Femtosecond X-ray protein nanocrystallography (original) (raw)
. Author manuscript; available in PMC: 2012 Aug 28.
Published in final edited form as: Nature. 2011 Feb 3;470(7332):73–77. doi: 10.1038/nature09750
Abstract
X-ray crystallography provides the vast majority of macromolecular structures, but the success of the method relies on growing crystals of sufficient size. In conventional measurements, the necessary increase in X-ray dose to record data from crystals that are too small leads to extensive damage before a diffraction signal can be recorded1-3. It is particularly challenging to obtain large, well-diffracting crystals of membrane proteins, for which fewer than 300 unique structures have been determined despite their importance in all living cells. Here we present a method for structure determination where single-crystal X-ray diffraction ‘snapshots’ are collected from a fully hydrated stream of nanocrystals using femtosecond pulses from a hard-X-ray free-electron laser, the Linac Coherent Light Source4. We prove this concept with nanocrystals of photosystem I, one of the largest membrane protein complexes5. More than 3,000,000 diffraction patterns were collected in this study, and a three-dimensional data set was assembled from individual photosystem I nanocrystals (~200 nm to 2 μm in size). We mitigate the problem of radiation damage in crystallography by using pulses briefer than the timescale of most damage processes6. This offers a new approach to structure determination of macromolecules that do not yield crystals of sufficient size for studies using conventional radiation sources or are particularly sensitive to radiation damage.
Radiation damage has always limited resolution in biological imaging using electrons or X-rays2. With the recent invention of the femtosecond X-ray laser, an opportunity has arisen to break the nexus between radiation dose and spatial resolution. It has been proposed that femtosecond X-ray pulses can be used to outrun even the fastest damage processes by using single pulses so brief that they terminate before the manifestation of damage to the sample6. Experiments at the FLASH free-electron laser (FEL), Germany, confirmed the feasibility of ‘diffraction before destruction’ at resolution lengths down to 60 Å on test samples fixed on silicon nitride membranes7. It was predicted that the irradiance (or power density) of focused pulses from a hard-X-ray FEL such as the Linac Coherent Light Source (LCLS), USA, would be sufficient to produce diffraction patterns at near-atomic resolution6.
We demonstrate here that this notion of diffraction before destruction operates at subnanometre resolution, using the membrane protein photosystem I as a model system, and establish an approach to structure determination based on X-ray diffraction data from a stream of nanocrystals6,8. Membrane proteins have a central role in the functioning of cells and viruses, yet our knowledge of the structure and dynamics responsible for their functioning remains limited. Photosystem I is a large membrane protein complex (1-MD a molecular mass, 36 proteins, 381 cofactors) that acts as a biosolar energy converter in the process of oxygenic photosynthesis. Its crystals display the symmetry of space group _P_63, with unit-cell parameters a = b = 281 Å and c = 165 Å, and consist of 78% solvent by volume. We show that diffraction data can be recorded from these fragile protein nanocrystals before destruction occurs. Furthermore, we demonstrate that structure factors can be extracted from the ‘partial’ reflections of tens of thousands of single-crystal diffraction snapshots, showing that interpretable high-quality, three-dimensional (3D) structure factor data can be obtained from a suspension of submicrometre crystals.
Our experimental set-up (Fig. 1 and Methods) records single-crystal diffraction data from a stream of crystals carried in a 4-μm-diameter, continuous liquid water jet9 that flows across the focused LCLS X-ray beam in vacuum at 10 μl min−1. In contrast to cryo-electron microscopy10,11 or standard crystallography on microcrystals3, which require cryogenic cooling, these data were collected on fully hydrated, 3D nanocrystals. The crystal located in the interaction region when an X-ray pulse arrives gives rise to a diffraction pattern that is detected on a set of two low-noise, X-ray p–n junction charge-coupled device (pnCCD) modules12 and read out before the arrival of the next pulse at the FEL repetition rate of 30 Hz, or 1,800 patterns per minute. The photon energy of the X-ray pulses was 1.8 keV (6.9-Å wavelength), with more than 1012 photons per pulse at the sample and pulse durations of 10, 70, and 200 fs (ref. 13). An X-ray fluence of 900 J cm−2 was achieved by focusing the FEL beam to a full-width at half-maximum of 7 μm, corresponding to a sample dose of up to 700 MGy per pulse (calculated using the program RADDOSE14) and a peak power density in excess of 1016 W cm−2 at 70-fs duration. In contrast, the typical tolerable dose in conventional X-ray experiments is only about 30 MGy (ref. 1). A single LCLS X-ray pulse destroys any solid material placed in this focus, but the stream replenishes the vaporized sample before the next pulse.
Figure 1. Femtosecond nanocrystallography.
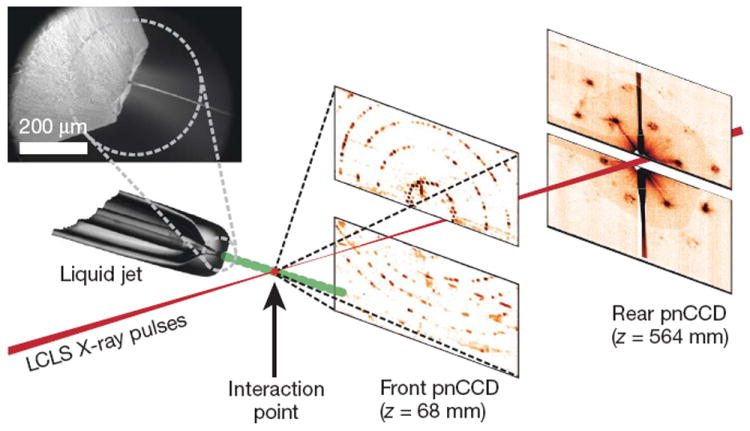
Nanocrystals flow in their buffer solution in a gas-focused, 4-μm-diameter jet at a velocity of 10 m s−1 perpendicular to the pulsed X-ray FEL beam that is focused on the jet. Inset, environmental scanning electron micrograph of the nozzle, flowing jet and focusing gas30. Two pairs of high-frame-rate pnCCD detectors12 record low-and high-angle diffraction from single X-ray FEL pulses, at the FEL repetition rate of 30 Hz. Crystals arrive at random times and orientations in the beam, and the probability of hitting one is proportional to the crystal concentration.
The front detector module, located close to the interaction region, recorded high-angle diffraction to a resolution of 8.5 Å, whereas the rear module intersected diffraction at resolutions in the range of 4,000 to 100 Å. We observed diffraction from crystals smaller than ten unit cells on a side, as determined by examining the data recorded on the rear pnCCDs (Fig. 2). A crystal with a side length of N unit cells gives rise to diffraction features that are finer by a factor of 1/N than the Bragg spacing (that is, with N − 2 fringes between neighbouring Bragg peaks), providing a simple way to determine the projected size of the nanocrystal. Images of crystal shapes obtained using an iterative phase retrieval method15,16 are shown in Fig. 2. The 3D Fourier transform of the crystal shape is repeated on every reciprocal lattice point. However, the diffraction condition for lattice points is usually not exactly satisfied, so each recorded Bragg spot represents a particular ‘slice’ of the Ewald sphere through the shape transform, giving a variety of Bragg spot profiles in a pattern; these are apparent in Fig. 2. The sum of counts in each Bragg spot underestimates the underlying structure factor square modulus, representing a partial reflection.
Figure 2. Coherent crystal diffraction.
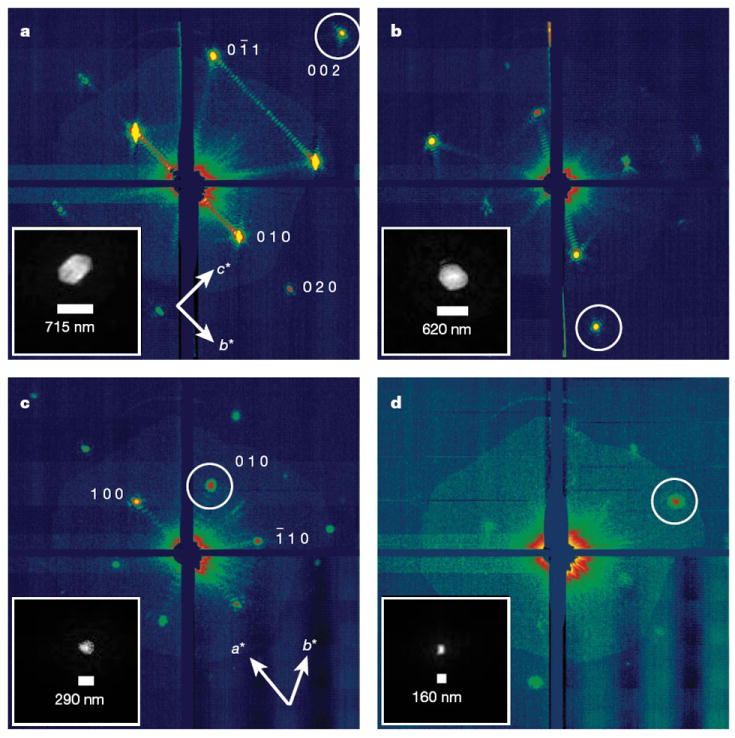
Low-angle diffraction patterns recorded on the rear pnCCDs, revealing coherent diffraction from the structure of the photosystem I nanocrystals, shown using a logarithmic, false-colour scale. The Miller indices of the peaks in a were identified from the corresponding high-angle pattern. In c we count seven fringes in the b* direction, corresponding to nine unit cells, or 250 nm. Insets, real-space images of the nanocrystal, determined by phase retrieval (using the Shrinkwrap algorithm15) of the circled coherent Bragg shape transform.
Figure 3a shows strong single-crystal diffraction to the highest angles of the front detector. The nanocrystal shape transform is also apparent in many patterns at the high angles detected by the front detector, giving significant measured intensities between Bragg peaks as is noticeable in Supplementary Fig. 3a. These mid-Bragg intensities oversample the molecular transform, providing a potential route to phasing of the pattern17,18.
Figure 3. Diffraction intensities and electron density of photosystem I.
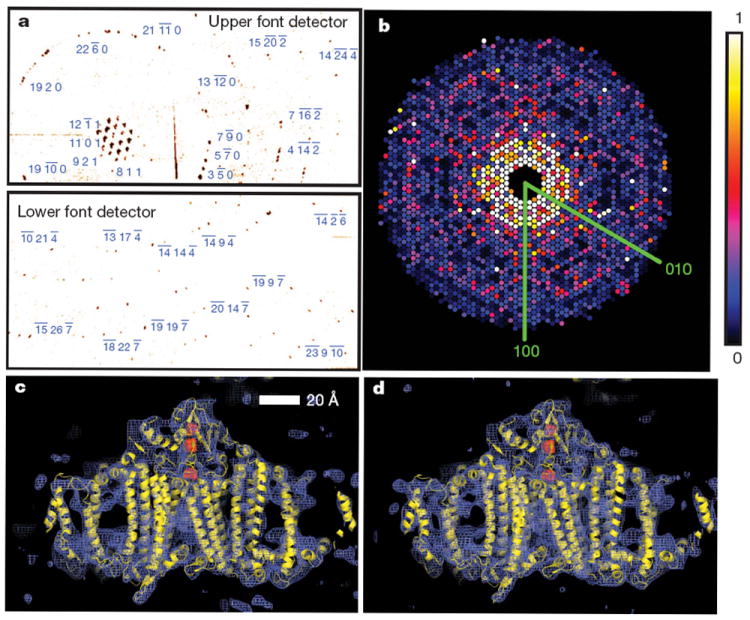
a, Diffraction pattern recorded on the front pnCCDs with a single 70-fs pulse after background subtraction and correction of saturated pixels. Some peaks are labelled with their Miller indices. The resolution in the lower detector corner is 8.5 Å. b, Precession-style pattern of the [001] zone for photosystem I, obtained from merging femtosecond nanocrystal data from over 15,000 nanocrystal patterns, displayed on the linear colour scale shown on the right. c, d, Region of the 2_mFo_ − DFc electron density map at 1.0_σ_ (purple mesh), calculated from the 70-fs data (c) and from conventional synchrotron data truncated at a resolution of 8.5 Å and collected at a temperature of 100 K (d) (Methods). The refined model is depicted in yellow.
In conventional crystallography, the ‘full’ Bragg reflection is determined to high precision, for example by integrating counts as the crystal is rotated such that these reflections pass through the diffraction condition. By indexing individual patterns and then summing counts in all partial reflections for each index, we performed a Monte Carlo integration over the reciprocal-space volume of the Bragg reflection and the distribution of crystal shapes and orientations and variations in the X-ray pulse fluence. The result of this procedure converges to the square of the structure factor moduli18. We found that over 13% of diffraction patterns with ten or more spots could be consistently indexed using the programs MOSFLM19 and DirAx20 (Methods). Merged intensities at 70-fs pulse duration are presented as a precession-style image of the [001]-zone axis in Fig. 3b (see also Supplementary Figs 3 and 4). We tested the reliability of this approach by comparing the LCLS merged data with data collected at 100 K with 12.4-keV synchrotron radiation from a single crystal of photosystem I cryopreserved in 2 M sucrose. These data sets show good agreement, with a difference metric, _R_iso, of 22.1% computed over the entire resolution range and of less than 13% in the middle resolution shells; see Supplementary Table 1 for detailed statistics.
To complete our proof of principle, we conducted a rigid-body refinement of the published photosystem I structure (Protein Data Bank ID, 1JB0) against the nanocrystal structure factors, yielding _R/R_free = 0.25/0.23. A representative region of the 2_mF_o − _DF_c electron density map at 8.5 Å (Methods) from the LCLS data set is shown in Fig. 3c. This map shows the details expected at this resolution, including transmembrane helices, membrane extrinsic features and some loop structures. For comparison, the electron density refined from the 12.4-keV, single-crystal data set truncated to a resolution of 8.5 Å is given in Fig. 3d.
The dose of 700 MGy corresponds to a K-shell photoabsorption of 3% of all carbon atoms in the protein. This energy is subsequently released by photoionization and Auger decay, followed by a cascade of lower-energy electrons caused by secondary ionizations, taking place on the 10–100-fs timescale21. Using a model of the plasma dynamics22,23, we calculated that by the end of a 100-fs pulse each atom of the crystal was ionized once, on average, and that motion of nuclei had begun. This is expected to give rise to a decrease in Bragg amplitudes, similar to an increase in a Debye–Waller temperature factor24. We studied the effects of the initial ionization damage on the diffraction of photosystem I nanocrystals by collecting a series of data sets at pulse durations of 10, 70 and 200 fs. The 10-fs pulses were produced with lower pulse energy: ~10% of the total number of photons of the longer pulses13, or a 70-MGy dose. Plots of the scattering strength of the crystals versus resolution, generated by selecting and summing Bragg spots from more than 66,000 patterns for each of the three pulse durations measured, are shown in Fig. 4. The 10- and 70-fs traces are very similar, indicating that these pulses are short enough to overcome radiation damage at the observed resolution, 8.5 Å. For 200-fs pulses, there is a decrease in scattering strength at resolutions beyond 25 Å, indicating disordering on this longer timescale. The highest-resolution Bragg peaks for the 200-fs pulses were not broadened or shifted relative to the short-duration data sets, which indicates there was no strain or expansion of the lattice, respectively.
Figure 4. Pulse-duration dependence of diffraction intensities.
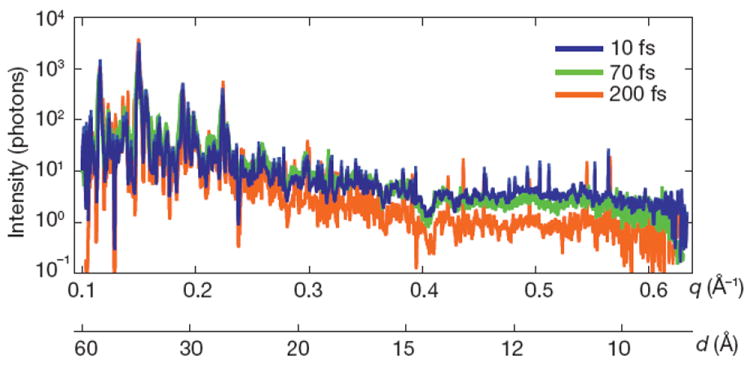
Plot of the integrated Bragg intensities of photosystem I nanocrystal diffraction as a function of photon momentum transfer, q = (4π/λ)sin(θ) = 2π/d (wavelength, λ; scattering angle 2_θ_; resolution, d) for pulse durations of 10, 70 and 200 fs. Averages were obtained by isolating Bragg spots from 97,883, 805,311 and 66,063 patterns, respectively, normalized to pulse fluence. The error in each plot is indicated by the thickness of the line. The decrease in irradiance for 200-fs pulses and d < 25 Å indicates radiation damage for these long pulses, which is not apparent for 70-fs pulses and shorter.
Our next step is to improve resolution by using shorter-wavelength X-rays. Resolution may ultimately be limited by X-ray pulse fluence, the ultrafast radiation damage and the intrinsic disorder within the nanocrystals themselves. Recent experiments21 at LCLS indicate a brief saturation of the X-ray photoabsorption of atoms in a tightly focused pulse, resulting in a decrease in photoionization damage on a 20-fs timescale without a reduction in the scattering cross-sections that give rise to the diffraction pattern22. Planned beamlines at LCLS aim to achieve up to a 105-fold increase in pulse irradiance by tighter focusing, allowing data collection with low-fluence, 10-fs pulses or pulses of even shorter duration25. This provides a route to further reducing radiation damage and may allow measurements on even smaller nanocrystals, down to a single unit cell6 (that is, a single molecule). As this limit is approached, the ordering of the nanocrystals will become increasingly irrelevant, as each crystal may be treated as a single object and the ‘disorder’ that conventionally leads to reduced resolution will simply manifest itself as shot-to-shot variability, providing information about not just the average structure but also the range of dynamically accessible conformations.
Data are collected on fully hydrated nanocrystals without cryogenic cooling. We expect that the results presented here will open new avenues for crystallography using X-ray laser pulses that are so short that only negligible X-ray-induced radiation damage occurs during data collection. Significant improvements in sample utilization are expected by exploiting higher X-ray repetition rates or by slowing the liquid flow. For example, the generation, using inkjet technologies, of liquid droplets at a rate that matches the LCLS X-ray pulses would dramatically decrease the total required sample volume by a factor of 25,000, meaning that less than 0.4 μl of nanocrystal suspension would be needed in our particular case, of photosystem I. Further efficiency gains would result from indexing and merging a greater proportion of patterns into the 3D data set, which may be achieved by applying methods for merging continuous diffraction patterns of single molecules26,27 or by using ‘post-refinement’28 to obtain accurate structure factor estimates from fewer diffraction patterns. These methods will also remove the twinning ambiguity that exists in our current indexing scheme. Our method also has potential application to the study of chemical reactions, such as the processes in photosynthesis or enzymatic reactions.
METHODS
Experimental set-up
The experiments were performed at LCLS4, at SLAC, at the AMO beamline29 in vacuo using the CAMP end station12. X-ray pulses, generated at a repetition rate of 30 Hz, were focused to a spot with a full-width at half-maximum of 7 μm (full-width of 13 μm at 10% maximum irradiance) and a pulse fluence of 900 J cm−2, corresponding to a peak power density (irradiance) in excess of 1016 W cm−2 at 70-fs duration. The pnCCD detectors were read out, digitized and stored at the 30-Hz rate of the delivered LCLS pulses. Each detector panel consists of 512 × 1,024 pixels 75 × 75 μm2 in area. The rear detectors, located 564 mm from the jet, record low-angle scattering from 0.1° to 4.0° in the vertical scattering plane, and the front detectors, located 68 mm from the jet, cover 4.6° to 40.5° in the same vertical plane. The largest scattering-angle magnitude accepted by the front detector was 47.9°, corresponding to a resolution, d, of 8.5 Å at a wavelength of 6.9 Å. X-ray fluorescence from the water jet was filtered by an 8-μm-thick polyimide film in front of the pnCCDs.
A liquid microjet8,9 was used to inject the nanocrystal suspension into the FEL beam at a flow rate of 10 μl min−1. The microjet was emitted from a 40-μm-diameter capillary and focused to a 4-μm-diameter column by a coaxial flow of helium. The X-ray attenuation in the water was at most 30%. The interaction region of the X-rays and crystals is located in the continuous liquid column, before the Rayleigh break-up of the jet into drops, such that most of the X-ray scattering from the liquid is confined to a narrow vertical streak in reciprocal space.
Crystallization conditions of photosystem I nanocrystals were established by determining the phase diagrams31,32. Nanocrystals were grown in batches at 10 mg ml−1 protein concentration (30 μM P700, or 10 μM photosystem I trimer) and low ionic strength (8 mM MgSO4, 5 mM MES, pH 6.4, and 0.02% β-dodecylmaltoside) at 4 °C. The photosystem I nanocrystals were then suspended in harvesting buffer (5 mM MES, pH 6.4, and 0.02% β-dodecylmaltoside) to establish a protein concentration of 1 mg ml−1. The crystal suspension was filtered through 2-μm cut-off filters (In-line Filter, Upchurch) and stored at 4 °C until use in the experiment.
The nanocrystals are needles of hexagonal cross-section, with the long axis of the needle along the c axis and an aspect ratio (length to maximum diameter of hexagon) ranging from 1:1 to 2:1, as determined from reconstructing single-shot views of the whole crystal from their shape transforms. For example, Fig. 2a shows a view of the crystal almost perpendicular to the c axis, where we reconstruct a shape of aspect ratio 1.6:1. A view along the c axis (Fig. 2c) shows the hexagonal profile. Large, millimetre-sized, crystals of photosystem I have an aspect ratio of up to 5:1, which is seen to decrease with decreasing crystal size.
The nanocrystal suspension was introduced directly into the microjet through a sample loop (Supplementary Fig. 1). A micropore filter in the fluid delivery line was used to restrict the size of the nanocrystals to less than 2 μm. The suspension was diluted to observe a crystal ‘hit rate’ of 20% (Supplementary Fig. 2), to minimize the occurrence of double hits. The observed concentration of crystals was therefore 0.2 per illuminated volume of 4 × 4 × 13 μm3, or 109 crystals per millilitre. The overall photosystem I protein concentration after dilution was 1 mg ml−1, and a complete set of structure factors was obtained from 1,850,000 X-ray pulses, or 10 mg of protein. With the current set-up, at the 30-Hz X-ray pulse rate less than 0.004% of the continuously flowing solution was exposed to the X-ray beam, so only one in 25,000 nanocrystals was actually hit by an X-ray pulse.
Details of the acquisition of diffraction patterns and the primary data reduction are given in Supplementary Methods.
3D merging of intensities
Peaks in the processed patterns were located in each pattern using the algorithm of ref. 33, and their locations were mapped into three dimensions according to the curvature of the Ewald sphere, the calibrated detector geometry and the X-ray wavelength. The 3D peak locations for each pattern in turn were presented to the auto-indexing program DirAx20. If DirAx succeeded in finding a unit cell for the peaks, linear combinations of the cell basis vectors were checked for correspondence with the photosystem I unit cell5 from the Protein Data Bank (ID, 1JB0). If a match was found, pixel intensities were summed within a circle of ten-pixel radius centred on the pixel closest to each located Bragg condition. Patterns were rejected if fewer than 10% of the detected peaks were accounted for by unit-cell parameters from DirAx. From 1,850,000 recorded patterns, we identified 112,725 as hits (more than ten detected peaks) and 15,445 were successfully indexed. New peak-finding and -indexing algorithms are under development and are expected to increase significantly the number of patterns that can be indexed, thereby further reducing the number of protein crystals required for a useful data set. The variation of pixel solid angle across the detector plane was accounted for, as was polarization of the X-ray beam assuming complete horizontal polarization. A list of reflection indices and intensities was produced for each individual diffraction pattern, and merging was performed by taking the mean value for the intensity of each unique reflection. Because the indexing algorithm makes use of the positions of the peaks but not their intensities, it was unable to distinguish between crystal orientations related by the symmetry of the lattice. As the symmetry of the lattice is higher than that of the actual structure of photosystem I, an ambiguity exists in that each pattern could correspond to one of two possible orientations. For programmatic convenience, these data (with actual space group symmetry _P_63) were merged as _P_6322 and treated as though merohedrally twinned during refinement (see below). A 3D rendering of the final full data set is shown in Supplementary Fig. 4.
Data quality
Metrics of the merged data quality are shown in Supplementary Table 1 and discussed in Supplementary Information. We carried out a rigid-body refinement of the published photosystem I structure (Protein Data Bank ID, 1JB0) to the merged structure factors using the program REFMAC34 in twin mode. The refinement R and R_free values were 0.25 and 0.23, respectively. The 2_mFo − DFc electron density map35 at a resolution of 8.5 Å is shown in Fig. 3c. The corresponding 2_mFo_ − DFc electron density map from the conventional synchrotron data, truncated to a resolution of 8.5 Å, is shown in Fig. 3d. The electron density maps show the large subunits PsaA and PsaB, as well as the membrane extrinsic subunits. The transmembrane helices, and even some loop structures, are clearly visible. In these figures, the ribbon representation of the protein model is shown in yellow and the atoms of three iron–sulphur clusters are depicted in red.
Supplementary Material
Femtosecond supp data
Acknowledgments
Experiments were carried out at the Linac Coherent Light Source and the Advanced Light Source, both National User Facilities operated respectively by Stanford University and the University of California on behalf of the US Department of Energy (DOE), Office of Basic Energy Sciences. We acknowledge support from the DOE through the PULSE Institute at the SLAC National Accelerator Laboratory; the Lawrence Livermore National Laboratory under contract DE-AC52-07NA27344; the Center for Bio-Inspired Solar Fuel Production, an Energy Frontier Research Center funded by the DOE, Office of Basic Energy Sciences (award DE-SC0001016); the Hamburg Ministry of Science and Research and the Joachim Herz Stiftung, as part of the Hamburg Initiative for Excellence in Research (LEXI); the Hamburg School for Structure and Dynamics; the Max Planck Society, for funding the development and operation of the CAMP instrument within the ASG at CFEL; the US National Science Foundation (awards 0417142 and MCB-1021557); the US National Institutes of Health (awards 1R01GM095583-01 (ROADMAP) and 1U54GM094625-01 (PSI:Biology)); the Swedish Research Council; the Swedish Foundation for International Cooperation in Research and Higher Education; Stiftelsen Olle Engkvist Byggmästare; the DFG Cluster of Excellence at the Munich Centre for Advanced Photonics; and the CBST at the University of California under cooperative agreement no. PHY 0120999. We acknowledge discussions with M. Rossmann, E. Snell, R. Stroud and A. Brunger, thank B. Hedman, E. Gullikson, F. Filsinger, A. Berg, H. Mahn and C. Kaiser for technical help and thank the staff of the LCLS for their support in carrying out these experiments.
Footnotes
Author Contributions H.N.C. and J.C.H.S. conceived the experiment, which was designed with P.F., A.B., R.A.K., J.S., D.P.D., U.W., R.B.D., S. Boutet, M.J.B., D.S., I.S., S.M. and J.H. The CAMP instrument was the responsibility of S.W.E., R.H., D. Rolles, A. Rudenko, C.S., L.F., N.K., P.H., B.R., B.E., A.H., Ch.R., D.P., G.W., L.S., G.H., H. Gorke, J.U., I.S., S.H., G.S., F.S., H.S., K.-U.K., R.A., C.-D.S., F.K., M. Bott, S. Schorb, D. Rupp, M.A., T.G., H.H., L.G., G.P., H. Graafsma and B.N., who designed and set up the instrument and/or developed and operated the pnCCD detectors. C.B., J.D.B. and M.M. set up and aligned the beamline. P.F., M.S.H. and I.G. prepared samples; R.B.D., D.P.D., U.W., J.C.H.S., P.F., L.L. and R.L.S. developed and operated the sample delivery system; H.N.C., A.B., A.A., J.S., D.P.D., U.W., R.B.D., S. Bajt, M.J.B., L.G., J.H., M.M.S., N.T., J.A., S. Stern and J.C.H.S. developed diffraction instrumentation; and M. Barthelmess, M.L., A.B. and K.N. designed and/or fabricated calibration samples. J.K., S.P.H.-R., A.B., H.N.C., J.S. and A.V.M. characterized the focus. H.N.C., J.C.H.S., P.F., A.B., T.A.W., R.A.K., A.A., J.S., D.P.D., U.W., R.B.D., I.S., N.C., R.L.S., M.S.H., L.L., M. Bott, S.W.E., R.H., D. Rolles, A. Rudenko, M.L., C.B., J.U., L.F., J.D.B., M.M., M.F., C.Y.H., R.G.S., G.J.W., A. Rocker, M.S., O.J., I.A. and J.H. carried out the experiment. A.B., T.A.W., R.A.K., A.A., F.R.N.C.M., A.V.M., L.L., T.R.M.B., N.C., L.F., N.K., R.N., G.W., P.H., C.C., J.M.H., I.S., J.H., H.N.C. and J.C.H.S. analysed the data. A.V.M. performed the Bragg shape phase retrieval. T.A.W. and R.A.K. merged the 3D data. R.F. collected and evaluated the reference data set; R.A.K., T.A.W., J.M.H. and R.F. refined the structure and calculated the electron density maps; and H.N.C., P.F., J.C.H.S. and I.S. wrote the manuscript with discussion and improvements from all authors.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
The authors declare no competing financial interests.
References
- 1.Owen RL, Rudino-Pinera E, Garman EF. Experimental determination of the radiation dose limit for cryocooled protein crystals. Proc Natl Acad Sci USA. 2006;103:4912–4917. doi: 10.1073/pnas.0600973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henderson R. The potential and limitations of neutrons, electrons and X-rays for atomic resolution microscopy of unstained biological molecules. Q Rev Biophys. 1995;28:171–193. doi: 10.1017/s003358350000305x. [DOI] [PubMed] [Google Scholar]
- 3.Riekel C. Recent developments in microdiffraction on protein crystals. J Synchr Radiat. 2004;11:4–6. doi: 10.1107/s090904950302541x. [DOI] [PubMed] [Google Scholar]
- 4.Emma P, et al. First lasing and operation of an ångstrom-wavelength free-electron laser. Nature Photon. 2010;4:641–647. [Google Scholar]
- 5.Jordan P, et al. Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature. 2001;411:909–917. doi: 10.1038/35082000. [DOI] [PubMed] [Google Scholar]
- 6.Neutze R, Wout R, van der Spoel D, Weckert E, Hajdu J. Potential for biomolecular imaging with femtosecond X-raypulses. Nature. 2000;406:752–757. doi: 10.1038/35021099. [DOI] [PubMed] [Google Scholar]
- 7.Chapman HN, et al. Femtosecond time-delay X-ray holography. Nature. 2007;448:676–679. doi: 10.1038/nature06049. [DOI] [PubMed] [Google Scholar]
- 8.Spence JCH, Doak RB. Single molecule diffraction. Phys Rev Lett. 2004;92:198102. doi: 10.1103/PhysRevLett.92.198102. [DOI] [PubMed] [Google Scholar]
- 9.DePonte DP, et al. Gas dynamic virtual nozzle for generation of microscopic droplet streams. J Phys D. 2008;41:195505. [Google Scholar]
- 10.Henderson R, et al. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J Mol Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- 11.Wang DN, Kühlbrandt W. High-resolution electron crystallography of light-harvesting chlorophyll a/b-protein complex in three different media. J Mol Biol. 1991;217:691–699. doi: 10.1016/0022-2836(91)90526-c. [DOI] [PubMed] [Google Scholar]
- 12.Strüder L, et al. Large-format, high-speed, X-ray pnCCDs combined with electron and ion imaging spectrometers in a multipurpose chamber for experiments at 4th generation light sources. Nucl Instrum Methods Phys Res A. 2010;614:483–496. [Google Scholar]
- 13.Ding Y, et al. Measurements and simulations of ultralow emittance and ultrashort electron beams in the Linac Coherent Light Source. Phys Rev Lett. 2009;102:254801. doi: 10.1103/PhysRevLett.102.254801. [DOI] [PubMed] [Google Scholar]
- 14.Paithankar KS, Owen RL, Garman EF. Absorbed dose calculations for macromolecular crystals: improvements to RADDOSE. J Synchr Radiat. 2009;16:152–162. doi: 10.1107/S0909049508040430. [DOI] [PubMed] [Google Scholar]
- 15.Marchesini S, et al. X-ray image reconstruction from a diffraction pattern alone. Phys Rev B. 2003;68:140101. [Google Scholar]
- 16.Robinson IK, Harder R. Coherent X-ray diffraction imaging of strain at the nanoscale. Nature Mater. 2009;8:291–298. doi: 10.1038/nmat2400. [DOI] [PubMed] [Google Scholar]
- 17.Sayre D. Some implications of a theorem due to Shannon. Acta Crystallogr. 1952;5:843. [Google Scholar]
- 18.Kirian R, et al. Femtosecond protein nanocrystallography—data analysis methods. Opt Express. 2010;18:5713–5723. doi: 10.1364/OE.18.005713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leslie AG. The integration of macromolecular diffraction data. Acta Crystallogr D. 2006;62:48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- 20.Duisenberg AJM. Indexing in single-crystal diffractometry with an obstinate list of reflections. J Appl Cryst. 1992;25:92–96. [Google Scholar]
- 21.Young L, et al. Femtosecond electronic response of atoms to ultra-intense X-rays. Nature. 2010;466:56–61. doi: 10.1038/nature09177. [DOI] [PubMed] [Google Scholar]
- 22.Hau-Riege SP, London RA, Szoke A. Dynamics of biological molecules irradiated by short X-ray pulses. Phys Rev E. 2004;69:051906. doi: 10.1103/PhysRevE.69.051906. [DOI] [PubMed] [Google Scholar]
- 23.Bergh M, Huldt G, Timneanu N, Maia FRNC, Hajdu J. Feasibility of imaging living cells at subnanometer resolutions by ultrafast X-ray diffraction. Q Rev Biophys. 2008;41:181–204. doi: 10.1017/S003358350800471X. [DOI] [PubMed] [Google Scholar]
- 24.Willis B, Pryor A. Thermal Vibrations in Crystallography. Cambridge Univ. Press; 1975. p. 92. [Google Scholar]
- 25.Emma P, et al. Femtosecond and sub femtosecond X-ray pulses from a self-amplified spontaneous-emission based free-electron laser. Phys Rev Lett. 2004;92:074801. doi: 10.1103/PhysRevLett.92.074801. [DOI] [PubMed] [Google Scholar]
- 26.Loh N-TD, Elser V. Reconstruction algorithm for single-particle diffraction imaging experiments. Phys Rev E. 2009;80:026705. doi: 10.1103/PhysRevE.80.026705. [DOI] [PubMed] [Google Scholar]
- 27.Fung R, Shneerson V, Saldin DK, Ourmazd A. Structure from fleeting illumination of faint spinning objects in flight. Nature Phys. 2008;5:64–67. [Google Scholar]
- 28.Rossmann MG, Leslie AG, Sherin SA, Tsukihara T. Processing and post-refinement of oscillation camera data. J Appl Cryst. 1979;12:570–581. [Google Scholar]
- 29.Bozek JD. AMO instrumentation for the LCLS X-ray FEL. Eur Phys J Spec Top. 2009;169:129–132. [Google Scholar]
- 30.DePonte DP, et al. SEM imaging of liquid jets. Micron. 2009;40:507–509. doi: 10.1016/j.micron.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 31.Fromme P, Grotjohann I. In: Membrane Protein Crystallization. DeLukas L, editor. Elsevier; 2009. pp. 192–224. Curr. Top. Membr. 63. [Google Scholar]
- 32.Hunter MS, et al. X-ray diffraction from membrane protein nanocrystals. Biophys J. doi: 10.1016/j.bpj.2010.10.049. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaefferer S. New developments of computer-aided crystallographic analysis in transmission electron microscopy. J Appl Cryst. 2000;33:10–25. [Google Scholar]
- 34.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likehood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 35.Praznikar J, Afonine PV, Guncar G, Adams PD, Turk D. Averaged kick maps: less noise, more signal…and probably less bias. Acta Crystallogr D. 2009;65:921–931. doi: 10.1107/S0907444909021933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Femtosecond supp data