences in surface expression. Although we used stochastically selected “seed” cells to initiate the tree generation along with local similarity clustering and minimum-spanning trees, the approach is amenable to incorporation of other more deterministic partitioning approaches that might allow for other standardized tree structure formation.
Ex vivo analysis of healthy human bone marrow signaling
Historically, by detailing immune functions in vivo and in vitro a model of specialized cell types in immunology and hema-topoiesis were mapped primarily on the basis of expressed cell surface antigens—many of which were codified by using single-cell analysis and fluorescence-based cytometry (31-34). Because cell-surface proteins represent only a small proportion of the repertoire of gene products governing cell behavior, intracellular proteins (33) are also critical in defining cell types. Because surface and intracellular molecules work together in concert to support different cellular roles, it might be expected that proteins governing specialized immunological cell functions (T cell receptor, B cell receptor, or cytokine receptors) are modulated in a coordinated manner as cells transit developmental pathways from stem cell precursors to differentiated endpoints.
We monitored 13 surface markers to identify immune cell types and 18 intracellular epitopes in order to interrogate intracellular signaling biology in healthy human bone marrow. We examined the signaling dynamics of these 18 intracellular markers in response to 13 ex vivo stimulation conditions (such as IL-7 or GM-CSF), including those shown to have prognostic value in leukemia, lymphoma, and myeloproliferative disorders [such as granulocyte colony stimulating factor (G-CSF)] (10, 35-37). Cell populations were first defined on the basis of conventional surface expression gates, ultimately identifying 24 immunological populations in human bone marrow (fig. S5). The induced intracellular signaling responses (changes in phosphorylation state) in these populations, as compared with those of an untreated control, were summarized as a heat-map (Fig. 3A). Unsupervised, hierarchical clustering of the phosphorylation responses allowed distinction of biologically related cell types (T cell subsets) by their signaling behavior alone, demonstrating that signaling capacities are closely tied to cellular lineage (fig. S6). Several canonical signaling responses that mapped to manually determined cell types are shown in Fig. 3B. These extremely specialized responses, such as the tight restriction of IL-7–mediated pSTAT5 responsiveness in T cells (Fig. 3B, arrow 4) (38) or lipopolysaccharide (LPS)–stimulated phosphorylation of the mitogen-activated protein kinase (MAPK) p38 (p-p38) responsiveness in monocytes (Fig. 3B, arrow 5) (39), suggest the existence of correlations between signaling events and surface marker–defined boundaries, thus presenting an opportunity to establish a unified view of immune signaling during hematopoiesis.
Fig. 3.
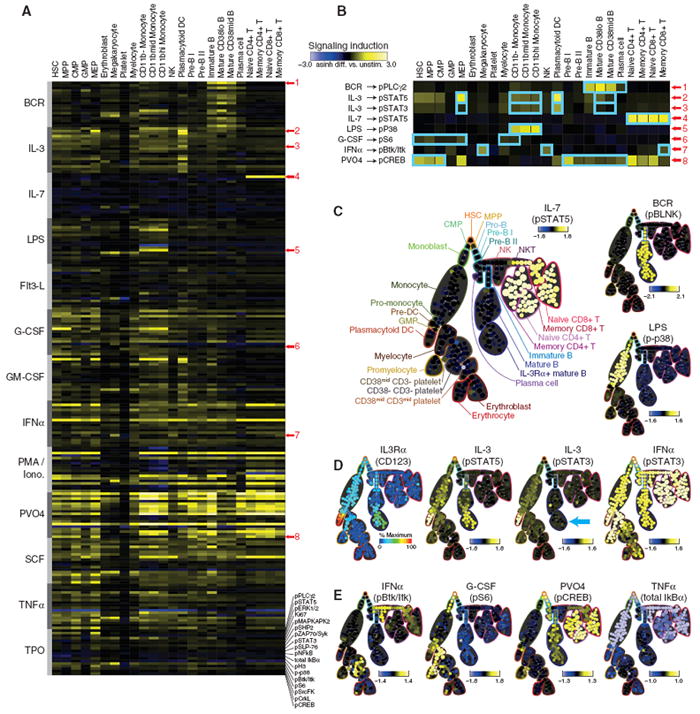
Signaling functions mark developmental transitions in hematopoietic progression. (A) A heatmap summary, ordered developmentally by cell type and stimulation condition, of the status of 18 intracellular functional markers in cells treated with 1 of 13 biological and chemical stimuli. (Left) Abbreviations refer to recombinant human proteins, except BCR, B cell receptor cross-linking; LPS, lipopolysaccharide; PMA/Iono, phorbol-12-myristate-13-acetate with ionomycin; and PVO4, pervanadate. Single-cell data from healthy human bone marrow were manually divided (“gated”) into 24 conventional cell populations (fig. S5) according to 13 surface markers and DNA content. Signaling induction was calculated as the difference of inverse hyperbolic sine (arcsinh) medians of the indicated ex vivo stimulus compared with the untreated control for each manually assigned cell type (21). Each row within a given stimulus group (gray bars) indicates the signaling induction of 1 of 18 intracellular functional markers (bottom). A subset of conditions (red numbers) was highlighted for further discussion in (B). (B) Magnified view of the conditions marked in (A). A subset of these signaling responses (blue boxes) are shown as SPADE plots [(C) to (E)] to investigate correlations between signaling function and differences in immunophenotype as discussed in the text. (C) Canonical, cell type–specific signaling functions. Stimulation by IL-7, BCR, or LPS each induced phosphorylation of STAT5 in T cells, BLNK (SLP-65) [detected with an antibody raised against pSLP-76 (21)] in B cells, and p38 MAPK in monocytes, respectively. Signaling induction for each node in the SPADE diagram was calculated as the difference of arcsinh median intensity of the indicated ex vivo stimulus compared with the untreated control. (D) Correlation of IL-3–mediated induction of pSTAT3 and pSTAT5 with IL-3Ra expression [(Left) color scale as described in Fig. 2B] in myeloid and B cells. The B cell population that did not phosphorylate STAT3 in response to IL-3 stimulation is indicated (blue arrow). All nucleated cell subsets, including IL-3Rα+ B cells, exhibited pSTAT3 induction in response to IFNα stimulation. Signaling induction calculated as in (C). (E) Examples of phosphorylation responses that paralleled immunophenotypic progression identified by the SPADE algorithm. Changes in Btk/Itk, S6, CREB phosphorylation, and total IκBα are shown in response to IFNα, G-CSF, PVO4, and TNFα, respectively. Signaling induction is calculated as in (C).
With ~104 signaling observations (Fig. 3A and fig. S10A) for each replicate bone marrow, it was necessary to filter the data set in order to arrive at the most significant and potentially novel observations. Using a one-sample _t_-test, over 500 observations were observed with a Bonferroni-adjusted significance of P < 0.05 in each replicate bone marrow for a total of 860 unique responses (fig. S7 and table S3). Of the 248 observations overlapping between patient marrows, 28 belonged exclusively to cells residing in the human hematopoietic progenitor cell compartment [hematopoietic stem cells (HSCs), multipotent progenitors (MPPs), granulocyte/macrophage progenitors (GMPs), and megakaryocyte-erythroid progenitors (MEPs)], including G-CSF induction of pSTAT3 in the most primitive cell types, HSC and MPP (40). This same signaling behavior correlated with negative prognosis in acute myeloid leukemia (10), suggesting that, as in the case of other malignancies, there may be a selective advantage for cells to mimic the properties of their most primitive counterparts.
For a more objective and fine-grained view of these cell type–specific responses, free of the biases of conventional 1D and 2D surface marker categorization, we overlaid the signaling behavior of the 18 functional epitopes on the tree structure using a similar approach as described for the immunophenotype staining panel (Fig. 2), allowing the intracellular signaling status to be visualized on the previously annotated tree structure (Fig. 3C). Nodes were colored according to the magnitude of the difference in their median responses relative to the untreated control. This effectively eliminated the subjectivity of manual classification and improved the resolution of the heatmap (Fig. 3A), separating the 24 manually assigned cell types into 282 logically connected nodes of phenotypically distinct, but locally similar, cell clusters.
The stimuli that corresponded closely with cell types identified manually in the heatmap also exhibited appropriately specific responses when overlaid on the tree structure—specifically, IL-7/pSTAT5 in T cells, B cell receptor (BCR)/phosphorylated B cell linker protein (pBLNK) exclusively in immature and mature B cells, and LPS/p-p38 restricted to the monocyte compartments (Fig. 3C), with the latter corresponding to the expression of the LPS co-receptor (CD14) (Fig. 2F). A complete set of the effects of 13 stimuli on 18 different functional markers is presented as tree plots (fig. S8) along with a confirmatory analysis of a second bone marrow (fig. S9).
With multiple matching canonical signaling pathways to validate the approach, we examined the data set for previously unidentified or unexpected signaling behaviors. For example, although pSTAT5 activation by IL-3 (Fig. 3D) was commensurate with IL-3Rα (CD123) expression levels (Fig. 3D) in myeloid cells, IL-3–mediated pSTAT3 activation was unexpectedly absent in mature B cells in spite of abundant presence of the receptor (Fig. 3D, blue arrow). This suggests that mature B cells share some, but not all, IL-3 signaling mechanisms with myeloid cell types.
Other responses, such as phosphorylation of the protein tyrosine kinases Btk and Itk mediated by IFNα or ribosomal protein S6 by G-CSF, were less tightly confined, exhibiting a range of activity that spanned multiple cell types (Fig. 3E). Yet other responses showed a signaling “gradient,” as exemplified by pervanadate (PVO4)–mediated disruption of the kinase/phosphatase balance upstream of the adenosine 3′,5′-monophosphate (cAMP) response element–binding protein (CREB) transcription factor. A gradient of responses, highest in HSCs, decreased gradually along the path of B cell maturation (Fig. 3E). A range of NFκB signaling responses, as measured by monitoring total IκBα levels, were observed across monocyte, NK and T cell subsets following TNFα stimulation (Fig. 3E, light blue nodes). As in the CREB response to PVO4 described above (Fig. 3E), the consistency of responses within the different T cell subsets suggests tightly regulated differences in signaling molecules that underlie the discrete functional roles of these related cell types. Together, these varied signaling responses across algorithmically defined partitions dictated solely by surface marker immunophenotype imply the existence of different classes of developmental transition points: (i) precise transitions, which are characterized by coordinated changes in cell signaling, such as the IL-7/pSTAT5 response in T cells and the LPS/p-p38 response in monocytes (Fig. 3C), and (ii) continuous developmental progressions, which are characterized by gradual gain or loss of expression of certain kinases or phosphatases, as highlighted by PVO4/pCREB (Fig. 3E) in B cells (28). The latter is indicative of fine-grained changes in regulatory architecture that track with immuno-phenotype within conventionally defined hema-topoietic compartments and provides an opportunity to explore the mechanisms that define these distinctive regulatory phenomena.
Confirmation of progression-specific signaling in hematopoietic development
To investigate more closely the signaling transitions and the observed signaling heterogeneity inside seemingly homogeneous cell compartments, we mapped changes in B cell signaling as they coincided with the progression of B cell maturation. Using the cell events comprising the Pre-B II through IL-3Rα+ mature B cell subsets defined in the SPADE plot (Fig. 4A) (see table S2 for surface marker definitions of B lineage stages), we used an independent statistical method—principal component analysis (PCA)—to distill the dimensionality of 13-parameter surface marker data to a single linear “progression axis.” Principal component analysis found that the first principal component—the axis of greatest variation in the data (explaining 23% of the variation)—followed known markers of B cell maturation. This progression axis was primarily defined by increasing CD20 expression and decreasing CD38 expression, with smaller contributions from increasing CD45RA (Fig. 4B). The match between the first principal component and the established sequence of B cell development (26, 41) is further supported by additional markers, such as the increase in CD19 and CD123 along this axis of progression. Projecting cells onto this progression trajectory defines a continuum of cells, rather than distinct subsets (Fig. 4A, gray line).
Fig. 4.
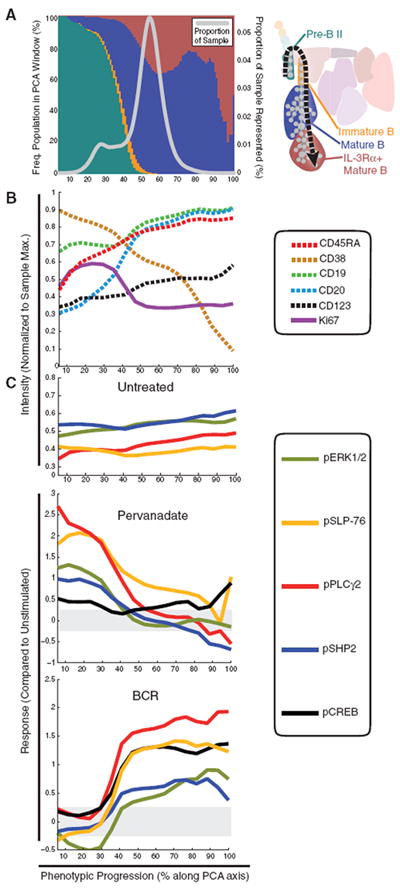
PCA confirms that cellular signaling potentially tracks with the immunophenotypic continuum in B cell subsets. (A) Using the SPADE representation (right), cells assigned to pre-B II, immature B, mature B, and IL-3Rα+ mature B cell populations were selected for PCA in 13 dimensions defined by the core immunophenotypic markers used in both panels. The relative frequencies of the four B cell populations are shown as stacked bars in 1% windows along the phenotypic progression axis (colors correspond to key at right); the number of cells in each window is expressed as a proportion of the sample subjected to PCA (gray line). (B) The measured intensities of five immunophenotypic markers (CD45RA, CD19, CD20, CD38, and CD123) along the phenotypic progression axis. These markers captured the majority of the phenotypic changes observed here during B cell maturation. Intracellular Ki67 expression, an indicator of cellular proliferation, was not used in defining the PCA axis but was among the 18 functional markers that were measured concurrently at thesingle-cell level.(C) Phosphorylation of ERK1/2, SLP-76(BLNK/SLP-65), PLCγ2, CREB, and SHP2 overlaid on the PCA progression axis. These and other functional epitopes were not used in the PCA axis construction. The top plot displays the basal levels (untreated) of these phosphorylated epitopes in the untreated sample. Subsequent plots display induced changes in phosphorylation in response to PVO4 and B cell receptor cross-linking relative to the untreated control.
Although no intracellular parameters were used in defining the PCA progression, many intracellular markers demonstrated a smooth and gradual change along this axis of progression, providing the opportunity to explore how signaling changes during B cell development. Abundance of the cell cycle–associated nuclear protein Ki67 at basal state revealed a transition point characterized by a peak in the amount of Ki67 (cycling cells) followed by a concomitant increase in CD20 expression (Fig. 4B). The inverse relationship between these parameters suggests that the continuous production of B cells in bone marrow is paused when CD20 is gained, thus coinciding with the exit of immature B cells from the marrow (42). The dramatic increase in the abundance of CD20+ B cells shortly after this transition point (Fig. 4, A and B) may indicate the presence of a reservoir of dormant CD20+ mature B cells in bone marrow awaiting antigen activation (43).
Overlaying the basal (untreated) intensities of phosphorylated extracellular signal–regulated kinase 1 and 2 (pERK1/2), Src homology protein tyrosine phosphatase 2 (SHP2), SLP-76/BLNK (SLP-65), pPLCγ2, and CREB onto the PCA axis revealed that basal phosphorylation of these molecules at the measured sites was relatively constant across differentiating B cell types (Fig. 4C). However, induced phosphorylation of these sites by PVO4 and BCR (Fig. 4C) clarified the heterogeneity of PLCγ2 phosphorylation observed after PVO4 treatment in the tree plots (Fig. 5B), revealing instead a gradual decline that tracked with maturation. An opposing trend was observed in pPLCγ2 responsiveness to BCR activation, which exhibited an increase that closely tracked with CD20 expression (Fig. 4C). The same trend was observed for pCREB pERK1/2, pSLP-76, and pSHP2, suggesting a parallel and coordinated change in signaling pathways. In contrast, mature B cells lacked a PVO4-sensitive mechanism, but the same set of signaling mediators appeared to be repurposed in a coordinated phosphorylation response to BCR activation (Fig. 4C). These results suggest that in pre-B II and immature B cells, but not mature B cells, pPLCγ2 activation is increased by an upstream tyrosine kinase with tightly regulated activity. This example supports the existence of parallel signaling mechanisms affecting these nodes (SLP-76, SHP2, PLCγ2, and ERK1/2) that gradually switches along with the expression continuums determined by external immunophenotypic markers. Together, these parallel continuums could define each cell’s functional role within the greater hematopoietic system.
Fig. 5.
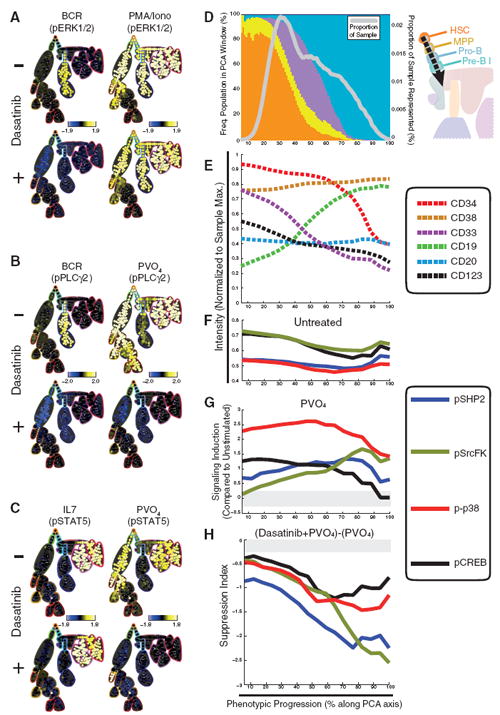
Multiplexed mass cytometry analysis reveals diverse signaling dynamics in response to the kinase inhibitor agent dasatinib. (A) SPADE plots of exemplary cell type–specific inhibitory effects of dasatinib. Phosphorylation of ERK1/2 was sensitive to dasatinib when induced with BCR cross-linking but not when induced with PMA/ionomycin. Signaling induction for each node in the SPADE diagram was calculated as the difference of arcsinh median of the indicated ex vivo stimulus compared with the untreated control. (B) T lymphocytes exhibited STAT5 phosphorylation in response to IL-7 in the presence of dasatinib with similar magnitudes as the response observed without drug. PVO4 induction of STAT5 phosphorylation was inhibited by dasatinib in all but plasmacytoid dendritic cells. Calculated as in (A). (C) Blymphocytes exhibited specific PLCγ2 phosphorylation in response to receptor cross-linking that was completely abolished in the presence of dasatinib. PLCγ2 phosphorylation was relatively large in all but B lymphocyte lineages in the presence of PVO4 but was inhibited completely by dasatinib treament in all cells. Calculated as in (A). (D) Using the SPADE representation, cells corresponding to HSC, MPP, pro-B, and pre-B I cell populations were selected for PCA of the 13 core immunophenotypic markers. The relative frequencies of the four progenitor cell populations are shown as stacked bars in 1% windows along the phenotypic progression axis (colors correspond to key at right); the number of cells in each window is expressed as a proportion of the sample subjected to PCA (gray line). (E) The measured intensities of six immunophenotypic markers (CD34, CD33, CD19, CD20, CD38, and CD123) along the progression axis. These markers captured the majority of the phenotypic changes observed here during progenitor cell maturation. (F) Basal (untreated) phosphorylation levels of p38, SrcFK, CREB, and SHP2 overlaid on the phenotypic progression axis. These and other functional epitopes were not used in the PCA axis construction. (G) Induced changes in phosphorylation of p38, SrcFK, CREB, and SHP2 in response to PVO4 compared with untreated control. Signaling induction is calculated as in (A). (H) Suppression of normal PVO4 response by dasatinib. Suppression index is calculated as the signaling induction by PVO4 with dasatinib pretreatment, minus the signaling induction by PVO4 alone.
Effect of pharmacologic kinase inhibition on normal hematopoietic signaling
Having established a baseline of healthy signaling responses to a panel of stimuli, we examined cell type–specific pharmacologic effects of some well-characterized kinase inhibitors. These included the Janus kinase (JAK) I inhibitor and MAPK kinase (MEK) inhibitor U0126. Predictably, when combined respectively with G-CSF and PMA/Ionomycin treatments of human BM (figs. S8 and S9) reliable and specific inhibition, respectively, of STAT3 and ERK1/2 phosphorylation are observed, which is consistent with previously reported observations that used conventional single-cell analysis platforms (44). Although interesting results were obtained with these inhibitors, we expanded to focus on dasatinib, a clinically relevant small-molecule kinase inhibitor. Dasatinib was originally introduced as a second-line BCR-ABL kinase inhibitor for imatinib-resistant chronic myelogenous leukemia (CML) (45). Unlike imatinib, dasatinib is estimated to inhibit over 100 kinases besides Abl—particularly, Src family kinases (SrcFKs) (46). This promiscuity is credited for dasatinib’s therapeutic efficacy in other malignancies (47). However, both malignant and healthy cells must integrate the effects of a drug with myriad other environmental inputs. We postulated that assessing drug activity in the presence of ex vivo stimuli may reveal interactions that underlie side effects for patients or expose new opportunities for pathway intervention.
Using the same healthy human bone marrow, we selected a panel of ex vivo stimuli that induced signaling in cell subsets either broadly [phorbol 12-myristate 13-acetate (PMA)/ionomycin and PVO4] or specifically (Flt3-L, IL-7, and BCR), after 30 min of pretreatment with dasatinib. The results showed several examples of pathway-specific inhibition that fit with expected roles of dasatinib (fig. S10). For instance, activation of pERK1/2 in immature and mature B cells through BCR cross-linking was completely suppressed by dasatinib (Fig. 5A), most likely through inhibition of Lyn, a critical SrcFK downstream of the B cell receptor (48). In contrast, PMA/ionomycin–mediated activation of pERK1/2 was unaffected by dasatinib, thus confirming the observation that protein kinase C (PKC) signaling is mediated by dasatinib-insensitive kinases (Fig. 5A) (49).
Phosphorylation patterns of PLCγ2 after PVO4 induction (Fig. 5B) were similar to PMA/ionomycin induction of pERK1/2 in the absence of dasatinib. However, dasatinib had a uniformly suppressive effect upon PVO4 induction of PLCγ2 in all cell types (Fig. 5B), whereas it minimally inhibited induction of Erk1/2 upon PMA/ionomycin stimulation (Fig. 5A). Thus, in contrast to the dasatinib-insensitive PKC pathway described above (Fig.5A) the PVO4-sensitive cascade upstream of PLCγ2 was inhibited by dasatinib in all cell types, including platelets. This result may reconcile a clinical observation of platelet dysfunction in CML patients treated with dasatinib (50), in which the inhibition of the SFKs upstream of PLCγ2 (Lyn and Fyn) is the proposed mechanism of dysregulation.
In B cells, dasatinib prevented phosphorylation of all measured components of the BCR signaling cascade (Syk, SHP2, Btk, BLNK, and PLCγ2) regardless of whether activation was through BCR crosslinking or PVO4 treatment (Fig. 5B and fig. S8C). This off-target activity may underlie the efficacy reported in a patient with chronic lymphocytic leukemia, a B cell malignancy (51). However, these effects may also have undesirable consequences. For example, suppression of subtle pro-survival (“tonic”) B cell signaling (52) may account for the decline of circulating B cells observed in CML patients undergoing high-dose dasatinib therapy (53).
Disruption of the tyrosine kinase/phosphatase equilibrium with PVO4 also caused potent phosphorylation of STAT5 in nearly all cell types, but this was completely abrogated by dasatinib in all but the plasmacytoid dendritic cells (Fig. 5C). This time, dasatinib had no effect on the exclusive induction of STAT5 phosphorylation shown in T cells by IL-7 (Fig. 5C). The suppression of pSTAT5 in PVO4-treated myeloid cells (Fig. 5C) supports the alternative mechanism of SrcFK activation of STAT5 activity and resembles the effect of dasatinib in a BCR-ABL–positive CML cell line (54).
That these pleiotropic downstream signaling molecules (ERK, STAT5, and PLCγ2) can be potently activated via both dasatinib-sensitive and -insensitive pathways highlights the drug’s context and cell-type specificity for different signaling mediators. Thus, the unchecked endogenous tyrosine kinase activity revealed by PVO4 may unveil differences in druggable signaling architecture between cell types by mimicking the dysregulated signaling of cells susceptible to disease or dysfunction. Additionally, as underlined by these limited examples, the dasatinib data set provided a mechanistic blueprint of regulatory cell signaling events that could potentially be exploited in later clinical applications.
Given the diverse cell type–specific effects of dasatinib, we investigated whether drug sensitivity would follow immunophenotypic progressions as observed above (Fig. 4). Cells representing HSC through pre-B I phenotypes were selected from the SPADE plot and subjected to principal component analysis (Fig. 5D). Unsupervised PCA detected the phenotypic progression in early B cell development as a single linear progression axis defined primarily by CD19, CD34, and CD33 intensities (Fig. 5E). This axis recapitulated the expected developmental sequence and independently verified the ordering identified by the SPADE algorithm (Figs. 2 and 5D). The basal intensities of four intracellular signaling mediators (pSHP2, pCREB, pSrcFK, and p-p38) were unchanged throughout the PCA progression axis (Fig. 5F) and exhibited similar potential for activation by PVO4 (Fig. 5G). In contrast, these same signaling mediators exhibited a gradual increase in sensitivity to dasatinib correlated with maturation (Fig. 5H). This observation may be attributable to high expression of drug efflux pumps in the most immature cell types (HSCs and MPPs) (55). Alternatively, because PVO4 reveals endogenous kinase activity by repressing tyrosine phosphatases these observations may indicate a gradual shift in the phosphatase/kinase balance during early B cell maturation. Altogether, this approach provides insight into how a high dimensional analysis can call attention to potential off-target effects and new therapeutic opportunities that can be leveraged at all stages of the drug development pipeline.
Discussion
In this study of the immune system, coupling classical phenotypic organization to cellular functional responses was unrestricted by the inherent limitations imposed by fluorescence. This merging provided a systems-level view of human hematopoiesis and immunology from the perspective of immunophenotype and coupled it to underlying events as measured through receptor engagement and small-molecule drug actions. Although yielding both qualitatively and quantitatively identical measurements when compared (Fig. 1 and fig. S1), current mass cytometry detection may not be as sensitive as fluorescent detection of the most quantum-efficient dyes (Fig. 1, D to E). However, the differences in sensitivity between mass cytometry isotopes used here are within a twofold range (19), whereas quantum yields of routinely used fluorescence dyes vary across a 10-fold range and require compensation for spectral overlap. This combined with the lack of background signal (autofluorescence) and the substantially greater number of parameters that can be simultaneously analyzed makes mass cytometry an attractive platform currently available for highly multiplexed single-cell analysis.
The single-cell functional outcome data set (free to explore and available online) both confirmed expected immunological phenomena and yielded unexpected observations related to the spectrum of cell type–specific signaling faculties and drug responses that arise during hematopoietic development. For instance, there was a lack of IL-3 regulation of STAT3 in mature B cells, despite the presence of CD123 (IL-3 receptor–α chain) (Fig. 3). Additionally, both precise and more gradual continuous signaling transitions observed in the data set across developing cell subtypes (Fig. 3) represent some of the most interesting biological insights. Many of these precise transitions correlate well with receptor expression measured here [IL3/CD123 and LPS/CD14 (Fig. 3 and figs. S4 and S8) and known from previous work (IL7/pSTAT5 based on IL7 receptor expression on T cells (56)]. As for the continuous transitions, more broadly acting conditions such as PVO4 treatment revealed more subtle phosphorylation changes (Figs. 3 to 5) that probably reflect equally subtle changes in the kinase/phosphatase expression levels upstream of each of these measured targets, paralleling the phenotypic transitions.
These observations not only offer an opportunity to investigate the mechanism underlying the differences but may also provide a possibility to design drugs that might more precisely modulate disease states. There were many examples of signaling that corresponded with known distinct hematopoietic stages as well as multiple examples of progressive signaling responses across continuums of related cell types. We expect that a deeper mining of this and additional data sets will reveal many unexpected, system-wide correlations that could initiate new forms of mechanistic inquiry beyond what is currently possible with conventional techniques.
The extension of this analysis pipeline to preclinical settings can provide new insights into the mechanisms of diseases that perturb hematological function and could help pinpoint the true specificity and efficacy of drugs designed to restore the system to homeostasis. Expansion of this technology to additional parameters per cell (18, 19) can be enabled by the use of other isotopes and binding agents—such as with isotopically enriched nanocrystals and new metal chelators. Combination of the increased availability of parameters in this platform with the high-throughput methods previously demonstrated in fluorescence flow cytometry [fluorescent cell barcoding (57 ) and drug screening (44)] opens avenues for massively multiplexed single-cell assays. Opportunities exist to extend the repertoire of transition element isotope reporter–enabled reagents to mimic (and potentially improve on) many of the assayable capabilities of fluorophores. Together, these advances offer an opportunity to delve deeper into signaling, studying entire pathways in cellulo, and thus explore the developmental functions of the immune system as a whole. Such studies of normal immune function can act as a backdrop to better understand how cancer, inflammatory, and autoimmune diseases affect or disable system-wide immune functions. An important next step will be the unification of these single-cell systems studies with other “-omic” (such as genomic, epigenomic, metabolomic, and proteomic) approaches to lead to an integrated view of how disease manifests and the ways we can precisely correct pathologic processes.
Supplementary Material
Supplementary Data
Acknowledgments
The authors thank W. J. Fantl for critical reading of the manuscript. S.C.B. is supported by the Damon Runyon Cancer Research Foundation Fellowship (DRG-2017-09). This work was supported by NIH grants, U19 AI057229, P01 CA034233, 272200700038C, 1R01CA130826, U54 CA149145, 5U54 CA143907, RB2-01592, PN2 EY018228, NOI-HV-00242, and HEALTH.2010.1.2-1 (European Commission grant to the Sweden Diatools Consortium), as well as California Institute for Regenerative Medicine (DR-01477, RB2-01592) to G.P.N. G.P.N. is supported by an endowed chair from Rachtford and Carlota A. Harris. S.D.T. was supported by Genome Canada via the Ontario Genomics Institute for Cancer Research and by the Ontario Research Fund ORF-GL2-01-003. D.P. holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund and Packard Fellowship for Science and Engineering. Some antibodies were a gift from Becton Dickinson Biosciences. B.B. is a paid employee of Becton Dickinson, and G.P.N. and P.K. are paid consultants for Becton Dickinson Biosciences. G.P.N. is a member of the Board of Directors and a consultant for DVS Sciences. O.O., G.P.N., and S.T. have equity holdings in DVS Biosciences, and S.T. is an employee of DVS Sciences. A patent has been applied for on the SPADE algorithm on behalf of Stanford University. Raw data and SPADE trees can be downloaded open access at www.cytobank.org/nolanlab.
Footnotes
References
- 1.Kondo M, et al. Annu Rev Immunol (Paris) 2003;21:759. doi: 10.1146/annurev.immunol.21.120601.141007. [DOI] [PubMed] [Google Scholar]
- 2.Doulatov S, et al. Nat Immunol. 2010;11:585. doi: 10.1038/ni.1889. [DOI] [PubMed] [Google Scholar]
- 3.Chattopadhyay PK, et al. Nat Med. 2006;12:972. doi: 10.1038/nm1371. [DOI] [PubMed] [Google Scholar]
- 4.Majeti R, Park CY, Weissman IL. Cell Stem Cell. 2007;1:635. doi: 10.1016/j.stem.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tárnok A, Ulrich H, Bocsi J. Cytometry A. 2010 Jan;77:6. doi: 10.1002/cyto.a.20844. [DOI] [PubMed] [Google Scholar]
- 6.Roederer M, De Rosa SC, Watanabe N, Herzenberg LA. Semin Immunol. 1997;9:389. doi: 10.1006/smim.1997.0097. [DOI] [PubMed] [Google Scholar]
- 7.O’Brien CA, Kreso A, Dick JE. Semin Radiat Oncol. 2009;19:71. doi: 10.1016/j.semradonc.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Krasnow MA, Cumberledge S, Manning G, Herzenberg LA, Nolan GP. Science. 1991;251:81. doi: 10.1126/science.1898782. [DOI] [PubMed] [Google Scholar]
- 9.Tsien RY. Annu Rev Biochem. 1998;67:509. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 10.Irish JM, et al. Cell. 2004;118:217. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 11.Sachs K, Perez O, Pe’er D, Lauffenburger DA, Nolan GP. Science. 2005;308:523. doi: 10.1126/science.1105809. [DOI] [PubMed] [Google Scholar]
- 12.Frisa PS, Jacobberger JW. PLoS ONE. 2009;4:e7064. doi: 10.1371/journal.pone.0007064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kornblau SM, et al. Clin Cancer Res. 2010;16:3721. doi: 10.1158/1078-0432.CCR-10-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosen DB, et al. PLoS ONE. 2010;5:e13543. doi: 10.1371/journal.pone.0013543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perfetto SP, Chattopadhyay PK, Roederer M. Nat Rev Immunol. 2004;4:648. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- 16.Autissier P, Soulas C, Burdo TH, Williams KC. Cytometry A. 2010;77:410. doi: 10.1002/cyto.a.20859. [DOI] [PubMed] [Google Scholar]
- 17.Lou X, et al. Angew Chem Int Ed Engl. 2007;46:6111. doi: 10.1002/anie.200700796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bandura DR, et al. Anal Chem. 2009;81:6813. doi: 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- 19.Ornatsky O, et al. J Immunol Methods. 2010;361:1. doi: 10.1016/j.jim.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 20.Ornatsky OI, et al. Anal Chem. 2008;80:2539. doi: 10.1021/ac702128m. [DOI] [PubMed] [Google Scholar]
- 21.Materials and methods are available as supporting material on Science Online
- 22.Krutzik PO, Nolan GP. Cytometry A. 2003;55:61. doi: 10.1002/cyto.a.10072. [DOI] [PubMed] [Google Scholar]
- 23.Albeck JG, et al. Mol Cell. 2008;30:11. doi: 10.1016/j.molcel.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janes KA, et al. Mol Cell Proteomics. 2003;2:463. doi: 10.1074/mcp.M300045-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Janes KA, et al. Cell. 2006;124:1225. doi: 10.1016/j.cell.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 26.van Lochem EG, et al. Cytom B Clin Cytom. 2004;60:1. doi: 10.1002/cyto.b.20008. [DOI] [PubMed] [Google Scholar]
- 27.Lemischka IR. Ann N Y Acad Sci. 2005;1044:132. doi: 10.1196/annals.1349.017. [DOI] [PubMed] [Google Scholar]
- 28.Fraser IDC, Germain RN. Nat Immunol. 2010;10:327. doi: 10.1038/ni.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takano K, et al. Int J Hematol. 2000;72:48. [PubMed] [Google Scholar]
- 30.Chang JT, et al. Science. 2007;315:1687. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 31.Fulwyler MJ. Science. 1965;150:910. doi: 10.1126/science.150.3698.910. [DOI] [PubMed] [Google Scholar]
- 32.Herzenberg LA, Chan EL, Ravitch MM, Riblet RJ, Herzenberg LA. J Exp Med. 1973;137:1311. doi: 10.1084/jem.137.6.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shapiro HM. Practical Flow Cytometry. Vol. 4. Wiley-Liss; Hoboken, NJ: 2003. [Google Scholar]
- 34.Fulwyler M. Cytometry A. 2005;67:61. doi: 10.1002/cyto.a.20183. [DOI] [PubMed] [Google Scholar]
- 35.Oh ST, et al. Blood. 2010;116:988. doi: 10.1182/blood-2010-02-270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Irish JM, et al. Proc Natl Acad Sci U S A. 2010;107:12747. doi: 10.1073/pnas.1002057107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kotecha N, et al. Cancer Cell. 2008;14:335. doi: 10.1016/j.ccr.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Surh CD, Sprent J. Immunity. 2008;29:848. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 39.Ulevitch RJ, Tobias PS. Annu Rev Immunol. 1995;13:437. doi: 10.1146/annurev.iy.13.040195.002253. [DOI] [PubMed] [Google Scholar]
- 40.Gibbs KD, Jr, et al. Blood. published online 28 February 2011, 10.1182/blood-2010-07-298232. [Google Scholar]
- 41.Hardy RR, Kincade PW, Dorshkind K. Immunity. 2007;26:703. doi: 10.1016/j.immuni.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 42.LeBien TW, Tedder TF. Blood. 2008;112:1570. doi: 10.1182/blood-2008-02-078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurosaki T, Shinohara H, Baba Y. Annu Rev Immunol. 2010;28:21. doi: 10.1146/annurev.immunol.021908.132541. [DOI] [PubMed] [Google Scholar]
- 44.Krutzik PO, Crane JM, Clutter MR, Nolan GP. Nat Chem Biol. 2008;4:132. doi: 10.1038/nchembio.2007.59. [DOI] [PubMed] [Google Scholar]
- 45.Druker BJ. Blood. 2008;112:4808. doi: 10.1182/blood-2008-07-077958. [DOI] [PubMed] [Google Scholar]
- 46.Karaman MW, et al. Nat Biotechnol. 2008;26:127. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 47.Lindauer M, Hochhaus A. Recent Results Cancer Res. 2010;184:83. doi: 10.1007/978-3-642-01222-8_7. [DOI] [PubMed] [Google Scholar]
- 48.Yang C, et al. Leukemia. 2008;22:1755. doi: 10.1038/leu.2008.163. [DOI] [PubMed] [Google Scholar]
- 49.Schade AE, et al. Blood. 2008;111:1366. doi: 10.1182/blood-2007-04-084814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quintás-Cardama A, Han X, Kantarjian H, Cortes J. Blood. 2009;114:261. doi: 10.1182/blood-2008-09-180604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Russwurm G, et al. Blood. 2010;116:2617. doi: 10.1182/blood-2010-04-279786. [DOI] [PubMed] [Google Scholar]
- 52.Monroe JG. Nat Rev Immunol. 2006;6:283. doi: 10.1038/nri1808. [DOI] [PubMed] [Google Scholar]
- 53.Sillaber C, et al. Eur J Clin Invest. 2009;39:1098. doi: 10.1111/j.1365-2362.2009.02206.x. [DOI] [PubMed] [Google Scholar]
- 54.Nam S, et al. Mol Cancer Ther. 2007;6:1400. doi: 10.1158/1535-7163.MCT-06-0446. [DOI] [PubMed] [Google Scholar]
- 55.Dohse M, et al. Drug Metab Dispos. 2010;38:1371. doi: 10.1124/dmd.109.031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tan JT, et al. Proc Natl Acad Sci U S A. 2001;98:8732. doi: 10.1073/pnas.161126098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krutzik PO, Nolan GP. Nat Methods. 2006;3:361. doi: 10.1038/nmeth872. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data