Pathological Stress Granules in Alzheimer’s Disease (original) (raw)
. Author manuscript; available in PMC: 2015 Oct 10.
Abstract
A feature of neurodegenerative disease is the accumulation of insoluble protein aggregates in the brain. In some conditions, including Amyotrophic Lateral Sclerosis and Frontotemporal lobar degeneration, the primary aggregating entities are RNA binding proteins. Through regulated prion-like assembly, RNA binding proteins serve many functions in RNA metabolism that are essential for the healthy maintenance of cells of the central nervous system. Those RNA binding proteins that are the core nucleating factors of Stress Granules (SGs), including TIA-1, TIAR, TTP and G3BP1, are also found in the pathological lesions of other neurological conditions, such as Alzheimer’s disease, where the hallmark aggregating protein is not an RNA binding protein. This discovery suggests that the regulated cellular pathway, which utilizes assembly of RNA binding proteins to package and silence mRNAs during stress, may be integral in the aberrant pathological protein aggregation that occurs in numerous neurodegenerative conditions.
Keywords: Pathological stress granules, tau, TIA-1, G3BP1, Alzheimer’s Disease
Introduction
In 2012 we proposed an important mechanistic role for regulated protein aggregation of stress granule RNA binding proteins (RBPs) in the development of neurodegenerative disorders (Wolozin, 2012). Aggregation of misfolded proteins is a key pathological feature of neurodegenerative disorders including Alzheimer’s Disease and Parkinson’s Disease. The model of deleterious protein aggregation in neurodegenerative disease is classically viewed as an unregulated and random process that arises from the uncoordinated interaction of misfolded proteins. In this model, random misfolding of protein monomers occurs resulting in non-functional forms. Whereas these misfolded proteins would normally undergo repair by chaperone-mediated refolding or targeted degradation, in disease-affected neurons these processes are impaired. Consequently, the accumulation of misfolded protein monomers leads to their assembly into oligomers and further aggregation into fibrils through the principle of mass action and energy minimization (Dobson, 2003; Jarrett and Lansbury, 1993). The rate of aggregation is dependent upon the respective concentrations and hydrophobicities of the misfolded monomers, oligomers and fibrils. It is also dependent upon their rates of proteosomal and autophagic degradation. As such a high abundance of a highly hydrophobic misfolded protein will lead to more aggressive rate of aggregation.
However, assembly of hydrophobic proteins into aggregates is not a uniquely unfavorable event. Prion-like proteins in yeast, such as the elongation initiation factor Sup35, undergo conformational change to confer adaptability to changing environmental conditions (True and Lindquist, 2000). In eukaryotes, it is a normal physiological role of RNA binding proteins to aggregate in a regulated and reversible manner. This is achieved through the interaction of hydrophobic inherently aggregation-prone domains which show striking sequence homology to the Sup35 prion-related domain (PRD); these PRDs are notable for containing glycine-rich and low complexity sequences (Han et al., 2012; Kato et al., 2012). In particular, the assembly of non-membrane bound RNA granules, such as stress granules (SGs), processing bodies (P-bodies) and RNA transport granules, are dependent upon the co-aggregation of numerous RBPs (Gilks et al., 2004; Kedersha and Anderson, 2007). Each of these processes plays an essential role in healthy maintenance of a neuron’s transcriptome. SGs translationally suppress non-essential proteins to favor cytoprotective protein expression in response to an environmental stress. P-bodies are centers of mRNA silencing and ultimately degradation. RNA transport granules shuttle translationally repressed mRNAs to distinct cellular locations, a process that is particularly important for activity-dependent localized translation in neurons.
Nonetheless, RBPs are particularly relevant to the biology of neurodegenerative disease. TAR DNA binding protein-43 (TDP-43), a nuclear RNA binding protein with many RNA metabolic functions (reviewed in Lagier-Tourenne et al., 2010), is the core component of pathological inclusions in sporadic and familial forms of Amyotrophic Lateral Sclerosis (ALS) and tau-negative Frontotemporal Lobar Degeneration with TDP-43 lesions (FTLD-TDP; Neumann et al., 2006). Mutation of the RNA binding proteins TDP-43, Fused in Sarcoma (FUS), survival of motor neuron (SMN1), ataxin-2 (ATXN2), optineurin (OPTN), angiogenin (ANG; Chen et al., 2013) and the heterogeneous Ribonucleoproteins A1 and A2/B1 (hnRNP A1 and hnRNP A2/B1; Kim et al., 2013) all lead to heritable motor neuron diseases.
Importantly, we have demonstrated a role in neurodegenerative disease for the core RNA binding proteins of SG assembly, T-cell-restricted intracellular antigen-1 (TIA-1); TIA-like-1 (TIAR); Tristetraprolin (TTP) and GTPase Activating Protein (SH3 Domain) Binding Protein 1 (G3BP1). These core SG markers colocalize with TDP-43 positive lesions in ALS and FTLD-TDP (Liu-Yesucevitz et al., 2010) and are observed in the hallmark tau lesions of AD and FTLD with tau lesions (FTLD-tau; Vanderweyde et al., 2012). SG markers have also found to associate with pathological lesions in other neurological disorders, such as Huntington’s Disease (HD) and Creutzfeldt-Jakob Disease (CJD; Goggin et al., 2008; Waelter et al., 2001).
This observation suggests an important role for the development of pathological stress granules in the pathobiology of both neurological diseases commonly associated with dysfunction of RNA binding proteins (like FTLD-TDP and FTLD-FUS, ALS and related motor disorders) and in diseases where the primary aggregating protein is not an RBP (such as FTLD-tau, AD, HD and CJD). Although initially the formation of SGs in affected neurons likely occurs as a cytoprotective mechanism, the chronic stress associated with neurodegeneration may lead to deposition of “pathological SGs” into the detergent-insoluble pathological lesions common to these conditions.
Stress granules
Stress granules are non-membrane bound cytoplasmic complexes of protein and RNA. They form as a means for a cell to rapidly and reversibly alter its proteome to respond to an environmental stressor. This is achieved through the regulated aggregation of core SG RNA binding proteins (TIA-1, TIAR, TTP and G3BP1), which bind to and sequester mRNA transcripts of non-essential proteins (Gilks et al., 2004; Kedersha et al., 2000). By this means, select transcripts are translationally suppressed allowing for the upregulation of stress-responsive cytoprotective proteins.
As shown in Figure 1, SG formation is initiated by the phosphorylation of eIF2α at Serine 51 (S51). The phosphorylation of eIF2α at S51 is mediated by a number of kinases acting in response to variable environmental stresses. Protein kinase R (PKR) is a double-stranded RNA-dependent kinase that responds to viral infection, heat shock and UV irradiation. PKR-like endoplasmic reticulum kinase (PERK) is an endoplasmic reticulum localized kinase that is activated as part of the unfolded protein response to accumulation of misfolded protein in the ER lumen. General control non-derepressible 2 (GCN2) kinase activity is triggered upon amino acid deprivation, thereby sensing nutritional starvation. Heme-regulated initiation factor 2α kinase (HRI) is responsive to oxidative stress. Z-DNA kinase is activated as part of the anti-viral response.
Figure 1. Development of pathological stress granules.
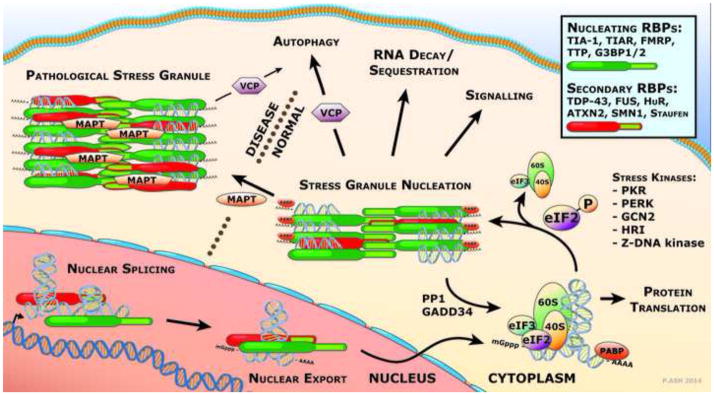
RNA binding proteins facilitate many aspects of RNA metabolism including both nuclear and cytoplasmic functions in mRNA splicing, maturation and cellular localization. Under stress conditions, responsive kinases phosphorylate eIF2α leading to the disassembly of the polysome and the induction of SG formation. SGs are nucleated through the binding of core RBPs to mRNA and their self-assembly through the association of prion-related domains. Maturation of SGs follows with the recruitment of secondary RBPs. In disease, prolonged stress, mutations that favor the pro-aggregation state or the inability to recover and disassemble the RBPs leads to the deposition of abundant, large pathological stress granules.
Transcripts targeted to SGs initially exist as capped mRNAs bound to translation pre-initiation 40S ribosomal complexes containing other translation factors, such as elongation factors EF-4A, E and G, eIF3 and poly-A binding protein. This eIF2α phospho-S51 signal inhibits the formation of the eIF2, GTP and tRNAMet initiation complex (Kedersha et al., 1999) resulting in the stalling of translation initiation and the dissociation of the ribosomal units. The mRNAs are then bound by the SG-nucleating RNA binding proteins TIA-1, TIAR, TTP and G3BP1. SGs form through consolidation of these RNA binding proteins through association of the glycine-rich prion-related domains (Kedersha et al., 2000).
As SGs mature they recruit additional RNA binding proteins from both the cytoplasm and the nucleus. During the process of secondary maturation, a number of disease-relevant RBPs including TDP-43 (Colombrita et al., 2009; Liu-Yesucevitz et al., 2010), FUS (Dormann and Haass, 2011), ataxin 2 (Hart and Gitler, 2012) and SMN1 (Hua and Zhou, 2004) associate with the stress granules in cultured cells. These new recruits bring with them additional transcripts for sequestration in the SG, depending upon the RNA binding consensus of each RNA binding protein. The composition of proteins, and therefore RNA transcripts, differs depending on the nature of the environmental stressor so that the resulting stress response is correctly tailored (Munchel et al., 2011).
The formation of stress granules in response to an acute stress is a transient event. SGs can be observed in severely stressed cells within minutes of exposure, and once the stress is removed, the SGs will dissipate after 1-3 hours (Kedersha et al., 1999). As SGs are designed to mediate recovery from stress, they are also capable of inhibiting apoptosis. Pro-apoptotic proteins including RACK1, ROCK1 and TRAF2 are sequestered in SGs, preventing their activity (Arimoto et al., 2008; Kim et al., 2005). Dissociation of SGs is mediated by the dephosphorylation of eIF2α S51, which has been most extensively studied in relation to the Growth arrest and DNA damage-inducible protein 34 (GADD34). This factor associates with Protein Phosphatase 1 (PP1) and recruits it to eIF2α for the dephosphorylation of Serine 51 (Brush et al., 2003). Indeed, GADD34 is maintained at low levels in the cells under basal conditions, but is induced upon exposure to various forms of stress (Brush and Shenolikar, 2008), thereby establishing a feedback mechanism to limit eIF2α phosphorylation and allow recovery from transient stress.
SGs in Neurodegenerative disease
Because stress granules develop as a cellular response to an environmental stress, their prevalence in neurological disorders suggests that their formation in affected neurons is, at least initially, a cytoprotective response to stresses associated with the disease. Blocking SG formation by either depleting TIA-1 or inhibiting phosphorylation of eIF2α renders cells more vulnerable to stress (Jiang et al., 2003; Phillips et al., 2004). We have observed that tau pathology, such as that which occurs in many neurodegenerative diseases, is associated with very large stress granules. This raises the possibility that stress granules, which are normally transient structures, form abnormally stable structures in neurodegenerative diseases. We hypothesize that these large, stable SGs are pathological, and contribute to the neurodegenerative process, perhaps by sequestering RNA binding proteins, much like what is thought to occur in ALS or Myotonic Dystrophy.
Aggregation of TDP-43 and FUS, both of which are stress granule associated RNA binding proteins, play a causative role neurodegenerative disease and drive pathological lesions detectable in post-mortem human tissue that incorporate canonical markers of SGs (Dormann and Haass, 2011; Liu-Yesucevitz et al., 2010). In addition, most of the RNA binding proteins mutated in disease associate with SGs in cultured cells, including hnRNP A1 and A2/B1, ataxin-2, SMN1, optineurin and angiogenin (Hart and Gitler, 2012; Hua and Zhou, 2004; Kim et al., 2013). Similar to the conventional model of protein aggregation, the kinetics of regulated protein assembly that contributes to pathological SG are also dependent upon the localized concentrations of monomeric RNA binding proteins, the “prionogenicity” of their self-assembly domains and the ability of the cell to recover from a stress and clear SGs. Human mutations in RNA binding proteins that lead to ALS, FTD and related neurological disorders have been shown to affect aspects of this system. The majority of ALS-causative mutations in the nuclear RNA binding protein FUS occur in the PY-nuclear localization signal. These mutations disrupt Transportin-mediated nuclear import, which increases the localized cytoplasmic concentration of mutant FUS enhancing likelihood of incorporation into SGs (Dormann and Haass, 2011). Nearly all of mutations in TDP-43, certain mutations in FUS and those mutations in hnRNP A1 and A2/B1 modify the prion-related domain of each protein. These mutations alter local protein hydrophobicity, thereby increasing “prionogenicity”, the propensity of the mutant protein to self-assemble and form aggregates with increased stability, and enhance the recruitment of each of these RBPs into SGs (Dewey et al., 2011; Johnson et al., 2008; Kim et al., 2013; King et al., 2012).
The examples above demonstrate how mutations leading to dysfunction of RBPs that contribute to the maturation of SGs play a causative role in the development of neurodegeneration, particularly in familial forms of motor neuron disease. However, SG-positive pathology also occurs in many cases of sporadic neurological disease, which are not explicitly associated with mutations in an RBP. For instance, inclusions of normal TDP-43 with concomitant SG-positive markers are the major pathology of sporadic ALS, they are also prevalent in FTLD-TDP due to non-sense mutations in progranulin (PGRN; Kumar-Singh, 2011), and due to hexanucleotide repeat expansion-mutations in C9ORF72 (DeJesus-Hernandez et al., 2011).
In sporadic disease an as yet uncharacterized environmental cue may induce SG formation. Dysfunctional proteostasis and enhanced oxidative stress are well documented in neurodegenerative disease (Harris and Rubinsztein, 2012; Powers et al., 2009) and maybe responsible for inducing or prolonging the SG-inductive signal. The unfolded-protein-response (UPR) is activated in response to accumulation of misfolded protein oligomers, such tau, and may trigger the phosphorylation of eIF2α via activated PERK (Abisambra et al., 2013). The oxidative damage to cellular components observed in AD (Perry et al., 2002) and other neurodegenerative disease could activate SG formation through HRI kinase. Alternately, treatment of cultured cells with the double-stranded RNA mimetic, Polyinosinic-polycytidylic acid (Poly(I:C)), results in the activation of PKR leading to eIF2α S51 phosphorylation (Weissbach and Scadden, 2012), perhaps indicting viruses in the induction of SG formation.
Ultimately, the kinetics of SG assembly and disassembly are controlled by those signals created by the onset of stress and the recovery from it. Disassembly is orchestrated by molecular-chaperones and by autophagy. In post-mortem tissue, the detection of abundant, large SGs suggests a reduced ability of affected neurons to clear SGs. It is likely that the persistent protein aggregates transform over time acquiring post-translational modifications such as phosphorylation, ubiquitination, glycosylation and cross-linking that further prevents the regulated dissolution of the pathological SG.
Clearance of SGs is mediated in part by the autophagic pathway and depends on the function of valosin containing protein (VCP). VCP is an ATP-dependent segregase, which facilitates removal of ubiquitinated proteins from larger complexes (Stolz et al., 2011). Mutations in VCP result in Multisystem Proteinopathy (MSP; or Inclusion Body Myopathy with Paget’s Disease and Frontotemporal Dementia), a TDP-43 proteinopathy that may present clinically as ALS, Frontotemporal Dementia or myopathy (Watts et al., 2004). By expressing VCP carrying disease-relevant mutations in stressed cultured cells, Buchan et al. (2013) demonstrated that the mutations prolonged the duration of SGs during recovery from the stressor. This observation suggests that the mechanism by which VCP mutations cause pathological aggregation of TDP-43 resulting in MSP could be through the prolonged duration of stress granules, and further demonstrates the importance of pathological stress granules in neurological disease.
The primary nucleating RNA binding proteins of stress granules, TIA-1, TIAR, TTP and G3BP1, are also present in those neurological diseases in which the signature aggregating protein is not an RNA binding protein. We have shown that TIA-1 interacts and colocalizes with the aggregated phosphorylated and conformationally altered tau observed in AD and in FTLD-tau (Figure 2). Furthermore, using constitutively expressing P301L tau mice, we observed that as the severity of tau pathology increased in aged animals, there is a concomitant increase in accumulation and colocalization of TIA-1 in affected neurons (Vanderweyde et al., 2012). Tau directly interacts with TIA-1 and the related SG-nucleating RNA binding protein TTP, when co-immunoprecipitated from tau transgenic mouse and human FTLD-tau tissue. The affinity of this interaction in increased in affected tissue with severe pathology, where TIA-1 and TTP more strongly interact with phosphorylated forms of tau (Vanderweyde et al., 2012).
Figure 2. TIA-1-positive tau pathology.
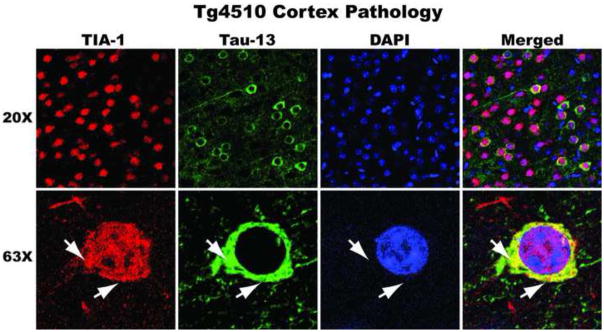
8 month old P301L tau transgenic mice display co-localization of TIA-1 (red; SantaCruz) and tau (green; Tau13) in cortical neurons. Nuclei counterstained with DAPI.
Under basal conditions, expression of tau or TIA-1 in cultured SH-SY5Y induced neither the formation of TIA-1-positive SGs nor the conversion of tau to PHF-1 immunoreactive phospho-tau. However, co-expression of tau and TIA-1 is sufficient to trigger formation of TIA-1-positive SGs. The induction of SGs is greatly increased when tau and TIA-1 co-expressing cells are treated with sodium arsenite and more importantly PHF-1 tau was observed to accumulate in these SGs. It is particularly interesting to note that expression of wild-type or P301L tau in cultured cells led to moderate formation of TIA-1-positive SGs when treated with arsenite when compared to either a) untransfected cells treated with arsenite or b) cells transfected with TIA-1 treated with arsenite (Vanderweyde et al., 2012). This evidence indicated reciprocal effects of TIA-1 and tau on each other, in which accumulation of phosphorylated tau is stimulated by TIA-1 and tau expression may play a role in SG formation.
Conversely, the SG protein G3BP1 accumulates in severely affected tau transgenic animals, but does not accumulate in those neurons with severe tau pathology. This differential pattern of SG marker association with pathological tau was reproducible in FTLD-tau affected human tissue. Thus, it appears that G3BP1-positive SGs may reduce tau pathology. The presence of G3BP in neurons that appear to lack tau pathology provides a concrete example of pathology occurring in tauopathies that are not explicitly coupled to the accumulation of pathological tau. Work performed by the Luebke and Ashe laboratories both suggest that the tau pathology in specific neurons is not correlated with that neuron’s demise (Crimins et al., 2012; Santacruz et al., 2005). Thus, the accumulation of G3BP in neurons that lack tau pathology complements the presence of tau pathology in functional neurons and emphasizes the possibility that neurofibrillary tangles are not the principle agent causing death in tauopathy. It is therefore possible that RBPs represent one of the previously unrecognized components of tauopathy. In this context, it is notable that in each of these diseases, the signature pathologies include the core component RNA binding proteins of SG nucleation and assembly, although the exact compliment of SG RNA binding proteins that associate with the pathology varies from one condition to another.
Conclusions
The question that remains is whether or not stress granule development and prolonged duration represents a protective mechanism throughout the course of the disease. The disease process increases the propensity of stress granule RBPs to enter into a pro-aggregation state and the progressive nature of the disease appears to prevent the recovery from stress and the disassembly of SGs. The importance of SG in maintaining healthy neurons will likely resemble the “Goldilocks effect”, in that a basal but responsive level of function is required. Transient reversible SG formation is necessary for a cytoprotective response to an acute stresses, but the hyperactive response to chronic stresses in neurodegenerative disease leads to robust persistent SGs that are deleterious. The pathological SGs may prolong the sequestration of transiently non-essential mRNA transcripts that in the longer-term are required for the healthy functioning of a neuron, analogous to what occurs in myotonic dystrophy (Udd and Krahe, 2012).
The current data indicates that the development of pathological stress granules occurs in conjunction with the disease process and integrates into the pathological lesions. Our data indicates a reciprocal effect of tau and TIA-1 on each other. Tau is able to facilitate the assembly of TIA-1-positive SG, just as TIA-1 facilitates the assembly of phosphorylated and conformationally altered tau (Vanderweyde et al., 2012). We suggest that the pathological aggregation of protein observed in numerous neurodegenerative conditions may initiate through the regulated self-assembly of core SG-associated RNA binding proteins, such as TIA-1.
The importance of this concept is in the reversibility of RNA granules and SGs through regulated signaling cascades and, therefore, the potential benefits of small molecule modulation of protein aggregation and disaggregation. Already, some exciting work has been presented detailing the effect on neurodegeneration in a Prion-protein infected mouse model of relieving the signal for the formation of SGs, namely the phosphorylation of eukaryotic Initiation Factor 2α (eIF2α). This was achieved through expression of GADD34, which mediates the dephosphorylation of eIF2α (Moreno et al., 2012), and through inhibition of the kinase of eIF2α, PERK (Moreno et al., 2013). Neuroprotection was observed in PrP-infected mice through alleviation of the unfolded protein response and re-initiation of stalled translation of critical neuronal proteins including synaptic proteins.
Highlights.
- In Alzheimer’s disease, pathological tau is associated with RNA binding proteins
- The RNA binding proteins fulfill established criteria of stress granules
- TIA-1 appears capable of inducing the appearance of inclusions with misfolded tau
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abisambra JF, Jinwal UK, Blair LJ, O’Leary JC, Li Q, Brady S, Wang L, Guidi CE, Zhang B, Nordhues BA, Cockman M, Suntharalingham A, Li P, Jin Y, Atkins CA, Dickey CA. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J Neurosci. 2013;33:9498–507. doi: 10.1523/JNEUROSCI.5397-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, Takekawa M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat Cell Biol. 2008;10:1324–32. doi: 10.1038/ncb1791. [DOI] [PubMed] [Google Scholar]
- Brush MH, Shenolikar S. Control of cellular GADD34 levels by the 26S proteasome. Mol Cell Biol. 2008;28:6989–7000. doi: 10.1128/MCB.00724-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brush MH, Weiser DC, Shenolikar S. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol Cell Biol. 2003;23:1292–303. doi: 10.1128/MCB.23.4.1292-1303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic Stress Granules Are Cleared by Autophagy and Cdc48/VCP Function. Cell. 2013;153:1461–74. doi: 10.1016/j.cell.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Sayana P, Zhang X, Le W. Genetics of amyotrophic lateral sclerosis: an update. Mol Neurodegener. 2013;8:28. doi: 10.1186/1750-1326-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E, Silani V, Ratti A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111:1051–61. doi: 10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- Crimins JL, Rocher AB, Luebke JI. Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathol. 2012;124:777–95. doi: 10.1007/s00401-012-1038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GYR, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey CM, Cenik B, Sephton CF, Dries DR, Mayer P, Good SK, Johnson BA, Herz J, Yu G. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol. 31:1098–108. doi: 10.1128/MCB.01279-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–90. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- Dormann D, Haass C. TDP-43 and FUS: a nuclear affair. Trends Neurosci. 2011;34:339–348. doi: 10.1016/j.tins.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell. 2004;15:5383–98. doi: 10.1091/mbc.E04-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goggin K, Beaudoin S, Grenier C, Brown AA, Roucou X. Prion protein aggresomes are poly(A)+ ribonucleoprotein complexes that induce a PKR-mediated deficient cell stress response. Biochim Biophys Acta. 2008;1783:479–91. doi: 10.1016/j.bbamcr.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G, McKnight SL. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell. 149:768–79. doi: 10.1016/j.cell.2012.04.016. [DOI] [PubMed] [Google Scholar]
- Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. 2012;8:108–17. doi: 10.1038/nrneurol.2011.200. [DOI] [PubMed] [Google Scholar]
- Hart MP, Gitler AD. ALS-associated ataxin 2 polyQ expansions enhance stress-induced caspase 3 activation and increase TDP-43 pathological modifications. J Neurosci. 2012;32:9133–42. doi: 10.1523/JNEUROSCI.0996-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Zhou J. Survival motor neuron protein facilitates assembly of stress granules. FEBS Lett. 2004;572:69–74. doi: 10.1016/j.febslet.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Jarrett JT, Lansbury PT. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol Cell Biol. 2003;23:5651–63. doi: 10.1128/MCB.23.16.5651-5663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BS, McCaffery JM, Lindquist S, Gitler AD. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2008;105:6439–6444. doi: 10.1073/pnas.0802082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R, McKnight SL. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–67. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Anderson P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007;431:61–81. doi: 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P. Dynamic Shuttling of Tia-1 Accompanies the Recruitment of mRNA to Mammalian Stress Granules. J Cell Biol. 2000;151:1257–1268. doi: 10.1083/jcb.151.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol. 1999;147:1431–42. doi: 10.1083/jcb.147.7.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–73. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WJ, Back SH, Kim V, Ryu I, Jang SK. Sequestration of TRAF2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol Cell Biol. 2005;25:2450–62. doi: 10.1128/MCB.25.6.2450-2462.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. doi: 10.1016/j.brainres.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar-Singh S. Progranulin and TDP-43: mechanistic links and future directions. J Mol Neurosci. 2011;45:561–73. doi: 10.1007/s12031-011-9625-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19:R46–64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Vanderwyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, Petrucelli L, Wolozin B. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One. 2010;5:e13250. doi: 10.1371/journal.pone.0013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno Ja, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori Ca, Barrett Da, Tsaytler P, Bertolotti A, Willis AE, Bushell M, Mallucci GR. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature. 2012;485:507–11. doi: 10.1038/nature11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, Mallucci GR. Oral Treatment Targeting the Unfolded Protein Response Prevents Neurodegeneration and Clinical Disease in Prion-Infected Mice. Sci Transl Med. 2013;5:206ra138–206ra138. doi: 10.1126/scitranslmed.3006767. [DOI] [PubMed] [Google Scholar]
- Munchel SE, Shultzaberger RK, Takizawa N, Weis K. Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol Biol Cell. 2011;22:2787–95. doi: 10.1091/mbc.E11-01-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VMY. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Perry G, Cash AD, Smith MA. Alzheimer Disease and Oxidative Stress. J Biomed Biotechnol. 2002;2:120–123. doi: 10.1155/S1110724302203010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips K, Kedersha N, Shen L, Blackshear PJ, Anderson P. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor alpha, cyclooxygenase 2, and inflammatory arthritis. Proc Natl Acad Sci U S A. 2004;101:2011–6. doi: 10.1073/pnas.0400148101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–91. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–81. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Hilt W, Buchberger A, Wolf DH. Cdc48: a power machine in protein degradation. Trends Biochem Sci. 2011;36:515–23. doi: 10.1016/j.tibs.2011.06.001. [DOI] [PubMed] [Google Scholar]
- True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature. 2000;407:477–83. doi: 10.1038/35035005. [DOI] [PubMed] [Google Scholar]
- Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11:891–905. doi: 10.1016/S1474-4422(12)70204-1. [DOI] [PubMed] [Google Scholar]
- Vanderweyde T, Yu H, Varnum M, Liu-Yesucevitz L, Citro A, Ikezu T, Duff K, Wolozin B. Contrasting Pathology of the Stress Granule Proteins TIA-1 and G3BP in Tauopathies. J Neurosci. 2012;32:8270–83. doi: 10.1523/JNEUROSCI.1592-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts GDJ, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–81. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- Weissbach R, Scadden ADJ. Tudor-SN and ADAR1 are components of cytoplasmic stress granules. RNA. 2012;18:462–71. doi: 10.1261/rna.027656.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B. Regulated protein aggregation: stress granules and neurodegeneration. Mol Neurodegener. 2012;7:56. doi: 10.1186/1750-1326-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]