PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure (original) (raw)
. Author manuscript; available in PMC: 2013 Apr 16.
Abstract
Purpose of the review
PGC-1α has been extensively described as a master regulator of mitochondrial biogenesis. However, PGC-1α activity is not constant, and can be finely tuned in response to different metabolic situations. From this point of view, PGC-1α could be described as a mediator of the transcriptional outputs triggered by metabolic sensors, providing the idea that these sensors, together with PGC-1α, might be weaving a network controlling cellular energy expenditure. In this review we will focus on how pathologies such as type 2 diabetes and the metabolic syndrome might be related to an abnormal and improper function of this network.
Recent findings
Two metabolic sensors, AMPK and SIRT1 have been described to directly affect PGC-1α activity through phosphorylation and deacetylation, respectively. While the physiological relevance of these modifications and their molecular consequences are still largely unknown, recent insight from different in vivo transgenic models clearly suggests that AMPK, SIRT1 and PGC-1α might act as an orchestrated network to improve metabolic fitness.
Summary
Metabolic sensors such as AMPK and SIRT1, gatekeepers of the activity of the master regulator of mitochondria, PGC1α, are vital links in a regulatory network for metabolic homeostasis. Together these players explain many of the beneficial effects of physical activity and dietary interventions in our battle against type 2 diabetes and related metabolic disorders. Hence, understanding the mechanisms by which they act could guide us to identify and improve preventive and therapeutic strategies for metabolic diseases.
Keywords: energy expenditure, PGC-1α, SIRT1, AMPK
Introduction
There is an undeniable inner beauty in how intracellular signalling and metabolic pathways respond to environmental challenges as a coordinated network involving distinct cellular compartments to generate an integrated response that allows proper adaptation. A paradigmatic and illustrative example of these coordinated actions can be found in the regulation of energetic metabolism. Energy homeostasis requires the coordinated regulation of energy intake, storage and expenditure. In healthy individuals, fluctuations in any of these processes are normally counterbalanced by regulation of the other two. In contrast, abnormalities in the proper equilibrium of the caloric equation lead to metabolic malfunctions.
Obesity, type 2 diabetes mellitus (T2DM) and the metabolic syndrome are among the most frequent pathological consequences induced by a misbalance of energy homeostasis. Insulin resistance, defined as the failure to respond to normal circulating concentrations of insulin, constitutes a characteristic hallmark preceding the overt manifestation of the above-mentioned disorders, as evidenced by cross-sectional studies demonstrating insulin resistance in virtually all patients with T2DM [1], as well as prospective studies demonstrating the presence of insulin resistance one to two decades before the onset of the disease [2]. While the exact molecular mechanisms responsible for the development of insulin resistance and how it paves the path to T2DM are enigmas yet to be deciphered, major progress has been achieved in recent years indicating that the regulation of energy expenditure takes center stage in this scenario.
Defective energy expenditure is linked to metabolic disease
Evidence gathered during the last decade indicates that alterations in lipid metabolism may have a central role in the onset of T2DM. First, insulin resistant states are commonly linked with a decreased efficiency to use fatty acids as an energy source in skeletal muscle [3-5]. This, in turn, redirects the fatty acid flux toward storage, leading to the increased ectopic lipid deposition. Evidence for this has been obtained in a wide array of experimental models of human insulin resistance [6-8], to the point that intramuscular triglyceride accumulation has been recognized as one of the most consistent markers of whole-body insulin resistance [9]. Together, these observations led to the hypothesis that defects in mitochondrial oxidative function and the subsequent decrease in energy expenditure may contribute to the metabolic dysfunctions observed both in insulin resistant states, T2DM and aging. Confirming this hypothesis, two seminal studies by Shulman’s lab using magnetic resonance spectroscopy to asses mitochondrial function in vivo, demonstrated that two different populations with high susceptibility to develop T2DM, i.e. lean, healthy offspring of type 2 diabetic parents and an elderly population, displayed impaired mitochondrial function in skeletal muscle [10, 11]. Subsequent studies reporting compromised mitochondrial function in T2DM patients further consolidated these observations [12, 13]. To date, however, it is not fully clear whether the impaired mitochondrial oxidative capacity in T2DM is due to diminished mitochondrial content, to intrinsic defects in mitochondrial functionality or both (see [14] for review).
At the molecular level, the oxidative dysfunction displayed in T2DM subjects may find an explanation in a coordinated decrease in the expression of genes involved in lipid oxidation and mitochondrial metabolism in skeletal muscle, as demonstrated by gene-clustering approaches [15, 16]. Furthermore, changes in gene expression are also correlated with changes in muscle fiber-type phenotype [11, 17]. Hence, obese and diabetic individuals display lower ratios of type I muscle fibers, which display high mitochondrial content and oxidative rates, relative to type IIb fibers, which have a glycolytic nature. The latter observation is important since insulin sensitivity is positively correlated with the oxidative capacity of the muscle [18]. Thus, increased glycolytic muscle mass may contribute into decreased whole-body insulin sensitivity, considering that skeletal muscle is responsible for up to 80% of post-prandial insulin-stimulated glucose disposal in healthy individuals [19]. For these reasons, it seems logical that regulation of mitochondrial oxidative capacity may hold promise as a preventive and therapeutic strategy to reduce the burden of T2DM and its associated diseases.
PGC-1α: fueling the oxidative fire
Interestingly, both fiber-type switching and the expression of many genes related to lipid oxidation and mitochondrial metabolism are under the transcriptional control of the peroxisome proliferator-activated receptor gamma (PPARγ) coactivator-1α (PGC-1α). PGC-1α was originally cloned as a cold-inducible coactivator of PPARγ in brown adipose tissue [20], but it has emerged as a potent coactivator of a plethora of transcription factors impacting on whole body energy expenditure (see [21] for review).
Early studies demonstrated that overexpression of PGC-1α in cultured cells was enough to increase energy expenditure [22-24]. To do so, PGC-1α coordinately increases mitochondrial biogenesis and respiration rates, as well as the uptake and utilization of substrates for energy production. In order to exert such a wide array of functions PGC-1α directly coactivates multiple transcription factors, including nuclear receptors – such as the PPARs [25, 26], or the thyroid hormone receptor (TR) [20], glucocorticoid receptors (GRs) [25], estrogen receptors (ERs) [20, 25] and estrogen-related receptors (ERRs) [27, 28] among others – and non-nuclear receptor transcription factors, such as myocyte enhancer factor-2 (MEF-2) [29] and the family of forkhead O-box (FOXO) transcription factors [30]. By simultaneously coactivating these transcriptional factors PGC-1α can quickly and coordinately modulate a transcriptional program that governs energy metabolism. While PGC-1α coactivation can change in response to different stimuli or in a tissue-specific manner, the molecular mechanisms determining how PGC-1α partners can be selectively targeted are still largely unknown.
Arguably, the most compelling data on the key role of PGC-1α in energy metabolism comes from in vivo studies. PGC-1α is mainly expressed in tissues with high energy oxidative capacity, like heart, skeletal muscle, liver, brown adipose tissue and brain, and is robustly induced in conditions that require energy, such as cold, fasting and exercise [20, 31]. Further evidence supporting a key role of PGC-1α came from tissue-specific genetically engineered mouse models, since germline PGC-1α knock-out mice have pronounced central nervous system perturbations that complicate the interpretation of the phenotype. Muscle-specific PGC-1α transgenic animals display increased mitochondrial number and function, as well as a higher relative amount of type I oxidative fibers [32]. Conversely, mice with a muscle-specific deletion of PGC-1α show abnormal glucose homeostasis linked to a moderate reduction in the number of oxidative type I fibers, decreased endurance capacity and mitochondrial gene expression [33]. Altogether, these data provide compelling evidence that PGC-1α is a key regulator of mitochondrial biogenesis in muscle.
Given the potential action of PGC-1α as a signalling amplifier of specific gene sets, minor disturbances in the activity of this coactivator might strongly impact on whole-body energy homeostasis and on the pathogenesis of the metabolic syndrome. Consistent with the compromised bioenergetic capacity and coordinated reduction in the expression of mitochondrial genes, decreased PGC-1α levels has been reported in skeletal muscle from insulin resistant [34] and T2DM [15, 16] subjects. Similarly, correlations between muscular PGC-1α expression and insulin-stimulated glucose uptake and oxidation have been found in studies on monozygotic and dizygotic twins [35]. PGC-1α expression is also reduced with aging, providing a possible explanation for the link between aging and an increased susceptibility to develop T2DM [35]. Conversely, PGC-1α expression is restored by treatments known to normalize body weight and/or glucose homeostasis [36]
While these findings are mostly corelational, genetic association studies have supported a potential causal role for PGC-1α in the susceptibility to develop T2DM. The PGC-1α gene (PPARGC1A) is localized on chromosome 4p15.1-2 [37], a region that has been associated with basal insulin levels [38], abdominal subcutaneous fat [39] and obesity [40, 41] in different populations. Furthermore, single nucleotide polymorphisms on the PPARGC1A show a strong association to diabetes-related phenotypes [42-44]. Particularly, a possible association of a Gly482Ser PPARGC1A polymorphism with diabetes and its complications has drawn attention lately. Studies on diverse populations indicated that the Ser variant rendered individuals susceptible to develop T2DM in [42, 43, 45], but, even if in most populations studied there was a tendency to find a higher representation of the Ser variant in type 2 diabetic patients, not all studies have found significant associations [46-48]. The functional meaning of this substitution is currently unknown, even though it has been shown that the Ser variant was associated with an age-dependent reduction in muscle PGC-1α expression [35]. While the genetic evidence collected points towards a potential causal role of PGC-1α in the pathogenesis of T2DM, further research will be required to consolidate this possibility and provide a link between human genetic studies and PGC-1α biology.
Metabolic sensors and PGC-1a: sparks to the fire
The biology of PGC-1α and the regulation of its activity, however, are far from totally understood. The data presented above does not always take into account that PGC-1α activity is not only determined by its expression levels, but also by a number of post-translational modifications, such as phosphorylation [49-51], acetylation [52-54] and methylation [55], amongs others. As recently reviewed [56], these modifications can affect the intrinsic activity of PGC-1α, the stability of the PGC-1α protein or might regulate the interaction with other proteins that determine its activity, such as the corepressor p160MBP [57]. It has also been proposed that PGC-1α might be forming part of different complexes which would provide also ways to influence its activity [58-60]. Hence, understanding this regulatory conundrum of signals and complexes that regulate PGC-1α activity and specificity holds strong promise to improve the pharmacological efficiency in order to selectively target PGC-1α action.
Since PGC-1α has a prominent role in the metabolic adaptations to the energetic status, it should not be surprising that its activity might be targeted by cellular mechanisms capable of sensing perturbations in the metabolic status of the cell as well as the availability of substrates, and transform these inputs into a specific metabolic fate. These sensors would act as integrative nodes of cellular metabolism determining which enzymatic and transcriptional responses will be exerted in order to adapt to the environmental conditions. Such crucial sensing functions are normally performed by enzymes, that are conserved all along the evolutionary scale. Two of them, SIRT1 and AMPK play major roles in metabolic regulation and have recently been shown to impact on PGC-1α to transcriptionally regulate energy expenditure.
SIRT1 and the control of PGC-1α activity
SIRT1 is one of the mammalian homologs of yeast the Sir2 protein, the founding member of the sirtuin gene family. SIRT1 is an enzyme that mediates NAD+-dependent deacetylation of target substrates. Since the cellular redox balance of NAD+ and NADH is highly related to catabolic fluxes, it has been postulated that SIRT1 could act as a sensor that directly connects metabolic perturbations with transcriptional outputs, as it was initially characterized as a histone deacetylase [61]. The regulatory role of SIRT1 may, however, be far more complex, as its activity also highly depends on the intracellular levels of nicotinamide, the natural product of the deacetylase reaction catalyzed by sirtuins [62], as well as on post-translational modifications [63] or interaction with other proteins [64-66]. Additionally, the regulation of the intracellular localization of SIRT1 and how NAD+/NADH variations in distinct cellular compartments may affect its activity are issues yet to be fully understood.
While our understanding of mammalian SIRT1 biology is still surprisingly weak, there are a vast number of evidences suggesting that this enzyme has a major role in metabolic homeostasis. First of all, during the last decade, a number of reports have shown that SIRT1 is not just a histone deacetylase. In fact, SIRT1 can directly interact and regulate the activity of transcription factors and coregulators, including PPARγ [67], p53 [68] and the FOXO family of transcription factors [69], all of which are key regulators of metabolism in a variety of tissues. While there is a tendency to associate SIRT1-mediated deacetylation with transcriptional repression, this is not always the case. For example, in the case of FOXO transcription factors, deacetylation by SIRT1 seems to confer target gene specificity [69]. In other cases, such as with Tat-mediated transcription of the HIV long terminal repeats, SIRT1-mediated deacetylation is associated with positive regulation of gene expression [70]. Moreover, the realm of SIRT1 substrates might expand beyond transcriptional regulators, as has been demonstrated by the work showing that SIRT1 can directly regulate the activity of AcetylCoA syntethases through deacetylation [71]. Interestingly, SIRT1 can also directly interact and deacetylate PGC-1α [52].
The regulation of PGC-1α acetylation merits some discussion, as the deacetylation of PGC-1α is tightly linked with enhanced PGC-1α transcriptional activation and mutation of the acetylation sites to arginine, which mimics the deacetylated state, markedly increases basal PGC-1α transcriptional activity [52]. Conversely, acetyltransferase enzymes such as GCN5 [72] and SRC-3 [73] have been demonstrated to inhibit PGC-1α activity by increasing its acetylation. This yin yang between acetyltransferases and deacetylases in the control of PGC-1α activity seems a recurring theme (Figure 1). In fact, the expression of SRC-3 and GCN5 increases upon high fat feeding, while SIRT1 expression diminishes, providing a plausible explanation to why PGC-1α is hyperacetylated and the expression of PGC-1α–dependent genes is downregulated in this state [73]. The converse situation, with an induction of SIRT1 and reduction of GCN5 and SRC3, occurs upon fasting and dietary restriction, now inducing PGC-1α activity [73]. The changes in the expression levels of deacetylases and acetyltransferases will result in a convergent regulation of PGC-1α acetylation status in conditions when energy levels change. Together, these data indicate that acetylation of PGC-1α has a major influence on its activity, and, consequently, the acetyltransferase and deacetylase enzymes modulating this process may heavily impact on whole-body metabolism.
Figure 1. A metabolic sensor network regulating energy expenditure.
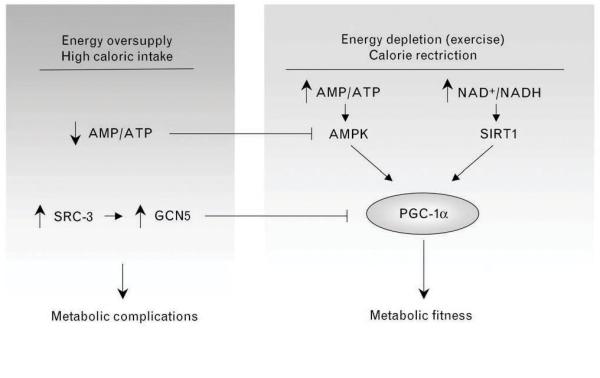
Situations of energy depletion and/or decreased catabolic rates can be sensed by different enzymes, such as AMPK and SIRT1, whose activation enhance PGC-1α-dependent transcription. Upon calorie rich diets or situations when energy is not limited, AMPK activity is shut down by the high intracellular ATP levels. Similarly, high fat diets increase SRC-3, who positively regulates the protein levels of the acetyltransferase GCN5, which, in turn, plays the opposite role of SIRT1 action on PGC-1α acetylation, hence diminishing PGC-1α transcriptional activity. Perturbations in this metabolic network controlling PGC-1α activity may importantly contribute to whole-body metabolic complications.
In vivo experiments support a major role for SIRT1 as a metabolic regulator. Several lines of evidence indicate that SIRT1-mediated regulation of PGC-1α activity may play a major role in the metabolic adaptations to energy metabolism in different tissues [52, 54, 74, 75]. In liver, SIRT1 is known to control gluconeogenic activity by modulation of PGC-1α [75] and CREB regulated trancription coactivator 2 (CRTC2) [76], In vivo treatment with SIRT1 agonists promotes deacetylation of PGC-1α in skeletal muscle and brown adipose tissue, which translates into enhanced mitochondrial activity, which, in turn, improves exercise performance and thermogenic activity [54]. Unfortunately, the current SIRT1 knock-out models display several developmental defects [77, 78], they are smaller at birth and show elevated postnatal lethality, making it impossible to evaluate the metabolic role of SIRT1 and its impact on PGC-1α activity and energy expenditure. However, mice models having a mild-overexpression of SIRT1 present ameliorated glucose tolerance when insulin resistance and/or diabetes are induced [79]. A second model mice overexpressing SIRT1 also displayed enhanced glucose tolerance linked to increased metabolic rates [80]. Similarly, treatment with different SIRT1 agonists prevents weight gain and insulin resistance when mice are challenged with high-fat diets [54, 81]. Altogether, these results highlight a role for SIRT1 in the control of metabolic homeostasis. Given that situations of energy deficiency, such as fasting, promote increased intracellular NAD+ levels [52, 74, 82], it seems plausible that such conditions might affect SIRT1 activity, which would result in the deacetylation of PGC-1α and an increase its transcriptional activity. This mechanism would allow the cell to increase mitochondrial respiration and meet the energetic requirements of the cell in circumstances of energy stress. While the finding of a link between SIRT1 and PGC-1α represented a promising breakthrough in our understanding of how the cell transcriptionally regulates energy metabolism, the edges of most of the concepts underlying this link need to be mechanistically polished. For example, the SIRT1/PGC-1α axis is not likely to respond similarly in all tissues upon energy stress [82]. Similarly, how SIRT1 interacts with PGC-1α upon energy stress and whether SIRT1 displays any regulation at the level of substrate specificity upon activation, are major issues yet to be solved.
AMPK and the link of PGC-1a activity to energy status
As stated in a recent review [83], AMP-activated protein kinase (AMPK) is a conserved fuel-gauge that has probably played a major role in the maintainance of intracellular energy balance during eukaryotic evolution. Mammalian AMPK is a Ser/Thr kinase that is activated upon alterations in the cellular AMP/ATP ratio. Hence, perturbations in this ratio due to either defects in energy production or increased energy consumption will activate the kinase. Once activated, AMPK switches on catabolic pathways to produce ATP while simultaneously shutting down energy-consuming anabolic processes. In order to perform these actions, AMPK can quickly regulate metabolic enzymes through direct phosphorylation, but, additionally, AMPK also has long-term effects at the transcriptional level in order to adapt gene expression to energy demands. Hence, upon energy deficiency, AMPK will enhance the expression of genes related to glucose transport and glycolysis [83, 84] and mitochondrial respiration [85] while down-regulating lipid synthesis genes [86].
AMPK activation is highly relevant for the transcriptional adaptation to physiological situations of energy demand. Mice expressing a dominant-negative form of AMPK cannot increase mitochondrial biogenesis in response to energy deprivation in skeletal muscle [87]. Similarly, mice where the predominantly muscular γ3 subunit of AMPK has been knocked-out, hence blunting AMPK activation in muscle, show impaired fasting-induced expression of lipid oxidative genes [88], as well as impaired expression of genes induced by exercise [89]. In contrast, in mice overexpressing an activated form of the AMPKγ3 subunit the expression of genes controlling lipid oxidation and mitochondrial activity is induced [88-90]. Furthermore, muscles from mice that overexpress the active form of the AMPKγ3 subunit were protected from diet-induced insulin resistance [91] and fatigue resistance [89]. Finally, it has recently been shown that mice fed with a AMPK agonists display increased oxidative gene expression, enhancement in their endurance capacity and protection against metabolic disease [92, 93]. Therefore, AMPK constitutes a major regulator of basal mitochondrial gene expression as well as mitochondrial gene expression upon energy stress.
Interestingly, there is a strong overlap in the genes transcriptionally regulated by AMPK and those by PGC-1α, hence suggesting that PGC-1α might be an important mediator AMPK-induced gene expression. Supporting this hypothesis, AMPK activation leads to increased PGC-1α expression [94, 95], and AMPK requires PGC-1α activity to modulate the expression of several key players in mitochondrial and glucose metabolism [50]. However, a closer link has been provided by recent findings showing that AMPK can directly interact and phosphorylate PGC-1α [50]. Direct phosphorylation of PGC-1α by AMPK seems to increase transcriptional activity of PGC-1α, even though the reasons why, where, and how that happens are still elusive. Phosphorylation of PGC-1α by AMPK may, hence, be part of the link between the sensing of the energetic status and the induction of transcriptional programs that control energy expenditure, even though how it interacts with other modifications, such as the above-mentioned PGC-1α acetylation, is still unknown.
Conclusion
A decade after its cloning, and with hundreds of research articles on its back, we might still be at the tip of the iceberg in our understanding of PGC-1α biology. There are major caveats on how PGC-1α transcriptional activity and target gene-sets are specifically regulated upon different circumstances. While a few possible mechanisms have been described, whether and how PGC-1α might act is largely unknown. Nonetheless, the finding that SIRT1 and AMPK impact on PGC-1α furthers our knowledge on how information on the cellular metabolic status is transmitted to PGC-1α and how it adapts transcriptional outputs (Figure 1). Albeit promising, these links are still weak. While there is considerable data supporting that SIRT1 interacts with PGC-1α and promotes its transcriptional activity through deacetylation, the evidence that SIRT1 is a true physiological metabolic sensor is far from conclusive. Furthermore, it is very plausible that additional acetyltransferases and deacetylases, yet to be identified, affect PGC-1α activity. As to AMPK, which is unequivocally defined as a metabolic sensor, the evidence showing that AMPK can activate PGC-1α through direct phosphorylation derives mostly from in vitro assays and further validation in vivo will be important. Given the possible influence of phosphorylation on PGC-1α activity, it will similarly be important to identify the mechanisms governing dephosphorylation of PGC-1α. Additionally, any further implications caused by these post-translational modifications of PGC-1α, such as selective interaction with transcription factors and cofactors or modifying its intracellular localization, still require further study. Similarly, whether perturbations in the acetylation or phosphorylation levels of PGC-1α could be relevant in the development of insulin resistance and T2DM is a question yet to be answered. The combination of such promising links and an exhaustive list of unexplored questions, hence, warrant exciting research for the upcoming years.
Acknowledgements
The work in the laboratory of the authors is sponsored by the Ecole Polytechnique Fédérale de Lausanne and an advanced research award by the European Research Council “Ideas” program (231138-Sirtuins).
References and recommended reading
- [1].DeFronzo RA, Simonson D, Ferrannini E. Hepatic and peripheral insulin resistance: a common feature of type 2 (non-insulin-dependent) and type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1982;23:313–9. doi: 10.1007/BF00253736. [DOI] [PubMed] [Google Scholar]
- [2].Lillioja S, Mott DM, Howard BV, et al. Impaired glucose tolerance as a disorder of insulin action. Longitudinal and cross-sectional studies in Pima Indians. N Engl J Med. 1988;318:1217–25. doi: 10.1056/NEJM198805123181901. [DOI] [PubMed] [Google Scholar]
- [3].Kelley DE, Simoneau JA. Impaired free fatty acid utilization by skeletal muscle in non-insulin-dependent diabetes mellitus. J Clin Invest. 1994;94:2349–56. doi: 10.1172/JCI117600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].He J, Watkins S, Kelley DE. Skeletal muscle lipid content and oxidative enzyme activity in relation to muscle fiber type in type 2 diabetes and obesity. Diabetes. 2001;50:817–23. doi: 10.2337/diabetes.50.4.817. [DOI] [PubMed] [Google Scholar]
- [5].Simoneau JA, Kelley DE. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. J Appl Physiol. 1997;83:166–71. doi: 10.1152/jappl.1997.83.1.166. [DOI] [PubMed] [Google Scholar]
- [6].Bachmann OP, Dahl DB, Brechtel K, et al. Effects of intravenous and dietary lipid challenge on intramyocellular lipid content and the relation with insulin sensitivity in humans. Diabetes. 2001;50:2579–84. doi: 10.2337/diabetes.50.11.2579. [DOI] [PubMed] [Google Scholar]
- [7].Boden G, Lebed B, Schatz M, et al. Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes. 2001;50:1612–7. doi: 10.2337/diabetes.50.7.1612. [DOI] [PubMed] [Google Scholar]
- [8].Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–11. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- [9].Kelley DE, Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes. 2000;49:677–83. doi: 10.2337/diabetes.49.5.677. [DOI] [PubMed] [Google Scholar]
- [10].Petersen KF, Befroy D, Dufour S, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–2. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Petersen KF, Dufour S, Befroy D, et al. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–71. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Szendroedi J, Schmid AI, Chmelik M, et al. Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. PLoS Med. 2007;4:e154. doi: 10.1371/journal.pmed.0040154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schrauwen-Hinderling VB, Kooi ME, Hesselink MK, et al. Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and BMI-matched control subjects. Diabetologia. 2007;50:113–20. doi: 10.1007/s00125-006-0475-1. [DOI] [PubMed] [Google Scholar]
- [14].Rabol R, Boushel R, Dela F. Mitochondrial oxidative function and type 2 diabetes. Appl Physiol Nutr Metab. 2006;31:675–83. doi: 10.1139/h06-071. [DOI] [PubMed] [Google Scholar]
- [15].Patti ME, Butte AJ, Crunkhorn S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–71. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- [17].Tanner CJ, Barakat HA, Dohm GL, et al. Muscle fiber type is associated with obesity and weight loss. Am J Physiol Endocrinol Metab. 2002;282:E1191–6. doi: 10.1152/ajpendo.00416.2001. [DOI] [PubMed] [Google Scholar]
- [18].Hom FG, Goodner CJ. Insulin dose-response characteristics among individual muscle and adipose tissues measured in the rat in vivo with 3[H]2-deoxyglucose. Diabetes. 1984;33:153–9. doi: 10.2337/diab.33.2.153. [DOI] [PubMed] [Google Scholar]
- [19].DeFronzo RA, Jacot E, Jequier E, et al. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes. 1981;30:1000–7. doi: 10.2337/diab.30.12.1000. [DOI] [PubMed] [Google Scholar]
- [20].Puigserver P, Wu Z, Park CW, et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–39. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- [21].Feige JN, Auwerx J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol. 2007;17:292–301. doi: 10.1016/j.tcb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- [22].Wu Z, Puigserver P, Andersson U, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–24. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- [23].St-Pierre J, Lin J, Krauss S, et al. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J Biol Chem. 2003;278:26597–603. doi: 10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- [24].Lehman JJ, Barger PM, Kovacs A, et al. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–56. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–76. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang YX, Lee CH, Tiep S, et al. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell. 2003;113:159–70. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- [27].Huss JM, Kopp RP, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J Biol Chem. 2002;277:40265–74. doi: 10.1074/jbc.M206324200. [DOI] [PubMed] [Google Scholar]
- [28].Schreiber SN, Knutti D, Brogli K, et al. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha) J Biol Chem. 2003;278:9013–8. doi: 10.1074/jbc.M212923200. [DOI] [PubMed] [Google Scholar]
- [29].Michael LF, Wu Z, Cheatham RB, et al. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci U S A. 2001;98:3820–5. doi: 10.1073/pnas.061035098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Puigserver P, Rhee J, Donovan J, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–5. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- [31].Mootha VK, Handschin C, Arlow D, et al. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci U S A. 2004;101:6570–5. doi: 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lin J, Wu H, Tarr PT, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- *[33].Handschin C, Chin S, Li P, et al. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J Biol Chem. 2007;282:30014–21. doi: 10.1074/jbc.M704817200. Avoiding all the complications derived from the whole-body PGC-1α knock-out mice, the muscle-specific approach described in this manuscript clearly highlights the relevance of PGC-1α in the regulation of skeletal muscle oxidative capacity.
- [34].Richardson DK, Kashyap S, Bajaj M, et al. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J Biol Chem. 2005;280:10290–7. doi: 10.1074/jbc.M408985200. [DOI] [PubMed] [Google Scholar]
- [35].Ling C, Poulsen P, Carlsson E, et al. Multiple environmental and genetic factors influence skeletal muscle PGC-1alpha and PGC-1beta gene expression in twins. J Clin Invest. 2004;114:1518–26. doi: 10.1172/JCI21889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mensink M, Hesselink MK, Russell AP, et al. Improved skeletal muscle oxidative enzyme activity and restoration of PGC-1 alpha and PPAR beta/delta gene expression upon rosiglitazone treatment in obese patients with type 2 diabetes mellitus. Int J Obes (Lond) 2007;31:1302–10. doi: 10.1038/sj.ijo.0803567. [DOI] [PubMed] [Google Scholar]
- [37].Esterbauer H, Oberkofler H, Krempler F, Patsch W. Human peroxisome proliferator activated receptor gamma coactivator 1 (PPARGC1) gene: cDNA sequence, genomic organization, chromosomal localization, and tissue expression. Genomics. 1999;62:98–102. doi: 10.1006/geno.1999.5977. [DOI] [PubMed] [Google Scholar]
- [38].Pratley RE, Thompson DB, Prochazka M, et al. An autosomal genomic scan for loci linked to prediabetic phenotypes in Pima Indians. J Clin Invest. 1998;101:1757–64. doi: 10.1172/JCI1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Perusse L, Rice T, Chagnon YC, et al. A genome-wide scan for abdominal fat assessed by computed tomography in the Quebec Family Study. Diabetes. 2001;50:614–21. doi: 10.2337/diabetes.50.3.614. [DOI] [PubMed] [Google Scholar]
- [40].Stone S, Abkevich V, Hunt SC, et al. A major predisposition locus for severe obesity, at 4p15-p14. Am J Hum Genet. 2002;70:1459–68. doi: 10.1086/340670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Arya R, Duggirala R, Jenkinson CP, et al. Evidence of a novel quantitative-trait locus for obesity on chromosome 4p in Mexican Americans. Am J Hum Genet. 2004;74:272–82. doi: 10.1086/381717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ek J, Andersen G, Urhammer SA, et al. Mutation analysis of peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC-1) and relationships of identified amino acid polymorphisms to Type II diabetes mellitus. Diabetologia. 2001;44:2220–6. doi: 10.1007/s001250100032. [DOI] [PubMed] [Google Scholar]
- [43].Andrulionyte L, Zacharova J, Chiasson JL, Laakso M. Common polymorphisms of the PPAR-gamma2 (Pro12Ala) and PGC-1alpha (Gly482Ser) genes are associated with the conversion from impaired glucose tolerance to type 2 diabetes in the STOP-NIDDM trial. Diabetologia. 2004;47:2176–84. doi: 10.1007/s00125-004-1577-2. [DOI] [PubMed] [Google Scholar]
- [44].Bhat A, Koul A, Rai E, et al. PGC-1alpha Thr394Thr and Gly482Ser variants are significantly associated with T2DM in two North Indian populations: a replicate case-control study. Hum Genet. 2007;121:609–14. doi: 10.1007/s00439-007-0352-0. [DOI] [PubMed] [Google Scholar]
- [45].Hara K, Tobe K, Okada T, et al. A genetic variation in the PGC-1 gene could confer insulin resistance and susceptibility to Type II diabetes. Diabetologia. 2002;45:740–3. doi: 10.1007/s00125-002-0803-z. [DOI] [PubMed] [Google Scholar]
- [46].Oberkofler H, Linnemayr V, Weitgasser R, et al. Complex haplotypes of the PGC-1alpha gene are associated with carbohydrate metabolism and type 2 diabetes. Diabetes. 2004;53:1385–93. doi: 10.2337/diabetes.53.5.1385. [DOI] [PubMed] [Google Scholar]
- [47].Lacquemant C, Chikri M, Boutin P, et al. No association between the G482S polymorphism of the proliferator-activated receptor-gamma coactivator-1 (PGC-1) gene and Type II diabetes in French Caucasians. Diabetologia. 2002;45:602–3. doi: 10.1007/s00125-002-0783-z. author reply 604. [DOI] [PubMed] [Google Scholar]
- [48].Stumvoll M, Fritsche A, t’Hart LM, et al. The Gly482Ser variant in the peroxisome proliferator-activated receptor gamma coactivator-1 is not associated with diabetes-related traits in non-diabetic German and Dutch populations. Exp Clin Endocrinol Diabetes. 2004;112:253–7. doi: 10.1055/s-2004-817972. [DOI] [PubMed] [Google Scholar]
- [49].Puigserver P, Rhee J, Lin J, et al. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol Cell. 2001;8:971–82. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- *[50].Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–22. doi: 10.1073/pnas.0705070104. This manuscript describes for the first time a possible effect of AMPK on PGC-1α activity through direct interaction and phosphorylation.
- [51].Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447:1012–6. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- [52].Rodgers JT, Lerin C, Haas W, et al. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–8. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- [53].Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–60. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- [54].Lagouge M, Argmann C, Gerhart-Hines Z, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–22. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- [55].Teyssier C, Ma H, Emter R, et al. Activation of nuclear receptor coactivator PGC-1alpha by arginine methylation. Genes Dev. 2005;19:1466–73. doi: 10.1101/gad.1295005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fan M, Rhee J, St-Pierre J, et al. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: modulation by p38 MAPK. Genes Dev. 2004;18:278–89. doi: 10.1101/gad.1152204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Cooper MP, Qu L, Rohas LM, et al. Defects in energy homeostasis in Leigh syndrome French Canadian variant through PGC-1alpha/LRP130 complex. Genes Dev. 2006;20:2996–3009. doi: 10.1101/gad.1483906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Picard F, Gehin M, Annicotte J, et al. SRC-1 and TIF2 control energy balance between white and brown adipose tissues. Cell. 2002;111:931–41. doi: 10.1016/s0092-8674(02)01169-8. [DOI] [PubMed] [Google Scholar]
- [60].Puigserver P, Adelmant G, Wu Z, et al. Activation of PPARgamma coactivator-1 through transcription factor docking. Science. 1999;286:1368–71. doi: 10.1126/science.286.5443.1368. [DOI] [PubMed] [Google Scholar]
- [61].Lin SJ, Ford E, Haigis M, et al. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004;18:12–6. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Anderson RM, Bitterman KJ, Wood JG, et al. Manipulation of a nuclear NAD+ salvage pathway delays aging without altering steady-state NAD+ levels. J Biol Chem. 2002;277:18881–90. doi: 10.1074/jbc.M111773200. [DOI] [PubMed] [Google Scholar]
- [63].Yang Y, Fu W, Chen J, et al. SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat Cell Biol. 2007;9:1253–62. doi: 10.1038/ncb1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–6. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- [65].Zhao W, Kruse JP, Tang Y, et al. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–90. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell. 2007;28:277–90. doi: 10.1016/j.molcel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- [67].Picard F, Kurtev M, Chung N, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–6. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Vaziri H, Dessain SK, Ng Eaton E, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–59. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- [69].Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–5. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- [70].Pagans S, Pedal A, North BJ, et al. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 2005;3:e41. doi: 10.1371/journal.pbio.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A. 2006;103:10230–5. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lerin C, Rodgers JT, Kalume DE, et al. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006;3:429–38. doi: 10.1016/j.cmet.2006.04.013. [DOI] [PubMed] [Google Scholar]
- **[73].Coste A, Louet JF, Lagouge M, et al. The genetic ablation of SRC-3 protects against obesity and improves insulin sensitivity by reducing the acetylation of PGC-1{alpha} Proc Natl Acad Sci U S A. 2008;105:17187–92. doi: 10.1073/pnas.0808207105. This work describes how mice lacking SRC-3 have a major metabolic phenotype due to an impact on PGC-1α action, in a mechanism implying GCN5. Similarly, it shows how modifications in the acetylation status of PGC-1α upon high fat diet and fasting periods may rely on the opposite actions of SRC-3/GCN5 and SIRT1.
- [74].Gerhart-Hines Z, Rodgers JT, Bare O, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. Embo J. 2007;26:1913–23. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Rodgers JT, Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci U S A. 2007;104:12861–6. doi: 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *[76].Liu Y, Dentin R, Chen D, et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456:269–73. doi: 10.1038/nature07349. This paper identifiying how a cofactor network controlled by SIRT1 modulates gluconeogenesis.
- [77].Cheng HL, Mostoslavsky R, Saito S, et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794–9. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].McBurney MW, Yang X, Jardine K, et al. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003;23:38–54. doi: 10.1128/MCB.23.1.38-54.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *[79].Banks AS, Kon N, Knight C, et al. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–41. doi: 10.1016/j.cmet.2008.08.014. Together with the reference below, this work describes how a moderate SIRT1 gain of function may protects against metabolic disease in various models of insulin resistance.
- [80].Bordone L, Cohen D, Robinson A, et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007;6:759–67. doi: 10.1111/j.1474-9726.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- [81].Milne JC, Lambert PD, Schenk S, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–6. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *[82].Chen D, Bruno J, Easlon E, et al. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753–7. doi: 10.1101/gad.1650608. This is an interesting study that shows how SIRT1 expression is differentially regulated in a tissue-specific manner upon calorie restriction, which may reflect tissue-specific roles of SIRT1 and how SIRT1 targeting should be refined for optimal pharmacological use.
- [83].Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–85. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- [84].Ojuka EO, Nolte LA, Holloszy JO. Increased expression of GLUT-4 and hexokinase in rat epitrochlearis muscles exposed to AICAR in vitro. J Appl Physiol. 2000;88:1072–5. doi: 10.1152/jappl.2000.88.3.1072. [DOI] [PubMed] [Google Scholar]
- [85].Winder WW, Holmes BF, Rubink DS, et al. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol. 2000;88:2219–26. doi: 10.1152/jappl.2000.88.6.2219. [DOI] [PubMed] [Google Scholar]
- [86].Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Zong H, Ren JM, Young LH, et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–7. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Long YC, Barnes BR, Mahlapuu M, et al. Role of AMP-activated protein kinase in the coordinated expression of genes controlling glucose and lipid metabolism in mouse white skeletal muscle. Diabetologia. 2005;48:2354–64. doi: 10.1007/s00125-005-1962-5. [DOI] [PubMed] [Google Scholar]
- [89].Barnes BR, Long YC, Steiler TL, et al. Changes in exercise-induced gene expression in 5′-AMP-activated protein kinase gamma3-null and gamma3 R225Q transgenic mice. Diabetes. 2005;54:3484–9. doi: 10.2337/diabetes.54.12.3484. [DOI] [PubMed] [Google Scholar]
- *[90].Garcia-Roves PM, Osler ME, Holmstrom MH, Zierath JR. Gain-of-function R225Q mutation in AMP-activated protein kinase gamma 3 subunit increases mitochondrial biogenesis in glycolytic skeletal muscle. J Biol Chem. 2008 doi: 10.1074/jbc.M805078200. This manuscript describes how an AMPK gain of function mutation is enough to basally increase mitochondrial content in vivo.
- [91].Barnes BR, Marklund S, Steiler TL, et al. The 5′-AMP-activated protein kinase gamma3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J Biol Chem. 2004;279:38441–7. doi: 10.1074/jbc.M405533200. [DOI] [PubMed] [Google Scholar]
- **[92].Narkar VA, Downes M, Yu RT, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–15. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **[93].Cool B, Zinker B, Chiou W, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–16. doi: 10.1016/j.cmet.2006.05.005. Together with the above manuscript, these works provide evidence that treatment with AMPK agonists in mice can protect agains metabolic syndrome, increase mitochondrial content and enhance endurance capacities.
- [94].Suwa M, Nakano H, Kumagai S. Effects of chronic AICAR treatment on fiber composition, enzyme activity, UCP3, and PGC-1 in rat muscles. J Appl Physiol. 2003;95:960–8. doi: 10.1152/japplphysiol.00349.2003. [DOI] [PubMed] [Google Scholar]
- [95].Terada S, Goto M, Kato M, et al. Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochem Biophys Res Commun. 2002;296:350–4. doi: 10.1016/s0006-291x(02)00881-1. [DOI] [PubMed] [Google Scholar]