NFAT Gene Family in Inflammation and Cancer (original) (raw)
. Author manuscript; available in PMC: 2014 May 1.
Abstract
Calcineurin-NFAT signaling is critical for numerous aspects of vertebrate function during and after embryonic development. Initially discovered in T cells, the NFAT gene family, consisting of five members, regulates immune system, inflammatory response, angiogenesis, cardiac valve formation, myocardial development, axonal guidance, skeletal muscle development, bone homeostasis, development and metastasis of cancer, and many other biological processes. In this review we will focus on the NFAT literature relevant to the two closely related pathological systems: inflammation and cancer.
Keywords: NFAT, calcineurin, nuclear import, nuclear export, inflammation, cancer
Regulation of calcineurin-NFAT signaling
NFAT (Nuclear Factor of Activated T cell) proteins were first discovered in T-cells as transcriptional activators of interleukin-2 [1, 2], a key regulator of T cell immune response. The immunosuppressant cyclosporine A (CsA) and FK506 (Tacrolimus) inhibit the NFAT pathway to reduce rejection in patients receiving organ transplantation. More than two decades after the discovery, the NFAT gene family is found to play critical roles in many other biological systems of vertebrates.
NFAT proteins are regulated by the phosphatase calcineurin that dephosphorylates NFAT proteins to expose their nuclear localization signals, thus triggering the transport of NFAT proteins from the cytoplasm to the nucleus. Once in the nucleus, NFAT proteins collaborate with other factors to control target gene expression, essential for many biological functions. Calcineurin responds to sustained rise of intracellular calcium level, and NFAT proteins are rapidly imported into or exported from the nucleus, depending on the calcium level and activity of calcineurin. Such mechanism determines that NFAT responds only to a sustained elevation of intracellular calcium level, but not to a transient rise of calcium, to maintain its nuclear presence for the duration needed for transcriptional action. This sets NFAT apart from many regulators that respond to transient calcium signaling [3–6]. Furthermore, the weak DNA binding property of NFAT requires that NFAT partners with other factors to execute transcription regulation [4–6]. For example, NFAT partners with GATA to control heart development, with FOXP3 to regulate immune tolerance, with AP-1 to trigger T cell response, and with MEF to control muscle development [4]. Because of the ability to respond to calcium/calcineurin and to partner with a variety of transcription regulators, NFAT provides a powerful, versatile and delicate tool to control many aspects of the developmental and cellular events.
There are four classic members in the NFAT gene family: NFATc1 (NFATc or NFAT2), NFATc2 (NFATp or NFAT1), NFATc3 (NFAT4), and NFATc4 (NFAT3). In contrast to the classic NFATc proteins, NFAT5 (also known as tonicity enhancer binding protein) does not require calcineurin or a nuclear partner for its activity. In this review, we will focus on the classic NFAT proteins. The classic NFAT protein is composed of a number of functional modules that dictate the protein’s phosphorylation, nuclear localization, DNA binding, and transactivation of target genes (Figure 1). The N-terminal region contains regulatory domains, including the casein kinase 1 (CK1) docking site, the transactivation domain (TAD), and a calcineurin (Cn) docking site. The C-terminus contains the DNA-binding Rel Homology Domain (RHD) and an additional calcineurin docking site. In the middle of the protein are several serine-rich domains (SRR, SP1-3) that provide phosphorylation sites for kinases targeting NFAT. Furthermore, the NFAT protein contains two signal sequences that regulate its subcellular localization: the nuclear localization signal sequences (NLS1 and NLS2) and the nuclear export signal (NES).
Figure 1.
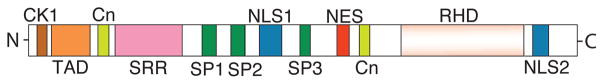
Sketch of NFAT Structural Features. The structure is based on NFATc1 which contains an N-terminal transactivation domain (TAD), an N-terminal Casein Kinase 1 (CK1) docking site, two calcineurin (Cn) docking domains (one in the N-terminus and another in the C-terminus), one Serine-Rich Region (SRR), three SP repeat motifs, two Nuclear Localization Signal (NLS) sequences, one Nuclear Export Signal (NES), and a Rel Homology Domain (RHD) at the C-terminus.
When NFAT proteins are heavily phosphorylated on the serine residues in the SRR and SP regions, the proteins are confined to the cytoplasm [5, 6]. The activation of cell surface receptors such as T cell receptor (TCR), receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs), leads to a signaling cascade (Figure 2) that activates phosphoinositide phospholipase C (PLC) for the cleavage of membrane-bound phosphatidylinositol 4,5-bisphoaphate (PIP2) to generate diacyl glycerol (DAG) and inositol-1,4,5-trisphosphate (IP3). DAG activates protein kinase C (PKC), whereas IP3 interacts with its receptor (IP3R) on the surface of the endoplasmic reticulum (ER). IP3R activity causes calcium efflux from the ER and subsequent depletion of the ER-stored calcium, thus activating the calcium sensor protein STIM1 [7–9]. STIM1 proteins then form oligomers and move to the junction between ER and the plasma membrane, where the STIMI oligomers bind to the calcium release-activated calcium (CRAC) channel Orai1, inducing a sustained influx of calcium and the activation of calcineurin. Activated calcineurin then dephosphorylates the cytoplasmic NFAT proteins, causing them to rapidly move into the nucleus [10–13]. The calcineurin activity can be inhibited by the immunosuppressant CsA and FK506, which form CsA–cyclophilin A and FK506–FK506 binding protein 12 (FKBP12) complexes, respectively, to bind and competitively inhibit calcineurin phosphatase activity [14].
Figure 2.

Activation of the calcineurin-NFAT pathway. Upon receptor activation (TCR, RTKs, GPCRs, etc.), phospholipase C (PLC) is activated. Activated PLC catalyzes the hydrolysis of phosphatidylinositol-4,5-bisphosphonate (PIP2) to inisitol-1,4,5-triphosphate (IP3) and diacyl glycerol (DAG). IP3 binds to IP3R on the surface of the endoplasmic reticulum (ER), resulting in a brief efflux of calcium from the ER to the cytoplasm, transiently raising the calcium level in the cytoplasm but depleting the calcium store in the ER. The depletion of store calcium causes the calcium sensor STIM on the ER to form oligomers and translocate to the ER-plasma membrane junction where they bind to and activate Orai (CRAC channel) on the plasma membrane. Orai forms a tetramer channel and opens a sustained oscillating low amplitude influx of calcium that binds to the calcium sensor calmodulin (CAM) in the cytoplasm, forming a calcium/calmodulin/calcineurin complex, and activating calcineurin [7–13]. Calcineurin has a catalytic (CnA) and a regulatory subunit (CnB). Activated CnA dephosphorylates NFAT proteins and unmasks their NLSs, leading to their nuclear translocation [7–13].
There are fourteen phosphorylation sites in the NFAT regulatory domain, and this high number of phosphorylation sites illustrates the importance of phosphorylation and dephosphorylation for controlling NFAT function through nuclear shuttling [15]. Dephosphorylation of serine residues by calcineurin unmasks the nuclear localization signals (NLS) [16], whereas re-phosphorylation of these serine residues masks the NLS sequences and exposes the nuclear export signal (NES). Calcineurin dephosphorylates 13 of the 14 phosphorylation sites of NFAT, thus triggering NFAT to translocate to the nucleus. Calcineurin activity is regulated by several physiological inhibitors, including calcineurin homologous protein (CHP), a CnB homolog [17], DSCR, Down Syndrome’s Candidate Region (also called RCAN, regulator of calcineurin) [18–20], DYRK 1 and 2 (dual-specificity tyrosine (Y)-phosphorylation-regulated protein kinase 1 and 2) [20], AKAP79 that binds to CnA subunit [21], and Cabin (CAIN) [22] (Figure 3). To counteract NFAT dephosphorylation and nuclear localization, several kinases act to phosphorylate NFAT proteins to reduce their nuclear occupancy by either decreasing their nuclear import or increasing their nuclear export. For example, Protein kinase A (PKA) serves as a priming kinase by phosphorylating NFATc1 on Ser269 to prepare its regulatory domain for further phosphorylation by GSK3, and this two-step phosphorylation process is crucial for the efficient nuclear export of NFAT proteins to terminate their transcriptional activity [23, 24]. PKA also phosphorylates NFATc4 on Ser289 to prepare it for forming a complex with 14-3-3 which maintains it in the cytosol in an inactive state, thus also functioning as a maintenance kinase of NFAT [25]. DYRK1 and 2 are priming kinases that phosphorylate NFATc2 on its SP3 region, preparing its subsequent phosphorylation by GSK3 and casein kinase 1 (CK1) on the SRR motif for export [26, 27]. CK1 and MEKK1 phosphorylate NFATc3 to mask its NLS and suppress its nuclear import [28], thus CK1 could serve as an export and a maintenance kinase. JNK phosphorylates NFATc1 and NFATc3 in T cells to facilitate nuclear export [29, 30]. while p38MAPK phosphorylates NFAT c2 and NFATc4 in adipocyte to block its nuclear accumulation [31,32].
Figure 3.

Regulation of the calcineurin-NFAT pathway. Calcium influx activates calcineurin. Calcineurin is negatively regulated by several endogenous inhibitors including the calcineurin homologous protein (CHP), a CnB homolog [17], DSCR, Down Syndrome’s Candidate Region (also called RCAN, regulator of calcineurin) [18–20], DYRK 1 and 2 (dual-specificity tyrosine (Y)-phosphorylation-regulated protein kinase 1 and 2) [20], AKAP79 that binds to the CnA subunit [21], and Cabin (CAIN) [22]. The immunosuppressant CsA and FK506 form CsA–cyclophilin A (Cy) and FK506–FB506 binding protein (FKBP) complexes, respectively, to bind and competitively inhibit calcineurin phosphatase activity [14]. NFAT is negatively regulated by several kinases including Protein kinase A (PKA) that serves as a priming kinase to phosphorylate NFATc1 to prepare its regulatory domain for further phosphorylation by GSK3 [23,24]. PKA also functions as a maintenance kinase to phosphorylates NFATc4 for forming a complex with 14-3-3 and maintain it in the cytosol in an inactive state [25]. DYRK1 and 2 are priming kinases that phosphorylate NFATc2 on its SP3 region and prepare its subsequent phosphorylation by GSK3 and casein kinase 1 (CK1) on the SRR motif for export [26,27]. CK1 and MEKK1 phosphorylate NFATc3 to mask its NLS and suppress its nuclear import [28], while JNK and p38MAPK serve as export kinases to phosphorylate NFAT for facilitating nuclear export [29–32]. NFATc1 can autoamplify its own transcription and also stimulate the transcription of NFATc2 [50]. NFAT proteins partner with other transcription factors to stimulate the expression of their target genes.
NFAT proteins are also regulated by several other mechanisms including sumoylation, proteasome degradation and RNA-protein scaffolding complex [33–35]. Several NFAT review articles over the past few years have covered many areas in details [3–6]. Here we will focus on the literature relevant to NFAT in two pathological aspects that impact human health and are closely related to each other: inflammation and cancer.
NFAT and inflammation
Although the role of NFAT in immune regulation is well established, our knowledge of NFAT in human diseases remains limited. The best clinical knowledge of NFAT resides in the regulation of T cells in organ rejection and the critical importance of cyclosporine and FK506 in preventing rejection. The functions of NFAT in other aspects of human immune or inflammatory diseases are largely unknown.
NFAT and inflammatory bowel disease (IBD)
There are two types of IBD, Crohn’s Disease and Ulcerative Colitis, both of which are associated with genetic susceptibility and environmental factors. The mainstay of treatment is the control of bowel inflammation to minimize symptoms through the use of immunosuppressants (such as steroid, azathioprine, and 6-mercaptopurine) and anti-TNFa therapy [36].
NFAT is important in modulating the inflammation of IBD. Genome-wide association studies have identified more than 70 susceptibility loci for IBD, including one residing in the gene that encodes LRRK2 (leucine-rich repeat kinase 2) [37]. LRRK2 inhibits nuclear translocation of NFATc2 by increasing the association of NFATc2 with its negative regulator NRON (Negative non-coding RNA Repressor of NFAT), which is a long non-coding RNA that holds NFATc2 protein in the cytoplasm [37]. In the LRRK2-deficient mice, NFATc2 has higher nuclear occupancy in macrophages, leading to increased activation of NFAT-dependent cytokines that trigger severe colitis [37]. Furthermore, NFATc2-deficient mice are resistant to colitis induced by oxazolone, due to reduced production of IL-6, IL-13, and IL-17 cytokines [38].
NFATc2 may control the transcription of major IBD susceptibility gene NKX2-3 variant rs11190140 [39]. In the intestinal tissues of IBD patients NKX2-3 is expressed in endothelial cells and muscularis mucosa to regulate the expression of endothelin-1 and vascular endothelial growth factor (VEGF) [40], which may contribute to the inflammation and angiogenesis of IBD. NFAT c1, c3 and c4 may also be involved in the pathogenesis of IBD. The level of nuclear NFATc1 in mononuclear cells of the lamina propria of colon epithelium is increased in patients with ulcerative colitis [41]. NFATc1 activates the expression of TRAIL (a member of the TNFα family) [42], whereas NFATc1 and c4 activate the expression of PTEN [43]—a lipid phosphatase that inhibits mTOR signaling whose activity is enhanced in IBD (44). In addition, NFATc3 inhibits mTOR signaling by activating the expression of its negative regulator REDD1 in human intestinal cells [45]. Further studies of NFAT’s role in IBD could be conducted by deleting each individual NFAT gene in the colonic epithelium or inflammatory cells to investigate their precise pathophysiological functions.
Gene deletion experiments in mice have shown that NFATc1 and NFATc2 have redundant functions in lymphocytes but are individually indispensable for cytokine production [45, 46]. Mice lacking Nfatc2 develops lymphoproliferative disorder [47], whereas mice with Nfatc2 and c3 double deletion develop spontaneous differentiation of T cells into Th2 cells and excessive production of IgE [48]. Conversely, mice with Nfatc3 deletion exhibit loss of DP (double positive) cells, likely caused by a failure of immature thymocytes to induce the expression of anti-apoptotic protein Bcl2 during thymic development [49]. In the transgenic mice carrying a constitutively active NFATc1 mutant (NFATc1nuc) a severe global inflammatory response was observed without altering thymocyte development [50]. Despite the above roles of NFAT in immune regulation, it is unclear how NFAT plays into the development of autoimmune diseases in humans. In Figure 4, we outline the unbalanced positive feedback pathway by NFATc1 as a potential mechanism for the run-away autoimmune reaction to its own antigens. In the normal T cells, the ratio of nuclear and cytoplasmic NFATc1 is maintained in a delicate balance in response to TCR stimulation, depending on the level of calcium and calcineurin activity. This regulation is constant and tightly monitored by the several opposing proteins that activate or inhibit calcineurin-NFAT pathway. GSK3 appears to be the central molecule responsible for removing NFATc1 from the nucleus to the cytoplasm, allowing only appropriate duration of NFATc1 in the nuclei of T cells. The perturbation of such tightly regulated feedback loop can lead to uncontrolled NFATc1 activation and subsequent T cell activation, cytokine production, and other serious events.
Figure 4.

NFATc1nuc provokes a positive feedback loop. Because NFATc1nuc is not phosphorylable by GSK3 and other NFAT kinases, it remains in the nucleus constitutively and not exportable, therefore bypassing the critical step of negative regulation by GSK3 and other kinases for removing it back to the cytoplasm. This small 1/7th of physiologic level of NFATc1 tips the balance in response to the receptor occupancy and initiates an unopposed positive feedback mechanism. NFATc1 activates it own transcription, though the newly synthesized NFATc1 proteins are subjected to the regulation by NFAT kinases, the signal coming from the hyperactivated TCR, continuously send the activated NFATc1 to the nucleus, causing the imbalance of the immune regulation [50, 78, 79].
NFAT in Rheumatoid Arthritis (RA) and Systemic Lupus Erythematosus (SLE)
RA and SLE are two classic autoimmune diseases challenging to treat. Recent evidence shows that TNFα activates the calcineurin-NFAT pathway in macrophages, which may contribute to the pathogenesis of autoimmune diseases [51]. CsA and FK506 block the activation of calcineurin and TNFα expression in the synoviocytes of patients with RA [52, 53]. TNFα is one of the many pro-inflammatory cytokines implicated in the inflammatory pathology of RA and is stimulated by NFAT activation [50]. Therefore, the dynamic interaction between calcineurin-NFAT and TNFα pathway likely plays an important role in the pathogenesis of RA.
Clinically, CsA has been shown to ameliorate the disease activity of RA [54]. Also, the combination of L-type calcium channel blocker nifedipine and low dose CsA have additive effects on the inhibition on T cell and NFATc1 activation in patients with RA [55]. Moreover, CsA is as effective as intravenous cyclophosphamide in the treatment of proliferative lupus nephritis for preserving renal function and maintaining remission [56]; CsA is also effective for refractory SLE [56]. Other randomized trials and retrospective studies have shown the efficacy of CsA and FK506 in the treatment of lupus nephritis [57–59]. The toxicities of CsA and FK506 have limited their clinical application in RA and SLE. In a mouse model of SLE, dipyridamole (a platelet and calcineurin inhibitor) reduces the disease activity, inhibits T cell and NFAT activation, and blocks the production of CD40 ligand (CD154) and IL-6 [60, 61]. CD40 ligand is a member of TNF family primarily expressed in the CD4 T cells, important for B cell development, antibody production, and CD8 effector cell function. CD40 ligand is overexpressed in CD4 T cells in patients with SLE and is correlated with the severity of nephritis and NFAT activity [62, 63]. Nuclear NFATc1 activates CD40 ligand in the T cells in our NFATc1nuc transgenic mice [50]. In addition, the T cells of SLE patients show hyper-responsiveness to TCR stimulation with a robust calcium influx and nuclear translocation of NFATc2 which may result in the consequence of increased cytokine production and subsequent tissue and organ damages [64]. The mTOR pathway is involved in the inflammation of joints in RA and its inhibition decreases the invasion of synovial fibroblasts that is a cause of the joint erosion [65]. The interaction between mTOR pathway and NFAT may be an important element in the inflammatory process of RA. The enhanced mTOR activity mediates basal and rephosphorylation of NFATc4 on the serine residues 168 and 170, which removes NFATc4 from the nucleus and terminates its signaling, though it is not clear if NFATc4 is involved in the joint inflammation of RA. ERK5/MEK5 which is involved in the pathogenesis of RA joints, also rephosphorylates the two same residues to export NFATc4 from the nucleus to the cytoplasm [66]. Future studies to examine the nuclear presence of each individual NFAT protein in the synovial cells of the joints of RA patients and of the animal models may provide clearer insight into the role each NFAT may play in the disease process. Quantitative examination of the activity of each individual NFAT in the macrophages of synovial fluid of RA joints can be valuable as well. If specific gene deletion of individual NFAT in the synoviocytes can be established, it would provide enormously valuable models for understanding the role of NFAT in joint diseases including RA.
NFAT and glomerulosclerosis
Glomerulus of the kidneys is a filtration unit consisting of endothelial cells, mesangial cells, podocytes, and the glomerular basement membrane. Disruption of this highly specialized structure can lead to many types of glomerular pathologies, including nephrotic syndrome and glomerular sclerosis with the risk of progressing to renal failure. Many autoimmune diseases, especially SLE, involve kidneys as a common disease manifestation and cause secondary nephritis. Steroid and other immunosuppressive drugs remain the standard of care for immune-related glomerular diseases and many forms of idiopathic nephrotic syndromes.
Idiopathic focal segmental glomerulosclerosis (FSGS) occurs in both adults and children. Several cytokines, including TNFα, TGF-β1, and IL-10, have been implicated in its pathogenesis [67]. IL-10 gene promoter polymorphism has been shown to be associated with FSGS [68]. Treatment with the inhibitors of calcineurin-NFAT pathway (CsA, FK506) ameliorates proteinuria and improves symptoms in these patients [69].
Contrary to the CRAC channels essential for T cell response and to the L-type channels critical in the central nervous system, the canonical transient receptor potential channel (TRPC6) responds to store-induced calcium influx and is important in regulating renal podocyte function. Gain-of-function mutations of TRPC6 cause human hereditary autosomal dominant FSGS, and overexpressing TRPC6 in the podocytes causes FSGS in mice [70, 71]. Such gain-of-function TRPC6 leads to increased intracellular calcium influx and enhanced NFAT signaling [72], suggesting that hyperactive NFAT may be causal for FSGS. Indeed, when constitutively nuclear NFATc1 (NFATc1nuc) was expressed in the murine podocytes, mice developed proteinuria and FSGS similar to the phenotypes observed the patients with TRPC6 gain-of-function mutation [73]. These studies thus implicate a crucial role of NFATc1 in a non-immune associated kidney disorder and suggest that NFAT activation may be a key intermediate step in the pathogenesis of mutant TRPC6-mediated FSGS.
Angiotensin II (AT-II) is implicated in kidney injuries in patients with uncontrolled hypertension, diabetes, and other diseases that can lead to renal failure. Drugs that block the AT-II pathway have been the milestones of treatment for hypertension and heart failure. AT-II was recently found to activate TRPC6 expression in podocytes both in vitro and in vivo via the NFAT signaling pathway, possibly forming a positive feedback loop in a disease condition in which AT-II level is increased [74]. CsA inhibits TRPC6 expression, while the constitutively active NFATc1nuc stimulates TRPC6 expression [69, 73]. Certain NFATc1-activated-cytokines are likely involved in this AT-II-mediated kidney injury. CAML, a calcium-modulating cyclophilin ligand mediates the AT-II activation of NFATc1 [75]. These findings suggest that specific inhibition of NFAT activity may be effective in ameliorating the progression of certain secondary renal insufficiency caused by diabetes, hypertension and other primary diseases. CsA and FK506 cannot accomplish such task due to its toxicities of triggering kidney vessel spasm and hypertension. Clinically effective specific NFAT inhibitor is still lacking.
NFATc1 and global inflammation
In addition to its critical role in a wide range of T and B cell functions, NFAT signaling regulates the function of other hematopoietic cells including dendritic cells [76], megakaryocytes [77], and osteoblasts [78]. NFAT pathway plays an important role in innate immunity and is activated by LPS (lipopolysaccharide) via CD14 to cause apoptosis of terminally differentiated dendritic cells, and by doing so, to help maintain self-tolerance and prevent autoimmunity [76]. The broad range of functions exerted by NFAT molecules predict that deregulation of NFAT expression can be associated with severe immune and inflammatory disorders.
The transgenic mice that express constitutively nuclear NFATc1 (NFATc1nuc) in T cells show a wide range of pathology involving multiple organs due to severe global inflammation [50, 79]. These mice only have 1/7 increased nuclear level of NFATc1 in the T cells yet display some of the most severe inflammatory pathology. In an unstimulated state, the T cells of mutant mice produce Th1 and Th2 cytokines (IL-4, IL-5, IL-2, interferon-gamma, TNF-α, IgG2a) by several folds higher than the wild-type T cells. However, when stimulated with TCR ligation, the mutant T cells generates cytokines by 10–100 folds higher than the control, accompanied by increased CD25-, CD69-, and CD40L-positive cells. This nuclear NFATc1 triggers a positive feedback loop that leads to the destabilization of T cell regulation and to the hypersensitivity of T cells in response to external stimulation. The hyperactive feedback loop is associated with many autoimmune diseases, including SLE. The mutant NFATc1nuc mice show immune cell infiltrations to the lungs, liver, kidneys, muscle, and joints. The serology titers for autoimmune disease are elevated in the NFATc1nuc mice and the glomeruli contain immune complex deposits. In Figure 4 we illustrate the unopposed positive feedback loop triggered by the constitutively active NFATc1 mutant that represents a possible mechanism for immune-related inflammatory diseases such as SLE and others. Such a destabilized positive feedback mechanism may also be a key pathogenic trigger of certain variants of FSGS.
NFAT and cancer
The evidence is accumulating that NFAT genes are involved in the development and metastasis of cancer. It is speculated that the low rate of cancer incidence (except a slightly increased risk of leukemia) in Down Syndrome patients may be related to the 1.5 fold increase in the gene dosage of DSCR (Down Syndrome Critical Region) and DYRK1 [20, 80], which synergistically inhibit the calcineurin/NFAT signaling. How NFAT genes are involved in cancer development and metastasis is far from being understood. There is also evidence that NFAT genes can be tumor suppressive as well. No viral homolog of NFAT has been identified. However, a gene translocation involving NFATc2 was recently identified in four cases of a variant of Ewing’s sarcoma (an aggressive sarcoma that occurs most commonly in pediatric population), in which EWSR1 was fused to the N-terminally truncated and C-terminally intact active domain of NFATc2 [81]. The role of NFATc2 in Ewing’s sarcoma, an aggressive sarcoma that most commonly occurs in pediatric population, is not clear and remains to be investigated. Although NFAT proteins are involved in inflammatory and immune responses, there is no evidence at the present time that NFAT is involved in inflammation-associated malignancies, unlike that of NF-kB [82, 83]. Although CsA inhibits cell growth in some cell lines, its clinical application in cancer therapy is not expected due to its immunosuppression and other toxicities. In Table 1, we have summarized the literature showing NFAT’s impact on cancer development, cell proliferation, and drug resistance.
Table 1.
Summary of NFAT regulation in cancer
| NFAT Subtype | Cell/Tissue type | Tumor/Phenotype | Mechanism | Ref |
|---|---|---|---|---|
| NFATc1 | 3T3-L1 fibroblasts | Transformed phenotype | Stimulating c-Myc expression and activating JAK-Stat pathway | 8485 |
| NFATc4 | Cl41 epidermal cells | Transformed phenotype | Enhanced Cox2 expression | 87 |
| NFATc1 and c3 | A375, CHL-1 and WM266-4 | Human melanoma cell lines | Upregulation of NFATc1 and c3 by oncogenic BRAF mutation via the MEK/ERK signaling | 88 |
| NFAT | Endometrial adenocarcinoma | Increased cell growth | Upregulating CXCL8 and Interleukin-11 | 8990 |
| NFAT | LNCaP prostate cancer cell line | Enhanced proliferation | TRPC6-mediated Ca2+ influx activates NFAT promoter | 91 |
| NFATc2 | NFATc2−/− mice | Lower transplanted melanoma growth | Changing cytokine profile and reducing expression of TGF-β | 92 |
| NFATc1 | Panc-1, S2-028, IMIM-PC | Pancreatic cancer cell lines | Stimulating c-Myc expression, recruiting Elk to c-Myc promoter | 9394 |
| NFATc1 and c2 | Panc-1, PaTu8988t, HT-29 | Pancreatic and colon cancer lines | Displacing Smad3 repressor complex on the c-Myc promoter to activate c-Myc expression | 95 |
| NFATc1 | CML cell line | Increased resistance to imatinib | Wnt/Ca2+/NFAT pathway mediates resistance to imatinib | 96 |
| NFATc1 | LBCL-MS cells | Human B-cell lymphoma | Enhanced resistance to apoptosis by stimulating expression of CD154 ligand and BLYS | 99 |
| NFATc2NFAT5 | Human ductal breast carcinomas | Enhanced invasiveness of human breast carcinoma | Mediating α6β4 integrin signaling and stimulating Cox2 expression | 105107 |
| NFATc2 | Breast carcinoma cell line | Enhanced invasive migration phenotype | Activating JNK and p38MAPK via GPC6 and Wnt5A signaling | 109 |
| NFATc3 | angiosarcoma | Stimulates angiogenesis | NFATc3 inhibition reduces SFRP2-mediated angiogenesis | 113 |
| NFATc3 | MMTV-Neu cells | Suppresses angiogenesis | NFATc3 mediates SFRP2 stimulated angiogenesis | 114 |
| NFATc2NFATc1 | NIH3T3NIH3T3 | Tumor suppressive Oncogenic | Induces cell cycle arrest, apoptosis Stimulates cell proliferation | 120120 |
| NFATc3 | Murine T cell lymphoma | Suppresses cell growth | Inactivated by murine lymphomagenic virus SL3-3 | 121 |
| NFATc2 | Breast cancer | Inhibits Stat-5 activity | 122 |
NFAT’s cross talk with other major oncogenic pathways
A constitutively active and nuclear NFATc1 transforms a preadipocyte cell line via activation of the JAK-Stat3 pathway, and the transformed cells are capable of forming tumors in athymic mice [84, 85]. This is the first laboratory evidence that NFAT is involved in promoting cellular transformation. NFATc1 induces the expression of TNFα and cyclooxygenase 2 (COX2). COX2 is implicated in the progression and angiogenesis of several cancers and has been shown in many studies to mediate the oncogenic effect of NFAT [86]. For example, NFATc4 mediates COX2 expression and the transformation of the Cl41 epidermal cells induced by TNFα [87]. In a malignant melanoma cell line, the BRAF-MEK-ERK pathway activates NFATc1 and c3 which in turn directly activates COX2 [88]. In endometrial adenocarcinoma, NFAT mediates prostaglandin-induced expression of chemokine CXCL8 and cytokine Interleukin-11 (IL-11) [89, 90]. NFAT signaling is activated by TRPC channel to maintain high proliferation rate in prostate cancer cells [91]. In NFATc2-deficient mice, less tumor masses grow in the lungs with the inoculation of malignant melanoma cells compared to the wild-type mice [92]. In addition, NFATc2-deficient mice have different cytokine profile in the lung tissues surrounding the tumor masses in the lungs [92]. These findings suggest that COX2, certain chemokines and cytokines are downstream of NFAT to create an environment for promoting cancer growth.
NFATc1 is overexpressed and activated in many pancreatic cancers. Also, NFATc1 activates c-Myc, a potent proto-oncogene, in pancreatic cancer cells. Inhibiting NFAT activity with CsA or knock-down of NFATc1 by siRNA attenuates c-Myc expression, cell growth, and cell cycle progression [93]. NFAT proteins can bind to a serum responsive element within the c-Myc promoter to initiate a p300-dependent histone acetylation which creates a permissive chromosomal environment for recruiting Ets-like gene 1 (ELK-1) to maximally activate c-Myc [94]. Remarkably, NFAT signaling is found to control the switch of TGF-β pathway from a repressor in the early stage cancer to a promoter of cell proliferation at the advanced stage. TGF-β stimulates a 3–5 fold increase in NFATc1 and c2 expression and also triggers NFAT accumulation in the nucleus, displacing Smad3 repressor from the gene promoter of c-Myc and thus activating c-Myc transcription [95]. Knockdown of NFATc1 or c2 by siRNA in two pancreatic cell lines partially restores the cell growth inhibitory effect of TGF-β [95]. These findings are interesting because NFATc1 is not physiologically expressed in pancreatic cells. How NFATc1 is activated in pancreatic cancer cells will be an important question to investigate.
A recent study using synthetic lethal screen with a shRNA library shows that the activation of NFATc1 is associated with the resistance to imatinib therapy in chronic myeloid leukemia (CML) [96]. The Wnt receptor FZD-8 is also identified in the same lethal synthetic screening, suggesting that the non-canonical Wnt/Ca2+/NFAT pathway supports the survival of CML cells and their resistance to the tyrosine kinase inhibtor (TKI) therapy. IL-4 appears to be the main target gene of this Wnt/Ca2+/NFAT pathway because IL-4 down-regulation renders CML cells sensitive to TKI. Furthermore, down-regulating NFATc1 with RNAi or blocking calcineurin-NFAT pathway with CsA sensitizes CML cells to the BCR-ABL kinase inhibitor dasatinib. This is reminiscent of our study in which the expression of NFATc1nuc in osteoblasts results in coordinated expression of the proteins that underlie the positive and negative regulation of Wnt-Frizzled pathway [78]. NFAT and Wnt pathways may therefore reciprocally regulate each other in certain cells and tissues for coordinating their effects on cell growth and differentiation.
Dephosphorylated and aberrantly activated NFATc1 and c2 can be found in many human and mouse lymphomas, including a Notch- and a TEL-JAK-induced mouse model of acute T-lymphoblastic leukemia [97, 98]. Treatment with CsA inhibits leukemia cell growth, induces apoptosis and prolongs mouse survival [98]. NFATc1 is found to be constitutively active in large B-cell lymphoma where it cooperated with NF-κB to activate the expression of CD40 ligand and to maintain lymphoma cell survival [99]. Since NFATc1 is normally expressed in many lymphoid tissues, its activation in lymphoma and leukemia may be viewed as tumor cells taking advantage of an inherent pathway for growth. The unexpected finding of ectopic activation of NFATc1 in pancreatic cancers suggests that NFAT activation may contribute to a much wider set of tumors originated from cells that have no physiological expression of these proteins [94–98]. In large B cell lymphoma, ectopically expressed NFATc1 also recruits chromatin remodeling complex proteins Brg-1 and Brm to the promoter of its target genes, including c-Myc [100]. This is interesting as Brg1 is considered a tumor suppressor gene, which is mutated in many malignancies with deletion of one copy of Brg1 causing epithelial cancer in mice [101, 102] though evidence is accumulating that Brg1 can have an opposite role by suppressing p53 to promote cell growth in certain cellular context [103]. In our preliminary study in 150 human invasive breast cancer cases, we find that NFATc1 is expressed in 22% of the cases, and its expression correlates with that of Brg1 and Brm [104]. These data suggest that there are additional roles for NFAT genes in the chromatin remodeling for regulating cell growth and differentiation that remain to be explored.
NFAT in metastasis and angiogenesis
In addition to enhancing cell growth and proliferation, NFAT has been shown to play roles in tumor cell migration that is intimately linked to tumor invasion and metastasis. NFATc2 and NFAT5 are expressed in invasive human ductal breast carcinomas. In both breast and colon cancer cell lines, NFATc2 and NFAT5 promote cell migration, and their expression correlates with that of α6β4 integrin, which promotes cancer metastasis. Interestingly, α6β4 integrin activates NFAT5 transcription [105]. Further more, Akt promotes E3 ubiquitin ligase HDM2-mediated degradation of NFATc2 to block breast cancer mobility and invasion [106]. There is also evidence that NFATc2 promotes breast cancer cell invasion through upregulation of Cox2 [107]. NFATc2 can bind to the promoter of glypican-6 (GPC6) and directly regulates its transcription to promote invasive migration of breast cancer cells [108]. Also in breast cancer cells, mutations in casein kinase 1 epsilon (CK1ε) activates NFAT pathway through the non-canonical Wnt signaling to reduce cell adhesion and to enhance cell migration [109]. The potential roles of NFAT in tumor invasion are not limited to breast cancer cells. In Notch-driven glioblastoma cells, hypoxia induces the expression of the calcium channel protein TRPC6, thus activating Ca2+ entry and NFAT to enhance tumor cell invasiveness [110], The interactions between Notch, Wnt and NFAT pathways in cancer development will be interesting to explore further.
Angiogenesis has been the target for cancer drug development over the past two decades. The role of calcineurin-NFAT signaling in angiogenesis was first revealed in the double NFATc3/c4 knockout mice and in the calcineurin B (Cnb1) knockout mice [111]. Mice with deletion of Cnb1 or both NFATc3/c4 genes died at midgestation due to disorganized vasculature, caused by ectopic expression of VEGF (vascular endothelial growth factor) that is normally suppressed by NFAT. Calcineurin-NFAT signaling plays an important role in tumor vasculature as well. NFAT controls the angiogenic functions of SFRP2 (secreted frizzled-related protein 2). A modulator of Wnt signaling, SFRP2 is expressed in the vasculature of a wide range of tumors, including approximately 85% of human breast carcinomas [112]. Inhibition of NFATc3 reduces the SFRP2-stimulated angiogenesis in vitro, and inhibition of calcineurin with FK506 also blocks SFRP2-stimulated angiogenesis and angiosarcoma growth [113]. In a MMTV-neu breast cancer transgenic mouse model, FK506 treatment results in the reduction of tumor microvascular density and tumor growth rate [114]. However, the involvement of calcineurin-NFAT signaling in tumor vasculature appears to be a complex one. Although one isoform of the endogenous calcineurin inhibitor DSCR1 (DSCR1.Ex4) blocks angiogenesis through suppressing calcineurin-NFAT signaling, the other isoform DSCR.Ex1 appears to promote angiogenesis. In the study by Ryeom et al., deletion of both DSCR1 isoforms in mice causes hyperactivation of calcineurin-NFAT signaling and premature endothelial apoptosis, leading to inhibition of tumor angiogenesis [115]. Treatment of these mutant mice with the calcineurin inhibitor CsA rescues the endothelial defects and restored tumor growth. On the other hand, Baek et al. show that modest increase of DSCR1 expression from an extra transgenic copy of DSCR1 contributes to deficient tumor angiogenesis due to calcineurin inhibition, resulting in significant suppression of tumor growth [80]. Results from these studies suggest that the exact effects of calcineurin-NFAT are context dependent, and the relative strength of the signals may dictate the phenotypic outcome. Furthermore, DSCR1 is induced by VEGF in the endothelial cells to suppress tissue factor (TF), E-selectin, and Cox2 expression. Knockdown of DSCR1 attenuates NFAT activity and reduces the expression of these genes [116]. The paradoxical interaction is also observed between NFAT and VEGF signaling pathways. Though NFAT suppresses VEGF for guiding vascular formation, VEGF can induce NFAT transcriptional activity in HUVEC (human umbilical vein endothelial cells) to upregulate the expression of TF [117]. Using CsA to suppress calcineurin-NFAT inhibits VEGF-induced Cox2 expression in endothelial cells and angiogenesis [118]. Zaichuk et al. has proposed that NFAT balances its effect on angiogenesis by using c-FLIP to modulate endothelial cell apoptosis [119]. NFAT signaling induces the expression of c-FLIP, a caspase-8 inhibitor, while pigment epithelial derived factor (PEGF) recruits JNK kinases to phosphorylate and confine NFATc2 to the cytoplasm to inhibit angiogenesis. Therefore, NFAT targets different array of genes in different cellular and tissue context to regulate vessel growth. Dysregulation of NFAT signaling in human and animal malignancies can thus affect tumor angiogenesis.
Tumor suppression by NFAT
Different NFAT proteins appear to have distinct roles in the development of cancer. Although NFATc1 has been consistently found to be pro-transformation and oncogenic, constitutively active NFATc2 was found to induce cell cycle arrest and apoptosis in NIH3T3 fibroblasts [120]. NFATc3 expression is repressed in T cell lymphomas induced by the murine lymphomagenic virus SL3-3, whereas mice with the germline deletion of NFATc3 develop SL3-3 virus-induced lymphoma in shorter latency and with higher frequency than wild-type and NFATc2-deficient mice. These studies suggest a tumor suppressor role for NFATc3 [121]. In breast cancer cells, NFATc2 inhibits Stat5-dependent gene expression and appears to be inversely related to cancer progression [122]. The context-dependent mechanism of tumor suppression by NFAT remains to be further understood. It is possible that the differential combination of downstream target genes of NFAT determines growth versus differentiation, setting an intracellular or intercellular environment for responding to external oncogenic stimuli.
In summary, we have reviewed the recent literature of NFAT concerning two closely connected pathological processes: inflammation and cancer. The literature of NFAT in inflammation is more extensive than that of NFAT in cancer. Studies in these areas will continue to expand and hopefully lead to therapeutic intervention targeting the NFAT pathway.
Figure 5.

Figure 5A. NFATc1 as part of the non-canonical Wnt-Frizzled pathway. Wnt-Frizzled pathway is implicated in many malignancies, most notably colorectal cancer in which Wnt activates its canonical β-catenin-TCF pathway for signal transduction and nuclear transcription. In CML cells, non-canonical Wnt pathway activates NFATc1 pathway, causing activation of IL-4, leading to the resistance to TKI therapy [96].
Figure 5B. NFAT promotes cancer metastasis and angiogenesis. In many cancer cell types, NFAT can activate COX2, c-Myc, Wnt, Frizzled, SFRP2 and others to cause increased cell migration, metastasis, and angiogenesis. This NFAT action is context dependent and is responsive to the external stimuli such as the activation of RTKs, integrin, and Wnt pathway (also see Table 1).
Acknowledgments
M.-G.P. is supported by the Kaiser Foundation Community Benefit research grant; F.C. by NIH grants DK081592 and DK087960; and Y.X. by the Oak foundation. We thank Drs. Piyush Tripathi and Ching-Pin Chang for comments.
ABBREVIATIONS
NFAT
Nuclear factor of activated T cell
NFATc
Cytoplasmic NFAT
NFATn
Nuclear NFAT (NFATc partner)
Cn
Calcineurin
CsA
Cyclosporine A
FKBP
FK binding protein
NES
Nuclear export signal
NLS
Nuclear localization signal
RANKL
Receptor activator of NF-κB ligand
NF-κB
Nuclear factor of κB promoter
CRAC channel
Calcium release-activated calcium channel
RTK
Receptor tyrosine kinase
GPGR
G-protein coupled receptor
Stat
Signal transducers and activators of transcription
Footnotes
Conflict of interest: authors declare no conflict of interest.
References
- 1.Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA, Crabtree GR. Identification of a putative regulator of early T cell activation genes. Science. 1988;241:202–205. doi: 10.1126/science.3260404. [DOI] [PubMed] [Google Scholar]
- 2.Durand DB, Shaw JP, Bush MR, Replogle RE, Belagaje R, Crabtree GR. Characterization of antigen receptor response elements within the interleukin-2 enhancer. Mol Cell Biol. 1988;8:1715–1724. doi: 10.1128/mcb.8.4.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu H, Peisley A, Graef IA, Crabtree GR. NFAT signaling and the invention of vertebrates. Trends in Cell Biol. 2007;17:251–260. doi: 10.1016/j.tcb.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Crabtree GR, Olsen EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109:S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 5.Hogan PG, Chen L, Nardone J, Rao A. Transcription regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 6.Graef IA, Chen F, Crabtree GR. NFAT signaling in vertebrate development. Curr Opinion Genet Dev. 2001;11:505–512. doi: 10.1016/s0959-437x(00)00225-2. [DOI] [PubMed] [Google Scholar]
- 7.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Meyer T. STIM Is a Ca2+ Sensor Essential for Ca2+-Store-Depletion-Triggered Ca2+ Influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a calcium sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membranes. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stathopulos PB, Zheng L, Ikura M. Stromal Interaction Molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J Biol Chem. 2009;284:728–732. doi: 10.1074/jbc.C800178200. [DOI] [PubMed] [Google Scholar]
- 10.Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454:538–542. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, Tang XJ, Gill DL. The Calcium Store Sensor, STIM1, Reciprocally Controls Orai and CaV1.2 Channels. Science. 2010;330:105–109. doi: 10.1126/science.1191086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan P. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 13.Park CY, Shcheglovitov A, Dolmetsch R. The CRAC channel activator binds and inhibits L-type voltage-gated calcium channels. Science. 2010;330:101–105. doi: 10.1126/science.1191027. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 15.Okamura H, Aramburu J, García-Rodríguez C, Viola JP, Raghavanm A, Tahiliani M, Zhang X, Qin J, Hogan PG, Rao A. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol Cell. 2000;6:539–550. doi: 10.1016/s1097-2765(00)00053-8. [DOI] [PubMed] [Google Scholar]
- 16.Beals CR, Clipstone NA, Ho SN, Crabtree GR. Nuclear localization of NF-ATc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes Dev. 1997;11:824–834. doi: 10.1101/gad.11.7.824. [DOI] [PubMed] [Google Scholar]
- 17.Lin X, Sikkink RA, Rusnak F, Barber DL. Inhibition of calcineurin phosphatase activity by a calcineurin B homologous protein. J Biol Chem. 1999;274:36125–36131. doi: 10.1074/jbc.274.51.36125. [DOI] [PubMed] [Google Scholar]
- 18.Fuentes JJ, Genescà L, Kingsbury TJ, Cunningham KW, Pérez-Riba M, Estivill X, de la Luna S. DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin-mediated signaling pathways. Hum Mol Genet. 2000;9:1681–1690. doi: 10.1093/hmg/9.11.1681. [DOI] [PubMed] [Google Scholar]
- 19.Mulero MC, Aubareda A, Schlüter A, Pérez-Riba M. RCAN3, a novel calcineurin inhibitor that down-regulates NFAT-dependent cytokine gene expression. Biochim Biophys Acta. 2007;1773:330–341. doi: 10.1016/j.bbamcr.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 20.Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK, Yamasaki N, Miyakawa T, Francke U, Graef IA, Crabtree GR. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441:595–600. doi: 10.1038/nature04678. [DOI] [PubMed] [Google Scholar]
- 21.Jurado S, Biou V, Malenka RC. A calcineurin/AKAP complex is required for NMDA receptor-dependent long-term depression. Nat Neurosci. 2010;13:1053–55. doi: 10.1038/nn.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai MM, Burnett PE, Wolosker H, Blackshaw S, Snyder SH. Cain, a novel physiologic protein inhibitor of calcineurin. J Biol Chem. 1998;273:18325–31. doi: 10.1074/jbc.273.29.18325. [DOI] [PubMed] [Google Scholar]
- 23.Sheridan CM, Heist EK, Beals CR, Crabtree GR, Gardner P. Protein kinase A negatively modulates the nuclear accumulation of NF-ATc1 by priming for subsequent phosphorylation by glycogen synthase kinase-3. J Biol Chem. 2002;277:48664–76. doi: 10.1074/jbc.M207029200. [DOI] [PubMed] [Google Scholar]
- 24.Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997;275:1930–34. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- 25.Chow CW, Davis RJ. Integraiton of calcium and cyclic AMP signaling pathways by 14-3-3. Mol Cell Biol. 2000;20:702–12. doi: 10.1128/mcb.20.2.702-712.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gwack Y, Sharma S, Nardone J, Tanasa B, Iuga A, Srikanth S, Okamura H, Bolton D, Feske S, Hogan PG, Rao A. A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature. 2006;441:646–50. doi: 10.1038/nature04631. [DOI] [PubMed] [Google Scholar]
- 27.Okamura H, Garcia-Rodriguez C, Martinson H, Qin J, Virshup DM, Rao A. A conserved docking motif for CK1 binding controls the nuclear localization of NFAT1. Mol Cell Biol. 2004;24:4184–95. doi: 10.1128/MCB.24.10.4184-4195.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu J, Shibasaki F, Price R, Guillemot JC, Yano T, Dotsch V, Wagner G, Ferraraa P, Meckeon F. Intramolecular masksing of nuclear import signal on NFAT4 by casein kinase I and MEKK1. Cell. 1998;93:851–61. doi: 10.1016/s0092-8674(00)81445-2. [DOI] [PubMed] [Google Scholar]
- 29.Chow CW, Rincón M, Cavanagh J, Dickens M, Davis RJ. Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science. 1997;278:1638–41. doi: 10.1126/science.278.5343.1638. [DOI] [PubMed] [Google Scholar]
- 30.Chow CW, Chen D, Flavell RA, Davis RJ. c-Jun NH2-terminal kinase inhibits targeting of the protein phosphotase calcineurin to NFATc1. Mol Cell Biol. 2000;20:5227–34. doi: 10.1128/mcb.20.14.5227-5234.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang TC, Xiong Q, Enslen H, Davis RJ, Chow CW. Phosphorylation of NFATc4 by p38 mitogen-activated protein kinase. Mol Cell Biol. 2002;22:3892–04. doi: 10.1128/MCB.22.11.3892-3904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gómez del Arco P, Martínez-Martínez S, Maldonado JL, Ortega-Pérez I, Redondo JM. A role for the p38 MAP kinase pathway in the nuclear shuttling of NFATp. J Biol Chem. 2000;275:13872–78. doi: 10.1074/jbc.275.18.13872. [DOI] [PubMed] [Google Scholar]
- 33.Terui Y, Saad N, Jia S, McKeon F, Yuan J. Dual role of sumoylation in the nuclear localization and transcriptional activation of NFAT1. J Biol Chem. 2004;279:28257–65. doi: 10.1074/jbc.M403153200. [DOI] [PubMed] [Google Scholar]
- 34.Nayak A, Glöckner-Pagel J, Vaeth M, Schumann JE, Buttmann M, Bopp T, Schmitt E, Serfling E, Berberich-Siebelt F. Sumoylation of the transcription factor NFATc1 leads to its subnuclear relocalization and interleukin-2 repression by histone deacetylase. J Biol Chem. 2009;284:10935–46. doi: 10.1074/jbc.M900465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma S, Findlay GM, Bandukwala HS, Oberdoerffer S, Baust B, Li Z, Schmidt V, Hogan PG, Sacks DB, Rao A. Dephosphorylation of the nuclear factor of activated T cells (NFAT) transcription factor is regulated by an RNA-protein scaffold complex. Proc Natl Acad Sci USA. 2011;108:11381–86. doi: 10.1073/pnas.1019711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colombel JF, et al. Infliximab, azathioprine, or combination for Crohn’s disease. N Engl J Med. 2010;362:1383–95. doi: 10.1056/NEJMoa0904492. [DOI] [PubMed] [Google Scholar]
- 37.Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol. 2011;12:1063–70. doi: 10.1038/ni.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weigmann B, et al. The transcription factor NFATc2 controls IL-6-dependent T cell activation in experimental colitis. J Exp Med. 2008;209:2099–110. doi: 10.1084/jem.20072484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.John G, Hegary JP, Yu W, Berg A, Pastor DM, Kelly AA, Wang YY, Poritz LS, Schreiber S, Koltun WA, Lin Z. NKX2-3 variant rs11190140 is associated with IBD and alters binding of NFAT. Mol Genet Metab. 2011;104:174–9. doi: 10.1016/j.ymgme.2011.06.023. [DOI] [PubMed] [Google Scholar]
- 40.Yu W, Hegarty JP, Berg A, Chen X, West G, Kelly AA, Wang Y, Poritz LS, Koltun WA, Lin Z. NKX2-3 transcriptional regulation of endothelin-1 and VEGF signaling in human intestinal microvascular endothelial cells. PLoS One. 2011;6:e20454. doi: 10.1371/journal.pone.0020454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shih TC, Hsieh SY, Hsieh YY, Chen TC, Yeh CY, Lin CY, Lin DY, Chiu CT. Aberrant activation of nuclear factor of activated T cell 2 in lamina propria mononuclear cells in ulcerative colitis. World J Gastroenterol. 2008;14:1759–67. doi: 10.3748/wjg.14.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q, Zhou Y, Weiss HL, Chow CW, Evers BM. NFATc1 regulation of TRAIL expression in human intestinal cells. PLoS One. 2011;6:e19882. doi: 10.1371/journal.pone.0019882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Q, Zhou Y, Jackson LN, Johnson SM, Chow CW, Evers BM. Nuclear factor of activated T cells (NFAT) signaling regulates PTEN expression and intestinal differentiation. Mol Biol Cell. 2012;22:412–20. doi: 10.1091/mbc.E10-07-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deng L, et al. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis. Am J Pathol. 2010;176:952–67. doi: 10.2353/ajpath.2010.090622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou Y, Wang Q, Guo Z, Weiss HL, Evers BM. Nuclear factor of activated T-cells (NFAT) c3 inhibition of mTOR signaling through induction of REDD1 in human intestinal cells. Mol Biol Cell. 2012 doi: 10.1091/mbc.E12-01-0037. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peng SL, Gerth AJ, Ranger AM, Glimcher LH. NFATc1 and NFATc2 together control both T and B cell activation and differentiation. Immunity. 2001;14:13–20. doi: 10.1016/s1074-7613(01)00085-1. [DOI] [PubMed] [Google Scholar]
- 47.Xanthoudakis S, Viola JP, Shaw KT, Luo C, Wallace JD, Bozza PT, Luk DC, Curran T, Rao A. An enhanced immune response in mice lacking the transcription factor NFAT1. Science. 1996;272:892–5. doi: 10.1126/science.272.5263.892. [DOI] [PubMed] [Google Scholar]
- 48.Ranger AM, Oukka M, Rengarajan J, Glimcher LH. Inhibitory function of two NFAT family members in lymphoid homeostasis and Th2 development. Immunity. 1998;9:627–35. doi: 10.1016/s1074-7613(00)80660-3. [DOI] [PubMed] [Google Scholar]
- 49.Oukka M, Ho IC, de la Brousse FC, Hoey T, Grusby MJ, Glimcher LH. The transcription factor NFAT4 is involved in the generation and survival of T cells. Immunity. 1998;9:295–304. doi: 10.1016/s1074-7613(00)80612-3. [DOI] [PubMed] [Google Scholar]
- 50.Pan M, Winslow M, Chen L, Kuo A, Felsher D, Crabtree GR. Enhanced NFATc1 nuclear occupancy causes T cell activation independent of CD28 costimulation. J Immunol. 2007;178:4315–21. doi: 10.4049/jimmunol.178.7.4315. [DOI] [PubMed] [Google Scholar]
- 51.Yarilina A, Xu K, Chen J, Ivashkiv LB. TNF activates calcium–nuclear factor of activated T cells NFATc1 signaling pathways in human macrophages. Proc Natl Aca Sci USA. 2011;108:1573–8. doi: 10.1073/pnas.1010030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cho ML, et al. Cyclosporine differentially regulates interleukin-10, interleukin-15, and tumor necrosis factor a production by rheumatoid synoviocytes. Arthritis Rheum. 2002;46:42–51. doi: 10.1002/1529-0131(200201)46:1<42::AID-ART10026>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 53.Yoo SA, et al. Calcineurin is expressed and plays a critical role in inflammatory arthritis. J Immunol. 2006;177:2681–90. doi: 10.4049/jimmunol.177.4.2681. [DOI] [PubMed] [Google Scholar]
- 54.van Rijthoven AW, Dijkmans BA, Goei, The HS, Hermans J, Montnor-Beckers ZL, Jacobs PC, Cats A. Cyclosporin treatment for rheumatoid arthritis: a placebo controlled, double blind, and multicentre study. Ann Rheum Dis. 1986;45:726–31. doi: 10.1136/ard.45.9.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lai NS, Yu CL, Yin WY, Yu HC, Huang HB, Tung CH, Lu MC. Combination of nifedipine and subtherapeutic dose of cyclosporin additively suppresses mononuclear cells activation of patients with rheumatoid arthritis and normal individuals via Ca(2+) -calcineurin-nuclear factor of activated T cells pathway. Clin Exp Immunol. 2012;168:78–86. doi: 10.1111/j.1365-2249.2012.04563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zavada J, et al. Cyclosporine A or intravenous cyclophosphomide for lupus nephritis: the Cyclofa-Lune study. Lupus. 2010;19:1281–9. doi: 10.1177/0961203310371155. [DOI] [PubMed] [Google Scholar]
- 57.Ogawa H, Kameda H, Amano K, Takeuchi T. Efficacy and safety of cyclosporine A in patients with refractory systemic lupus erythematosus in a daily clinical practice. Lupus. 2010;19:162–9. doi: 10.1177/0961203309350320. [DOI] [PubMed] [Google Scholar]
- 58.Austin HA, III, Illei GG, Braun M, Balow JE. Randomized, Controlled Trial of Prednisone, Cyclophosphamide, and Cyclosporine in Lupus Membranous Nephropathy. J Am Soc Nephrol. 2009;20:901–11. doi: 10.1681/ASN.2008060665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Szeto CC, Kwan CH, Lai FM, Tam LS, Li EK, Chow KM, Gang W, Li PK. Tacrolimus for the treatment of systemic lupus erythematosus with pure class V nephritis. Rheumatol. 2008;68:1678–81. doi: 10.1093/rheumatology/ken335. [DOI] [PubMed] [Google Scholar]
- 60.Vasileios CK, Zhang Z, Kampagiannni Q, Tsokos GC. Calcium signaling in systemic lupus erytematosus T cells. Arthritis Rheum. 2011;63:2058–66. doi: 10.1002/art.30353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsokos GC, Moulton VR. Abnormalities of T cell signaling in systemic lupus erythematosus. Arthr Res Ther. 2011;13:207–15. doi: 10.1186/ar3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mehta J, Genin A, Brunner M, Scalzi LV, Mishra N, Beukelman T, Cron RQ. Prolonged CD154 expression on pediatric Lupus CD4 T Cells Correlates with increased CD154 Transcription, increased NFAT activity, and glomerulonephritis. Arthritis Rheum. 2010;62:2499–509. doi: 10.1002/art.27554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kyttaris VC, Wang Y, Juang YT, Weinstein A, Tsokos GC. Increased levels of NF-ATc2 differentially regulate CD154 and IL-2 genes in T cells from patients with systemic lupus erythematosus. J Immunol. 2007;178:1960–6. doi: 10.4049/jimmunol.178.3.1960. [DOI] [PubMed] [Google Scholar]
- 64.Fujii Y, Fujii K, Iwata S, Suzuki K, Azuma T, Saito K, Tanaka Y. Abnormal intracellular distribution of NFAT1 in T lymphocytes from patients with systemic lupus erythematosus and characteristic clinical features. Clin Immunol. 2006;119:297–306. doi: 10.1016/j.clim.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 65.Lagagione T, Gulko PS. mTOR regulates the invasive properties of synovial fibroblasts in rheumatoid arthritiss. Mol Med. 2010;16:352–358. doi: 10.2119/molmed.2010.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Singh K, Deshpande P, Pryshchep S, Colmegna I, Liarski V, Weyand CM, Goronzy JJ. ERK-dependent T cell receptor threshold calibration in rheumatoid arthritis. J Immunol. 2009;183:8258–67. doi: 10.4049/jimmunol.0901784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Strehlau J, Schachter AD, Pavlakis M, Singh A, Tejani A, Strom TB. Activated intrarenal transcription of CTL-effectors and TGF-1 in children with focal segmental glomerulosclerosis. Kidney Int. 2002;61:90–5. doi: 10.1046/j.1523-1755.2002.00090.x. [DOI] [PubMed] [Google Scholar]
- 68.Bantis C, Heering PJ, Aker S, Klein-Vehne N, Grabensee B, Ivens K. Association of interleukin-10 gene G-1082A polymorphism with the progression of primary glomerulonephritis. Kidney Int. 2004;66:288–94. doi: 10.1111/j.1523-1755.2004.00730.x. [DOI] [PubMed] [Google Scholar]
- 69.Raafat RH, Kalia A, Travis LB, Diven SC. High-dose oral cyclosporin therapy for recurrent focal segmental glomerulosclerosis in children. Am J Kidney Dis. 2004;44:50–6. doi: 10.1053/j.ajkd.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 70.Winn MP, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–4. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 71.Reiser J, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37:739–44. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pollak MR, Schlondorff J, del Camino D, Carrasquillo R, Lacey V. TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am J Physiol-Cell Physiol. 2009;296:C558–C569. doi: 10.1152/ajpcell.00077.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Y, Jarad G, Tripathi P, Pan M, Cuningham J, Martin DR, Liapis H, Miner JH, Chen F. Activation of NFAT signaling in podocytes causes glomerulosclerosis. J Am Soc Nephrol. 2010;21:1657–66. doi: 10.1681/ASN.2009121253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nijenhuis T, et al. Angiotensin II contributes to podocyte injury by inceasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am J Pathol. 2011;179:1719–32. doi: 10.1016/j.ajpath.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guo S, Lopex-Ilasaca M, Dzau VJ. Identification of calcium-modulating cyclophilin ligand (CAML) as transducer of angiotensin II-mediated nuclear factor of activated T cells (NFAT) activation. J Biol Chem. 2005;280:12536–41. doi: 10.1074/jbc.M500296200. [DOI] [PubMed] [Google Scholar]
- 76.Zanoni I, Ostuni R, Capuano G, et al. CD14 regulates the dendritic cell life cycle after LPS exposure through NFAT activation. Nature. 2009;460:264–8. doi: 10.1038/nature08118. [DOI] [PubMed] [Google Scholar]
- 77.Crist SA, Sprague DL, Ratliff TL. Nuclear factor of activated T cells (NFAT) mediates CD154 expression in megakaryocytes. Blood. 2008;111:3553–61. doi: 10.1182/blood-2007-05-088161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Winslow MM, Pan M, Starbuck M, Gallo EM, Karsentry G, Crabtree GR. Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev Cell. 2006;10:771–82. doi: 10.1016/j.devcel.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 79.Pan M, Winslow M, Keum JS, Crabtree GR. Stringent control of NFAT nuclear occupancy is critical for maintaining balanced immune response. Gene Ther Mol Biol. 2007;11:171–6. [Google Scholar]
- 80.Baek KH, et al. Down’s syndrome suppression of tumour growth and the role of the calcineurin inhibitor. Nature. 2009;459:1126–30. doi: 10.1038/nature08062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Szuhai K, Ijszenga M, de Jong D, Karseladze A, Tanke HJ, Hogendoorn PC. The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res. 2009;15:2259–68. doi: 10.1158/1078-0432.CCR-08-2184. [DOI] [PubMed] [Google Scholar]
- 82.Greten FR, Eckman L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 83.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 84.Neal JW, Clipstone NA. A constitutively active NFATc1 mutant induces a transformed phenotype in 3T3-L1 fibroblasts. J Biol Chem. 2003;278:17246–54. doi: 10.1074/jbc.M300528200. [DOI] [PubMed] [Google Scholar]
- 85.Lagunas L, Clipstone NA. Deregulated NFATc1 activity transforms murine fibroblasts via an autocrine growth factor-mediated Stat3-dependent pathway. J Cell Biochem. 2009;108:237–48. doi: 10.1002/jcb.22245. [DOI] [PubMed] [Google Scholar]
- 86.Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30:377–86. doi: 10.1093/carcin/bgp014. [DOI] [PubMed] [Google Scholar]
- 87.Yan Y, Li J, Ouyang W, Ma Q, Hu Y, Zhang D, Ding J, Qu Q, Subbaramaiah K, Huang C. NFAT3 is specifically required for TNF-alpha-induced cyclooxygenase-2 (COX-2) expression and transformation of Cl41 cells. J Cell Sci. 2006;119:2985–94. doi: 10.1242/jcs.03014. [DOI] [PubMed] [Google Scholar]
- 88.Flockhart RJ, Armstrong JL, Reynolds NJ, Lovat PE. NFAT signalling is a novel target of oncogenic BRAF in metastatic melanoma. Br J Cancer. 2009;101:1448–55. doi: 10.1038/sj.bjc.6605277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sales KJ, Maldonado-Perez D, Grant V, Catalano R, Wilson MR, Brown P, Williams AR, Anderson RA, Thompson EA, Jabbour HN. Prostaglandin F(2alpha)-F-prostanoid receptor regulates CXCL8 expression in endometrial adenocarcinoma cells via the calcium-calcineurin-NFAT pathway. Biochim Biophys Acta. 2009;1793:1917–28. doi: 10.1016/j.bbamcr.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sales KJ, Grant V, Cook IH, Maldonado-Perez D, Anderson RA, Williams AR, Jabbour HN. Interleukin-11 in endometrial adenocarcinoma is regulated by prostaglandin F2alpha-F-prostanoid receptor interaction via the calcium-calcineurin-nuclear factor of activated T cells pathway and negatively regulated by the regulator of calcineurin-1. Am J Pathol. 2010;176:435–45. doi: 10.2353/ajpath.2010.090403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lehen’kyi V, Flourakis M, Skryma R, Prevarskaya N. TRPV6 channel controls prostate cancer cell proliferation via Ca(2+)/NFAT-dependent pathways. Oncogene. 2007;26:7380–5. doi: 10.1038/sj.onc.1210545. [DOI] [PubMed] [Google Scholar]
- 92.Werneck MB, Vieira-de-Abreu A, Chammas R, Viola JP. NFAT1 transcription factor is central in the regulation of tissue microenvironment for tumor metastasis. Cancer Immunol Immunother. 2011;60:537–46. doi: 10.1007/s00262-010-0964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Buchholz M, Schatz A, Wagner M, Michl P, Linhart T, Adler G, Gress TM, Ellenrieder V. Overexpression of c-myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBO J. 2006;25:3714–24. doi: 10.1038/sj.emboj.7601246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Köenig A, et al. NFAT-induced histone acetylation relay switch promotes c-Myc-dependent growth in pancreatic cancer cells. Gastroenterol. 2010;138:1189–99. doi: 10.1053/j.gastro.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Singh G, Singh SK, Konig A, Reutlinger K, Nye MD, Adhikary T, Eilers M, Gress TM, Fernandez-Zapico ME, Ellenrieder V. Sequential activation of NFAT and c-Myc transcription factors mediates the TGF-beta switch from a suppressor to a promoter of cancer cell proliferation. J Biol Chem. 2010;285:27241–50. doi: 10.1074/jbc.M110.100438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gregory MA, et al. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell. 2010;18:74–87. doi: 10.1016/j.ccr.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marafioti T, et al. The NFATc1 transcription factor is widely expressed in white cells and translocates from the cytoplasm to the nucleus in a subset of human lymphomas. Br J Haematol. 2005;128:333–42. doi: 10.1111/j.1365-2141.2004.05313.x. [DOI] [PubMed] [Google Scholar]
- 98.Medyouf H, Alcalde H, Berthier C, Guillemin MC, dos Santos NR, Janin A, Decaudin D, de The H, Ghysdael J. Targeting calcineurin activation as a therapeutic strategy for T-cell acute lymphoblastic leukemia. Nat Med. 2007;13:736–41. doi: 10.1038/nm1588. [DOI] [PubMed] [Google Scholar]
- 99.Pham LV, Tamayo AT, Yoshimura LC, Lin-Lee YC, Ford RJ. Constitutive NF-kappaB and NFAT activation in aggressive B-cell lymphomas synergistically activates the CD154 gene and maintains lymphoma cell survival. Blood. 2005;106:3940–7. doi: 10.1182/blood-2005-03-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pham LV, Tamayo AT, Li C, Bueso-Ramos C, Ford RJ. An epigenetic chromatin remodeling role for NFATc1 in transcriptional regulation of growth and survival genes in diffuse large B-cell lymphomas. Blood. 2010;116:3899–906. doi: 10.1182/blood-2009-12-257378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree GR, Magnuson T. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell. 2000;6:1287–95. doi: 10.1016/s1097-2765(00)00127-1. [DOI] [PubMed] [Google Scholar]
- 102.Bultman SJ, Herschkowitz JI, Godfrey V, Gebuhr TC, Yaniv M, Perou CM, Magnuson T. Characterization of mammary tumors from Brg1 heterozygous mice. Oncogene. 2008;27:460–8. doi: 10.1038/sj.onc.1210664. [DOI] [PubMed] [Google Scholar]
- 103.Naidu SR, Love IM, Imbalzano AN, Grossman SR, Androphy EJ. The SWI/SNF chromatin remodeling subunit BRG1 is a critical regulator of p53 necessary for proliferation of malignant cells. Oncogene. 2009;28:2492–501. doi: 10.1038/onc.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pan M. Expression of calcium/calcineurin pathway-regulated transcription factor NFATc1 and chromatin-remodelling genes BRG1 and BRM in invasive breast cancer. Eur J Cancer. 2011;9:18. [Google Scholar]
- 105.Jauliac S, Lopez-Rodriguez C, Shaw LM, Brown LF, Rao A, Toker A. The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nat Cell Biol. 2002;4:540–44. doi: 10.1038/ncb816. [DOI] [PubMed] [Google Scholar]
- 106.Yoeli-Lerner M, Yiu GK, Rabinovitz I, Erhardt P, Jauliac S, Toker A. Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Mol Cell. 2005;20:539–50. doi: 10.1016/j.molcel.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 107.Yiu GK, Toker A. NFAT induces breast cancer cell invasion by promoting the induction of cyclooxygenase-2. J Biol Chem. 2006;281:12210–7. doi: 10.1074/jbc.M600184200. [DOI] [PubMed] [Google Scholar]
- 108.Yiu GK, Kaunisto A, Chin YR, Toker A. NFAT promotes carcinoma invasive migration through glypican-6. Biochem J. 2011;440:157–66. doi: 10.1042/BJ20110530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Foldynova-Trantirkova S, Sekyrova P, Tmejova K, Brumovska E, Bernatik O, Blankenfeldt W, Krejci P, Kozubik A, Dolezal T, Trantirek L, et al. Breast cancer-specific mutations in CK1epsilon inhibit Wnt/beta-catenin and activate the Wnt/Rac1/JNK and NFAT pathways to decrease cell adhesion and promote cell migration. Breast Cancer Res. 2010;12:R30. doi: 10.1186/bcr2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chigurupati S, Venkataraman R, Barrera D, Naganathan A, Madan M, Paul L, Pattisapu JV, Kyriazis GA, Sugaya K, Bushnev S, et al. Receptor channel TRPC6 is a key mediator of Notch-driven glioblastoma growth and invasiveness. Cancer Res. 2010;70:418–27. doi: 10.1158/0008-5472.CAN-09-2654. [DOI] [PubMed] [Google Scholar]
- 111.Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Signals transduced by Ca(2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell. 2001;105:863–75. doi: 10.1016/s0092-8674(01)00396-8. [DOI] [PubMed] [Google Scholar]
- 112.Bhati R, et al. Molecular characterization of human breast tumor vascular cells. Am J Pathol. 172:1381–90. doi: 10.2353/ajpath.2008.070988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Courtwright A, et al. SFRP2 Stimulates Angiogenesis via a Calcineurin/NFAT Signaling Pathway. Cancer Res. 2009;69:4621–8. doi: 10.1158/0008-5472.CAN-08-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Siamakpour-Reihani S, et al. The role of calcineurin/NFAT in SFRP2 induced angiogenesis--a rationale for breast cancer treatment with the calcineurin inhibitor tacrolimus. PLoS ONE. 2011;6:e20412. doi: 10.1371/journal.pone.0020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ryeom S, Greenwald RJ, Sharpe AH, McKeon F. The threshold pattern of calcineurin-dependent gene expression is altered by loss of the endogenous inhibitor calcipressin. Nat Immunol. 2003;4:874–81. doi: 10.1038/ni966. [DOI] [PubMed] [Google Scholar]
- 116.Hesser BA, Liang XH, Camenisch G, Yang S, Lewin DA, Scheller R, Ferrara N, Gerber HP. Down syndrome critical region protein 1 (DSCR1), a novel VEGF target gene that regulates expression of inflammatory markers on activated endothelial cells. Blood. 2004;104:149–58. doi: 10.1182/blood-2004-01-0273. [DOI] [PubMed] [Google Scholar]
- 117.Armesilla AL, Lorenzom E, Gómez del Arco P, Martínez-Martínez S, Alfranca A, Redondo JM. Vascular endothelial growth factor activates nuclear factor of activated T cells in human endothelial cells: a role for tissue factor gene expression. Mol Cell Biol. 1999;19:2032–43. doi: 10.1128/mcb.19.3.2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hernández GL, Volpert OV, Iñiguez MA, Lorenzo E, Martínez-Martínez S, Grau R, Fresno M, Redondo JM. Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin A: roles of the nuclear factor of activated T cells and cyclooxygenase 2. J Exp Med. 2001;193:607–20. doi: 10.1084/jem.193.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zaichuk TA, Shroff EH, Emmanuel R, Filleur S, Nelius T, Volpert OV. Nuclear activator of T cells balances angiogenesis activation and inhibition. J Exp Med. 2004;199:1513–22. doi: 10.1084/jem.20040474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Robbs BK, Cruz AL, Werneck MB, Mognol GP, Viola JP. Dual roles for NFAT transcription factor genes as oncogenes and tumor suppressors. Mol Cell Biol. 2008;28:7168–81. doi: 10.1128/MCB.00256-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Glud SZ, Sørensen AB, Andrulis M, Wang B, Kondo E, Jessen R, Krenacs L, Stelkovics E, Wabl M, Serfling E, Palmetshofer A, Pedersen FS. A tumor-suppressor function for NFATc3 in T-cell lymphomagenesis by murine leukemia virus. Blood. 2005;106:3546–52. doi: 10.1182/blood-2005-02-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zheng J, Fang F, Zeng X, Medler TR, Fiorillo AA, Clevenger CV. Negative cross talk between NFAT1 and Stat5 signaling in breast cancer. Mol Endocrinol. 2011;25:2054–64. doi: 10.1210/me.2011-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]