Interactions between β-amyloid and central cholinergic neurons: implications for Alzheimer's disease (original) (raw)
. 2004 Nov;29(6):427–441.
Abstract
Alzheimer's disease is an age-related neurodegenerative disorder that is characterized by a progressive loss of memory and deterioration of higher cognitive functions. The brain of an individual with Alzheimer's disease exhibits extracellular plaques of aggregated β-amyloid protein (Aβ), intracellular neurofibrillary tangles that contain hyperphosphorylated tau protein and a profound loss of basal forebrain cholinergic neurons that innervate the hippocampus and the neocortex. Aβ accumulation may trigger or contribute to the process of neurodegeneration. However, the mechanisms whereby Aβ induces basal forebrain cholinergic cell loss and cognitive impairment remain obscure. Physiologically relevant concentrations of Aβ-related peptides have acute, negative effects on multiple aspects of acetylcholine (ACh) synthesis and release, without inducing toxicity. These data suggest a neuromodulatory influence of the peptides on central cholinergic functions. Long-term exposure to micromolar Aβ induces cholinergic cell toxicity, possibly via hyperphosphorylation of tau protein. Conversely, activation of selected cholinergic receptors has been shown to alter the processing of the amyloid precursor protein as well as phosphorylation of tau protein. A direct interaction between Aβ and nicotinic ACh receptors has also been demonstrated. This review addresses the role of Aβ-related peptides in regulating the function and survival of central cholinergic neurons and the relevance of these effects to cholinergic deficits in Alzheimer's disease. Understanding the functional interrelations between Aβ peptides, cholinergic neurons and tau phosphorylation will unravel the biologic events that precede neurodegeneration and may lead to the development of more effective pharmacotherapies for Alzheimer's disease.
Medical subject headings: acetylcholine; Alzheimer disease; amyloid; models, animal; neurodegenerative diseases; neuromodulation
Abstract
La maladie d'Alzheimer est une maladie neurodégénérative reliée à l'âge caractérisée par une perte progressive de la mémoire et la détérioration des fonctions cognitives supérieures. Le cerveau d'une personne atteinte de la maladie d'Alzheimer contient des plaques extracellulaires de protéine β-amyloïde (Aβ) agrégée et des enchevêtrements neurofibrillaires intracellulaires qui contiennent de la protéine tau hyperphosphorylée, et présente une perte profonde de neurones cholinergiques du cerveau antérieur basal qui innervent l'hippocampe et le néocortex. L'accumulation de protéine Aβ peut déclencher le processus de neurodégénération ou y contribuer. Les mécanismes par lesquels la protéine Aβ provoque la perte de cellules cholinergiques du cerveau antérieur basal et la déficience de la cognition demeurent toutefois obscurs. Des concentrations physiologiquement pertinentes de peptides reliés à la protéine Aβont des effets négatifs aigus sur de multiples aspects de la synthèse et de la libération de l'acétylcholine (ACh) sans provoquer de toxicité. Ces données indiquent que les peptides ont peut-être une influence neuromodulatrice sur les fonctions cholinergiques centrales. L'exposition chronique à des protéines Aβmicromolaires provoque la toxicité des cellules cholinergiques, peut-être par l'hyperphosphorylation de la protéine tau. On a par ailleurs démontré que l'activation de certains récepteurs cholinergiques altère la transformation de la protéine précurseur amyloïde, ainsi que la phosphorylation de la protéine tau. On a aussi démontré l'existence d'un lien direct entre la protéine Aβ et les récepteurs de l'ACh nicotiniques. Cette étude porte sur le rôle des peptides reliés à la protéine Aβ dans la régulation de la fonction et de la survie des neurones cholinergiques centraux et la pertinence de ces effets pour les déficits cholinergiques dans les cas de maladie d'Alzheimer. La compréhension des liens fonctionnels entre les peptides Aβ, les neurones cholinergiques et la phosphorylation de la protéine tau précisera les éléments biologiques qui précèdent la neurodégénération et pourra peut-être déboucher sur la mise au point de pharmacothérapies plus efficaces contre la maladie d'Alzheimer.
Introduction
First described in 1906 by Alois Alzheimer,1 Alzheimer's disease (AD) is the most common cause of dementia today, accounting for about 50%–60% of all age-related dementia that affects individuals over 65 years of age. It is characterized clinically by progressive memory loss that begins early in the disease process. Other cognitive (disorientation, confusion and problems with reasoning) and behavioural (agitation, anxiety, delusions, depression and insomnia) disturbances appear as the disease progresses and impair function in activities of daily living.2 The average course of AD is a decade, but the rate of progression is variable. Epidemiological data have shown that AD afflicts about 8%–10% of the population over 65 years of age, and its prevalence doubles every 5 years thereafter.3 With the proportion of elderly people in the population increasing steadily, AD is expected to pose an increasing economic challenge to Western economies and be a burden to health care delivery systems over the coming decades.2,3
Both genetic and environmental factors can contribute to the development of AD. A minority of cases have an obvious genetic origin and demonstrate an autosomal dominant pattern of inheritance. Linkage studies indicate that point mutations in the gene for the amyloid precursor protein (APP), on chromosome 21, are associated with a subset of early onset (< 65 yr) familial AD cases. However, most early onset cases have been linked to alterations in 2 other genes: presenilin 1 (PS1) on chromosome 14 and presenilin 2 (PS2) on chromosome 1.4,5,6 Although these findings are of immense importance in elucidating the biologic pathogenesis of AD, it is important to recognize that mutations in these 3 genes may only account for 30%–50% of all autosomal dominant early onset cases. Familial clustering is also found in individuals with late-onset AD. Various factors including concomitant pathology and limited sample sizes make it difficult to identify genetic causes of late-onset disease by conventional linkage analysis. However, association studies have identified candidate genes that significantly increase the risk for late-onset disease. The ε4 allele of the apolipoprotein E gene, on chromosome 19, is one such risk factor. Possessing a single copy of the allele may increase the chance of developing AD 2–5 times, whereas having two ε4 alleles raises this probability more than 5 times. Conversely, expression of the ε2 allele appears to protect against development of AD.6,7,8 A polymorphism in an intronic region of the α-2 macroglobulin was found to segregate with the AD phenotype in some subjects with late-onset disease.9 However, follow-up study found no association in 2 independent familial AD data sets, both of which had shown earlier evidence for linkage with a locus on chromosome 12. Thus, it is now thought that the locus of interest is situated elsewhere in the chromosome.10,11 Given that the vast majority of cases of AD have not been associated with any of the genes implicated to date, it is highly likely that additional causative mutations and genetic risk factors remain to be identified.6,11 Other factors that may play an important role in the pathogenesis of AD include age, head injury and oxidative stress.12
Neuropathologic features of AD
Neurofibrillary tangles
Neuropathologic hallmarks of both familial and sporadic AD include intracellular neurofibrillary tangles, extracellular parenchymal and cerebrovascular amyloid deposits, and loss of neurons and synaptic integrity in specific brain areas. These features are also seen in the brains of individuals with Down's syndrome (age < 40 yr) and, to a limited extent, in the normal aging brain.11,13,14,15Neurofibrillary tangles are composed of paired helical filaments (PHF) and occasional single straight filaments, mainly containing an abnormal hyperphosphorylated form of the microtubule- associated protein, tau. In healthy neurons, tau binds and stabilizes microtubules, which make up the cytoskeleton of the cell, by reversible enzymatically mediated phosphorylation and dephosphorylation processes. If the phosphorylated tau is not dephosphorylated, it is unable to bind other microtubules. This results in polymerization of the phosphorylated tau into straight filaments, which are then cross-linked by glycosylation to form PHF–tau.13,15,16,17 Intraneuronal PHF–tau aggregates are often found in conjugation with ubiquitin.16 Neurofibrillary tangles in the brain of an individual with AD are particularly abundant in the entorhinal cortex, hippocampus, amygdala, association cortices of the frontal, temporal and parietal lobes, and certain subcortical nuclei that project to these regions. Formation of PHF–tau reduces the ability of tau to stabilize microtubules, leading to disruption of neuronal transport and eventually to the death of affected neurons.16,17,18 The extent of neurofibrillary pathology, and particularly the number of cortical neurofibrillary tangles, correlates positively with the severity of dementia. However, tangles are also found in a variety of other neurodegenerative diseases that exhibit neither amyloid deposits nor neuritic plaques.11,15,16,19,20
Neuritic plaques
Neuritic plaques are multicellular lesions that contain a compact deposit of amyloid peptides surrounded by dystrophic neurites, activated microglia and reactive astrocytes. The major amyloid peptides that are found in the plaques are β-amyloid1–42 (Aβ1–42) and Aβ1–40, peptides that are generated by proteolytic cleavage of APP. In the brain with AD, Aβ1–42 is deposited first and is the predominant form in senile plaques, whereas Aβ1–40 is deposited later in the disease process. Other proteins, such as components of the complement cascade, apolipoprotein E, α-1-antichymotrypsin, lysosomal proteases and antioxidant enzymes, constitute minor components of the neuritic plaques. These plaques are most prominent in areas affected by neurodegeneration, such as the entorhinal cortex, hippocampus and association cortices.11,21,22,23 Neuritic plaque number does not itself correlate with the severity of dementia, although a clinical correlation between elevated levels of Aβ peptide in the brain and cognitive decline has been reported.24 Recent investigations in animal models and human brain samples have placed a special emphasis on soluble Aβ.25,26
Several lines of evidence suggest that accumulation of Aβ peptide in the brain may, over time, initiate and/or contribute to the pathogenesis of AD. These include the association of some cases of AD with inherited APP mutations,11,13,15 the elevation of Aβ peptides and the appearance of amyloid plaques in advance of other pathology in the brains of individuals with AD or Down's syndrome,27,28 the increased production of Aβ1-42 in vivo and in vitro by pathogenic mutations in PS1 and PS2,11 and the in-vitro neurotoxic potential of fibrillar Aβ peptides.11,29,30 Overproduction or reduced clearance, or both, of Aβ peptides are likely key to amyloid aggregation, which in turn contributes to the development of neurofibrillary tangles and subsequent neuronal degeneration.11,31,32,33 Recent studies of APP transgenic mice34,35,36,37 and of intrathecally administered Aβ in nontransgenic adult animals38,39,40,41 reinforce the notion that overexpression of Aβ peptide, or injection of aggregated Aβ, induces subcellular alterations or neuronal loss in selected brain regions. It has been suggested that overexpression or injection of Aβ peptide may potentiate the formation of neurofibrillary tangles in tau transgenic mice,42,43 a relation first inferred from consideration of kindreds with familial AD. Although these results suggest a role for Aβ peptides in the neurodegenerative process, both the role of Aβ in the normal brain and the mechanisms by which it causes neuronal loss and tau abnormalities in AD remain poorly understood.
Loss of basal forebrain cholinergic neurons
Degenerating neurons and synapses in the brain of individuals with AD are located predominantly within regions that project to or from areas that display high densities of plaques and tangles. Severely affected regions include the hippocampus, entorhinal cortex, amygdala, neocortex, and some subcortical areas such as basal forebrain cholinergic neurons, serotonergic neurons of the dorsal raphe and noradrenergic neurons of the locus coeruleus.44,45,46,47 Biochemical investigations of tissues from biopsy and autopsy indicate that various neurotransmitters and modulators including acetylcholine (ACh), serotonin, noradrenaline and somatostatin are differentially altered in the brains of individuals with AD.14,45,48 The early and most consistently reproduced finding is a profound reduction in the activity of the ACh-synthesizing enzyme, choline acetyltransferase (ChAT), in the neocortex, which correlates positively with the severity of dementia.45,47,49 Reduced choline uptake, ACh release and loss of cholinergic neurons from the basal forebrain region further indicate a selective presynaptic cholinergic deficit in the hippocampus and neocortex of brains of individuals with AD.48,50Cholinergic neurons in the brain stem and striatum are either spared or affected only in late stages of the disease.45,47,48 Together with pharmacologic evidence of cholinergic involvement in the affected cognitive processes, these findings led to the development of a “cholinergic hypothesis” of AD. This hypothesis posits the degeneration of the cholinergic neurons in the basal forebrain and the loss of cholinergic transmission in the cerebral cortex and other areas as the principal cause of cognitive dysfunction in patients with AD.47,48,50,51,52 The hypothesis is supported by evidence that drugs that potentiate central cholinergic function (such as donepezil, rivastigmine and galantamine) have some value in symptomatic treatment during early stages of the disease.47,53
The loss of basal forebrain cholinergic neurons has prompted extensive study of ACh receptors in the brains of individuals with AD.45,47,48,50,54 ACh exerts effects on the central nervous system by interacting with G-protein-coupled muscarinic and ligand-gated cation channel nicotinic receptors. Five distinct muscarinic receptor subtypes, m1–m5, have been cloned and shown to correspond to 5 pharmacologically defined M1–M5 muscarinic receptors. It is generally believed that M2 receptors, most of which are located on presynaptic cholinergic terminals, are reduced in the brains of individuals with AD.47,54 The density of postsynaptic M1 receptors remains unaltered, but there is some evidence for disruption of the coupling between the receptors, their G-proteins and second messengers.54,55 The profiles of M3 and M4 receptors in the brains of individuals with AD remain equivocal.56,57
For the nicotinic receptor family, 11 genes that encode 8 alpha (α2–α9) and 3 beta (β2–β4) receptor subunits have been identified.47,58,59 High-affinity central nervous system binding sites of the agonist nicotine are mostly composed of α4β2 subunits, whereas homomers of the α7 receptor subunit contribute to the high-affinity binding of the antagonist α-bungarotoxin (α-BgTx).59,60 Epibatidine, a potent nicotine agonist, binds with high affinity to a subtype of nicotinic receptor containing the α3 subunit.59 Nicotinic receptors are predominantly located on cholinergic terminals. High-affinity nicotinic binding sites are markedly reduced in the hippocampus and cortex of postmortem brains of individuals with AD, and these observations have been confirmed by in-vivo positron emission tomography.48,61 There is also evidence of a significant decrease in α7 protein expression and α-BgTx binding sites in the hippocampus of brains of individuals with AD.62 However, a recent immunocytochemical study demonstrated an increase in the proportion of astrocytes expressing α7 immunoreactivity in the hippocampus and entorhinal cortex of the brain of individuals with AD relative to age-matched controls.63 Notwithstanding these data, cholinomimetics only delay the cognitive decline in a subset of patients at early stages of the disease. Whether the early changes in the cholinergic system might play a pathogenic role in further dysregulating APP processing or promoting tau phosphorylation are important issues that remain to be addressed.
Cholinergic system and APP processing
APP processing
Aβ peptides, the principal component of amyloid deposits, are a group of hydrophobic peptides of 39–43 amino acid residues. These peptides are derived by proteolytic cleavage of APP, a single transmembrane glycoprotein with a long N-terminal extracellular region and a short C-terminal cytoplasmic tail.11,14,23,64 Nine isoforms are produced from a single APP gene by alternative mRNA splicing and encode proteins ranging from 365 to 770 amino acids. Two of these isoforms (APP365 and APP563) do not contain Aβ peptides. Some variants of APP (e.g., APP770) contain a 56–amino acid insert that is homologous to the Kunitz family of serine protease inhibitors (KPI) along with a 19-residue segment homologous to the thymocyte OX2 antigen. Others contain only the KPI domain (e.g., APP751) or neither segment (e.g., APP695).11,22,23 APP expression occurs ubiquitously, and the primary isoform varies according to cell and tissue type. In the nervous system, APP695 is expressed predominantly in neurons, whereas APP770 and APP751 are found in neuronal as well as nonneuronal cells.11,22,23,65 Two additional genes encoding for the APP-like proteins (i.e., APLP1 and APLP2) have been identified. The N-terminal regions of these proteins are homologous with APP. However, they lack the Aβ C-terminal domain. Recent data from mice with combined ablations of APP and APLP genes revealed that these proteins serve both overlapping and distinct functions in vivo.66
Mature APP is processed proteolytically by distinct α-secretase or β-secretase pathways (Fig. 1). The α- secretase activity cleaves the Aβ domain within Lys16 and Leu17 residues to prevent formation of full-length Aβ peptide. This pathway yields a soluble N-terminal APPα and a 10-kd C-terminal APP fragment that can be further processed by γ-secretase to generate Aβ17–40 or Aβ17–42, which are also known as the P3 peptides. The α-secretase cleavage occurs mostly at the cell surface, although it can be mediated to some extent during the secretory intracellular trafficking of APP.11,22 The β- secretase pathway, which results in the formation of intact Aβ peptide, is mediated by the sequential actions of β-secretase (β-APP cleaving enzyme [BACE]) and γ-secretase enzymes (Fig. 1). The β-secretase cleavage generates a truncated soluble APPβ and a membrane-bound Aβ-containing C-terminal fragment. Further proteolysis of the C-terminal fragment by γ-secretase yields the full-length Aβ1–40/42 peptide.11,22,23,67,68 γ-Secretase activity resides in a multimeric protein complex that contains PS, which is considered to be a putative aspartyl protease,69 along with at least 3 components (nicastrin, PEN-2 and APH-1) that are required for substrate recognition, complex assembly and targeting the complex to its site of action.70 Cell culture studies have suggested that most Aβ1–40/1–42 is generated in the endosomal recycling pathway. A minority of Aβ1–40/1–42 is produced in the secretory pathway, within the endoplasmic reticulum and Golgi apparatus.11,22,23 Under normal conditions, about 90% of secreted Aβ peptides are Aβ1–40, which is a soluble form of the peptide that only slowly converts to an insoluble β-sheet configuration and thus can be eliminated from the brain. In contrast, about 10% of secreted Aβ peptides are Aβ1–42/43, species that are highly fibrillogenic and deposited early in individuals with AD and Down's syndrome.11,14,27,28 Relative activities of the discrete APP processing pathways can be influenced by a variety of factors, including the stimulation of receptors for ACh, serotonin, glutamate, estrogen, neuropeptides and growth factors.71,72 The influence of cholinergic stimulation on amyloid formation is of particular interest in view of the early targeting of the cholinergic basal forebrain in AD and the possibility that maintenance of this cholinergic tone might slow amyloid deposition in cholinergic terminal fields.
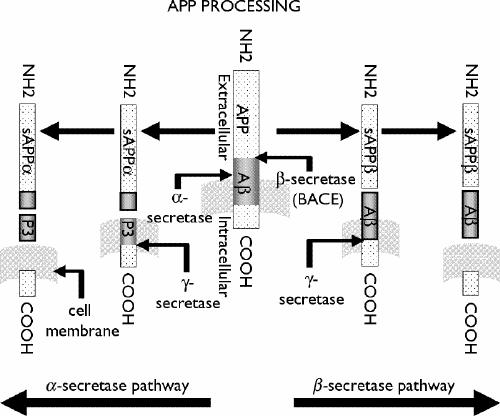
Fig. 1: Amyloid precursor protein (APP)-processing pathways. APP is a type I transmembrane protein that is processed by 2 distinct pathways. α-Secretase and β-secretase generate soluble APPα (sAPPα**) and sAPPβ, respectively. The remaining C-terminal fragment in the membrane is cleaved by** γ-secretase to yield P3 following α-secretase cleavage, or β-amyloid (Aβ**) subsequent to** β-secretase cleavage.
Cholinergic regulation of APP processing
A clear connection has been established between the cholinergic system and APP metabolism. Nitsch et al73 first demonstrated cholinergic regulation of APP processing in human embryonic kidney (HEK) 293 cell lines that were stably transfected with human muscarinic m1, m2, m3 and m4 receptors. Carbachol, a nonselective muscarinic receptor agonist, significantly increased the release of soluble APPα in cells expressing m1 and m3, but not in cells expressing m2 or m4 receptor subtypes. This response was both sensitive to atropine and blocked by staurosporine, indicating the mediation of intracellular protein kinases in receptor-controlled APPα secretion.73 Activation of muscarinic m1-receptor-transfected cells not only enhanced soluble APPα secretion but also reduced the secretion of Aβ. Similarly, muscarinic m1- and m3-receptor agonists stimulated soluble APPα release from rat cortical slices.74 Both m1 and m3 receptors activate signalling cascades involving phosphatidylinositol hydrolysis and protein kinase C (PKC). Treating cells with phorbol esters mimicked the effect of agonist administration on soluble APPα secretion, and this effect was blocked by PKC inhibitors.71,75 Moreover, other G-protein-coupled receptors that activate PKC-dependent signalling pathways, including the vasopressin, bradykinin, serotonin and metabotropic glutamate receptors, share this capacity to stimulate soluble APP secretion and inhibit Aβ formation.71,75 The mechanism whereby PKC activity increases soluble APPα secretion is still unknown, but it may involve additional kinase steps and the eventual activation of the proteases that mediate APP cleavage.71,72,75
In contrast to the muscarinic influence on APP processing, few studies have examined the contribution of nicotinic mechanisms. Treatment of PC12 cells with nicotine increases the release of soluble APPα in a concentration-dependent (> 50 μmol/L) and time- dependent (> 2 h) manner, without affecting the expression of APP mRNA or Aβ secretion.76 The relative increase in soluble APPα was attenuated by the α7-nicotinic receptor antagonist methyllycaconitine and also by EGTA, a Ca2+ chelator. The nicotine antagonist chlorisondamine blocked in-vivo elevation of total soluble APP induced by exposure to a high dose (8 mg/kg per day) of nicotine.77 A nicotine-induced increase in Ca2+ influx was found to correspond with the increase in soluble APP secretion, suggesting that Ca2+ influx through nicotinic receptors may be involved in enhanced secretion. This result is in agreement with the findings from several studies that show that increased cytoplasmic Ca2+ levels can stimulate soluble APP secretion.72,75,78
The effects of acetylcholinesterase (AChE) inhibitors on the level of soluble APPα differ between cell types and depend upon the specific drug, duration of treatment and the dose tested. For example, metrifonate did not alter soluble APP or Aβ levels in human SK-N-SH neuroblastoma cells,79 whereas short-term treatment with the inhibitor was able to increase the secretion of soluble APPα in SH-SY5Y neuroblastoma cells, presumably by increasing the availability of ACh and thereby stimulating muscarinic receptors.80,81 Physostigmine has been shown to elevate soluble APPα secretion in rat cortical slices82 but decreased soluble APP secretion without altering Aβ levels in SK-N-SH neuroblastoma cells.79 Tacrine, a potent cholinesterase inhibitor, was found to attenuate secretion of soluble APPα in glial, fibroblast, PC12 and neuroblastoma cells. Other AChE inhibitors such as phenserine, cymserine and tolserine decreased soluble APPα levels, whereas 3,4-diaminopyridine failed to affect soluble APPα levels in SK-N-SH neuroblastoma cells.79 The differential effects of the AChE inhibitors on APP processing appear to be unrelated to their selectivity for the cholinesterase enzymes but may depend upon other mechanisms, such as their influence on APP synthesis, expression, turnover or trafficking, or the regulation of APP-processing enzymes.75,79,83
Modulation of cholinergic functions by Aβ peptides
Short-term effects of Aβ on cholinergic neurons
Concentrations of Aβ peptides in the picomolar– nanomolar range can negatively regulate various steps of ACh synthesis and release, without apparent neurotoxicity (Table 1,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99 Fig. 2). The high potency and reversible nature of this effect, together with the fact that picomolar–nanomolar concentrations of Aβ peptides are found constitutively in normal brain cells, suggest that Aβ-related peptides may act as a modulator of cholinergic function under normal conditions.50,75,92,100,101,102 A 1-hour exposure to picomolar–nanomolar concentrations of Aβ can inhibit K+-evoked or veratridine-evoked endogenous ACh release from rat hippocampal and cortical slices. This effect is insensitive to tetrodotoxin, suggesting that the Aβ peptide may act at the level of cholinergic terminals.84,86Structure-activity studies reveal that several Aβ fragments, including Aβ1–42, Aβ1–40, Aβ1–28 and Aβ25–35, similarly inhibit ACh release from rat hippocampal slices, indicating that the activity resides within the sequence Aβ25–28 (GSNK, the C-terminal domain of the nontoxic Aβ1–28 fragment). Striatal ACh release is relatively insensitive to Aβ peptides.86 This regional selectivity indicates that transmitter phenotype expression does not fully explain the susceptibility of specific cell populations to effects of Aβ. Factors such as the distance over which cholinergic axons project to their terminal fields and regional variation in the expression of Aβ binding sites may contribute to the differences in cellular responsiveness to Aβ. However, the sensitivity to Aβ of cholinergic neurons in the cortex, hippocampus and striatum matches the pattern of regional vulnerability in AD.
Table 1
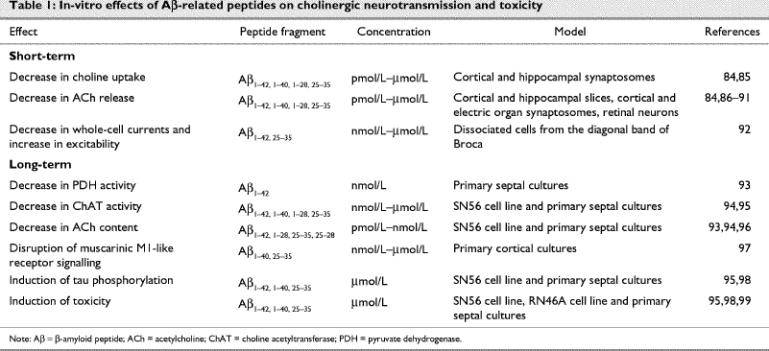
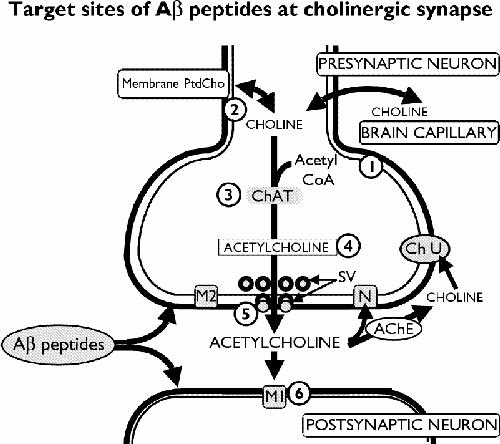
Fig. 2: Targets of Aβ that modulate cholinergic transmission: (1) Aβ reduces activity of pyruvate dehydrogenase, an enzyme that generates acetyl coenzyme A (CoA) from pyruvate; (2) Aβ reduces high-affinity uptake of choline; (3) long-term or high-dose exposure to Aβ reduces activity of the choline acetyltransferase (ChAT) enzyme; (4) Aβ reduces acetylcholine (ACh) content; (5) Aβ reduces ACh release from synaptic vesicles (SV); (6) Aβ impairs muscarinic M1-like signalling. AChE = acetylcholinesterase, Ch U = site of choline uptake, M2 = presynaptic muscarinic M2 receptor, N = presynaptic nicotinic receptor, PtdCho = phosphatidylcholine.
The inhibitory effects of Aβ on ACh release have been confirmed in rat and guinea pig cortical synaptosomes,87 rat retinal neurons88 and in cholinergic synaptosomes from the electric organ of the electric ray Narke japonica.89 These effects may be affected by aging. Higher levels of Aβ1–40 were observed in the aged rat hippocampus than were found in young adult rats, and the cholinergic neurons of aged, cognitively impaired rats may be more sensitive to Aβ-mediated inhibition of hippocampal ACh release than either cognitively unimpaired, aged rats or young adult rats.90 Lee et al91 reported that inhibition of ACh release by Aβ25–35 could be reversed by certain ginseng saponins at concentrations that did not by themselves alter ACh release. This effect was insensitive to tetrodotoxin, suggesting a direct interaction of ginseng at the level of the cholinergic synapse.
The cellular mechanisms by which Aβ-related peptides acutely attenuate ACh release from selected brain regions remain unclear. Steps that are critical for ACh synthesis and release, ranging from precursor recruitment to vesicular fusion, could be impaired by Aβpeptides (Table 1, Fig. 2). Turnover of ACh in the cholinergic terminals is regulated so that increased transmitter release is associated with increased synthesis. When brain slices are exposed to submaximal concentrations of depolarizing agents such as K+ or veratridine, ongoing synthesis of ACh keeps pace with transmitter release from the terminals.103 ACh synthesis under these conditions depends on the high-affinity uptake of choline from extracellular sources to intracellular acetyl CoA and ChAT. The availability of choline is a rate-limiting determinant of ACh biosynthesis, whereas ChAT activity is not.103 Under short-term treatment conditions, picomolar–nanomolar concentrations of Aβ1–40/1–42 do not affect ChAT activity in tissue homogenates or in slice preparations from the hippocampus, cortex or striatum.84 Similarly, Zambrzycka et al104 reported that soluble Aβ25–35 did not acutely affect ChAT activity in the adult or aged rat brain. Nanomolar Aβ1–42 (but not Aβ1–40) can acutely regulate the phosphorylation of the ChAT enzyme in IMR32 neuroblastoma cells expressing human ChAT.105 The significance of this phosphorylation regarding regulation of cholinergic transmission remains unclear.
Temperature-dependent high-affinity [3H]choline uptake is decreased by 20 minutes' preincubation with Aβ. This effect is particularly marked in tissues from the hippocampus and cortex, mirroring the effect of Aβ on ACh release in these regions.84 Acute incubation of hippocampal synaptosomes with low nanomolar Aβ1–40 suppresses depolarization-induced high-affinity choline uptake as well as [3H]hemicholinium-3 ([3H]HC-3) binding.85 Detailed analysis of these data indicate that changes in the transport are predominantly the result of an alteration of Vmax, whereas the changes in specific binding probably involve alterations of both Bmax and KD. Micromolar concentrations of Aβ1–40 decrease high-affinity choline uptake and the [3H]HC-3 binding under basal conditions in a time-dependent manner.85 These results indicate that Aβ can affect acute ACh release, at least in part, by regulating high-affinity choline uptake. The potential involvement of Aβ in the intracellular transport of ACh and the fusion of ACh vesicles with the presynaptic membrane remains to be investigated.
A variety of receptors (e.g., receptors for advanced glycation end products [RAGE], the class A scavenger receptor [SR], the 75-kd neurotrophin receptor [p75NTR] and serpin-enzyme complex receptors) interact with Aβ in vitro.106,107,108,109 These interactions have attracted attention both for the insights they may provide into linking amyloid accumulation to neurodegeneration and as potential targets for drug design. Recent evidence suggests that Aβ1–42 can also bind with high affinity to the α7**/**α-BgTx nicotinic receptor and with lower affinity to α4β2/cytisine nicotinic receptors (but not muscarinic) in the rat and guinea pig hippocampus and cerebral cortex.110 This is supported by whole- cell patch-clamp studies that showed that nanomolar Aβ1–40/Aβ1–42 can specifically and reversibly block the α7 nicotinic receptor current in cultured primary hippocampal neurons. The impairment of this current was noncompetitive, independent of voltage and mediated through the extracellular N-terminal length of the α7 subunit.111 In hippocampal slice preparations from rats that were 13–18 days old, Pettit et al112 reported that Aβ1–42 can reversibly inhibit carbachol-induced α7 and non-α7 nicotinic receptor currents.112 The broader range of effects in the slice study could relate to the concentration of Aβ peptide or to differences in cellular connectivity in the slice paradigm. The effects of Aβ on the nicotinic currents are consistent with receptor involvement in Aβ-mediated inhibition of ACh release. In support of this notion, the inhibitory effects of Aβ1–40 on cortical ACh release could be restored by the addition of a α7 agonist, such as nicotine and epibatidine, but not by the α4/β2 nicotinic receptor agonist cytisine.113 Further studies are needed to define the precise role of the α7 nicotinic receptor in regulating the inhibitory effects of Aβ peptides on endogenous ACh release.
In addition to affecting hippocampal and cortical terminals, Aβ peptide can act at the level of the cell body to increase neuronal excitability.92 Application of Aβ1–42/25–35, 1 μmol/L, to acutely dissociated rat neurons from the diagonal band of Broca decreased whole-cell voltage-sensitive currents in cholinergic neurons that were identified by single-cell reverse-transcriptase polymerase chain reaction.92 This reduction was associated with changes in several K+ currents, including the Ca2+-activated K+ currents (Bk or Ic), the delayed rectifier current (Ik) and transient outward current (Ia), but not in calcium or sodium currents. The responses were blocked by tyrosine kinase inhibitors, suggesting that Aβ induces phosphorylation-dependent cascades to alter these currents.92 Aβ effects on whole-cell currents can be replicated by human amylin (a 37-amino-acid pancreatic peptide that is deposited in the islet cells of patients with type 2 diabetes mellitus) and are not additive with those elicited by amylin and can be blocked by AC187, which is a specific amylin-receptor antagonist. These data raise the intriguing possibility that the amylin receptor may mediate certain effects of Aβ on basal forebrain cholinergic neurons.114
Long-term effects of Aβ peptide on cholinergic neurons
A 2-day exposure to picomolar–nanomolar concentrations of Aβ1–42, Aβ1–28, Aβ25–35 and, to a lesser extent, Aβ25–28 decreased intracellular ACh concentration in the cholinergic hybrid SN56 cell line (mouse septal neurons х neuroblastoma), without causing toxicity (Table 1, Fig. 2). The decrease in intracellular ACh could be attributed to reduced biosynthesis, because it was accompanied by a reduction in activity of ChAT but not of AChE. The decrease could be prevented by a co-treatment with trans-retinoic acid, a compound that increases ChAT mRNA expression in SN56 cells, or by co-administration of tyrosine kinase inhibitors.50,94,96 Inhibition of DNA synthesis or treatment with antioxidants did not alter the long-term effects of Aβ on ACh concentrations. Thus, gene transcription and free radical production are likely not involved in mediating the effect of Aβ on this cholinergic SN56 cell line.96 In keeping with these results, treatment of rat septal neurons with nanomolar concentrations of Aβ1–42 decreased ACh production and reduced activity of the acetyl-CoA biosynthesizing enzyme pyruvate dehydrogenase (PDH), without affecting ChAT activity or neuronal survival. The decreased PDH activity likely results from Aβ activation of the tau protein kinase I/glycogen synthase kinase-3β (GSK-3β), which can phosphorylate and inactivate PDH.93 This study suggests that long-term exposure to Aβ peptide may impair ACh processing by reducing the availability of acetyl CoA.
In addition to regulating ACh synthesis, solubilized Aβ peptide can disrupt transduction of the muscarinic M1-like receptor signal.97 A 4-hour exposure to nanomolar–micromolar Aβ1–40 reduced carbachol-induced GTPase activity in rat cortical cultured neurons without affecting muscarinic receptor ligand binding parameters. At higher concentrations, similar treatment with Aβ attenuated muscarinic M1 receptor signalling by decreasing intracellular Ca2+ and the accumulation of inositol phosphates Ins(1)P, Ins(1,4)P2, Ins(1,4,5)P3 and Ins(1,3,4,5)P4.97 Exposure of rat cortical cultured neurons to nanomolar Aβ1–42/Aβ25–35 inhibits the carbachol-induced, but not glutamate-induced, increase in intracellular Ca2+ and Ins(1,4,5)P3, indicating that selective disruption of muscarinic signalling is another means by which Aβ can affect the function of cholinergic neurons.115
Aβ-mediated toxicity, tau phosphorylation and cholinergic neurons
Long-term exposure to Aβ peptides can induce toxicity in a variety of cell lines, as well as in primary rat and human cultured neurons. The toxicity of the peptide, unlike its neuromodulatory effects, is related to its ability to form insoluble aggregates.29,30 However, recent evidence suggests that the most detrimental forms of Aβ peptides are the soluble oligomers and that the insoluble amorphous or fibrillar deposits represent a less harmful inactivated form of the peptide.116 Some neuronal phenotypes, such as γ-aminobutyric acid (GABA)-ergic and serotonergic neurons, appear resistant to Aβ toxicity, and various cell lines differ in their degree of sensitivity.99,117 Differentiated SN56 cholinergic cell lines are a susceptible line for toxicity studies, and when exposed to Aβ1–40 these cells exhibit retraction of neurites, cell shrinkage and death.98 When treated with ciliary neurotrophic factor, the RN46A cell line develops a cholinergic phenotype and is highly sensitive to Aβ peptides. In contrast, stimulation of RN46A differentiation with brain-derived neurotrophic factor yields an Aβ-insensitive cell population with a serotonergic transmitter phenotype.99 Prolonged exposure of rat primary septal culture neurons to micromolar Aβ peptides induces both cell death and a concomitant decrease in ChAT activity.95 Collectively, these results suggest that cells that express cholinergic transmitter phenotype are vulnerable to the toxic effects of the Aβ peptide.
The mechanisms by which Aβ induces cholinergic cell death may involve alteration in intracellular calcium and/or the production of toxic and inflammatory mediators such as nitric oxide, cytokines and reactive oxygen intermediates.118,119,120Studies of a variety of cell lines and primary cultured neurons suggest that Aβ toxicity might be mediated either by interaction with a hydroxysteroid dehydrogenase enzyme or by plasma membrane RAGE, SR, p75NTR or α7 nicotinic receptors.107,108,109,110 A role for the death domain of p75NTR in Aβ-induced cell death was observed in neuroblastoma (SK-N-BE) cells expressing full-length or truncated forms of p75NTR.121 Studies of transfected neuroblastoma (SK-N-MC) cells indicate that expression of the α7 nicotinic receptor may also have a critical role in the degeneration by facilitating internalization and accumulation of Aβ1–42 into neurons.122 Given the marked expression of p75NTR and of the α7 nicotinic receptor in the cholinergic basal forebrain, their role in cholinergic cell death bears further investigation.
Prolonged exposure to micromolar Aβ1–40 can increase choline conductance from PC12 cells.123 Should this also occur in cholinergic neurons, severe choline depletion could result as a consequence of decreased choline uptake and its increased leakage from neurons. Under conditions of choline depletion, cholinergic neurons can use choline from membrane phosphatidylcholine to synthesize ACh. Aβ-induced alteration in intracellular choline levels might thereby lead to an autocannibalistic process in which membrane turnover is disrupted to sustain neurotransmission.124
Tau phosphorylation can also contribute to the vulnerability of neurons by destabilizing microtubules and impairing axonal transport.98,125,126,127 Aggregated Aβ induces the phosphorylation of tau protein in SN56 cholinergic cell lines.98 Studies of cultured rat septal neurons have indicated that aggregated Aβ increases levels of tau and especially those of phosphorylated tau.95 Phosphorylated tau immunoreactivity could be detected primarily in the distal axons of untreated cells, whereas staining was evident in axons, soma and dendrites of neurons exposed to Aβ.95 Hyperphosphorylated tau protein can lead to neuronal death via disruption of the cytoskeletal network;16,17,18 it is likely that the increase in tau phosphorylation plays some role in Aβ-induced cell death.
How Aβ might induce the phosphorylation of the tau protein is unclear. Reactive oxygen species and the lipid peroxidation product 4-hydroxynonenal may be involved in Aβ neurotoxicity and cross-linking of tau proteins.128 However, Aβ might also affect tau phosphorylation by directly increasing relevant kinase activity or by decreasing phosphatase activity.98,125,129,130,131 Activation of GSK-3β127,130,132 and mitogen-activated protein (MAP) kinase129 induces tau protein phosphorylation and cell death in a variety of cultured neuron paradigms, and prolonged exposure of rat septal cultured neurons to micromolar Aβ peptide has been shown to induce tau phosphorylation by activating MAP kinase and GSK-3β.95 Various kinases phosphorylate tau at discrete sites, and it is likely that the phosphorylation of tau protein in cholinergic neurons is regulated by multiple kinases, including MAP kinase and GSK-3β. Thus, it is important to explore both the biochemical potential of additional tau kinases, such as cyclin- dependent kinase 5, PKC and calcium-calmodulin kinase, to phosphorylate tau17,18 and the particular cellular expression of these kinases by cholinergic neurons.
Tau phosphorylation can be regulated by cholinergic agonists, and control of tau hyperphosphorylation by muscarinic receptor activation may provide a side benefit of cholinomimetic therapeutics. Muscarinic agonists, carbachol and AF 102B, attenuate tau phosphorylation in cultured PC12 cells stably transfected with muscarinic m1 receptors.133 On the other hand, activation of the nicotinic receptor by nicotine and epibatidine increased the levels of phosphorylated as well as nonphosphorylated tau in SH-SY5Y human neuroblastoma cells.134 The mechanisms by which muscarinic m1 or nicotinic receptor activation modify tau phosphorylation remain unclear. These activities probably involve alteration of protein kinase/protein phosphatase systems.75
In-vivo effects of Aβ peptide on cholinergic neurons
Attempts have been made to measure the impact of intracerebroventricular or local administration of Aβ on the cholinergic system under in-vivo conditions. Several studies have reported that Aβ peptides can induce cholinergic hypofunction when administered to the brain.50,100 Injection of Aβ25–35/1–40 into the rat medial septum causes a reduction in ACh release from the hippocampus in the absence of toxicity.135 Using a similar approach, Harkany et al39 demonstrated that Aβ1–42 is toxic to cholinergic neurons, as indicated by the reduction in ChAT-immunoreactive cell bodies in the basal forebrain and fibres in the cerebral cortex. Other studies have shown that infusion of Aβ into the lateral ventricles of adult rats impairs performance on learning and memory tasks in a manner similar to the effect of cholinergic inhibition.38,40,100 Local injection of preaggregated Aβ1–42 into the nucleus basalis magnocellularis (NBM) produces congophilic deposits and a strong inflammatory response, characterized by activation of astrocytes and microglia and by induction of microglial p38MAP kinase activity.136 These changes were accompanied by a decrease in the number of cholinergic neurons around the congophilic amyloid deposit and hypofunction of the cortical cholinergic system.136 Clearly, the influence of these astrocytic and microglial responses must be considered in assessing the in-vivo effects of Aβ peptides on cholinergic function.
The cholinergic system in APP, PS1 and APP/PS1 transgenic mice
Over the past few years, the central cholinergic system has been examined extensively in a variety of mutant APP, PS1 or APP/PS1 transgenic mouse lines, all of which exhibit elevated Aβ levels.137,138,139,140,141,142,143,144,145,146
In mice that express the hAPPV642I London mutant transgene, a selective decrease was found in the size of medial septal cholinergic neurons, but not in NBM cholinergic neurons. At 17–22 months of age, this line exhibits both reorganization of AChE-positive fibres in the hippocampus and dystrophic AChE-positive fibres around amyloid plaques in the cortex.138
Cerebral amyloidosis was found to cause a significant cholinergic fibre loss and severe disruption of the neocortical cholinergic fibre network in aged APP23 mice that expressed the hAPPKM670/671NL Swedish mutant transgene.137 Although the cholinergic neurons of the medial septum and vertical limb of the diagonal band of Broca were smaller in APP23 transgenic mice than in nontransgenic controls, the number and volume of ChAT-positive neurons in the NBM complex were not affected. Hippocampal cholinergic fibre density in APP23 mice has yet to be reported.137
In another study, hAPPKM670/671NL mutant mice demonstrated an upregulation in the density of cholinergic synapses in the frontal cortex, parietal cortex and the hippocampus, whereas PS1M146L transgenic mice showed no changes in either the size or density of cholinergic synapses. When crossed to yield hAPPKM670/671NL/PS1M146L double transgenic mice, extensive amyloid plaques were found to be associated with decreased density and size of cholinergic synapses in the frontal cortex and hippocampus.139 One recent study showed a selective increase in immunostaining for p75NTR (a marker of basal forebrain cholinergic neurons) in the medial septum of 12-month-old hAPPKM670/671NL or PS1M146L single transgenic mice, but not in hAPPKM670/671NL/PS1M146L double transgenic mice. Staining of p75NTR-immunoreactive fibres in the hippocampus was more robust in single transgenic mice, relative to nontransgenic controls, whereas double transgenic mice displayed less intense p75NTR fibre staining.140 Whether the increased immunostaining in singly transgenic mice indicates a trophic effect on the cholinergic neurons as a consequence of either hAPPKM670/671NL or PS1M146L gene overexpression remains to be investigated. However, a separate study revealed no differences between hAPPKM670/671NL mice and nontransgenic controls in ChAT activity, AChE activity, vesicular ACh transporter binding or high-affinity choline uptake sites in the cortex, hippocampus, striatum or cerebellum, at multiple times up to 23 months of age.141
Densities of M1/[3H]pirenzepine, M2/[3H]AF-DX 384 or α7 nicotinic/[125I]α-BgTx receptor binding sites in all brain regions of mutant PS1L286V transgenic and wild-type PS1 transgenic mice are comparable with those found in nontransgenic controls.142 In hAPPKM670/671NL mutant mice, a decrease in M1/[3H]pirenzepine and α4β2 nicotinic/[3H]cytisine, but not M2/[3H]AF-DX 384, receptor binding was evident in the hippocampus and cortex compared with nontransgenic controls.146 However, in other studies, elevated hippocampal α7 nicotinic receptor levels have been reported in hAPPK670N/M671L single and 2 lines (i.e., hAPPK670N/M671L/PS1A246E and APPKM670/671NL+V717F/ PS1M146L+L286V) of double transgenic mice.143,145
In sum, increased expression of Aβ peptides produces a range of effects on the cholinergic systems of mutant APP, PS1 or APP/PS1 transgenic mice. Establishing which of these effects are robustly related to the type of pathogenic mutation, the level of transgene expression or to the intensity of amyloid deposits remains a work in progress.
Significance of amyloid interactions with cholinergic neurons
Aβ-related peptides are produced constitutively by brain cells and are found in the picomolar–nanomolar range in the cerebrospinal fluid of healthy individuals.11,31,32,33 These concentrations of Aβ can have a neuromodulatory role in the regulation of normal cholinergic functions through their negative effects on ACh biosynthesis and release. ACh may in turn reciprocally regulate APP synthesis and processing. For example, lesions of the basal forebrain cholinergic neurons or transient inhibition of cortical ACh release could elevate local APP synthesis,71,147,148,149 whereas agonist-induced activation of muscarinic m1 and m3 receptor subtypes increases the secretion of soluble APP derivatives and reduces the production of amyloidogenic Aβ peptides.71,72,73,74,75,150 These results suggest a mechanism whereby normal cholinergic innervation participates in the nonamyloidogenic maturation of APP via the α-secretase pathway, whereas the amyloidogenic Aβ-related peptides depress the activity of cholinergic neurons. A shift in the balance between these activities may be a key factor in the early targeting of cholinergic neurons in AD. Insults that reduce cholinergic transmission, increase Aβ generation or reduce peptide clearance may enhance vulnerability of neurons to direct toxicity of Aβ peptide11,29,30 or to choline limitation.84,85,100,102,123,124 Because Aβ deposits precede any other lesions in the brains of individuals with AD,27,28 it is likely that amyloid-induced tau phosphorylation plays a critical role in neuronal loss. This is supported by some in-vivo studies in which intrathecal administration, or transgene-delivered expression of Aβ peptides, was shown to induce a loss of neurons, or a change in presynaptic cholinergic markers, within selected brain regions.38,39,40,41,137,138,139 The selective interactions of Aβ with basal forebrain cholinergic neurons provide candidate mechanisms that may contribute, at least in part, to the vulnerability of these neurons and their projections in AD. It remains to be determined whether changes in cholinergic transmission alter APP processing pathways so as to further AD pathology. If so, appropriate cholinomimetic therapeutics might be expected both to provide symptomatic benefit and to abrogate AD pathogenesis. Although as yet unproven, this potential mechanism will drive further research into clarifying the precise interactions of amyloid with the elements of cholinergic neurotransmission.
Acknowledgments
We gratefully acknowledge the many contributions of Drs. Rémi Quirion, Elvire Vaucher, David Seto and Wen-Hua Zheng to this research program.
Footnotes
2002 CCNP Young Investigator Award Paper
We gratefully acknowledge the support of the Canadian Institutes of Health Research (S.K., D.W. and H.T.J.M.) and the Alzheimer Society of Ontario (D.W. and H.T.J.M.).
Competing interests: None declared.
Correspondence to: Dr. Satyabrata Kar, Neurochemical Research Unit, Departments of Medicine (Neurology) and Psychiatry, Walter Mackenzie Centre, University of Alberta, Edmonton AB T6G 2B7; fax 780 492-6841; skar@ualberta.ca
Submitted Aug. 29, 2003; Accepted Nov. 27, 2003
References
- 1.Whitehouse PJ. Genesis of Alzheimer's disease. Neurology 1997; 48(Suppl 7):S2-7. [DOI] [PubMed]
- 2.Terry RD, Katzman R. Senile dementia of the Alzheimer type. Ann Neurol 1983;14:497-506. [DOI] [PubMed]
- 3.Irizarry M, Hyman B. Alzheimer's disease. In: Batchlor T, Cudkowicz M, editors. Principles of neuroepidemiology. Boston: Butterworth-Heinemann; 2001. p. 69-98.
- 4.Levy-Lahad E, Wasco W, Pookraj P, Romano DM, Oshima J, Pettingell WH, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 1995;269:973-7. [DOI] [PubMed]
- 5.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 1995; 375: 754-60. [DOI] [PubMed]
- 6.Holmes C. Genotype and phenotype in Alzheimer's disease. Br J Psychiatry 2002;180:131-4. [DOI] [PubMed]
- 7.Strittmatter WJ, Saunders AM, Schmeckel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 1993;90:1977-81. [DOI] [PMC free article] [PubMed]
- 8.Poirier J, Davingnon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimer's disease. Lancet 1993;342:697-9. [DOI] [PubMed]
- 9.Blacker D, Wilcox MA, Laird NM, Rodes L, Horvath SM, Go RC, et al. Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nat Genet 1998;19:357-60. [DOI] [PubMed]
- 10.Wang X, Luedecking EK, Minster RL, Ganguli M, DeKosky ST, Kamboh MI. Lack of association between alpha2-macroglobulin polymorphisms and Alzheimer's disease. Hum Genet 2001;108:105-8. [DOI] [PubMed]
- 11.Selkoe DJ. Alzheimer's disease: genes, proteins and therapy. Physiol Rev 2001;81:741-66. [DOI] [PubMed]
- 12.Muller-Spahn F, Hock C. Risk factors and differential diagnosis of Alzheimer's disease. Eur Arch Psychiatry Clin Neurosci 1999; 249(Suppl 3):37-42. [DOI] [PubMed]
- 13.Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer's disease. Trends Neurosci 1993;16:460-5. [DOI] [PubMed]
- 14.Price DL, Sisodia SS. Mutant genes in familial Alzheimer's disease and transgenic models. Annu Rev Neurosci 1998;21:479-505. [DOI] [PubMed]
- 15.Lee VM. Disruption of the cytoskeleton in Alzheimer's disease. Curr Opin Neurobiol 1995;5:663-8. [DOI] [PubMed]
- 16.Iqbal K, Alonso AC, Gong CX, Khatoon S, Pei JJ, Wang JZ, et al. Mechanisms of neurofibrillary degeneration and the formation of neurofibrillary tangles. J Neural Transm Suppl 1998; 53: 169-80. [DOI] [PubMed]
- 17.Brion JP, Anderton BH, Authelet M, Dayanandan R, Leroy K, Lovestone S, et al. Neurofibrillary tangles and tau phosphorylation. Biochem Soc Symp 2001;67:81-8. [DOI] [PubMed]
- 18.Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneraion. Biochem J 1997;323:577-91. [DOI] [PMC free article] [PubMed]
- 19.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 1992;42: 631-9. [DOI] [PubMed]
- 20.Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, et al. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Arch Neurol 1995;52:81-8. [DOI] [PubMed]
- 21.Dickson DW. The pathogenesis of senile plaques. J Neuropathol Exp Neurol 1997;56:321-39. [DOI] [PubMed]
- 22.Clippingdale AB, Wade JD, Barrow CJ. The amyloid-β peptide and its role in Alzheimer's disease. J Pept Sci 2001;7:227-49. [DOI] [PubMed]
- 23.Wisniewski T, Ghiso J, Frangione B. Biology of Aβ amyloid in Alzheimer's disease. Neurobiol Dis 1997;4:313-28. [DOI] [PubMed]
- 24.Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, et al. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 2000;283:1571-7. [DOI] [PubMed]
- 25.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 1999;46:860-6. [DOI] [PubMed]
- 26.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 1999; 155: 853-62. [DOI] [PMC free article] [PubMed]
- 27.Giaccone G, Tagliavini F, Linoli G, Bouras C, Frigerio L, Frangione B, et al. Down patients: extracellular preamyloid deposits precede neuritic degeneration and senile plaques. Neurosci Lett 1989;97:232-8. [DOI] [PubMed]
- 28.Tanzi RE. Neuropathology in the Down's syndrome brain. Nat Med 1996;2:31-2. [DOI] [PubMed]
- 29.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci 1993;13:1676-87. [DOI] [PMC free article] [PubMed]
- 30.Yankner BA. Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron 1996;16:921-32. [DOI] [PubMed]
- 31.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, et al. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 1992;359: 322-5. [DOI] [PubMed]
- 32.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, et al. Isolation and quantification of soluble Alzheimer β- peptide from biological fluids. Nature 1992;359:325-7. [DOI] [PubMed]
- 33.Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, et al. Production of Alzheimer's β protein by normal proteolytic processing. Science 1992;258:126-9. [DOI] [PubMed]
- 34.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, et al. Alzheimer type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995;373:523-7. [DOI] [PubMed]
- 35.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996;274:99-102. [DOI] [PubMed]
- 36.Calhoun M, Wiederhold K, Abramowski D, Phinney AL, Sturchler-Pierrat C, Staufenbiel M, et al. Neuron loss in APP transgenic mice. Nature 1998;395:755-6. [DOI] [PubMed]
- 37.Bondolfi L, Calhoun M, Ermini F, Kuhn HG, Wiederhold KH, Walker L, et al. Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. J Neurosci 2002;22:515-22. [DOI] [PMC free article] [PubMed]
- 38.Giovannelli L, Casamenti F, Scali C, Bartolini L, Pepeu G. Differential effects of amyloid peptides β-(1-40) and β-(25-35) injections into rat nucleus basalis. Neuroscience 1995;66:781-92. [DOI] [PubMed]
- 39.Harkany T, Lengyel Z, Soos K, Penke B, Luiten PG, Gulya K. Cholinotoxic effects of β-amyloid1-42 peptide on cortical projections of the rat nucleus basalis magnocellularis. Brain Res 1995; 695:71-5. [DOI] [PubMed]
- 40.Itoh A, Nitta A, Nadai M, Nishimura K, Hirose M, Hasegawa T, et al. Dysfunction of cholinergic and dopaminergic neuronal systems in β-amyloid protein-infused rats. J Neurochem 1996; 66:1113-7. [DOI] [PubMed]
- 41.Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yanker BA. Aging renders the brain vulnerable to amyloid β-protein neurotoxicity. Nat Med 1998;4:827-31. [DOI] [PubMed]
- 42.Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 2001;293:1491-5. [DOI] [PubMed]
- 43.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001;293:1487-91. [DOI] [PubMed]
- 44.Braak H, Braak E. Pathology of Alzheimer's disease. In: Calne DB, editor. Neurodegenerative diseases. Philadelphia: Saunders; 1994. p. 585-613.
- 45.Geula C, Mesulam MM. Cholinergic system and related neuropathological predilection patterns in Alzheimer's disease. In: Terry RD, Katzman R, Bick KL, editors. Alzheimer disease. New York: Raven Press; 1994. p. 263-91.
- 46.DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration 1996;5:417-21. [DOI] [PubMed]
- 47.Lander CJ, Lee JM. Pharmacological drug treatment of Alzheimer disease: the cholinergic hypothesis revisited. J Neuropathol Exp Neurol 1998;57:719-31. [DOI] [PubMed]
- 48.Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer's disease: a review of progress. J Neurol Neurosurg Psychiatry 1999;66:137-47. [DOI] [PMC free article] [PubMed]
- 49.Davies P, Maloney AJF. Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet 1976;2:1403. [DOI] [PubMed]
- 50.Blusztajn JK. Berse B. The cholinergic neuronal phenotype in Alzheimer's disease. Metab Brain Dis 2000;15:45-64. [DOI] [PubMed]
- 51.Perry EK, Tomlinson BE, Blessed G, Bergman K, Gibson PH, Perry RH. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br Med J 1978; 2:1457-9. [DOI] [PMC free article] [PubMed]
- 52.Bartus RT, Dean RL III, Beer B, Lipa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982; 217: 408-17. [DOI] [PubMed]
- 53.Trinh NH, Hoblyn J, Mohanty S, Yaffe K. Efficacy of cholinesterase inhibitors in the treatment of neuropsychiatric symptoms and functional impairment in Alzheimer disease: a meta-analysis. JAMA 2003;289:210-6. [DOI] [PubMed]
- 54.Nordberg A, Alafuzoff I, Winbald B. Nicotinic and muscarinic receptor subtypes in the human brain: changes with aging and dementia. J Neurosci Res 1992;31:103-11. [DOI] [PubMed]
- 55.Warpman U, Alafuzoff I, Nordberg A. Coupling of muscarinic receptors to GTP proteins in postmortem human brain – alterations in Alzheimer's disease. Neurosci Lett 1993;150:39-43. [DOI] [PubMed]
- 56.Rodriguez-Puertas R, Pascual J, Vilaro T, Pazos A. Autoradiographic distribution of M1, M2, M3, and M4 muscarinic receptor subtypes in Alzheimer's disease. Synapse 1997;26:341-50. [DOI] [PubMed]
- 57.Mulugeta E, Karlsson E, Islam A, Kalaria R, Mangat H, Winblad B, et al. Loss of muscarinic M4 receptors in hippocampus of Alzheimer patients. Brain Res 2003;960:259-62. [DOI] [PubMed]
- 58.McGehee DS, Role LW. Physiological diversity of nicotine acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol 1995;57:521-46. [DOI] [PubMed]
- 59.Colquhoun LM, Patrick JW. Pharmacology of neuronal nicotinic acetylcholine receptor subtypes. Adv Pharmacol 1997;39: 191-20. [DOI] [PubMed]
- 60.Drisdel RC, Green WN. Neuronal α-bungarotoxin receptors are α7 subunit homomers. J Neurosci 2000;20:133-9. [DOI] [PMC free article] [PubMed]
- 61.Nordberg A, Lundqvist H, Hartvig P, Lilja A, Langstrom B. Kinetic analysis of regional (S)(-)11C-nicotine binding in normal and Alzheimer brains – in vivo assessment using positron emission tomography. Alzheimer Dis Assoc Disord 1995;9:21-7. [DOI] [PubMed]
- 62.Court J, Martin-Ruiz C, Piggott M, Spurden D, Griffiths M, Perry E. Nicotinic receptor abnormalities in Alzheimer's disease. Biol Psychiatry 2001;49:175-84. [DOI] [PubMed]
- 63.Teaktong T, Graham A, Court J, Perry R, Jaros E, Johnson M, et al. Alzheimer's disease is associated with a selective increase in α7 nicotinic acetylcholine receptor immunoreactivity in astrocytes. Glia 2003;41:207-11. [DOI] [PubMed]
- 64.Kang J, Lemaire GH, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell surface receptor. Nature 1987; 325:733-6. [DOI] [PubMed]
- 65.Koo EH, Sisodia SS, Cork LC, Unterbeck A, Bayney RM, Price DL. Differential expression of amyloid precursor protein mRNAs in cases of Alzheimer's disease and in aged nonhuman primates. Neuron 1990;4(1):97-104. [DOI] [PubMed]
- 66.Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rulicke T. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci 2000;20:7951-63. [DOI] [PMC free article] [PubMed]
- 67.Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, et al. Cleavage of amyloid β-peptide during constitutive processing of its precursor. Science 1990;248:1122-4. [DOI] [PubMed]
- 68.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, et al. β-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999;286:735-41. [DOI] [PubMed]
- 69.Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc Natl Acad Sci U S A 2003;100:6382-7. [DOI] [PMC free article] [PubMed]
- 70.Haass C, Steiner H. Alzheimer disease γ-secretase: a complex story of GxGD-type presenilin proteases. Trends Cell Biol 2002; 12: 556-62. [DOI] [PubMed]
- 71.Roberson MR, Harrell LE. Cholinergic and amyloid precursor protein metabolism. Brain Res Rev 1997;25:50-69. [DOI] [PubMed]
- 72.Mills J, Reiner PB. Regulation of amyloid precursor protein cleavage. J Neurochem 1999;72:443-60. [DOI] [PubMed]
- 73.Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic cholinergic receptor. Science 1992;258:304-7. [DOI] [PubMed]
- 74.Pittel Z, Heldman E, Barg J, Haring R, Fisher A. Muscarinic control of amyloid precursor protein secretion in rat cerebral cortex and cerebellum. Brain Res 1996;742:299-304. [DOI] [PubMed]
- 75.Hellstrom-Lindahl E. Modulation of β-amyloid precursor protein processing and tau phosphorylation by acetylcholine receptors. Eur J Pharmacol 2000;393:255-63. [DOI] [PubMed]
- 76.Kim SH, Kim YK, Jeong SJ, Haass C, Kim YH, Suh YH. Enhanced release of secreted form of Alzheimer's amyloid precursor protein from PC12 cells by nicotine. Mol Pharmacol 1997;52:430-6. [DOI] [PubMed]
- 77.Lahiri DK, Utsuki T, Chen D, Farlow MR, Shoaib M, Ingram DK, et al. Nicotine reduces the secretion of Alzheimer's β-amyloid precursor protein containing β-amyloid peptide in the rat without altering synaptic proteins. Ann N Y Acad Sci 2002; 965:364-72. [DOI] [PubMed]
- 78.Buxbaum JD, Ruefli AA, Parker CA, Cypress AA, Greengard P. Calcium regulates processing of the Alzheimer amyloid protein precursor in a protein kinase C-independent manner. Proc Natl Acad Sci U S A 1994;91:4489-93. [DOI] [PMC free article] [PubMed]
- 79.Lahiri DK, Farlow MR, Hintz N, Utsuki T, Greig NH. Cholinesterase inhibitors, β-amyloid precursor protein and amyloid β-peptides in Alzheimer's disease. Acta Neurol Scand Suppl 2000;176:60-7. [DOI] [PubMed]
- 80.Racchi M, Sironi M, Caprera A, Konig G, Govoni S. Short- and long-term effect of acetylcholinesterase inhibition on the expression and metabolism of the amyloid precursor protein. Mol Psychiatry 2001;6:520-8. [DOI] [PubMed]
- 81.Racchi M, Govoni S. The pharmacology of amyloid precursor protein processing. Exp Gerontology 2003;38:145-57. [DOI] [PubMed]
- 82.Mori F, Lai CC, Fusi F, Giacobini E. Cholinesterase inhibitors increase secretion of APPs in rat brain cortex. NeuroReport 1995; 6:633-6. [DOI] [PubMed]
- 83.Shaw KT, Utsuki T, Rogers J, Yu QS, Sambamurti K, Brossi A, et al. Phenserine regulates translation of beta-amyloid precursor protein mRNA by a putative interleukin-1 responsive element, a target for drug development. Proc Natl Acad Sci U S A 2001; 98:7605-10. [DOI] [PMC free article] [PubMed]
- 84.Kar S, Issa AM, Seto D, Auld DS, Collier B, Quirion R. Amyloid β-peptide inhibits high-affinity choline uptake and acetylcholine release in rat hippocampal slices. J Neurochem 1998; 70:2179-87. [DOI] [PubMed]
- 85.Kristofikova Z, Tekalova H, Klaschka J. Amyloid beta peptide 1-40 and the function of rat hippocampal hemicholinium-3 sensitive choline carriers: effects of a proteolytic degradation in vitro. Neurochem Res 2001;26:203-12. [DOI] [PubMed]
- 86.Kar S, Seto D, Gaudreau P, Quirion R. β-amyloid-related peptides inhibit potassium-evoked acetylcholine release from rat hippocampal formation. J Neurosci 1996;16:1034-40. [DOI] [PMC free article] [PubMed]
- 87.Wang HY, Wild KD, Shank RP, Lee DHS. Galanin inhibits acetylcholine release from rat cerebral cortex via a pertussis toxin-sensitive Gi protein. Neuropeptides 1999;33:197-205. [DOI] [PubMed]
- 88.Melo JB, Agostinho P, Oliveira CR. Amyloid beta-peptide 25-35 reduces [3H]acetylcholine release in retinal neurons. Involvement of metabolic dysfunction. Amyloid 2002;9:221-8. [DOI] [PubMed]
- 89.Satoh Y, Hirakura Y, Shibayama S, Hirashima N, Suzuki T, Kirino Y. Beta-amyloid peptides inhibit acetylcholine release from cholinergic nerve endings isolated from an electric ray. Neurosci Lett 2001;302:97-100. [DOI] [PubMed]
- 90.Vaucher E, Aumont N, Pearson D, Rowe W, Poirier J, Kar S. Amyloid β peptide levels and its effects on hippocampal acetylcholine release in aged, cognitively-impaired and -unimpaired rats. J Chem Neuroanat 2001;21:323-9. [DOI] [PubMed]
- 91.Lee TF, Shiao YJ, Chen CF, Wang LC. Effect of ginseng saponins on beta-amyloid-suppressed acetylcholine release from rat hippocampal slices. Planta Med 2001;67:634-7. [DOI] [PubMed]
- 92.Jhamandas JH, Cho C, Jassar B, Harris K, MacTavish D, Easaw J. Cellular mechanisms for amyloid β-protein activation of rat basal forebrain neurons. J Neurophysiol 2001;86:1312-20. [DOI] [PubMed]
- 93.Hoshi M, Takashima A, Murayama M, Yasutake K, Yoshida N, Ishiguro K, et al. Nontoxic amyloid β peptide1-42 supresses acetylcholine synthesis. J Biol Chem 1997;272:2038-41. [DOI] [PubMed]
- 94.Pedersen WA, Kloczewiak MA, Blusztajn JK. Amyloid β- protein reduces acetylcholine synthesis in a cell line derived from cholinergic neurones of the basal forebrain. Proc Natl Acad Sci U S A 1996;93:8068-71. [DOI] [PMC free article] [PubMed]
- 95.Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S. Amyloid β peptide induces tau phosphorylation and neuronal degeneration in rat primary septal cultured neurons. Neuroscience 2002;115:201-11. [DOI] [PubMed]
- 96.Pedersen WA, Blusztajn JK. Characterization of the acetylcholine reducing effect of the amyloid-beta peptide in mouse SN56 cells. Neurosci Lett 1997;239:77-80. [DOI] [PubMed]
- 97.Kelly JF, Furukawa K, Barger SW, Rengen MR, Mark RJ, Blanc EM, et al. Amyloid β-peptide disrupts carbachol-induced muscarinic cholinergic signal transduction in cortical neurons. Proc Natl Acad Sci U S A 1996;93:6753-8. [DOI] [PMC free article] [PubMed]
- 98.Le W, Xie WJ, Kong R, Appel SH. β-amyloid-induced neurotoxicity of a hybrid septal cell line associated with increased tau phosphorylation and expression of β-amyloid precursor protein. J Neurochem 1997;69:978-85. [DOI] [PubMed]
- 99.Olesen OF, Dago L, Mikkelsen JD. Amyloid β neurotoxicity in the cholinergic but not in the serotonergic phenotype of RN46A cells. Mol Brain Res 1998;57:266-74. [DOI] [PubMed]
- 100.Auld DS, Kornecook TJ, Bastianetto S, Quirion R. Alzheimer's disease and the basal forebrain cholinergic system: relations to beta-amyloid peptides, cognition, and treatment strategies. Prog Neurobiol 2002;68:209-45. [DOI] [PubMed]
- 101.Dolezal V, Kasparova J. β-amyloid and cholinergic neurons. Neurochem Res 2003;28:499-506. [DOI] [PubMed]
- 102.Kar S. Role of amyloid β peptides in the regulation of central cholinergic function and its relevance to Alzheimer's disease pathology. Drug Res Dev 2002;56:248-63.
- 103.Wecker L. The synthesis and release of acetylcholine by depolarized hippocampal slices is increased by increased choline available in vitro prior to stimulation. J Neurochem 1991;57: 1119-27. [DOI] [PubMed]
- 104.Zambrzycka A, Alberghina M, Strosznajder JB. Effects of aging and amyloid-beta peptides on choline acetyltransferase activity in rat brain. Neurochem Res 2002;27:277-81. [DOI] [PubMed]
- 105.Dobransky T, Brewer D, Lajoie G, Rylett RJ. Phosphorylation of 69-kDa choline acetyltransferase at threonine 456 in response to amyloid-beta peptide 1-42. J Biol Chem 2003;278: 5883-93. [DOI] [PubMed]
- 106.Joslin G, Krause JE, Hershey AD, Adams SP, Fallon RJ, Perlmutter DH. Amyloid-β peptide, substance P, and bombesin bind to the serpin-enzyme complex receptor. J Biol Chem 1991;266: 21897-902. [PubMed]
- 107.El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature 1996;382:716-9. [DOI] [PubMed]
- 108.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature 1996;382:685-91. [DOI] [PubMed]
- 109.Kuner P, Schubenel R, Hertel C. β-amyloid binds to p75NTR and activates NFkB in human neuroblastoma cells. J Neurosci Res 1998;54:798-804. [DOI] [PubMed]
- 110.Wang HY, Lee DHS, Davis CB, Shank RP. Amyloid peptide Aβ1-42 binds selectively and with picomolar affinity to α7 nicotinic acetylcholine receptors. J Neurochem 2000;75:1155-61. [DOI] [PubMed]
- 111.Liu Q, Kawai H, Berg DK. β-amyloid peptide blocks the response of α7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci U S A 2001;98:4734-9. [DOI] [PMC free article] [PubMed]
- 112.Pettit DL, Shao Z, Yakel JL. β-amyloid1-42 peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci 2001;21:RC120-24. [DOI] [PMC free article] [PubMed]
- 113.Lee DHS, Wang HY. Differential physiologic responses of α7 nicotinic acetylcholine receptors to β-amyloid1-40 and β-amyloid1-42. J Neurobiol 2003;55:25-30. [DOI] [PubMed]
- 114.Jhamandas JH, Harris KH, Cho C, Fu W, MacTavish D. Human amylin actions on rat cholinergic basal forebrain neurons: antagonism of beta-amyloid effects. J Neurophysiol 2003; 89(6): 2923-30. [DOI] [PubMed]
- 115.Huang HM, Ou HC, Hsieh SJ. Amyloid beta peptide impaired carbachol but not glutamate-mediated phosphoinositide pathways in cultured rat cortical neurons. Neurochem Res 2000; 25: 303-12. [DOI] [PubMed]
- 116.Selkoe DJ, Schenk D. Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol 2003;43:545-84. [DOI] [PubMed]
- 117.Pike CJ, Cotman CW. Cultured GABA-immunoreactive neurons are resistant to toxicity induced by β-amyloid. Neuroscience 1993;56:269-74. [DOI] [PubMed]
- 118.Behl C, Cole GM, Schubert D. Vitamin E protects nerve cells from amyloid β protein toxicity. Biochem Biophys Res Commun 1992; 186:944-50. [DOI] [PubMed]
- 119.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. β-amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 1992;12:376-89. [DOI] [PMC free article] [PubMed]
- 120.Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, et al. A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci U S A 1994; 91: 3270-4. [DOI] [PMC free article] [PubMed]
- 121.Perini G, Della-Bianca V, Politi V, Della Valle G, Dal-Pra I, Rossi F, et al. Role of p75 neurotrophin receptor in the neurotoxicity by beta-amyloid peptides and synergistic effect of inflammatory cytokines [published erratum appears in J Exp Med 2002;195(9):1231]. J Exp Med 2002;195:907-18 [DOI] [PMC free article] [PubMed]
- 122.Nagele RG, D'Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience 2002;110:199-211. [DOI] [PubMed]
- 123.Allen DD, Galdzicki Z, Brining SK, Fukuyama R, Rapoport SI, Smith QR. Beta-amyloid induced increase in choline flux across PC12 cell membranes. Neurosci Lett 1997;234:71-3. [DOI] [PubMed]
- 124.Wurtman R. Choline metabolism as a basis for the selective vulnerability of cholinergic neurones. Trends Neurosci 1992; 15: 117-22. [DOI] [PubMed]
- 125.Busciglio J, Lorenzo A, Yeh J, Yankner BA. β-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 1995;14:879-88. [DOI] [PubMed]
- 126.Shea TB, Prabhakar S, Ekinci FJ. β-amyloid and ionophore A23187 evoke tau hyper-phosphorylation by distinct intracellular pathways: differential involvement of the calpain/ protein kinase C system. J Neurosci Res 1997;49:759-68. [DOI] [PubMed]
- 127.Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, Diaz-Nido J. Lithium protects cultured neurons against β-amyloid-induced neurodegeneration. FEBS Lett 1999;453:260-4. [DOI] [PubMed]
- 128.Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid β-peptide. J Neurochem 1997;68:255-64. [DOI] [PubMed]
- 129.Greenberg SM, Kosik KS. Secreted β-APP stimulates MAP kinase and phosphorylation of tau in neurons. Neurobiol Aging 1995;16:403-8. [DOI] [PubMed]
- 130.Takashima A, Honda T, Yasutake K, Michel G, Murayama O, Murayama M, et al. Activation of tau protein kinase I/ glycogen synthase kinase-3 beta by amyloid beta peptide (25-35) enhances phosphorylation of tau in hippocampal neurons. Neurosci Res 1998;4:317-23. [DOI] [PubMed]
- 131.Alvarez A, Toro R, Caceres A, Maccioni RB. Inhibition of tau phosphorylating protein kinase cdk5 prevents β-amyloid- induced neuronal death. FEBS Lett 2001;459:421-6. [DOI] [PubMed]
- 132.Hong M, Chen DCR, Klein PS, Lee VMY. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem 1997;40:25326-32. [DOI] [PubMed]
- 133.Sadot E, Gurwitz D, Barg J, Behar L, Ginzburg I, Fisher A. Activation of m1 muscarinic acetylcholine receptor regulates tau phosphorylation in transfected PC12 cells. J Neurochem 1996;66:877-80. [DOI] [PubMed]
- 134.Hellstrom-Lindahl E, Moore H, Nordberg A. Increased levels of tau protein in SH-SY5Y cells after treatment with cholinesterase inhibitors and nicotinic agonists. J Neurochem 2000; 74:777-84. [DOI] [PubMed]
- 135.Abe E, Casamenti F, Giovannelli L, Scali C, Pepeu G. Administration of amyloid β-peptides into the medial septum of rats decreases acetylcholine release from hippocampus in vivo. Brain Res 1994; 636:162-4. [DOI] [PubMed]
- 136.Giovannini MG, Scali C, Prosperi C, Bellucci A, Vannucchi MG, Rosi S, et al. Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: involvement of the p38MAPK pathway. Neurobiol Dis 2002;11:257-74. [DOI] [PubMed]
- 137.Boncristiano S, Calhoun ME, Kelly PH, Pfeifer M, Bondolfi L, Stalder M, et al. Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J Neurosci 2002;22:3234-43. [DOI] [PMC free article] [PubMed]
- 138.Bronfman FC, Moechars D, Van Leuven F. Acetylcholinesterase-positive fiber deafferentation and cell shrinkage in the septohippocampal pathway of aged amyloid precursor protein london mutant transgenic mice. Neurobiol Dis 2000; 7: 152-68. [DOI] [PubMed]
- 139.Wong TP, Debeir T, Duff K, Cuello AC. Reorganization of cholinergic terminals in the cerebral cortex and hippocampus in transgenic mice carrying mutated presenilin-1 and amyloid precursor protein transgenes. J Neurosci 1999;19:2706-16. [DOI] [PMC free article] [PubMed]
- 140.Jaffar S, Counts SE, Ma SY, Dadko E, Gordon MN, Morgan D, et al. Neuropathology of mice carrying mutant APPswe and/ or PS1M146L transgenes: alterations in the p75NTR cholinergic basal forebrain septohippocampal pathway. Exp Neurol 2001; 170: 227-43. [DOI] [PubMed]
- 141.Gau JT, Steinhilb ML, Kao TC, D'Amato CJ, Gaut JR, Frey KA, et al. Stable β-secretase activity and presynaptic cholinergic markers during progressive central nervous system amyloidogenesis in Tg2576 mice. Am J Pathol 2002;160:731-8. [DOI] [PMC free article] [PubMed]
- 142.Vaucher E, Fluit P, Chishti MA, Westaway D, Mount HTJ, Kar S. Alteration in working memory but not cholinergic receptor binding sites in transgenic mice expressing human presenilin 1 transgenes. Exp Neurol 2002;21:323-9. [DOI] [PubMed]
- 143.Dineley KT, Xia X, Bui D, Sweatt JD, Zheng H. Accelerated plaque accumulation, associative learning deficits and upregulation of α7 nicotinic receptor protein in transgenic mice co- expressing mutant human presenilin 1 and amyloid precursor proteins. J Biol Chem 2002;227:22768-80. [DOI] [PubMed]
- 144.Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 2001;276:21562-70. [DOI] [PubMed]
- 145.Slowikowski SPM, Chishti MA, Zheng WH, Mount HTJ, Westaway D, Kar S. Alterations in cholinergic parameters in the hippocampus of transgenic mice expressing mutated amyloid precursor protein and/or presenilin-1 transgenes (poster). Program no. 295.18. 2002 Abstract Viewer/Itinerary Planner. Washington: Society for Neuroscience; 2002. Available: http://sfn.scholarone.com/itin2002/index.html (accessed 2004 Oct 15).
- 146.Apelt J, Kumar A, Schliebs R. Impairment of cholinergic neurotransmission in adult and aged transgenic Tg2576 mouse brain expressing the Swedish mutation of human beta-amyloid precursor protein. Brain Res 2002;953:17-30. [DOI] [PubMed]
- 147.Iverfeldt K, Walaas SI, Greengard P. Altered processing of Alzheimer amyloid precursor protein in response to neuronal degeneration. Proc Natl Acad Sci U S A 1993;90:4146-50. [DOI] [PMC free article] [PubMed]
- 148.Wallace W, Ahlers ST, Gotlib J, Bragin V, Sugar J, Gluck R, et al. Amyloid precursor protein in the cerebral cortex is rapidly and persistently induced by loss of subcortical innervation. Proc Natl Acad Sci U S A 1993;90:8712-6. [DOI] [PMC free article] [PubMed]
- 149.Lin L, Georgievska B, Mattsson A, Isacson O. Cognitive changes and modified processing of amyloid precursor protein in the cortical and hippocampal system after cholinergic synapse loss and muscarinic receptor activation. Proc Natl Acad Sci U S A 1999;96:12108-13. [DOI] [PMC free article] [PubMed]
- 150.Buxbaum JD, Oishi M, Chen HI, Pinkas-Kramarski R, Jaffe EA, Gandy SE, et al. Cholinergic agonists and interleukin 1 regulate processing and secretion of the β/A4 amyloid precursor protein. Proc Natl Acad Sci U S A 1992;89:10075-8. [DOI] [PMC free article] [PubMed]