Phosphorylation of XPB helicase regulates TFIIH nucleotide excision repair activity (original) (raw)
Abstract
Nucleotide excision repair (NER) removes damage from DNA in a tightly regulated multiprotein process. The xeroderma pigmentosum group B (XPB) helicase subunit of TFIIH functions in NER and transcription. The serine 751 (S751) residue of XPB was found to be phosphorylated in vivo. This phosphorylation inhibits NER and the microinjection of a phosphomimicking XPB-S751E mutant is unable to correct the NER defect of XP-B cells. Conversely, XPB-S751 dephosphorylation or its substitution with alanine (S751A) restores NER both in vivo and in vitro. Surprisingly, phospho/dephosphorylation of S751 spares TFIIH-dependent transcription. Finally, the phosphorylation of XPB-S751 does not impair the TFIIH unwinding of the DNA around the lesion, but rather prevents the 5′ incision triggered by the ERCC1-XPF endonuclease. These data support an additional role for XPB in promoting the incision of the damaged fragment and reveal a point of NER regulation on TFIIH without interference in its transcription activity.
Keywords: DNA damage response, GG-NER, helicase, phosphorylation, TFIIH
Introduction
One of the most versatile mammalian repair pathways is nucleotide excision repair (NER), in which two distinct modes of damage recognition are operational (Wood, 1997). Within transcription-coupled repair (TCR), the rapid removal of lesions located on the transcribed strand of a gene is intimately connected with their detection by the elongating transcription machinery (Mellon et al, 1987). Injuries located in the nontranscribed sequences are detected by the XPC-hHR23B complex, which initiates the process of global genome repair (GGR) (Sugasawa et al, 1998; Volker et al, 2001). Both subpathways then funnel into a common process by the recruitment of the multiprotein complex TFIIH. This transcription/repair factor contains several enzymatic activities including XPB and XPD helicases and cdk7 kinase (Zurita and Merino, 2003). Following recruitment of TFIIH, both XPB and XPD helicases unwind the DNA around the lesion, providing a three-dimensional structure ready to recruit XPA and RPA factors (Evans et al, 1997a; de Laat et al, 1998; Riedl et al, 2003). The margins of the resulting DNA bubble are recognized by XPG and ERCC1-XPF, two junction-specific endonucleases with opposite single-strand polarities, thereby generating 3′ and 5′ incisions relative to the damage, respectively (O'Donnovan et al, 1994; Sijbers et al, 1996). This dual incision precedes the excision of a 24–32-mer damaged oligonucleotide and the subsequent gap-filling DNA resynthesis (Shivji et al, 1995).
In protein coding gene transcription, following assembly of the preinitiation machinery including TFIIA, B, D, E, F and RNA polymerase II (RNA pol II), TFIIH opens DNA around the promoter (Holstege et al, 1996), phosphorylates the carboxyl-terminal (C-ter) domain of the largest RNA pol II subunit (Feaver et al, 1991; Lu et al, 1992) and allows promoter escape (Dvir et al, 1997).
The crucial role of TFIIH in preserving genome integrity is illustrated by the severe clinical consequences associated with inherited XPB and XPD mutations, as presented by patients suffering from the photohypersensitive disorders xeroderma pigmentosum (XP), Cockayne's syndrome (CS) and trichothiodystrophy (TTD) in which abnormal responses to DNA damage have been primarily identified (Lehmann, 2001). However, this original picture has been recently challenged by studies demonstrating that mutations in XPB and XPD impair proper control of activator- and repressor-specific target genes (Liu et al, 2001; Keriel et al, 2002), suggesting that symptoms displayed by XP-B and XP-D patients likely arise from a subtle mix of both transcription and repair defects.
The multiple functions of TFIIH raise the question of how the cell controls its different engagements in transcription and NER, a crucial regulation for maintaining genome stability. For instance, TFIIH transcription activity is inhibited during mitosis following the phosphorylation of cdk7 and p62, two subunits of TFIIH, by the mitotic cdc2/MPF kinase (Akoulitchev and Reinberg, 1998; Long et al, 1998). Alternatively, the cdk7 kinase activity, required for transcription (Wallenfang and Seydoux, 2002) but detrimental for repair (Araujo et al, 2000), is quickly reduced following UV irradiation (Adamczewski et al, 1996). To gain further insights into the regulation of TFIIH, here we focus on the C-ter part of XPB involved in both DNA repair and transcription (Weeda et al, 1990b; Hwang et al, 1996). We identify the serine 751 (S751) site within this region, and demonstrate that phosphorylation of S751 inhibits TFIIH repair activity but leaves its transcription function intact. The phosphorylation of S751 specifically prevents the 5′ incision of the lesion by ERCC1-XPF nuclease without modification of the damaged DNA opening or the 3′ incision by XPG nuclease.
Results
XPB is phosphorylated on S751
A frameshift altering the last 40 aa of XPB (from 741 to 782) caused severe NER and subtle transcription defects within the XP/CS patient XP11BE (Weeda et al, 1990b; Hwang et al, 1996). C-ter deletions suggested that a functional NER domain is located between residues 741 and 762 (Ma et al, 1994), a region that contains a consensus casein kinase II (CKII) motif (Figure 1A). To investigate whether XPB was phosphorylated in vivo, MRC5 fibroblasts were radiolabeled with orthophosphate and XPB was immunoprecipitated from cell lysates using Ab-hXPB antibody (Coin et al, 1999) and resolved by SDS–PAGE. Autoradiography and Western blotting revealed that a polypeptide corresponding to XPB was predominantly phospholabeled (Figure 1B).
Figure 1.
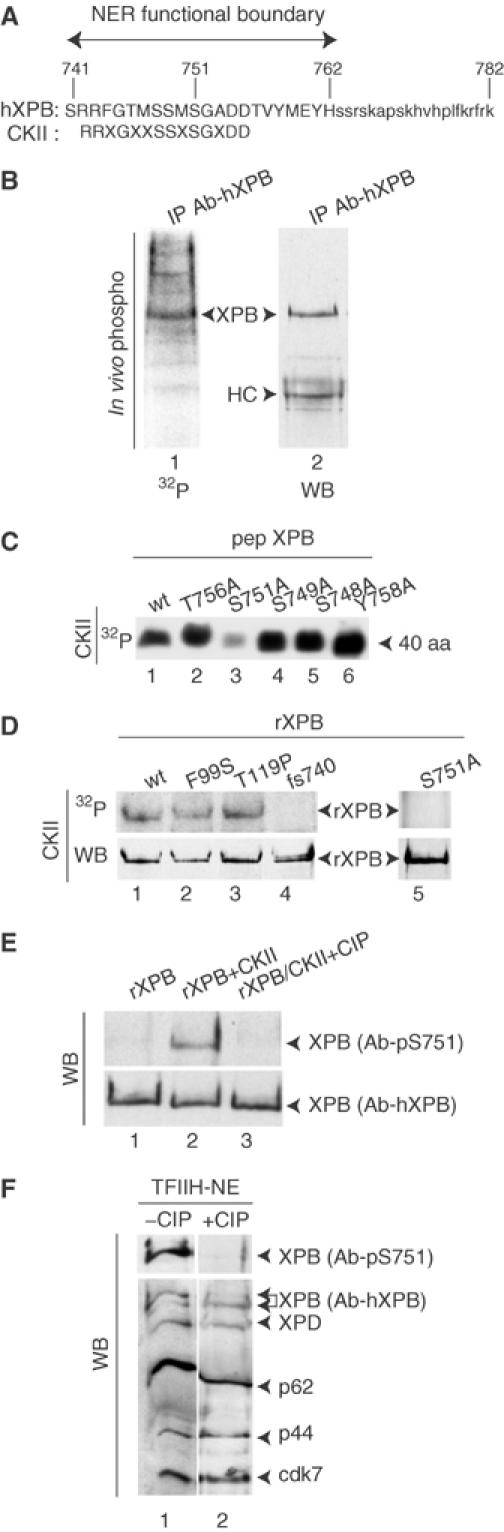
XPB is phosphorylated on S751 residue. (A) C-ter end of XPB. (B) Immunoprecipitated TFIIH from _in vivo_-labeled MRC5 cells, resolved by SDS–PAGE, autoradiography (lane 1) and subsequent Western blotting (lane 2). HC, γ-globulin heavy chain. (C) A 50 ng (10 pmol) portion of wild-type or mutated peptides encompassing the last 40 aa of XPB was treated with CKII, resolved by SDS–PAGE and autoradiographed. (D) A 50 ng (0.5 pmol) portion of wild-type and mutated rXPB was immunoprecipitated from Escherichia coli and treated with CKII. (E) A 50 ng (0.5 pmol) portion of immunoprecipitated wild-type rXPB (lane 1) was subjected to an in vitro kinase assay in the presence of CKII (lane 2) followed by treatment with CIP (lane 3). (F) Ab-hXPB immunoprecipitated TFIIH from 500 μg of HeLa NE was treated (lane 2) or not (lane 1) with CIP. Membranes were stripped following Ab-pS751 probing and reprobed with a batch of antibodies toward TFIIH subunits (Marinoni et al, 1997).
We synthesized oligopeptides representing the last 40 aa of XPB, in which the potential phosphorylated residues were changed to alanine. When tested in an in vitro CKII kinase assay, all of them except pepS751A incorporated radioactivity to a level similar to that of the wild-type sequence (Figure 1C). Accordingly, bacterially produced full-length XPBs were used as substrates. Wild-type XPB (XPB-wt), XPB-F99S and XPB-T119P, in which mutations found within XP-B patients have been introduced (Weeda et al, 1997), were phosphorylated (Figure 1D, lanes 1–3). In marked contrast, XPB-fs740, in which the altered XPB C-ter found in XP11BE cells has been introduced, and XPB-S751A, in which S751 has been changed to alanine, were not phosphorylated (lanes 4 and 5).
A mouse monoclonal antibody (Ab-pS751), raised against the phosphorylated S751 residue, recognized a bacterially produced XPB following phosphorylation by CKII (Figure 1E, upper panel). The corresponding band disappeared when phospholabeled XPB was treated with calf intestinal phosphatase (CIP) (upper and lower panels, lane 3). This antibody detected a band corresponding to XPB in a TFIIH immunoprecipitated fraction from HeLa nuclear extracts (NEs) (Figure 1F, upper panel, lane 1). Following CIP treatment, this band was no longer recognized by Ab-pS751 although XPB was clearly present in the fraction (upper and lower panels, lane 2).
XPB-S751 residue is essential for NER in vivo
To determine the biological significance of XPB-S751 phosphorylation in vivo, we introduced a glutamic acid (S751E) in position 751 to mimic the negative charge introduced by the phosphorylation of serine. XPB-wt, XPB-S751A or XPB-S751E were microinjected into the nucleus of fibroblasts from an NER-deficient XP-B patient (XPCS2BA) (Vermeulen et al, 1994). Cells were UV irradiated and their ability to restore DNA repair was monitored by incubation with [3H]thymidine and autoradiography. The unscheduled DNA synthesis (UDS) of the XP-B fibroblasts was restored almost to the normal level following microinjections of either XPB-wt or XPB-S751A cDNAs, indicating that expressions of the corresponding proteins were able to restore repair (Figure 2A, compare the polynuclear microinjected cell (1) with the noninjected one (2), upper and middle panels; Table I). In marked contrast, microinjection of XPB-S751E cDNA was unable to correct the NER defect (Figure 2A, lower panel; Table I).
Figure 2.
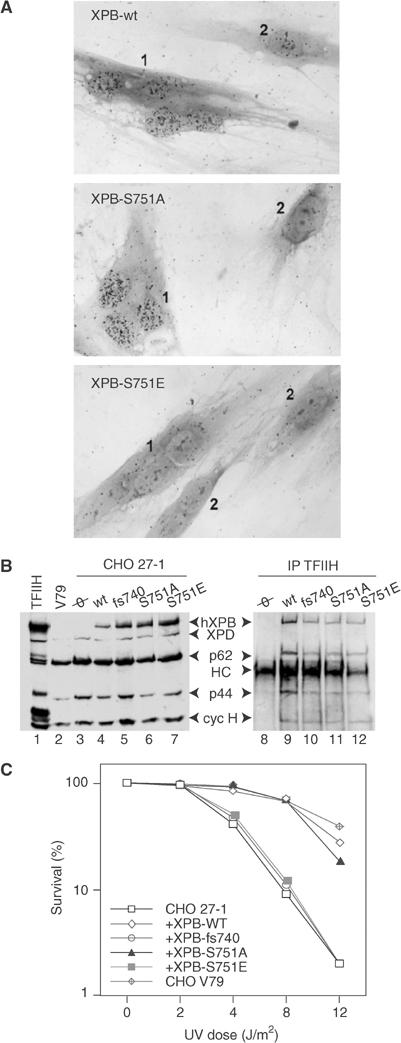
XPB-S751E expression prevents recovery of DNA repair. (A) Micrographs of XPCS2BA fibroblasts microinjected with either XPB-wt, XPB-S751A or XPB-S751E cDNAs showing induction of the UDS in the injected multinucleated cell (numbered 1) compared with the low residual UDS typical of the surrounding noninjected XPCS2BA mononuclear cells (numbered 2). (B) Detection of hXPB in transfected CHO 27-1 cells. Left panel: A 50 μg portion of extracts prepared from either CHO V79, CHO 27-1 or transfected CHO 27-1 cells expressing hXPB as indicated at the top of the panel was resolved by SDS–PAGE, followed by Western blotting with Ab-hXPB and a batch of antibodies raised toward TFIIH subunits. Lane 1: HeLa TFIIH. Right panel: TFIIHs from the corresponding transfected cell extracts were immunoprecipitated with Ab-hXPB and treated as above. (C) Quantitative UV survival analysis of G418-resistant CHO 27-1 cell lines. CHO V79 cells were used as controls. A total of 103 cells were plated per 6 cm Petri dishes, cultured overnight and UV irradiated at 254 nm at various doses (0.5 J/m2/s). Cells were stained by Trypan blue and counted after 14 days.
Table 1.
Microinjections of XPB cDNA into XPCS2BA fibroblasts
| Cell straina | cDNA injected | Grains/nucleusb | Mean UDS (%) |
|---|---|---|---|
| C5RO | Noninjected | 120±5 | 100 |
| XPCS2BA | Noninjected | 14±1 | 12 |
| XPCS2BA | XPB-wt | 96±4 | 80 |
| XPCS2BA | XPB-S751A | 113±4 | 94 |
| XPCS2BA | XPB-S751E | 22±2 | 18 |
| aInjections were performed in control fibroblasts (C5RO) or in XP-B XPCS2BA patient cells (Vermeulen et al, 1994). | |||
| bUDS, measured as the radioactive thymidine incorporation, is expressed as grains/nucleus±s.e.m. (>50 nuclei/sample). |
In another set of experiments, XPB-wt, XPB-S751A, XPB-S751E and XPB-fs740 were transfected into XPB-defective, UV-sensitive Chinese hamster ovary (CHO) 27-1 (Weeda et al, 1990a) and stably transfected mass population of cells were obtained. Protein expression analysis revealed that Ab-hXPB, directed against the N-terminal end of the human protein (hXPB), did not react with the hamster XPB in either V79 or 27-1 cell extracts (Figure 2B, lanes 2 and 3). Immunoprecipitations using Ab-hXPB demonstrated that the various hXPBs were efficiently incorporated into the hamster TFIIH complex (lanes 9 and 12). As expected, TFIIH containing the rodent XPB homolog was not pulled down (lane 8).
The stably transfected 27-1 cells were next UV irradiated at different doses and the cell survival was measured. Expressions of XPB-wt and XPB-S751A induced a substantial correction of the UV survival of 27-1 compared to wild-type CHO V79 cells (Figure 2C). On the contrary, the UV survival curve of XPB-S751E-transfected cells fell into the range of the parental 27-1, suggesting that expression of an artificially phosphorylated XPB protein does not restore repair, a fact that is also observed upon expression of XPB-fs740 (Figure 2C) and that correlates with the microinjection data.
C-ter end of XPB controls the 5′ incision by ERCC1-XPF
We next investigated how the various mutations modulate the removal of DNA damage in vitro. Three assays were monitored using a closed, circular plasmid containing a single 1,3-intrastrand d(GpTpG) cisplatin–DNA crosslink (Pt-DNA) as a template (Frit et al, 2002). The dual incision activity was followed by direct end-labeling of the excised 24–32 nt damaged oligonucleotide (Figure 3A). When added to the recombinant incision system containing purified XPC-HR23B, XPA, RPA, XPG and ERCC1-XPF proteins (Tapias et al, 2004), the Ab-hXPB immunoprecipitated TFIIH-wt and TFIIH-S751A trigger the excision of the damaged oligonucleotide (Figure 3B, upper panel, lanes 3, 4 and 7, 8). In contrast, both TFIIH-S751E and TFIIH-fs740 were unable to promote dual incision (lanes 5, 6 and 9, 10).
Figure 3.
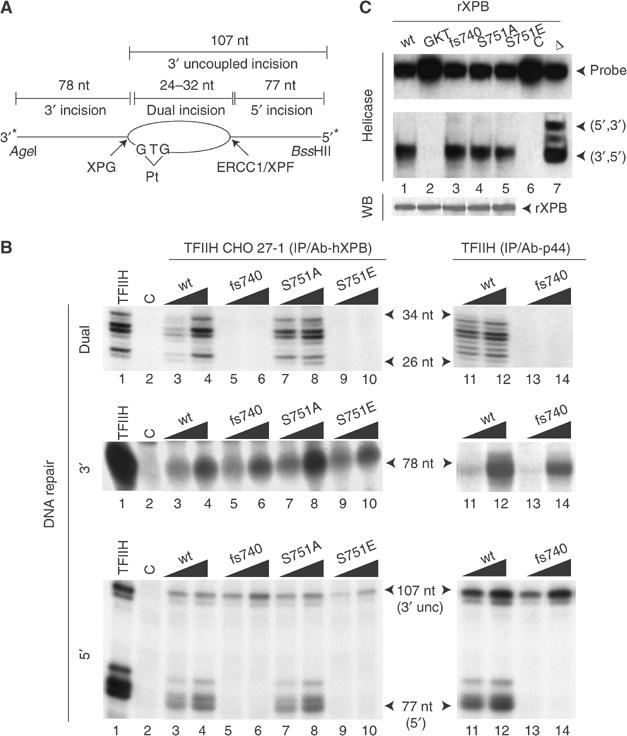
S751E mutation inhibits the 5′ incision. (A) Schematic diagram of assays used to detect dual and single incisions. (B) Left panel: Ab-hXPB immunoprecipitated TFIIH-wt (lanes 3 and 4), TFIIH-fs740 (lanes 5 and 6), TFIIH-S751A (lanes 7 and 8) and TFIIH-S751E (lanes 9 and 10) from 100 and 200 μg of 27-1 cell extracts were tested for their ability to promote either the dual (upper panel), 3′ (middle) or 5′ (lower) incisions. Right panel: A measure of 50 and 100 ng of either TFIIH-wt or TFIIH-fs740, immunopurified from GM1855 or GM2252 (XP11BE) cell extracts respectively, was similarly tested. Lane C contains all the NER factors except TFIIH. (C) A 200 ng portion of immunoprecipitated rXPB expressed in baculovirus-infected insect cells was tested either in helicase assay using a bidirectional probe (Coin et al, 1998) or resolved by SDS–PAGE followed by Western blotting (WB). XPB-GKT contains a mutation in the ATPase domain (Guzder et al, 1994). Δ: probe has been heated for 5 min at 100°C. C: control without XPB.
To examine the single DNA damage incisions by either XPG or ERCC1-XPF nucleases, an _Age_I/_Bss_HII linear DNA fragment of 185 nt (Figure 3A) was 3′- or 5′-end-labeled, respectively. The 3′ incision being made first, 3′ uncoupled incision by XPG (107 nt product) can be detected with the 5′-labeled probe (Mu et al, 1996). Both TFIIH-wt and TFIIH-S751A trigger the 3′ and 5′ incisions (Figure 3B, middle and lower panels, lanes 3, 4 and 7, 8). The 5′ incision of the damaged DNA by ERCC1-XPF was impaired with TFIIH-S751E, despite a functional 3′ incision by XPG (lanes 9 and 10). A selective inhibition of the 5′ incision promoted by ERCC1-XPF was also observed with TFIIH-fs740 purified from both 27-1 cells (lanes 5 and 6) and XP11BE cell extracts (lanes 13 and 14) (Evans et al, 1997b). Furthermore, inhibition of 5′ incision does not result from an impairment of XPB unwinding activity since XPB-fs740 and XPB-S751E exhibited a similar ATP-dependent 3′ to 5′ helicase activity, compared to XPB-wt and XPB-S751A (Figure 3C, compare lanes 3 and 5 with lanes 1 and 4).
S751A and S751E mutations spare TFIIH transcription activity
The TFIIHs expressed in 27-1 cells were then tested in an in vitro transcription assay containing purified RNA pol II, TFIIA, TFIIB, TFIID, TFIIE and TFIIF transcription factors (Tirode et al, 1999) and either the cdk7 kinase-independent adenovirus major late (AdML) or the cdk7 kinase-dependent dihydrofolate reductase (DHFR) promoters (Akoulitchev et al, 1995). In both cases, TFIIH-S751E exhibited a transcription activity similar to that achieved with either TFIIH-wt or TFIIH-S751A (Figure 4A, compare lanes 7–10 with 3 and 4). In contrast, TFIIH-fs740 was deficient in transcription of both templates (lanes 5 and 6). Using human recombinant TFIIH expressed and purified from insect cells (Tirode et al, 1999), we further confirmed that TFIIH-wt (Figure 4B, lanes 3 and 4), TFIIH-S751A and TFIIH-S751E (lanes 7–10) promoted an optimal RNA synthesis, whereas TFIIH-fs740 transcription activity was strongly affected (lanes 5 and 6).
Figure 4.
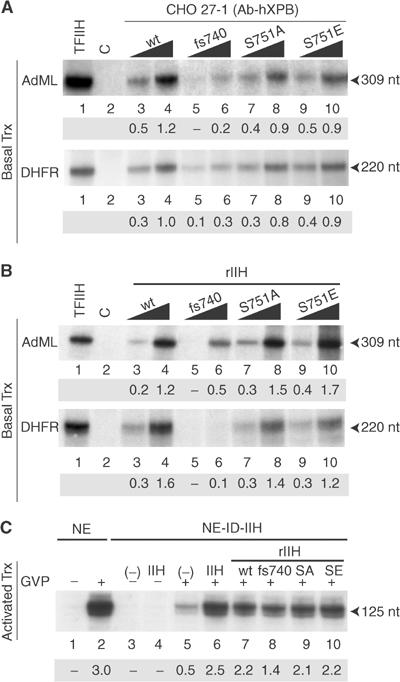
S751E mutation spares the TFIIH transcription activity. (A) Immunoprecipitated TFIIHs (as described in Figure 3B) containing either XPB-wt (lanes 3 and 4), XPB-fs740 (lanes 5 and 6), XPB-S751A (lanes 7 and 8) or XPB-S751E (lanes 9 and 10) were tested in an in vitro basal transcription assay using either AdML or DHFR promoters. The sizes of the corresponding transcripts are indicated. Lane 1: positive controls containing HeLa TFIIH fraction; lane 2: negative control without TFIIH. Values below the figures represent the mean from a triplicate experiment (optical densities). (B) A measure of 50 and 100 ng of recombinant TFIIH-wt (lanes 3 and 4), TFIIH-fs740 (lanes 5 and 6), TFIIH-S751A (lanes 7 and 8) or TFIIH-S751E (lanes 9 and 10) from baculovirus-infected insect cells was tested in a transcription assay as described above. (C) Chromatinized transcription templates containing GAL4 binding sites were incubated with either untreated (NE) or TFIIH-immunodepleted HeLa NEs (NE-ID-IIH), and transcription was performed as described by Frit et al (2002). When indicated at the top of the panel, purified HeLa or recombinant TFIIH was added.
Next, we tested the activity of the recombinant TFIIH-wt, TFIIH-fs740, TFIIH-S751A and TFIIH-S751E in an assay using a chromatinized template containing Gal4 binding sites upstream of the AdML promoter (Frit et al, 2002). Addition of the HeLa NE and Gal4-VP16 to the chromatin-packed template activated RNA synthesis (Figure 4C, lanes 1 and 2). The transcription was strongly reduced following depletion of TFIIH from NE (lane 5) and was rescued by adding purified HeLa TFIIH (lane 6). Complementation of the depleted extract by TFIIH-S751A and TFIIH-S751E restored the activated transcription (compare lanes 9 and 10 with lane 7), demonstrating that the phosphorylation of S751 spares TFIIH transcription activity. In contrast, addition of TFIIH-fs740 failed to restore it fully (lane 8).
Phosphorylation of S751 inhibits the 5′ incision formation
In another set of experiments, a HeLa TFIIH fraction (purified in the absence of phosphatase inhibitors; Gerard et al, 1991) was immunoprecipitated and further treated with CKII. Ab-pS751 recognized XPB only in the TFIIH-p+ fraction (CKII treated) (Figure 5A). The dual incision activity of TFIIH-p+ was strongly reduced when tested on a damaged DNA containing either a 1,3 d(GpTpG) cisplatin–DNA (Figure 5B, upper panel, lanes 4–6) or a natural UV-induced cyclobutane thymine dimer adduct (Ortiz Mayo et al, 2003) (Figure 5C, lanes 6–8), compared to TFIIH-p− (Figure 5B, upper panel, lanes 1–3, and Figure 5C, lanes 3–5). The 5′ incision by ERCC1-XPF was strongly inhibited with TFIIH-p+, but the 3′ incision by XPG was normal (Figure 5B, middle panel, lanes 4–6). Furthermore, transcription was fully active with both TFIIH-p+ and TFIIH-p− (lower panel, compare lanes 4–6 and 1–3).
Figure 5.
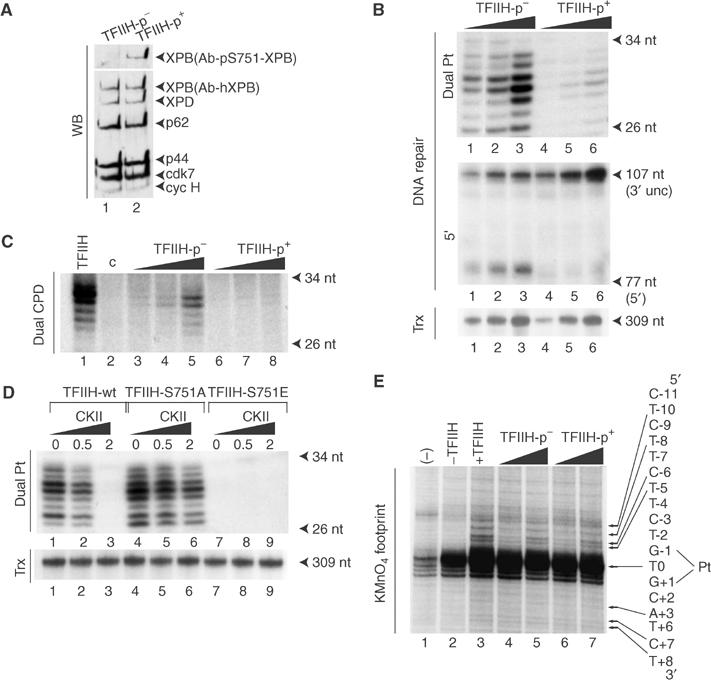
Phosphorylation of S751 by CKII inhibits 5′ incision. (A) Highly purified HeLa TFIIH fraction was immunoprecipitated with Ab-p44 antibody and subsequently treated with or without CKII (TFIIH-p+ and TFIIH-p−, respectively). Following extensive washing, both TFIIHs were eluted with the competitor peptide and resolved by SDS–PAGE and Western blotting. (B) TFIIH-p+ and TFIIH-p− (25, 50 and 100ng) were tested in dual, 5′ incision and transcription (using AdMLP as a template) assays. (C) TFIIH-p+ and TFIIH-p− (25, 50 and 100 ng) were tested in dual incision assay with a plasmid containing a unique cyclobutane pyrimidine dimer. (D) Immunoprecipitated TFIIH-wt, TFIIH-S751A and TFIIH-S751E from 27-1 cell extracts were incubated with increasing amounts (0, 0.5 and 2 units) of CKII. The kinase was removed by washing and the immunoadsorbed proteins were tested in dual incision and transcription assays. (E) Pt DNA was 3′-end-labeled and incubated with the NER factors and when indicated, with either HeLa TFIIH or increasing amounts (100 and 200 ng) of TFIIH-p− or TFIIH-p+. Lane 1: BSA; lane 2: reaction in the absence of TFIIH. Residues are numbered with the central thymine of the crosslinked GTG sequence designated T0. KMnO4-sensitive sites are indicated by arrows. Adducted strand residues to the 3′ and 5′ of T0 are denoted by positive and negative integers (+N, −N).
To obtain further evidences that the 5′ incision inhibition was a repercussion of S751 phosphorylation, TFIIH-wt, TFIIH-S751A and TFIIH-S751E were immunoprecipitated from 27-1 cell extracts and treated with increasing amounts of CKII. Phosphorylation of TFIIH-wt by CKII strongly inhibited dual incision (Figure 5D, upper panel, lanes 1–3), whereas the unphosphorylateable TFIIH-S751A mainly retained its repair activity (lanes 4–6). Moreover, we noticed that TFIIH-wt, TFIIH-S751A or TFIIH-S751E transcription activities were not affected by CKII treatment (lower panel).
We next investigated whether XPB-S751 phosphorylation could allow for accurate opening of the damaged DNA, a prerequisite for dual incision by XPG and ERCC1-XPF endonucleases. We monitored the formation of the single-stranded/double-stranded opening structure using a permanganate footprinting assay (Tapias et al, 2004). Upon addition of HeLa TFIIH to XPC-HR23B, XPA, RPA and XPG, piperidine-sensitive sites from position +8 to −10 (on the 3′ and 5′ sides of the lesion, respectively) on the damaged strand defined the optimal opening (Figure 5E, compare lanes 2 and 3). Such an optimal opening was promoted by TFIIH-p+ to the same extent as TFIIH-p− (lanes 4–7). Altogether, our data demonstrate that S751 phosphorylation prevents the 5′ incision by ERCC1-XPF without altering the DNA opening around the damage or the 3′ incision by XPG.
Dephosphorylation of S751 stimulates 5′ incision formation
Then, we speculated whether TFIIH from HeLa might regain its repair activity following dephosphorylation of S751. Increasing amounts of immunoprecipitated TFIIH from HeLa NE treated or not with CIP were tested in dual incision. The dephosphorylation of S751 (see Figure 1F) parallels the increase in dual incision but preserves the transcription from the AdML promoter (Figure 6A). We estimate from these results that 50% (±5) of TFIIH is phosphorylated on the S751 residue in our HeLa extracts. In parallel, CIP treatment of the immunoprecipitated TFIIH-wt from 27-1 cell extracts significantly stimulated dual incision (Figure 6B, upper panel, lanes 1–3). This increase was commensurate with a stimulation of the 5′ incision by ERCC1-XPF without modification of the 3′ incision by XPG (middle panel, lanes 1–3). When TFIIH-S751A was used, treatment with CIP did not modify either the dual or the 5′ incisions (lanes 4–6).
Figure 6.
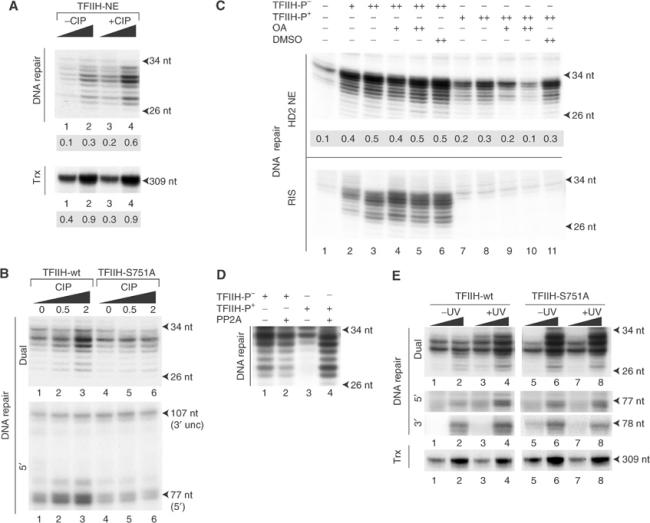
Reversible phosphorylation of S751 governs TFIIH NER activity. (A) A measure of 50 and 100 ng of CIP-treated (lanes 3 and 4) or untreated (lanes 1 and 2) TFIIH from HeLa NE (see Figure 1F) was tested in a dual incision (upper panel) or in a reconstituted transcription (lower panel) assay. (B) TFIIH-wt and TFIIH-S751A from 200 μg of 27-1 cell extracts was treated with increasing amounts of CIP as indicated (0, 0.5 and 2 U). After extensive washes, the various TFIIHs were tested for their dual and 5′ incisions. (C) HD2 NEs (20 μg protein, upper panel) or the reconstituted incision system (RIS, lower panel) was incubated in reaction buffer for 20 min at 30°C in the presence of 0.2 and 0.4 μM (lanes 4, 5 and 9, 10) of okadaic acid (OA). TFIIH-p− (lanes 2–6) or TFIIH-p+ (lanes 7–11) was then added for an additional 20 min. The reaction was then supplemented with damaged DNA. DMSO, in which OA was dissolved, was used as control (lanes 6 and 10). The values represent the intensities of the radioactive bands determined with a BioImager (arbitrary units). (D) Immunoprecipitated TFIIH-p− (lanes 1 and 2) or p+ (lanes 3 and 4) was treated with 2.5 mU of PP2A (lanes 2 and 4) and incision assay was performed with the reconstituted system. (E) A measure of 50 and 100 ng of TFIIH-wt or TFIIH-S751A immunopurified from irradiated or nonirradiated CHO 27-1 cells was tested in dual (upper panel), 5′ and 3′ incision assays (middle) or in a reconstituted transcription assay (lower) using the AdML promoter.
To investigate whether TFIIH in mammalian cells was subject to regulation by reversible protein phosphorylation, CKII-treated TFIIH-p+ or nontreated TFIIH-p− (as shown in Figure 5A) was preincubated with either NER-deficient HD2 nuclear extracts (HD2 NEs) (Johnson et al, 1985) or with all the purified recombinant NER factors before the addition of the damaged DNA. TFIIH-p+, which was unable to promote dual incision in the reconstituted system, regains its NER activity when preincubated with HD2 NE (Figure 6C, upper and lower panels, lanes 7 and 8). Previous work demonstrated that the ability of HeLa NE to carry out in vitro dual incision was highly sensitive to okadaic acid, suggesting the involvement of a type 2A protein phosphatase (PP2A) in NER (Ariza et al, 1996). Increasing amounts of okadaic acid, added to HD2 NE prior to the arrival of TFIIH-p+, prevent the recovery of NER (upper panel, lanes 9 and 10). The dual incision activity of TFIIH-p− was not affected by okadaic acid (upper and lower panels, lanes 4 and 5). These results suggest that NE contains a PP2A-like phosphatase activity, which can reversibly promote NER by dephosphorylating XPB-S751. We attempted to reverse the inhibition of incision with the purified catalytic subunit of PP2A. Addition of 2.5 mU of PP2A restored the repair activity of TFIIH-p+ (Figure 6D, lane 4) to the level seen with TFIIH-p− (lane 1).
Then, we asked whether UV irradiation of cells would stimulate TFIIH NER activity. Following UV irradiations of stably transfected 27-1 cells expressing either XPB-wt or XPB-S751A, their respective TFIIHs were immunopurified (Coin et al, 1999). In response to UV irradiation, the dual incision activity of TFIIH-wt, measured in a reconstituted system, increases significantly (Figure 6E, upper panel, lanes 1–4). In contrast, the dual incision activity of TFIIH-S751A remains unchanged, at the level seen with TFIIH-wt from irradiated cells (compare lanes 5 and 6 with 7 and 8). The increase in dual incision observed with the UV-irradiated TFIIH-wt parallels stimulation of the 5′ incision by ERCC1-XPF. The 3′ incision promoted by both TFIIHs (middle panels, lanes 1–4) and their ability to allow RNA synthesis from the AdML promoter (lower panel, lanes 1–8) remain stable following UV irradiation.
TFIIH-dependent recruitment of ERCC1-XPF to the damaged DNA
Then, we investigated whether such phosphorylation can prevent the recruitment of ERCC1-XPF to the intermediate incision/excision complex. We set up an assay (Figure 7A, upper panel) in which a biotinylated Pt DNA fragment was immobilized on magnetic beads (Riedl et al, 2003) and incubated (Incubation I) with a TFIIH-depleted HeLa NE, further supplemented with various TFIIH. Western blot of the depleted extract revealed that none of the other NER factors were codepleted with TFIIH (Figure 7A, left panel, and data not shown). Incubation I was performed in the presence of okadaic acid at 4°C to inhibit phosphatase activities and to prevent hydrolysis of the damaged DNA by XPG and ERCC1-XPF nucleases (Wakasugi and Sancar, 1998). The immobilized damaged DNA complexes were then washed with the incision buffer to remove unbound proteins, and the presence of ERCC1-XPF on the damaged DNA was analyzed either in a dual incision assay (Incubation II), containing all the recombinant incision factors except ERCC1-XPF, or by Western blotting. We first observed that depletion of TFIIH from NE prevents the recruitment of ERCC1-XPF to the damaged DNA (compare lanes 9 and 10). Complementations with either TFIIH-p+ or TFIIH-fs740 (in which the C-ter end is modified) allow a normal arrival of ERCC1-XPF compared to their respective controls (lanes 1–6).
Figure 7.
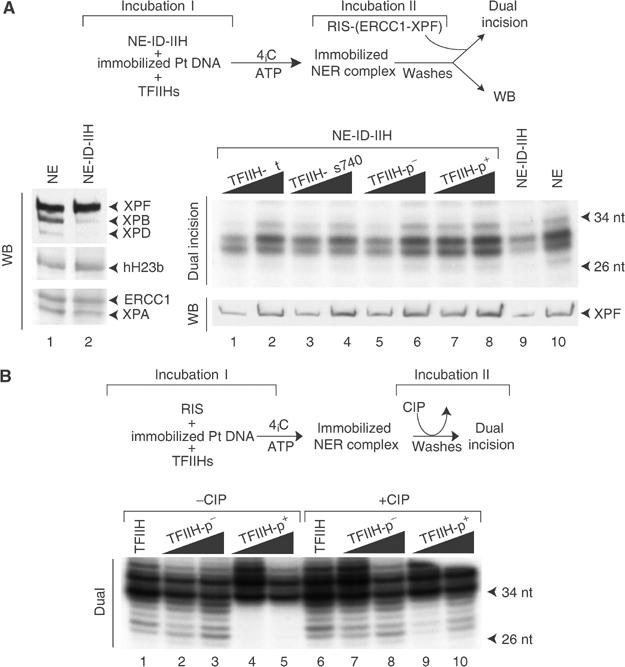
Recruitment of ERCC1-XPF to damaged DNA sites. (A) Left panel: A 50 μg portion of TFIIH nondepleted (lane 1) or depleted (lane 2) HeLa NEs. Right panel: Damage incision complexes were assembled on biotin-immobilized Pt damaged DNA by preincubation with HeLa NE (lane 10) or TFIIH-depleted HeLa NE (NE-ID-IIH) (lane 9) supplemented with either TFIIH-fs740 (lanes 3 and 4) or TFIIH-p+ (lanes 7 and 8) or their respective controls (lanes 1, 2 and 5, 6). Washed beads were then divided into two parts and either further incubated with the reconstituted incision system without ERCC1-XPF (dual incision) or loaded on an SDS–PAGE followed by Western blotting with anti-XPF antibody (WB). (B) Damage incision complexes were assembled on biotin-immobilized Pt damaged DNA by incubation of the reconstituted incision system with either TFIIH-p− (lanes 2 and 3) or TFIIH-p+ (lanes 4 and 5). Washed beads were then incubated with CIP (2 U) (lanes 7 and 8 for TFIIH-p− and lanes 9 and 10 for TFIIH-p+), washed and the reaction was continued for 40 min at 30°C. Reconstituted incision complex with HeLa TFIIH fraction from hydroxyapatite was assembled without (lane 1) or with (lane 6) subsequent CIP treatment. The strong band just above the 34 nt marker is not dependent on NER (Riedl et al, 2003); bands of dual incision are located below, between 26 and 34 nt.
We then investigated whether dephosphorylation of TFIIH, already bound to the damaged DNA, would restore ERCC1-XPF activity. All the NER factors including either TFIIH-p− or TFIIH-p+ were assembled on immobilized damaged DNA. After washing, the remaining protein/DNA complex was incubated with CIP, the phosphatase subsequently removed and the dual incision assay performed (Figure 7B, upper panel). Under these conditions, the activity of the repair complex containing TFIIH-p+ was restored (compare lanes 4 and 5 with 9 and 10) to a level almost similar to that achieved with TFIIH-p− (lanes 2 and 3). The CIP treatment of the repair complex containing an unphosphorylated TFIIH-p− had no effect on dual incision (compare lanes 1–3 with 6–8). Altogether, these data demonstrate that phosphorylation of S751 does not inhibit the assembly of any of the NER factors on the damaged DNA. Such inactive NER complex might regain its activity following dephosphorylation of S751.
Discussion
XPB S751 phosphorylation regulates TFIIH NER activity
The present study demonstrates that the cell can specifically regulate TFIIH NER function by modifying a single residue (XPB-S751) in one of its subunits. Several lines of evidences indicate that the phosphorylation of TFIIH/XPB-S751 is not constitutive and can be reversible. Kinases and phosphatases may then continuously add and remove phosphate groups on S751, thereby modulating TFIIH NER activity but leaving intact its transcription function. It is notable that XPB can be dephosphorylated by a PP2A-related enzyme following incubation with a crude extract, in which the subcellular compartmentalization has been destroyed indicating that the phosphatase activity involved might be the same or related to that induced by UV. Altogether, these data suggest that the phosphorylation of S751 specifically impaired the TFIIH NER activity, while its dephosphorylation, when the cell is damaged, will restore the capacity of TFIIH to repair the lesions on the DNA. In both cases, the capacity of TFIIH to fulfill its role in transcription is conserved.
S751 residue is crucial for the 5′ incision formation
How does the phosphorylation of XPB inhibit the multistep reaction of NER? It is accepted that TFIIH targets a lesion already recognized by XPC-HR23B and unwinds the DNA, resulting in an opened intermediate structure ready to recruit the successive NER factors (Volker et al, 2001; Riedl et al, 2003). Any change that will affect either TFIIH helicase activity, the margins and the range of the opening of the damaged DNA structure, the recruitment or the positioning of the other NER factors (i.e. XPA, RPA, XPG and ERCC1/XPF) could lead to an inhibition of the dual incision reaction. We investigated each step of the dual incision reaction and found that (i) neither XPB mutation nor its phosphorylation impairs its helicase activity per se, (ii) the phosphorylation of S751 (the present study), as well as the C-ter end frameshift mutation of XPB (Evans et al, 1997b), does not prevent the damaged DNA opening and (iii) ERCC1-XPF associates with the intermediate dual incision complex containing either TFIIH-p+ or fs740 as well as XPC, XPA and XPG although it does not initiate the 5′ incision of the damaged DNA. We therefore suggest a model whereby the phospho/dephosphorylation of S751 does not modify the composition of the dual incision complex but is rather used to remodel the complex. The fact that dual incision activity of a repair complex immobilized on the DNA can be regained following dephosphorylation of S751 strengthens our hypothesis and suggests an accurate positioning of the ERCC1-XPF nuclease to the 5′ site of incision once S751 is dephosphorylated. However, we cannot also exclude that the unphosphorylated C-ter end of XPB acts as a strong activator of ERCC1-XPF enzymatic activity, even if no interaction between TFIIH and ERCC1-XPF has been described.
Phosphorylation of S751: a switch to control 5′ incision formation?
The elimination of DNA damages proceeds through highly regulated steps involving several enzymatic activities. Firstly, to provide access to the NER machinery, the chromatin, a natural barrier that preserves genome integrity, should be remodeled in an ATP-dependent manner (Ura et al, 2001; Birger et al, 2003; Rubbi and Milner, 2003). Secondly, to eliminate rapidly the DNA lesions, the local concentration of the damage recognition factors XPC and XPE increases (Hwang et al, 1999; Ng et al, 2003). Our results demonstrated that NER is not only regulated in its first steps but also probably in the later steps. Through the phospho/dephosphorylation of XPB, the cell can control the different steps of dual incision and regulate the enzymatic activity of XPG and ERCC1-XPF endonucleases, where any introduction of inadvertent nicks in an undamaged DNA sequence could be deleterious and could interfere with other fundamental DNA transactions. Previous studies have already determined that the positioning and activity of ERCC1-XPF were regulated by the oriented binding of the replication protein A (RPA), another essential NER factor, to the single-stranded DNA (de Laat et al, 1998). An important issue will be to define the native protein phosphatases and kinases that modulate TFIIH DNA repair activity. Interestingly, inhibition of TFIIH dephosphorylation at low concentration of okadaic acid and recovery of TFIIH NER activity following treatment with PP2A catalytic subunit suggest that a type 2A protein phosphatase (PP2) (Bialojan and Takai, 1988) is a good candidate to regulate the phosphorylation of TFIIH. Another important issue could also be the investigation of the adverse effect of the expression of a constitutively active TFIIH-S751A in cell biology.
Sharing a common biochemical defect but not all the phenotypes
Our work also demonstrates the troubling discrepancy between the homogeneity of the biochemical repair defects harbored by some XP patients and the heterogeneity of their phenotypes. Indeed, in contrast to the strong clinical symptoms displayed by the XP11BE patient, including high incidence of skin cancer at an early age, neurodysmyelination, retarded growth and dimorphic faces (Weeda et al, 1990b), XP-F patients exhibit mild clinical features with no neurological abnormalities and slight sun sensitivity of the skin associated with a late onset of skin cancers (first tumors appear between 40 and 70 years old), which can be reminiscent of ageing (Matsumura et al, 1998). Both XP-F and ERCC1-defective cell extracts exhibit the same DNA repair defect as XP-B cell extracts. They are unable to promote the 5′ incision and consequently the removal of the damaged oligonucleotide; yet the opening of the DNA and the incision at the 3′ side of the lesion occur normally (Evans et al, 1997a). The similarity in the biochemical repair defect harbored by both groups of patients bolsters the idea that a failure in transcription could discriminate their phenotypes, illustrating the concept of transcription/repair syndromes associated with mutations in TFIIH (Bootsma and Hoeijmakers, 1993). In this respect, it is tempting to speculate that sun sensitivity of the skin harbored by the XP-B patient is one of the only hallmarks of NER phenotypes due to the inability of XPF to incise the damaged DNA (XP-F phenotype). The majority of XP-B patient phenotypes including the early onset of skin cancers could be assigned to a conjunction in the failure to transcribe specific genes and the persistence of lesions in the genome. Indeed, previous studies pointed out that XP11BE cells are specifically affected in the expression of c-myc, a fact that may explain the early development of malignancy in an XP-B patient (Liu et al, 2001).
Materials and methods
Stable cell lines
A 2 μg portion of pcDNA3.1(+) (Invitrogene) containing wild-type or mutated XPB cDNA was transfected in 106 27-1 cells in 10 cm Petri dishes using lipofectamine (Invitrogene). At 16 h post-transfection, cells were refed with fresh medium supplemented 48 h later with 500 μg/ml geneticin sulfate (G418, Gibco). After 2 weeks, G418-resistant clones were isolated and expanded.
DNA substrates for NER assays
Immobilized damaged DNA substrate was generated by a _Hin_cII/_Ban_I digestion of the single lesion (Pt-GTG) plasmids (Pt DNA) based on the 105.TS plasmid (Frit et al, 2002). Digestion gives rise to a 288 bp fragment with the cisplatin–DNA crosslink at position 87. The nondamaged strand of the 288 bp fragment was biotinylated at the _Ban_I site by Klenow fill-in reaction with Bio-dUTP and purified on agarose gel with the QIAEX gel extraction kit (Qiagen) in the absence of ethidium bromide. The biotinylated DNA (40 fmol) was bound to 2 μg of magnetic streptavidin beads (Dynabeads M-280, Dynal) and equilibrated in buffer B (45 mM Hepes–KOH (pH 7.8), 5 mM MgCl2, 1 mM DTT, 0.3 mM EDTA, 10% glycerol, 2.5 μg BSA, 50 mM KCl) prior to use. For 3′ and 5′ incision assays, the linear cisplatinated _Age_I/_Bss_HII fragment (185 bp) was radiolabeled at either the 3′ or 5′ end of the adducted strand. After purification, 20 fmol of the probe (20 000 cpm) was used per reaction. The pt-GTG plasmid was also modified to introduce a sequence of 32 bp, which allows, when single stranded, the annealing of a 32-mer (5′-GATCTCGGCGACATCGGTTCCGTCCTA ACTCG-3′) containing a cyclobutane thymine dimer adduct (in bold) (Ortiz Mayo et al, 2003).
In vitro kinase and phosphatase assays
Immunoprecipitated XPB or synthetic peptides were incubated for 30 min at 30°C in 20 μl of kinase buffer (20 mM Hepes (pH 7.9), 15 mM MgCl2, 30 mM KCl) supplemented with 1 μCi of [γ-32P]ATP (3000 Ci/mmol) and 5 U of casein kinase II (CKII; New England Biolabs). Reactions were stopped by protein loading buffer, followed by SDS–PAGE and autoradiography. Dephosphorylation by Calf intestinal phosphatase (CIP; New England Biolabs) was performed in 50 mM Tris–HCl (pH 7.9), 1 mM DTT, 10 mM MnCl2 and 100 mM NaCl on immunoprecipitated TFIIH from HeLa or hamster cell extracts washed twice with buffer A supplemented with 200 mM KCl and twice with 50 mM KCl.
Microinjections in XPCS2BA
Microinjections were performed as described by Vermeulen et al (1994).
Preparation of cell extracts and purification of TFIIH
Subconfluent cultures of exponentially growing cells were washed with PBS1x and glycerol 30% and lysed in buffer A supplemented with 400 mM NaCl, 0.25% Nonidet P-40, 1 × protease inhibitor cocktail, 2 μM okadaic acid and 20 mM sodium molybdate. Cell lysates were centrifuged for 30 min at 11 000 r.p.m. The supernatants (0.4 mg) were incubated overnight with Ab-hXPB bound to protein A–Sepharose. The beads are extensively washed with buffer A supplemented with 200 mM KCl and re-equilibrated in buffer A supplemented with 50 mM KCl. HeLa NEs were prepared as described (Frit et al, 2002). TFIIH depletion was performed by applying HeLa NE to protein A–Sepharose beads coupled to monoclonal Ab-p44 antibody.
Purification of TFIIH from UV-irradiated cells
Pure clones of CHO 27-1 expressing either XPB-wt or XPB-S751A were UV irradiated at 254 nm with 40 J/m2 and cultured for 1 h. Cells were then washed with PBS1x and proceeded as above. Immunopurifications of TFIIH-wt and TFIIH-S751A were performed with Ab-p44 antibody and its corresponding competitor peptide (Coin et al, 1999).
Dual and single incision NER assay
Dual incision assay (Araujo et al, 2000) was carried out in 25 μl of buffer B supplemented with 2 mM ATP. Each reaction contained 5 ng XPG, 15 ng XPF/ERCC1, 10 ng XPC-hHR23B, 50 ng RPA and 25 ng XPA. Following preincubation for 10 min at 30°C, 30 ng of Pt DNA was added and the reaction was continued for 90 min at 30°C. The excised fragment was detected on 14% urea–acrylamide after annealing with 9 ng of the complementary oligonucleotide and addition of four radiolabeled [α-32P]dCMP (3000 μCi/mmol) residues by Sequenase V2.1 (USB). Single incision assays were carried out similar to dual incision assays, except that radioactive DNA was extracted following incubations and resolved on an 8% urea–acrylamide gel.
Transcription assays
Run-off transcription was carried out as described by Gerard et al (1991). Activated transcription using Gal4-VP16 was performed as described by Frit et al (2002).
KMnO4 assay
The assay was performed as described by Evans et al (1997a) and Tapias et al (2004).
Protein binding studies on immobilized DNA
Immobilized DNA was incubated with TFIIH-depleted HeLa NE under dual incision assay conditions at 4°C for 40 min in the presence of 2 mM ATP. Upon incubation, magnetic beads were collected on a magnetic particle concentrator (Dynal MPC) and supernatants removed. Beads were then washed five times in four volumes of cold buffer B and resuspended in buffer B for functional analysis of bound ERCC1-XPF or in SDS–PAGE loading buffer for Western blotting. Studies were carried out with the equivalent of one dual incision reaction for the functional protein binding assay and with the equivalent of six dual incision reactions for Western blotting analysis (Riedl et al, 2003).
Antibody
A monoclonal mouse antibody (Ab-pS751) was raised against the polypeptide GTMSSMpSGADDTVY corresponding to residues 745–758 of XPB. Mouse monoclonal antibodies toward TFIIH subunits were used as described by Marinoni et al (1997). A 1/2000 dilution of monoclonal antibody Ab-5 (NeoMarkers) and a 1/100 dilution of rabbit polyclonal antibody (Volker et al, 2001) were used for XPF and ERCC1, respectively.
Acknowledgments
We thank A Larnicol for her excellent technical expertise, JL Weickert and I kolb for their technical assistance and P Gloekler for her critical reading of the manuscript. The IGBMC services (cells, antibodies and peptides) provided invaluable assistance. JA was supported by the Association pour la Recherche contre le Cancer. AT was supported by the EMBO Organization and Fondation pour la Recherche Medicale. This study was supported by funds from the Ligue contre le Cancer (équipe labelisée), the Institut National de la Santé et de la Recherche Médicale and the EEC (contract CDP51.1047).
References
- Adamczewski JP, Rossignol M, Tassan JP, Nigg EA, Moncollin V, Egly JM (1996) MAT1, cdk7 and cyclin H form a kinase complex which is UV light-sensitive upon association with TFIIH. EMBO J 15: 1877–1884 [PMC free article] [PubMed] [Google Scholar]
- Akoulitchev S, Mäkelä TP, Weinberg RA, Reinberg D (1995) Requirement for TFIIH kinase activity in transcription by RNA polymerase II. Nature 377: 557–560 [DOI] [PubMed] [Google Scholar]
- Akoulitchev S, Reinberg D (1998) The molecular mechanism of mitotic inhibition of TFIIH is mediated by phosphorylation of CDK7. Genes Dev 12: 3541–3550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo SJ, Tirode F, Coin F, Pospiech H, Syvaoja JE, Stucki M, Hubscher U, Egly JM, Wood RD (2000) Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev 14: 349–359 [PMC free article] [PubMed] [Google Scholar]
- Ariza RR, Keyse SM, Moggs JG, Wood RD (1996) Reversible protein phosphorylation modulates nucleotide excision repair of damaged DNA by human cell extracts. Nucleic Acids Res 24: 433–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialojan C, Takai A (1988) Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem J 15: 283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birger Y, West KL, Postnikov YV, Lim JH, Furusawa T, Wagner JP, Laufer CS, Kraemer KH, Bustin M (2003) Chromosomal protein HMGN1 enhances the rate of DNA repair in chromatin. EMBO J 22: 1665–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootsma D, Hoeijmakers JHJ (1993) DNA repair. Engagement with transcription. Nature 363: 114–115 [DOI] [PubMed] [Google Scholar]
- Coin F, Bergmann E, Tremeau-Bravard A, Egly JM (1999) Mutations in XPB and XPD helicases found in xeroderma pigmentosum patients impair the transcription function of TFIIH. EMBO J 18: 1357–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coin F, Marinoni JC, Rodolfo C, Fribourg S, Pedrini AM, Egly JM (1998) Mutations in the XPD helicase gene result in XP and TTD phenotypes, preventing interaction between XPD and the p44 subunit of TFIIH. Nat Genet 20: 184–188 [DOI] [PubMed] [Google Scholar]
- de Laat WL, Appeldoorn E, Sugasawa K, Weterings E, Jaspers NG, Hoeijmakers JH (1998) DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev 12: 2598–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvir A, Conaway RC, Conaway JW (1997) A role for TFIIH in controlling the activity of early RNA polymerase II elongation complexes. Proc Natl Acad Sci USA 94: 9006–9010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E, Fellows J, Coffer A, Wood RD (1997a) Open complex formation around lesion during nucleotide excision repair provides a structure for cleavage by human XPG protein. EMBO J 16: 625–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E, Moggs JG, Hwang JR, Egly J-M, Wood RD (1997b) Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J 16: 6559–6573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feaver WJ, Gileadi O, Li Y, Kornberg RD (1991) CTD kinase associated with yeast RNA polymerase II initiation factor b. Cell 67: 1223–1230 [DOI] [PubMed] [Google Scholar]
- Frit P, Kwon K, Coin F, Auriol J, Dubaele S, Salles B, Egly J (2002) Transcriptional activators stimulate DNA repair. Mol Cell 10: 1391–1401 [DOI] [PubMed] [Google Scholar]
- Gerard M, Fischer L, Moncollin V, Chipoulet JM, Chambon P, Egly JM (1991) Purification and interaction properties of the human RNA polymerase B(II) general transcription factor BTF2. J Biol Chem 266: 20940–20945 [PubMed] [Google Scholar]
- Guzder SN, Sung P, Bailly V, Prakash L, Prakash S (1994) RAD25 is a DNA helicase required for DNA repair and RNA polymerase II transcription. Nature 369: 578–581 [DOI] [PubMed] [Google Scholar]
- Holstege FC, van der Vliet PC, Timmers HT (1996) Opening of an RNA polymerase II promoter occurs in two distinct steps and requires the basal transcription factors IIE and IIH. EMBO J 15: 1666–1677 [PMC free article] [PubMed] [Google Scholar]
- Hwang BJ, Ford JM, Hanawalt PC, Chu G (1999) Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc Natl Acad Sci USA 96: 424–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JR, Moncollin V, Vermeulen W, Seroz T, van Vuuren H, Hoeijmakers JHJ, Egly JM (1996) A 3′–5′ XPB helicase defect in repair/transcription factor TFIIH of xeroderma pigmentosum group B affects both DNA repair and transcription. J Biol Chem 271: 15898–15904 [DOI] [PubMed] [Google Scholar]
- Johnson RT, Squires S, Ellion GC, Koch GLE, Rainbow AJ (1985) Xeroderma pigmentosum D-HeLa hybrids with low and high ultraviolet sensitivity associated with normal and diminished DNA repair ability, respectively. J Cell Sci 76: 115–133 [DOI] [PubMed] [Google Scholar]
- Keriel A, Stary A, Sarasin A, Rochette-Egly C, Egly JM (2002) XPD mutations prevent TFIIH-dependent transactivation by nuclear receptors and phosphorylation of RARalpha. Cell 109: 125–135 [DOI] [PubMed] [Google Scholar]
- Lehmann AR (2001) The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev 15: 15–23 [DOI] [PubMed] [Google Scholar]
- Liu J, Akoulitchev S, Weber A, Ge H, Chuikov S, Libutti D, Wang XW, Conaway JW, Harris CC, Conaway RC, Reinberg D, Levens D (2001) Defective interplay of activators and repressors with TFIH in xeroderma pigmentosum. Cell 104: 353–363 [DOI] [PubMed] [Google Scholar]
- Long JJ, Leresche A, Kriwacki RW, Gottesfeld JM (1998) Repression of TFIIH transcriptional activity and TFIIH-associated cdk7 kinase activity at mitosis. Mol Cell Biol 18: 1467–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Zawel L, Fisher L, Egly JM, Reinberg D (1992) Human general transcription factor IIH phosphorylates the C-terminal domain of RNA polymerase II. Nature 358: 641–645 [DOI] [PubMed] [Google Scholar]
- Ma L, Westbroek A, Jochemsen AG, Weeda G, Bosch A, Bootsma D, Hoeijmakers JHJ, Van Der EB AJ (1994) Mutational analysis of ERCC3, which is involved in DNA repair and transcription initiation: identification of domains essential for the DNA repair function. Mol Cell Biol 14: 4126–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinoni JC, Roy R, Vermeulen W, Miniou P, Lutz Y, Weeda G, Seroz T, Molina Gomez D, Hoeijmakers JHJ, Egly JM (1997) Cloning and characterization of p52, the fifth subunit of the core of the transcription/DNA repair factor TFIIH. EMBO J 16: 1093–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y, Nishigori C, Yagi T, Imamura S, Takebe H (1998) Characterization of molecular defects in xeroderma pigmentosum group F in relation to its clinically mild symptoms. Hum Mol Genet 7: 969–974 [DOI] [PubMed] [Google Scholar]
- Mellon I, Spivak G, Hanawalt PC (1987) Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell 51: 241–249 [DOI] [PubMed] [Google Scholar]
- Mu D, Hsu DS, Sancar A (1996) Reaction mechanism of human DNA repair excision nuclease. J Biol Chem 271: 8285–8294 [DOI] [PubMed] [Google Scholar]
- Ng JMY, Vermeulen W, van der Horst GTJ, Bergink S, Sugasawa K, Vrieling H, Hoeijmakers JHJ (2003) A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev 13: 1630–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnovan A, Davies AA, Moggs JG, West SC, Wood RD (1994) XPG endonuclease makes the 3′ incision in human DNA nucleotide excision repair. Nature 371: 432–435 [DOI] [PubMed] [Google Scholar]
- Ortiz Mayo JU, Thomas M, Saintomé C, Clivio P (2003) Facile synthesis of a cis–syn thymine dimer building block its incorporation into oligodeoxynucleotides. Tetrahedron 59: 7377–7383 [Google Scholar]
- Riedl T, Hanaoka F, Egly J (2003) The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J 22: 5293–5303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbi CP, Milner J (2003) p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J 22: 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivji MKK, Podust VN, Hubsher U, Wood RD (1995) Nucleotide excision repair DNA synthesis by DNA polymerase epsilon in the presence of PCNA, RFC, and RPA. Biochemistry 34: 5011–5017 [DOI] [PubMed] [Google Scholar]
- Sijbers AM, de Laat WL, Ariza RR, Biggerstaff M, Wei YF, Moggs JG, Carter KC, Shell BK, Evans E, de Jong MC, Rademakers S, de Rooij J, Jaspers NG, Hoeijmakers JH, Wood RD (1996) Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell 86: 811–822 [DOI] [PubMed] [Google Scholar]
- Sugasawa K, Ng J, Masutani C, Iwai S, van der Spek P, Eker A, Hanaoka F, Bootsma D, Hoeijmakers J (1998) Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell 2: 223–232 [DOI] [PubMed] [Google Scholar]
- Tapias A, Auriol J, Forget D, Enzlin J, Scharer O, Coin F, Coulombe B, Egly J (2004) Ordered conformational changes in damaged DNA induced by nucleotide excision repair factors. J Biol Chem 18: 19074–19083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirode F, Busso D, Coin F, Egly JM (1999) Reconstitution of the transcription factor TFIIH: assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol Cell 3: 87–95 [DOI] [PubMed] [Google Scholar]
- Ura K, Araki M, Saeki H, Masutani C, Ito T, Iwai S, Mizukoshi T, Kaneda Y, Hanaoka F (2001) ATP-dependent chromatin remodeling facilitates nucleotide excision repair of UV-induced DNA lesions in synthetic dinucleosomes. EMBO J 20: 2004–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen W, Scott RJ, Potger S, Müller HJ, Cole J, Arlett CF, Kleijer WJ, Bootsma D, Hoeijmakers JHJ, Weeda G (1994) Clinical heterogeneity within xeroderma pigmentosum associated with mutations in the DNA repair and transcription gene ERCC3. Am J Hum Genet 54: 191–200 [PMC free article] [PubMed] [Google Scholar]
- Volker M, Mone MJ, Karmakar P, van Hoffen A, Schul W, Vermeulen W, Hoeijmakers JH, van Driel R, van Zeeland AA, Mullenders LH (2001) Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell 8: 213–224 [DOI] [PubMed] [Google Scholar]
- Wakasugi M, Sancar A (1998) Assembly, subunit composition, and footprint of human DNA repair excision nuclease. Proc Natl Acad Sci USA 95: 6669–6674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallenfang MR, Seydoux G (2002) cdk-7 is required for mRNA transcription and cell cycle progression in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA 99: 5527–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeda G, Eveno E, Donker I, Vermeulen W, Chevallier-Lagente Taieb A, Stary A, Hoeijmakers JHJ, Mezzina M, Sarasin A (1997) A mutation in the XPB/ERCC3 DNA repair transcription gene, associated with trichothiodystrophy. Am J Hum Genet 60: 320–329 [PMC free article] [PubMed] [Google Scholar]
- Weeda G, van Ham R, Masurel R, Westerveld A, Odijk H, de Wit J, Bootsma D, van der Eb AJ, Hoeijmakers JHJ (1990a) Molecular cloning and biological characterization of the human excision repair gene ERCC3. Mol Cell Biol 10: 2570–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeda G, van Ham RCA, Vermeulen W, Bootsma D, van der Eb AJ, Hoeijmakers JHJ (1990b) A presumed DNA helicase encoded by ERCC-3 is involved in the human repair disorders xeroderma pigmentosum and Cockayne's syndrome. Cell 62: 777–791 [DOI] [PubMed] [Google Scholar]
- Wood RD (1997) Nucleotide excision repair in mammalian cells. J Biol Chem 272: 23465–23468 [DOI] [PubMed] [Google Scholar]
- Zurita M, Merino C (2003) The transcriptional complexity of the TFIIH complex. Trends Genet 19: 578–584 [DOI] [PubMed] [Google Scholar]