Early Diabetic Nephropathy in Type 1 Diabetes – New Insights (original) (raw)
. Author manuscript; available in PMC: 2015 Aug 1.
Published in final edited form as: Curr Opin Endocrinol Diabetes Obes. 2014 Aug;21(4):279–286. doi: 10.1097/MED.0000000000000074
Abstract
Purpose of review
Despite improvements in glycemic and blood pressure control in patients with T1D, diabetic nephropathy (DN) remains the most common cause of chronic kidney disease worldwide. A major challenge in preventing DN is the inability to identify high-risk patients at an early stage, emphasizing the importance of discovering new therapeutic targets and implementation of clinical trials to reduce DN risk.
Recent findings
Limitations of managing patients with DN with renin angiotensin aldosterone system (RAAS) blockade have been identified in recent clinical trials, including the failure of primary prevention studies in T1D and the demonstration of harm with dual RAAS blockade. Fortunately, several new targets, including serum uric acid, insulin sensitivity, vasopressin and sodium-glucose cotransporter-2 inhibition are promising in the prevention and treatment of DN.
Summary
DN is characterized by a long clinically silent period without signs or symptoms of disease. There is an urgent need for improved methods of detecting early mediators of renal injury, to ultimately prevent initiation and progression of DN. In this review, we will focus on early DN and summarize potential new therapeutic targets.
Keywords: Early diabetic nephropathy, glomerular filtration rate, serum uric acid, insulin sensitivity, sodium-glucose cotransporter-2
INTRODUCTION
Diabetic nephropathy (DN) is the leading cause of mortality in type 1 diabetes (T1D) (1–3). Microalbuminuria, generally recognized as the earliest clinical phenotype of DN, has a cumulative life-time incidence of approximately 50% in T1D, and develops at a rate of around 2 – 3 % annually (4). A quarter of T1D patients with DN progress to ESRD (5). In fact, the 2011 US Renal Data System showed that DN accounted for 44.5% of all cases of ESRD in the United States in 2009, making it the most common cause of ESRD (6). DN is also an important risk factor for coronary artery disease (CAD) (7–9) and overall mortality (7, 10). The natural history of DN is characterized by a long silent period without overt clinical signs and symptoms of nephropathy. This is further complicated by under-treatment in adolescents with T1D, as recently shown by data from the T1D Exchange registry which reported that only a third of subjects <20 years with a clinical diagnosis of microalbuminuria received ACE inhibitors (ACEi)/angiotensin-receptor blockers (ARB) (11).
By the time GFR <60mL/min/1.73m2 manifest, approximately 50% of renal function is lost, renal structural changes are well established and are usually refractory to therapeutic strategies including improved blood pressure and glycemic control (12, 13). Current therapeutic strategies may slow but do not prevent the progression of DN (4, 14). Earlier identification of GFR decline would allow interventions to decrease the rate of GFR loss and prolong the time to development of end stage renal disease (ESRD) (Figure 1).
Figure 1. Intervening early in the course of diabetic nephropathy.
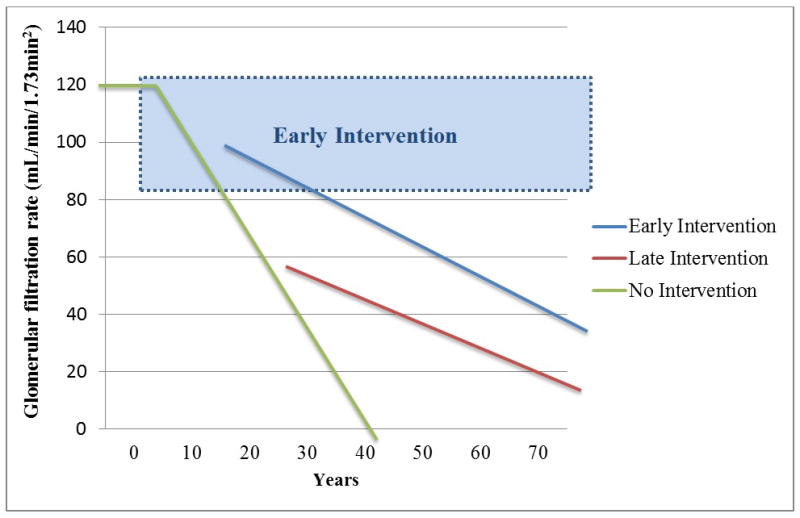
Adapted from a figure by Alessandro Doria M.D., Ph.D.
Doria A, Niewczas MA, Fiorina P. Can existing drugs approved for other indications retard renal function decline in patients with type 1 diabetes and nephropathy? Semin Nephrol. 2012 Sep;32(5):437–44. doi: 10.1016/j.semnephrol.2012.07.006.
Although there is strong evidence showing benefit of glycemic and blood pressure control in preventing microvascular complications in T1D (1, 15, 16), optimal control does not abolish the risk. In fact, DN outcomes have not changed significantly and continue to be a major concern for endocrinologists and nephrologists caring for patients with T1D (1, 2). Managing DN in patients with T1D has been further complicated by disappointing results of renin-angiotensin-aldosterone system (RAAS) inhibitor trials, including the failure of primary prevention studies (The Renin Angiotensin System Study [RASS]) (17) and the demonstration of harm with dual RAAS blockade (18). Furthermore, once promising renoprotective strategies for DN including protein kinase C-β antagonists, selective endothelin receptor-A antagonists, and the Nrf2 modulator bardoxolone have resulted in disappointing clinical trial results (19–21).
It is therefore important to identify novel and modifiable risk factors that contribute to the development and progression of early DN (22). Understanding these risk factors may enable us to identify subjects at high risk of early DN and intervene at a time when the renal lesions might be responsive to therapy. Accordingly, in this review, we examine the current evidence addressing novel mediators and therapeutic targets in T1D to prevent DN (Table 1).
Table 1.
Promising markers and therapeutic targets of DN
| Biomarker: | Important studies: | Randomized control trials: |
|---|---|---|
| Serum uric acid | (23, 24, 25) | The preventing early renal function loss (PERL) allopurinol study (NCT01575379) |
| Insulin sensitivity | (8, 26, 27) | Metformin Vascular Adverse Lesions in Type 1 Diabetes (REMOVAL) trial (NCT01483560) |
| Effects of Metformin On Cardiovascular Function In Adolescents with Type 1 Diabetes (EMERALD) study (NCT01808690) | ||
| Metformin Therapy for Overweight Adolescents With Type 1 Diabetes (NCT01881828) | ||
| Sodium glucose co-transporter 2 | (28, 29) | Safety and Efficacy of Empagliflozin (BI 10773) in Type 1 Diabetes Mellitus Patients With or Without Renal Hyperfiltration (NCT01392560) |
| Copeptin | (30, 31) | -- |
| Serum and urinary inflammatory markers | (32, 33) | -- |
Early diabetic nephropathy
In the conventional paradigm of DN, progressive pathological changes develop over a long silent period without evidence of proteinuria, hypertension or impaired GFR (34). In this paradigm, during the clinically silent phase, a significant proportion of patients exhibit renal hyperfiltration secondary to elevated intraglomerular pressure resulting in a glomerular injury, followed by microalbuminuria and progressive GFR decline, eventually resulting in ESRD (35). Renal hyperfiltration is the earliest hemodynamic abnormality seen in diabetes, and has been linked with an increased risk of DN in many, but not all, studies, reflected by microalbuminuria and declining GFR (35). In addition to controversy around hyperfiltration in the pathogenesis of DN, hyperfiltration remains difficult to detect clinically with current GFR estimating equations. The appearance of microalbuminuria is usually the earliest clinical sign of DN, but the paradigm of early DN in T1D has been further questioned over the past few years after the demonstration that microalbuminuria does not necessarily imply progressive nephropathy, and may in fact regress to normoalbuminuria (Figure 2) (36, 37). Early progressive renal decline defined as an annual eGFR decline greater than 3.3% (38) or 3mL/min/1.73m2 (39, 40) has been shown to occur prior to the onset of microalbuminuria (38), and thought to be more predictive of progression to impaired renal function and eventual ESRD than microalbuminuria (38, 41). Unfortunately, current understanding of determinants implicated in early GFR loss is limited (41). Recently identified modifiable biomarkers, including serum uric acid (23, 24) and insulin sensitivity (26), may provide insight into the design of more effective programs for preventing DN in T1D, reflected either by microalbuminuria and/or early GFR decline.
Figure 2. Progression of diabetic nephropathy.
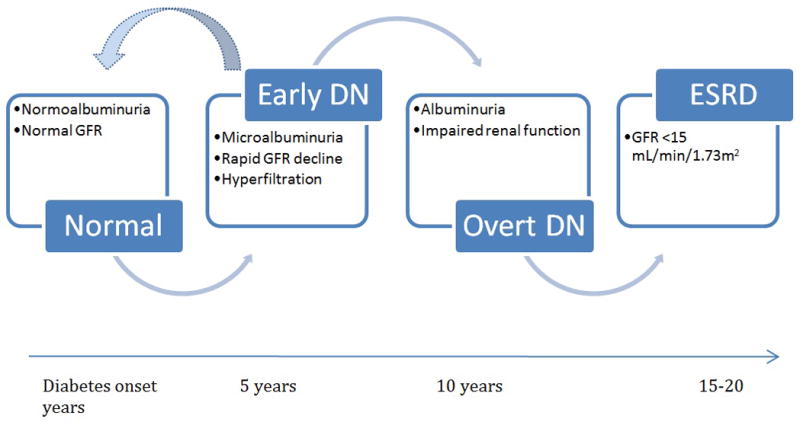
Adapted from a figure by Amy K. Mottl M.D., M.P.H. (not previously published)
Current methods of identifying early DN
The American Diabetes Association and International Society of Nephrology recommend annual screening for albuminuria and also measurement of estimated glomerular filtration rate (eGFR) to identify and monitor DN (42, 43). Microalbuminuria is defined as albumin excretion rate (AER) ≥20ug/min or albumin creatinine ratio (ACR) ≥30mg/g, and have both been shown to be associated with expansion of the glomerular basement membrane in T1D (44). The most state-of-the-art equations to estimate GFR for adults are the three recently published CKD-EPI equations: CKD-EPI Creatinine, CKD-EPI Cystatin C and CKD-EPI Creatinine and Cystatin C (45), and for children and adolescents are the CKiD Creatinine and Cystatin C equations (46). However, when eGFR is >90mL/min/1.73m2, concordance between CKD-EPI cystatin C eGFR and CKD-EPI creatinine eGFR has been reported to be as low as 56% (47, 48). These results echo the discordance between eGFR by CKD-EPI creatinine and CKD-EPI cystatin C reported by Inker et al in subjects with normal-to-high eGFR (45).
Overall, CKD-EPI cystatin C eGFR is considered to be less biased by age and weight compared to creatinine-based measurements, and correlates more closely with direct measures of GFR over a wide spectrum of plasma glucose levels compared to creatinine based measures in experimental studies (40, 49). These observations suggest that cystatin C more accurately reflects measured GFR in subjects with T1D, favoring its use as an estimate of GFR in this population. Moreover, GFR estimated by cystatin C appears to better predict micro- and macrovascular complications in subjects with T1D compared to creatinine-based equations (9, 26, 50, 51). Cystatin C more accurately detected rapid GFR decline than creatinine-based measurements in T1D subjects with normal renal function (51). Rapid GFR decline estimated by cystatin C is also associated with a higher risk for cardiovascular complications and mortality than creatinine based GFR estimated (39, 47). Furthermore, Skupien et al demonstrated that GFR staging with eGFRCYSTATIN C is superior for predicting ESRD and mortality compared to eGFRBOTH, which might suggest that serum creatinine counters the predictive effect of serum cystatin C (52). Finally, Shlipak et al demonstrated that the use of cystatin C compared to creatinine strengthens the association between eGFR and risk of death and ESRD in 11 diverse general-population studies (53).
Despite the possible superiority of cystatin C vs. creatinine, estimates of GFR by both serum creatinine and cystatin C, like measurements of urinary albumin excretion, remain imperfect (9, 54, 55). The CKD-EPI eGFR equation has not been validated in people with eGFR >80 mL/min/1.73m2, and it is associated with greater variability when eGFR >60 mL/min/1.73m2 (45). However, by the time eGFR is ≤60 mL/min/1.73m2 almost half of renal function has already been lost (56) (Figure 3).
Figure 3. Stages in Development of DN.

Adapted from David Maahs M.D., Ph.D. Early Detection of Kidney Disease in Type 1 Diabetes: What Do We Really Know? Diabetes Technology and Therapeutic. 2012. Vol 14. doi: 10.1089/dia.2012.0089 (Reprinted with permission from DIABETES TECHNOLOGY & THERAPEUTICS 14/7, 2012, published by Mary Ann Liebert, Inc., New Rochelle, NY)
Improved methods to easily and accurately measure GFR as well as changes in renal function in the normal and hyperfiltration range are therefore needed. Gold-standard measures of GFR with iothalamate, iohexol or inulin clearance in people with T1D are impractical and not routinely performed in clinical practice. Recently, a practical method of measuring GFR by iohexol clearance using dried capillary blood spots has been developed in non-diabetics (57, 58), which is ideally suited for people with T1D (59) in whom early detection of DN is imperative to prevent early morbidity and mortality (2, 60), which may hopefully allow the early identification of subjects at increased risk of early DN. Simultaneous hyperglycemia has an effect on renal hemodynamics and a difference of 4–6 mmol/L versus 9–11 mmol/L is associated with a 15–18 mL/min/1.73m2 difference in GFR measured by inulin (49). Furthermore, we’ve demonstrated that simultaneous blood glucose has an independent positive effect on eGFR by cystatin C, which could bias the accurate detection of early diabetic nephropathy (48, 61). Accounting for ambient blood glucose could improve intra-individual precision in GFR change over time in people with T1D.
Another promising method of identifying early DN is through the use of urinary proteomic techniques. Since many urinary proteomic markers remain stable for long enough to perform reliable polypeptide analysis, the emerging field of proteomics has given some recent insight into the development of practical and non-invasive novel biomarkers (62). The association between urinary proteomics and DN is well recognized (63–65, 32, 66). Both acute hyperglycemia and renal hyperfiltration are associated with increased urinary excretion of inflammatory cytokines/chemokines (e.g. IL-6, IL-8, IP-10, MCP-1) in T1D which may contribute to DN (67, 68), and in particular progressive renal decline (32, 33). In previous studies, we have also identified urinary biomarker panels for DN and coronary artery disease (CAD) in subjects with T1D in CACTI (66).
Novel targets for the prevention of DN
Inhibition of the renin-angiotensin-aldosterone system (RAAS) has been the mainstay therapy for the prevention and treatment of DN. However, the renoprotective role of ACEi/ARBs has recently been challenged by data from the Renin Angiotensin System Study (RASS) which demonstrated that early, primary prevention with ACE inhibition or angiotensin receptor blockade does not modify DN progression in adults with T1D (17). Moreover, large clinical trials including ONTARGET, ALTITUDE and VA-NEPHRON-D, have failed to demonstrate improved renal outcomes in diabetic subjects with dual RAAS inhibition using combined ACEi and ARB or using direct renin inhibitor-related regimens (18). Other classes of agents including protein kinase C-beta antagonists, selective endothelin receptor (A) antagonists and anti-oxidants such as bardoxolone have been associated with disappointing clinical trial results (19, 20).
There is therefore a need for novel methods of identifying and treating patients at high risk of developing DN at an early stage to prevent progression. Multiple studies have linked serum uric acid (SUA) levels to DN development and accumulating data have suggested that lowering SUA prevents renal function loss in animal models of diabetes and in patients with type 2 diabetes (23, 69, 70). To determine the role of SUA lowering in patients with T1D, the multi-center double-blind randomized clinical trial “Preventing Early Renal Function Loss - PERL”, will test the hypothesis that lowering SUA with allopurinol will prevent GFR decline measured by iohexol (24) (Table 1). An additional study design innovation in PERL is the use of GFR (measured by iohexol) as the study end point allowing for assessment of therapy earlier in the pathophysiologic pathway.
A second potential therapeutic target relates to insulin sensitivity. Reduced insulin sensitivity is well documented in both adolescents and adults with T1D, and is thought to contribute both to the initiation and progression of macro- and microvascular complications (71). We have previously demonstrated that insulin sensitivity predicts development of microalbuminuria and rapid eGFR decline by cystatin C over 6 years in patients with T1D (26), similar to data in the Epidemiology of Diabetes Complications study (72). Various hypotheses exist to explain reduced insulin sensitivity in T1D (73, 74), but we have shown that only 6% of the variance of insulin sensitivity is explained by SUA in adults with T1D, in contrast to 39% in non-diabetic adults (75). The absence of a clinically significant association between SUA and reduced insulin sensitivity in T1D may suggest that SUA and reduced insulin sensitivity are independent mediators of vascular pathology in T1D. Despite the findings from the BARI-2D study (76) which showed no benefit of insulin sensitizing strategy on DN in subjects with type 2 diabetes, modification of insulin sensitivity holds promise as a therapeutic target to reduce vascular complications in T1D, since both life style changes (diet and exercise) and drugs such as metformin can improve insulin sensitivity. The Metformin Vascular Adverse Lesions in Type 1 Diabetes (REMOVAL) trial was therefore designed to improve insulin sensitivity in T1D (NCT01483560) (Table 1).
Sodium glucose co-transporter 2 (SGLT2) inhibitors also hold promise as a therapeutic target to prevent progression of DN in T1D, after it was shown that SGLT2 inhibition with empagliflozin reduces HbA1c and significantly attenuates renal hyperfiltration to near normal GFR levels in patients with uncomplicated T1D (28) (Figure 4). Patients with T1D exhibited significant weight loss, HbA1c reductions and a decline in blood pressure (77–79, 29). In post-hoc analyses of clinical trials, this class of agents acutely reduces eGFR over 3–4 weeks, followed by preservation of renal function over 2 years, and also reduces albuminuria, suggesting a protective decline in intraglomerular pressure (29), similar to data from GFR inulin studies (28).
Figure 4. Tubuloglomerular feedback and SGLT-2 in DN.
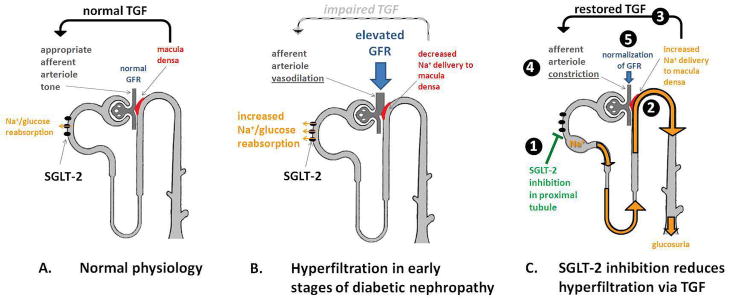
Reproduced with permission of the copyright owner: Cherney et al. The Renal Hemodynamic Effect of SGLT2 Inhibition in Patients with Type 1 Diabetes. Circulation. 2013 December 13
Emerging evidence also suggests that acute glycemic excursions may significantly contribute to microvascular end-organ injury in patients with diabetes, independent of long-term glycemic control (80). Although the mechanisms responsible for end-organ injury with increased glycemic variability are unclear, it has been demonstrated that acute hyperglycemia increases urinary excretion of inflammatory cytokines/chemokines in subjects with T1D (67). Acute hyperglycemia may also contribute to kidney injury via RAAS activation, as it has been demonstrated that acute clamped hyperglycemia activates the RAAS, which subsequently increases the urinary excretion of inflammatory cytokines/chemokines (81). Furthermore, the increased urinary excretion of inflammatory cytokines/chemokines in response to acute hyperglycemia is blunted by RAAS blockade by direct renin inhibitor in subjects with T1D (81).
Arginine vasopressin (AVP) plays an essential role in the regulation of volume status, and exerts important renal and cardiovascular effects. AVP modulates tubular cell growth thereby causing vasoconstriction of the renal microcirculation and in particular in the efferent arteriole (82, 83). Furthermore, AVP infusion induces hypertension, glomerular hyperfiltration and albuminuria (84, 85) and lowering the AVP concentration provides renal protection (86). There is also evidence linking increased fluid intake with decreased risk for developing CKD (87). Copeptin is a more stable peptide derived from the same precursor molecule as AVP, and appears to be a useful surrogate marker for AVP in the assessment of fluid and osmosis status in various diseases. Recently Boertien et al demonstrated copeptin predicts the estimated glomerular filtration rate decline in subjects with type 2 diabetes, however there is little if any data on its ability to predict DN in subjects with T1D (30). With the availability of AVP receptor antagonists (e.g. vaptans), AVP might also become a promising therapeutic target for DN in the future.
Conclusion
A major challenge in preventing DN relates to the accurate identification of high risk patients at an early stage. Identifying risk factors and biomarkers specifically associated with early DN will help us understand the mechanisms underlying the development and progression of DN. In this review we present novel methods of detecting early change in renal function in T1D, and also several promising new modifiable targets (insulin sensitivity, SUA, SGLT-2 and copeptin) to slow progression of early DN. The translation of these potential methods and therapies into clinical practice now requires investment in adequately powered clinical trials that will capture important renal and cardiovascular long-term outcomes.
Key Points.
- Diabetic nephropathy remains the most common cause of CKD worldwide
- There is a need for improved methods of detecting early diabetic nephropathy
- Novel targets to prevent diabetic nephropathy include; serum uric acid, insulin sensitivity, vasopressin and sodium-glucose cotransporter-2
Acknowledgments
NIH funded
Footnotes
References
- 1.Maahs DM, Rewers M. Editorial: Mortality and renal disease in type 1 diabetes mellitus--progress made, more to be done. J Clin Endocrinol Metab. 2006;91(10):3757–9. doi: 10.1210/jc.2006-1730. Epub 2006/10/10. [DOI] [PubMed] [Google Scholar]
- 2.Orchard TJ, Secrest AM, Miller RG, Costacou T. In the absence of renal disease, 20 year mortality risk in type 1 diabetes is comparable to that of the general population: a report from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetologia. 2010;53(11):2312–9. doi: 10.1007/s00125-010-1860-3. Epub 2010/07/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collins AJ, Foley RN, Chavers B, Gilbertson D, Herzog C, Johansen K, et al. United States Renal Data System 2011 Annual Data Report: Atlas of chronic kidney disease & end-stage renal disease in the United States. Am J Kidney Dis. 2012;59(1 Suppl 1):A7, e1–420. doi: 10.1053/j.ajkd.2011.11.015. Epub 2011/12/30. [DOI] [PubMed] [Google Scholar]
- 4**.Marshall SM. Diabetic nephropathy in type 1 diabetes: has the outlook improved since the 1980s? Diabetologia. 2012;55(9):2301–6. doi: 10.1007/s00125-012-2606-1. Epub 2012/06/15. An important review of the outlook of diabetic nephropathy in type 1 diabetes. [DOI] [PubMed] [Google Scholar]
- 5.Krolewski AS, Warram JH, Christlieb AR, Busick EJ, Kahn CR. The changing natural history of nephropathy in type I diabetes. The American journal of medicine. 1985;78(5):785–94. doi: 10.1016/0002-9343(85)90284-0. Epub 1985/05/01. [DOI] [PubMed] [Google Scholar]
- 6.Collins AJ, Foley RN, Herzog C, Chavers B, Gilbertson D, Ishani A, et al. US Renal Data System 2010 Annual Data Report. Am J Kidney Dis. 2011;57(1 Suppl 1):A8, e1–526. doi: 10.1053/j.ajkd.2010.10.007. Epub 2010/12/28. [DOI] [PubMed] [Google Scholar]
- 7.Borch-Johnsen K, Kreiner S. Proteinuria: value as predictor of cardiovascular mortality in insulin dependent diabetes mellitus. Br Med J (Clin Res Ed) 1987;294(6588):1651–4. doi: 10.1136/bmj.294.6588.1651. Epub 1987/06/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orchard TJ, Olson JC, Erbey JR, Williams K, Forrest KY, Smithline Kinder L, et al. Insulin resistance-related factors, but not glycemia, predict coronary artery disease in type 1 diabetes: 10-year follow-up data from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetes care. 2003;26(5):1374–9. doi: 10.2337/diacare.26.5.1374. Epub 2003/04/30. [DOI] [PubMed] [Google Scholar]
- 9**.Maahs DM, Jalal D, Chonchol M, Johnson RJ, Rewers M, Snell-Bergeon JK. Impaired Renal Function Further Increases Odds of 6-Year Coronary Artery Calcification Progression in Adults With Type 1 Diabetes: The CACTI study. Diabetes Care. 2013;36(9):2607–14. doi: 10.2337/dc12-2538. Epub 2013/07/10. Reports an important association between diabetic nephropathy and progression of coronary artery calcification in type 1 diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.International evaluation of cause-specific mortality and IDDM. Diabetes Epidemiology Research International Mortality Study Group. Diabetes care. 1991;14(1):55–60. doi: 10.2337/diacare.14.1.55. Epub 1991/01/01. [DOI] [PubMed] [Google Scholar]
- 11*.Daniels M, DuBose SN, Maahs DM, Beck RW, Fox LA, Gubitosi-Klug R, et al. Factors associated with microalbuminuria in 7,549 children and adolescents with type 1 diabetes in the T1D Exchange clinic registry. Diabetes Care. 2013;36(9):2639–45. doi: 10.2337/dc12-2192. Epub 2013/04/24. Reports under-treatment of microalbuminuria by ACEi/ARB in subjects < 20 years of age with type 1 diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mauer M, Drummond K. The early natural history of nephropathy in type 1 diabetes: I. Study design and baseline characteristics of the study participants. Diabetes. 2002;51(5):1572–9. doi: 10.2337/diabetes.51.5.1572. Epub 2002/04/30. [DOI] [PubMed] [Google Scholar]
- 13.Osterby R, Gall MA, Schmitz A, Nielsen FS, Nyberg G, Parving HH. Glomerular structure and function in proteinuric type 2 (non-insulin-dependent) diabetic patients. Diabetologia. 1993;36(10):1064–70. doi: 10.1007/BF02374500. Epub 1993/10/01. [DOI] [PubMed] [Google Scholar]
- 14.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. The New England journal of medicine. 1993;329(20):1456–62. doi: 10.1056/NEJM199311113292004. Epub 1993/11/11. [DOI] [PubMed] [Google Scholar]
- 15.Pambianco G, Costacou T, Ellis D, Becker DJ, Klein R, Orchard TJ. The 30-year natural history of type 1 diabetes complications: the Pittsburgh Epidemiology of Diabetes Complications Study experience. Diabetes. 2006;55(5):1463–9. doi: 10.2337/db05-1423. Epub 2006/04/29. [DOI] [PubMed] [Google Scholar]
- 16.Wood JR, Miller KM, Maahs DM, Beck RW, DiMeglio LA, Libman IM, et al. Most youth with type 1 diabetes in the T1D Exchange Clinic Registry do not meet American Diabetes Association or International Society for Pediatric and Adolescent Diabetes clinical guidelines. Diabetes care. 2013;36(7):2035–7. doi: 10.2337/dc12-1959. Epub 2013/01/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mauer M, Zinman B, Gardiner R, Suissa S, Sinaiko A, Strand T, et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. The New England journal of medicine. 2009;361(1):40–51. doi: 10.1056/NEJMoa0808400. Epub 2009/07/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18**.Mann JF, Anderson C, Gao P, Gerstein HC, Boehm M, Ryden L, et al. Dual inhibition of the renin-angiotensin system in high-risk diabetes and risk for stroke and other outcomes: results of the ONTARGET trial. J Hypertens. 2013;31(2):414–21. doi: 10.1097/HJH.0b013e32835bf7b0. Epub 2012/12/20. Demonstrates harm with dual RAAS blockade. [DOI] [PubMed] [Google Scholar]
- 19.Cherney DZ, Konvalinka A, Zinman B, Diamandis EP, Soosaipillai A, Reich H, et al. Effect of protein kinase Cbeta inhibition on renal hemodynamic function and urinary biomarkers in humans with type 1 diabetes: a pilot study. Diabetes Care. 2009;32(1):91–3. doi: 10.2337/dc08-1609. Epub 2008/10/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tuttle KR, McGill JB, Haney DJ, Lin TE, Anderson PW. Kidney outcomes in long-term studies of ruboxistaurin for diabetic eye disease. Clinical journal of the American Society of Nephrology: CJASN. 2007;2(4):631–6. doi: 10.2215/CJN.00840207. Epub 2007/08/21. [DOI] [PubMed] [Google Scholar]
- 21.de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. The New England journal of medicine. 2013;369(26):2492–503. doi: 10.1056/NEJMoa1306033. Epub 2013/11/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maahs DM. Cardiovascular disease (CVD) limbo: how soon and low should we go to prevent CVD in diabetes? Diabetes Technol Ther. 2012;14(6):449–52. doi: 10.1089/dia.2012.0078. Epub 2012/04/05. [DOI] [PubMed] [Google Scholar]
- 23.Jalal DI, Rivard CJ, Johnson RJ, Maahs DM, McFann K, Rewers M, et al. Serum uric acid levels predict the development of albuminuria over 6 years in patients with type 1 diabetes: findings from the Coronary Artery Calcification in Type 1 Diabetes study. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2010;25(6):1865–9. doi: 10.1093/ndt/gfp740. Epub 2010/01/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24**.Maahs DM, Caramori L, Cherney DZ, Galecki AT, Gao C, Jalal D, et al. Uric Acid Lowering to Prevent Kidney Function Loss in Diabetes: The Preventing Early Renal Function Loss (PERL) Allopurinol Study. Curr Diab Rep. 2013 doi: 10.1007/s11892-013-0381-0. Epub 2013/05/08. A clinical trial with allopurinol to prevent early diabetic nepropathy in type 1 diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodrigues TC, Maahs DM, Johnson RJ, Jalal DI, Kinney GL, Rivard C, et al. Serum uric acid predicts progression of subclinical coronary atherosclerosis in individuals without renal disease. Diabetes Care. 2010;33(11):2471–3. doi: 10.2337/dc10-1007. Epub 2010/08/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26*.Bjornstad P, Snell-Bergeon JK, Rewers M, Jalal D, Chonchol MB, Johnson RJ, et al. Early Diabetic Nephropathy: A complication of reduced insulin sensitivity in type 1 diabetes. Diabetes care. 2013 doi: 10.2337/dc13-0631. Epub 2013/09/13. Demonstrates an important association between insulin sensitivity and early diabetic nephropathy in type 1 diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27*.Specht BJ, Wadwa RP, Snell-Bergeon JK, Nadeau KJ, Bishop FK, Maahs DM. Estimated insulin sensitivity and cardiovascular disease risk factors in adolescents with and without type 1 diabetes. The Journal of pediatrics. 2013;162(2):297–301. doi: 10.1016/j.jpeds.2012.07.036. Epub 2012/08/28. Data demonstrating an important association between insulin sensitivity and cardiovascular disease risk in adolescents with type 1 diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28**.Cherney DZ, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A, et al. The Renal Hemodynamic Effect of SGLT2 Inhibition in Patients with Type 1 Diabetes. Circulation. 2013 doi: 10.1161/CIRCULATIONAHA.113.005081. Epub 2013/12/18. Attenuation of renal hyperfiltration to normal GFR with SGLT2 inhibitors in type 1 diabetes. [DOI] [PubMed] [Google Scholar]
- 29.Schernthaner G, Gross JL, Rosenstock J, Guarisco M, Fu M, Yee J, et al. Canagliflozin compared with sitagliptin for patients with type 2 diabetes who do not have adequate glycemic control with metformin plus sulfonylurea: a 52-week randomized trial. Diabetes Care. 2013;36(9):2508–15. doi: 10.2337/dc12-2491. Epub 2013/04/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30**.Boertien WE, Riphagen IJ, Drion I, Alkhalaf A, Bakker SJ, Groenier KH, et al. Copeptin, a surrogate marker for arginine vasopressin, is associated with declining glomerular filtration in patients with diabetes mellitus (ZODIAC-33) Diabetologia. 2013;56(8):1680–8. doi: 10.1007/s00125-013-2922-0. Epub 2013/04/30. Data demonstrating copeptin predicts the estimated glomerular filtration rate decline in subjects with type 2 diabetes. [DOI] [PubMed] [Google Scholar]
- 31.Riphagen IJ, Boertien WE, Alkhalaf A, Kleefstra N, Gansevoort RT, Groenier KH, et al. Copeptin, a Surrogate Marker for Arginine Vasopressin, Is Associated With Cardiovascular and All-Cause Mortality in Patients With Type 2 Diabetes (ZODIAC-31) Diabetes Care. 2013 doi: 10.2337/dc12-2165. Epub 2013/06/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rossing K, Mischak H, Dakna M, Zurbig P, Novak J, Julian BA, et al. Urinary proteomics in diabetes and CKD. Journal of the American Society of Nephrology: JASN. 2008;19(7):1283–90. doi: 10.1681/ASN.2007091025. Epub 2008/05/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zurbig P, Jerums G, Hovind P, Macisaac R, Mischak H, Nielsen SE, et al. Urinary Proteomics for Early Diagnosis in Diabetic Nephropathy. Diabetes. 2012 doi: 10.2337/db12-0348. Epub 2012/08/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drummond K, Mauer M. The early natural history of nephropathy in type 1 diabetes: II. Early renal structural changes in type 1 diabetes. Diabetes. 2002;51(5):1580–7. doi: 10.2337/diabetes.51.5.1580. Epub 2002/04/30. [DOI] [PubMed] [Google Scholar]
- 35.Magee GM, Bilous RW, Cardwell CR, Hunter SJ, Kee F, Fogarty DG. Is hyperfiltration associated with the future risk of developing diabetic nephropathy? A meta-analysis. Diabetologia. 2009;52(4):691–7. doi: 10.1007/s00125-009-1268-0. Epub 2009/02/10. [DOI] [PubMed] [Google Scholar]
- 36.Gross JL, de Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T. Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care. 2005;28(1):164–76. doi: 10.2337/diacare.28.1.164. Epub 2004/12/24. [DOI] [PubMed] [Google Scholar]
- 37.Perkins BA, Ficociello LH, Silva KH, Finkelstein DM, Warram JH, Krolewski AS. Regression of microalbuminuria in type 1 diabetes. The New England journal of medicine. 2003;348(23):2285–93. doi: 10.1056/NEJMoa021835. Epub 2003/06/06. [DOI] [PubMed] [Google Scholar]
- 38**.Krolewski AS, Niewczas MA, Skupien J, Gohda T, Smiles A, Eckfeldt JH, et al. Early progressive renal decline precedes the onset of microalbuminuria and its progression to macroalbuminuria. Diabetes care. 2014;37(1):226–34. doi: 10.2337/dc13-0985. Epub 2013/08/14. Demonstrates that rapid GFR decline (also known as early progressive renal decline) precedes the onset of microalbuminuria. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shlipak MG, Katz R, Kestenbaum B, Siscovick D, Fried L, Newman A, et al. Rapid decline of kidney function increases cardiovascular risk in the elderly. Journal of the American Society of Nephrology: JASN. 2009;20(12):2625–30. doi: 10.1681/ASN.2009050546. Epub 2009/11/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shlipak MG, Katz R, Kestenbaum B, Fried LF, Newman AB, Siscovick DS, et al. Rate of kidney function decline in older adults: a comparison using creatinine and cystatin C. American journal of nephrology. 2009;30(3):171–8. doi: 10.1159/000212381. Epub 2009/04/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perkins BA, Ficociello LH, Ostrander BE, Silva KH, Weinberg J, Warram JH, et al. Microalbuminuria and the risk for early progressive renal function decline in type 1 diabetes. Journal of the American Society of Nephrology: JASN. 2007;18(4):1353–61. doi: 10.1681/ASN.2006080872. Epub 2007/03/03. [DOI] [PubMed] [Google Scholar]
- 42.Standards of medical care in diabetes--2013. Diabetes Care. 2013;36 (Suppl 1):S11–66. doi: 10.2337/dc13-S011. Epub 2013/01/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stevens PE, Levin A. Evaluation and management of chronic kidney disease: synopsis of the kidney disease: improving global outcomes 2012 clinical practice guideline. Annals of internal medicine. 2013;158(11):825–30. doi: 10.7326/0003-4819-158-11-201306040-00007. Epub 2013/06/05. [DOI] [PubMed] [Google Scholar]
- 44.Steinke JM, Sinaiko AR, Kramer MS, Suissa S, Chavers BM, Mauer M. The early natural history of nephropathy in Type 1 Diabetes: III. Predictors of 5-year urinary albumin excretion rate patterns in initially normoalbuminuric patients. Diabetes. 2005;54(7):2164–71. doi: 10.2337/diabetes.54.7.2164. Epub 2005/06/29. [DOI] [PubMed] [Google Scholar]
- 45.Inker LA, Schmid CH, Tighiouart H, Eckfeldt JH, Feldman HI, Greene T, et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med. 2012;367(1):20–9. doi: 10.1056/NEJMoa1114248. Epub 2012/07/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwartz GJ, Munoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, et al. New equations to estimate GFR in children with CKD. Journal of the American Society of Nephrology: JASN. 2009;20(3):629–37. doi: 10.1681/ASN.2008030287. Epub 2009/01/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47**.Krolewski AS, Warram JH, Forsblom C, Smiles AM, Thorn L, Skupien J, et al. Serum concentration of cystatin C and risk of end-stage renal disease in diabetes. Diabetes Care. 2012;35(11):2311–6. doi: 10.2337/dc11-2220. Epub 2012/08/02. CKD staging by eGFRCYSTATIN C improved ESRD risk stratification based on eGFRCREATININE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bjornstad P, McQueen RB, Snell-Bergeon JK, Cherney DZ, Perkins BA, Rewers M, et al. Fasting blood glucose – a missing variable in GFR-estimating equations in type 1 diabetes? PloS one. 2013 doi: 10.1371/journal.pone.0096264. in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cherney DZ, Sochett EB, Dekker MG, Perkins BA. Ability of cystatin C to detect acute changes in glomerular filtration rate provoked by hyperglycaemia in uncomplicated Type 1 diabetes. Diabetic medicine: a journal of the British Diabetic Association. 2010;27(12):1358–65. doi: 10.1111/j.1464-5491.2010.03121.x. Epub 2010/11/10. [DOI] [PubMed] [Google Scholar]
- 50.Maahs DM, Ogden LG, Kretowski A, Snell-Bergeon JK, Kinney GL, Berl T, et al. Serum cystatin C predicts progression of subclinical coronary atherosclerosis in individuals with type 1 diabetes. Diabetes. 2007;56(11):2774–9. doi: 10.2337/db07-0539. Epub 2007/07/31. [DOI] [PubMed] [Google Scholar]
- 51.Premaratne E, MacIsaac RJ, Finch S, Panagiotopoulos S, Ekinci E, Jerums G. Serial measurements of cystatin C are more accurate than creatinine-based methods in detecting declining renal function in type 1 diabetes. Diabetes Care. 2008;31(5):971–3. doi: 10.2337/dc07-1588. Epub 2008/03/06. [DOI] [PubMed] [Google Scholar]
- 52*.Skupien J, Warram JH, Groop PH, Krolewski AS. Cystatin-based estimated GFR versus creatinine-based and creatinine- and cystatin-based estimated GFR for ESRD and mortality risk in diabetes. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2013;62(1):184–6. doi: 10.1053/j.ajkd.2013.03.029. Epub 2013/05/21. Demonstrates superior ESRD and mortality risk stratification by eGFRCYSTATIN C than eGFRCYSTATIN C AND CREATININE equations. [DOI] [PubMed] [Google Scholar]
- 53.Shlipak MG, Matsushita K, Arnlov J, Inker LA, Katz R, Polkinghorne KR, et al. Cystatin C versus creatinine in determining risk based on kidney function. The New England journal of medicine. 2013;369(10):932–43. doi: 10.1056/NEJMoa1214234. Epub 2013/09/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maahs DM, Jalal D, McFann K, Rewers M, Snell-Bergeon JK. Systematic shifts in cystatin C between 2006 and 2010. Clin J Am Soc Nephrol. 2011;6(8):1952–5. doi: 10.2215/CJN.11271210. Epub 2011/07/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maahs DM, Prentice N, McFann K, Snell-Bergeon JK, Jalal D, Bishop FK, et al. Age and sex influence cystatin C in adolescents with and without type 1 diabetes. Diabetes Care. 2011;34(11):2360–2. doi: 10.2337/dc11-0829. Epub 2011/09/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Diabetes and Chronic Kidney Disease. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2007;49(2 Suppl 2):S12–154. doi: 10.1053/j.ajkd.2006.12.005. Epub 2007/02/06. [DOI] [PubMed] [Google Scholar]
- 57.Niculescu-Duvaz I, D’Mello L, Maan Z, Barron JL, Newman DJ, Dockrell ME, et al. Development of an outpatient finger-prick glomerular filtration rate procedure suitable for epidemiological studies. Kidney international. 2006;69(7):1272–5. doi: 10.1038/sj.ki.5000240. Epub 2006/04/13. [DOI] [PubMed] [Google Scholar]
- 58.Mafham MM, Niculescu-Duvaz I, Barron J, Emberson JR, Dockrell ME, Landray MJ, et al. A practical method of measuring glomerular filtration rate by iohexol clearance using dried capillary blood spots. Nephron Clinical practice. 2007;106(3):c104–12. doi: 10.1159/000102997. Epub 2007/05/25. [DOI] [PubMed] [Google Scholar]
- 59**.Maahs D, Bushman L, Kerr B, Ellis S, McFann K, Bouffard A, et al. An accurate and convenient method to measure GFR in people with type 1 diabetes. Submitted to 2014 American Diabetes Association’s Scientific Sessions. 2014 Presents a novel and practical way to measure GFR by iohexol and dried blood spots in type 1 diabetes. [Google Scholar]
- 60.Groop PH, Thomas MC, Moran JL, Waden J, Thorn LM, Makinen VP, et al. The presence and severity of chronic kidney disease predicts all-cause mortality in type 1 diabetes. Diabetes. 2009;58(7):1651–8. doi: 10.2337/db08-1543. Epub 2009/04/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cherney D, Maahs DM. Cystatin C versus creatinine for kidney function-based risk. The New England journal of medicine. 2013;369(25):2458. doi: 10.1056/NEJMc1312801. Epub 2013/12/20. [DOI] [PubMed] [Google Scholar]
- 62.Theodorescu D, Wittke S, Ross MM, Walden M, Conaway M, Just I, et al. Discovery and validation of new protein biomarkers for urothelial cancer: a prospective analysis. The lancet oncology. 2006;7(3):230–40. doi: 10.1016/S1470-2045(06)70584-8. Epub 2006/03/03. [DOI] [PubMed] [Google Scholar]
- 63.von Zur Muhlen C, Schiffer E, Zuerbig P, Kellmann M, Brasse M, Meert N, et al. Evaluation of urine proteome pattern analysis for its potential to reflect coronary artery atherosclerosis in symptomatic patients. Journal of proteome research. 2009;8(1):335–45. doi: 10.1021/pr800615t. Epub 2008/12/05. [DOI] [PubMed] [Google Scholar]
- 64.Meier M, Kaiser T, Herrmann A, Knueppel S, Hillmann M, Koester P, et al. Identification of urinary protein pattern in type 1 diabetic adolescents with early diabetic nephropathy by a novel combined proteome analysis. Journal of diabetes and its complications. 2005;19(4):223–32. doi: 10.1016/j.jdiacomp.2004.10.002. Epub 2005/07/05. [DOI] [PubMed] [Google Scholar]
- 65.Zimmerli LU, Schiffer E, Zurbig P, Good DM, Kellmann M, Mouls L, et al. Urinary proteomic biomarkers in coronary artery disease. Molecular & cellular proteomics: MCP. 2008;7(2):290–8. doi: 10.1074/mcp.M700394-MCP200. Epub 2007/10/24. [DOI] [PubMed] [Google Scholar]
- 66.Snell-Bergeon JK, Maahs DM, Ogden LG, Kinney GL, Hokanson JE, Schiffer E, et al. Evaluation of urinary biomarkers for coronary artery disease, diabetes, and diabetic kidney disease. Diabetes Technol Ther. 2009;11(1):1–9. doi: 10.1089/dia.2008.0040. Epub 2009/01/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cherney DZ, Scholey JW, Sochett E, Bradley TJ, Reich HN. The acute effect of clamped hyperglycemia on the urinary excretion of inflammatory cytokines/chemokines in uncomplicated type 1 diabetes: a pilot study. Diabetes Care. 2011;34(1):177–80. doi: 10.2337/dc10-1219. Epub 2010/09/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68*.Har R, Scholey JW, Daneman D, Mahmud FH, Dekker R, Lai V, et al. The effect of renal hyperfiltration on urinary inflammatory cytokines/chemokines in patients with uncomplicated type 1 diabetes mellitus. Diabetologia. 2013;56(5):1166–73. doi: 10.1007/s00125-013-2857-5. Epub 2013/02/16. Presents evidence of an association between renal hyperfiltration and urinary inflammatory cytokines/chemokines. [DOI] [PubMed] [Google Scholar]
- 69.Hovind P, Rossing P, Tarnow L, Johnson RJ, Parving HH. Serum uric acid as a predictor for development of diabetic nephropathy in type 1 diabetes: an inception cohort study. Diabetes. 2009;58(7):1668–71. doi: 10.2337/db09-0014. Epub 2009/05/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ficociello LH, Rosolowsky ET, Niewczas MA, Maselli NJ, Weinberg JM, Aschengrau A, et al. High-normal serum uric acid increases risk of early progressive renal function loss in type 1 diabetes: results of a 6-year follow-up. Diabetes Care. 2010;33(6):1337–43. doi: 10.2337/dc10-0227. Epub 2010/03/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71**.Cleland SJ, Fisher BM, Colhoun HM, Sattar N, Petrie JR. Insulin resistance in type 1 diabetes: what is ‘double diabetes’ and what are the risks? Diabetologia. 2013;56(7):1462–70. doi: 10.1007/s00125-013-2904-2. Epub 2013/04/25. An important review of the importance of insulin resistance in type 1 diabetes and vascular pathology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Orchard TJ, Chang YF, Ferrell RE, Petro N, Ellis DE. Nephropathy in type 1 diabetes: a manifestation of insulin resistance and multiple genetic susceptibilities? Further evidence from the Pittsburgh Epidemiology of Diabetes Complication Study. Kidney Int. 2002;62(3):963–70. doi: 10.1046/j.1523-1755.2002.00507.x. Epub 2002/08/08. [DOI] [PubMed] [Google Scholar]
- 73.Perseghin G, Lattuada G, Danna M, Sereni LP, Maffi P, De Cobelli F, et al. Insulin resistance, intramyocellular lipid content, and plasma adiponectin in patients with type 1 diabetes. American Journal of Physiology - Endocrinology And Metabolism. 2003;285(6):E1174–E81. doi: 10.1152/ajpendo.00279.2003. [DOI] [PubMed] [Google Scholar]
- 74.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440(7086):944–8. doi: 10.1038/nature04634. Epub 2006/04/14. [DOI] [PubMed] [Google Scholar]
- 75*.Bjornstad P, Snell-Bergeon JK, McFann K, Wadwa RP, Rewers M, Rivard CJ, et al. Serum uric acid and insulin sensitivity in adolescents and adults with and without type 1 diabetes. Journal of diabetes and its complications. 2013 doi: 10.1016/j.jdiacomp.2013.12.007. Epub 2014/01/28. Data suggest independence of serum uric acid and insulin sensitivity in type 1 diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Frye RL, August P, Brooks MM, Hardison RM, Kelsey SF, MacGregor JM, et al. A randomized trial of therapies for type 2 diabetes and coronary artery disease. N Engl J Med. 2009;360(24):2503–15. doi: 10.1056/NEJMoa0805796. Epub 2009/06/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.List JF, Whaley JM. Glucose dynamics and mechanistic implications of SGLT2 inhibitors in animals and humans. Kidney international Supplement. 2011;(120):S20–7. doi: 10.1038/ki.2010.512. Epub 2011/03/05. [DOI] [PubMed] [Google Scholar]
- 78.Idris I, Donnelly R. Sodium-glucose co-transporter-2 inhibitors: an emerging new class of oral antidiabetic drug. Diabetes, obesity & metabolism. 2009;11(2):79–88. doi: 10.1111/j.1463-1326.2008.00982.x. Epub 2009/01/08. [DOI] [PubMed] [Google Scholar]
- 79.Vallon V, Sharma K. Sodium-glucose transport: role in diabetes mellitus and potential clinical implications. Current opinion in nephrology and hypertension. 2010;19(5):425–31. doi: 10.1097/MNH.0b013e32833bec06. Epub 2010/06/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brownlee M, Hirsch IB. Glycemic variability: a hemoglobin A1cindependent risk factor for diabetic complications. JAMA: the journal of the American Medical Association. 2006;295(14):1707–8. doi: 10.1001/jama.295.14.1707. Epub 2006/04/13. [DOI] [PubMed] [Google Scholar]
- 81**.Cherney DZ, Reich HN, Scholey JW, Daneman D, Mahmud FH, Har RL, et al. The effect of aliskiren on urinary cytokine/chemokine responses to clamped hyperglycaemia in type 1 diabetes. Diabetologia. 2013;56(10):2308–17. doi: 10.1007/s00125-013-3000-3. Epub 2013/07/31. Data suggest that hyperglycemia may contribute to kidney injury via RAAS activation. [DOI] [PubMed] [Google Scholar]
- 82.Torres VE. Vasopressin in chronic kidney disease: an elephant in the room? Kidney international. 2009;76(9):925–8. doi: 10.1038/ki.2009.325. Epub 2009/10/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bolignano D, Zoccali C. Vasopressin beyond water: implications for renal diseases. Current opinion in nephrology and hypertension. 2010;19(5):499–504. doi: 10.1097/MNH.0b013e32833d35cf. Epub 2010/08/07. [DOI] [PubMed] [Google Scholar]
- 84.Bardoux P, Martin H, Ahloulay M, Schmitt F, Bouby N, Trinh-Trang-Tan MM, et al. Vasopressin contributes to hyperfiltration, albuminuria, and renal hypertrophy in diabetes mellitus: study in vasopressin-deficient Brattleboro rats. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(18):10397–402. doi: 10.1073/pnas.96.18.10397. Epub 1999/09/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gellai M, Silverstein JH, Hwang JC, LaRochelle FT, Jr, Valtin H. Influence of vasopressin on renal hemodynamics in conscious Brattleboro rats. The American journal of physiology. 1984;246(6 Pt 2):F819–27. doi: 10.1152/ajprenal.1984.246.6.F819. Epub 1984/06/11. [DOI] [PubMed] [Google Scholar]
- 86.Bouby N, Bachmann S, Bichet D, Bankir L. Effect of water intake on the progression of chronic renal failure in the 5/6 nephrectomized rat. The American journal of physiology. 1990;258(4 Pt 2):F973–9. doi: 10.1152/ajprenal.1990.258.4.F973. Epub 1990/04/01. [DOI] [PubMed] [Google Scholar]
- 87.Strippoli GF, Craig JC, Rochtchina E, Flood VM, Wang JJ, Mitchell P. Fluid and nutrient intake and risk of chronic kidney disease. Nephrology (Carlton) 2011;16(3):326–34. doi: 10.1111/j.1440-1797.2010.01415.x. Epub 2011/02/24. [DOI] [PubMed] [Google Scholar]