New perspectives for targeting RAF kinase in human cancer (original) (raw)
. Author manuscript; available in PMC: 2018 Nov 1.
Published in final edited form as: Nat Rev Cancer. 2017 Oct 6;17(11):676–691. doi: 10.1038/nrc.2017.79
Abstract
The discovery that a subset of human tumours is dependent on mutationally deregulated BRAF kinase intensified the development of RAF inhibitors to be used as potential therapeutics. The US Food and Drug Administration (FDA)-approved second-generation RAF inhibitors vemurafenib and dabrafenib have elicited remarkable responses and improved survival of patients with BRAF-V600E/K melanoma, but their effectiveness is limited by resistance. Beyond melanoma, current clinical RAF inhibitors show modest efficacy when used for colorectal and thyroid BRAF-V600E tumours or for tumours harbouring BRAF alterations other than the V600 mutation. Accumulated experimental and clinical evidence indicates that the complex biochemical mechanisms of RAF kinase signalling account both for the effectiveness of RAF inhibitors and for the various mechanisms of tumour resistance to them. Recently, a number of next-generation RAF inhibitors, with diverse structural and biochemical properties, have entered preclinical and clinical development. In this Review, we discuss the current understanding of RAF kinase regulation, mechanisms of inhibitor action and related clinical resistance to these drugs. The recent elucidation of critical structural and biochemical aspects of RAF inhibitor action, combined with the availability of a number of structurally diverse RAF inhibitors currently in preclinical and clinical development, will enable the design of more effective RAF inhibitors and RAF-inhibitor-based therapeutic strategies, tailored to different clinical contexts.
The family of RAF kinases (ARAF, BRAF and CRAF (also known as RAF1)) constitute core components of the RAS–RAF–MEK–ERK signalling cascade (ERK signalling), a pathway that mediates signals from cell surface receptors to the nucleus to regulate cell growth, differentiation and survival1-4. Upon activation, RAF kinases phosphorylate and activate the kinases MEK1 and MEK2, which in turn phosphorylate and activate ERK1 and ERK2. Activated ERK promotes cell proliferation and survival by phosphorylating multiple substrates both in the cytosol and in the nucleus4.
Deregulation of ERK signalling commonly occurs in cancer, frequently due to mutations of components of the pathway5. It has been known for more than four decades that mutations in the genes encoding for the RAS family of proteins are often found in human tumours6,7. RAS protein has been extremely difficult to target, and thus the downstream kinases RAF, MEK and ERK remain attractive therapeutic targets in such tumours, although current inhibitors of these kinases have only showed limited efficacy in RAS-mutant cancers8. BRAF mutations are present in approximately 8% of human tumours9. The most frequent mutation in the BRAF gene is the 1799T>A substitution that results in an amino acid change from valine (V) to glutamic acid (E) in the activation segment of the kinase (V600E), promoting several-fold kinase hyperactivation9,10. In addition to V600E, and other substitutions at V600 (such as V600K, V600D and V600R), a number of non-V600 missense BRAF mutations have been found, mostly clustered in the activation segment or in the so-called glycine-rich loop of the kinase domain9-11. Point mutations are not the only alterations found in BRAF. Fusion proteins resulting from translocations containing the catalytic domain of BRAF, as well as in-frame deletions are also found in certain tumour types, resulting in constitutive activation of BRAF and downstream ERK signalling12,13.
BRAF is commonly mutated in melanomas (50%)14, papillary thyroid cancers (45%)15, colon cancers (10%)16, non-small-cell lung cancers (NSCLCs) (10%)17, in virtually 100% of hairy cell leukemias18 and in 50–60% of patients with the idiopathic disorder Langerhans cell histiocytosis (LCH)19. These findings, along with preclinical work demonstrating dependence of BRAF-V600E tumours on BRAF and ERK signalling activity20, supported the therapeutic benefit of targeting ERK signalling in cancer and intensified efforts for the development of selective ATP-competitive inhibitors of mutant BRAF kinase. Two second-generation RAF inhibitors, vemurafenib and dabrafenib, showed remarkable clinical activity in patients with BRAF-V600E/K melanoma and received US Food and Drug Administration (FDA) approval for the treatment of this disease21,22. However, despite prolonging patient survival, RAF inhibitor treatment is rarely curative and is limited in most cases by the development of drug resistance and tumour relapse. As preclinical and clinical evidence suggested that more potent inhibition of ERK signalling in the tumour would yield improved responses23, combinations of RAF inhibitors with MEK inhibitors were tested in subsequent trials. Both combinations (vemurafenib and cobimetinib (Roche/Genentech) and dabrafenib and trametinib (Novartis)) showed improved clinical efficacy compared with RAF inhibitor monotherapy24,25. Two combinations are now the standard of care for BRAF-V600E/K metastatic melanoma, although they are still not curative treatments for the majority of patients. Importantly, the most common mechanisms of resistance identified in tumours following relapse after combination RAF and MEK inhibitor therapy are associated with recovery of ERK signalling and are similar to the mechanisms of resistance detected after RAF inhibitor monotherapy26-28; this observation provides the rationale for the development of future therapeutic strategies that could achieve more potent inhibition of RAF–ERK signalling and may therefore yield deeper and more durable responses.
The remarkable clinical efficacy of currently approved RAF inhibitors has been so far confined to BRAF-V600E/K melanoma. Besides the most prevalent mutations in melanoma, BRAF-V600E (70–80%) and BRAF-V600K (5–12%), additional BRAF-V600 mutant forms have been identified in patients with melanoma but are less common. Among them, BRAF-V600R (3–7%) and the rare mutations BRAF-V600M, BRAF-V600D and BRAF-V600G (<5% in total) have been reported to be sensitive to approved RAF inhibitors in preclinical29-32 and clinical22,33-35 studies. By contrast, preclinical and clinical data suggested that colorectal and thyroid BRAF-V600E tumours, as well as tumours with BRAF mutations other than V600 substitutions, are less sensitive to current clinical RAF inhibitors36-39.
In this Review, we highlight the most recent developments in our understanding of the biochemical basis of sensitivity and resistance to second-generation RAF inhibitors in cancer therapy. We will also discuss the ongoing development of third-generation RAF inhibitors with new structural properties designed to both address certain toxicities elicited by current clinical inhibitors and provide more durable therapeutic responses in patients with BRAF-mutant tumours.
Structural insight into RAF activation
In normal cells and in the absence of upstream activity, cytosolic RAF is thought to adopt a monomeric, closed and inactive conformation due to the intramolecular interaction between the amino-terminal (regulatory) and the carboxy-terminal (kinase) domains40,41 (FIG. 1a). Canonical RAF activation is regulated by members of the RAS family of small GTPases (KRAS, NRAS and HRAS), which, upon growth factor stimulation of upstream receptors, are converted from the inactive (RAS-GDP) to the active (RAS-GTP) form in the plasma membrane. The increase in the levels of RAS-GTP results in RAF translocation to the membrane and formation of the RAF–RAS-GTP complex, due to the high affinity of RAS-GTP for the RAS-binding domain (RBD) in the N terminus of RAF42,43 (FIG. 1a). RAF kinases bound to RAS-GTP in the membrane become fully activated via priming (which includes phosphorylation at critical residues, such as S338 in CRAF) and homodimerization and heterodimerization through the kinase domain44,45.
Figure 1. RAF activation.
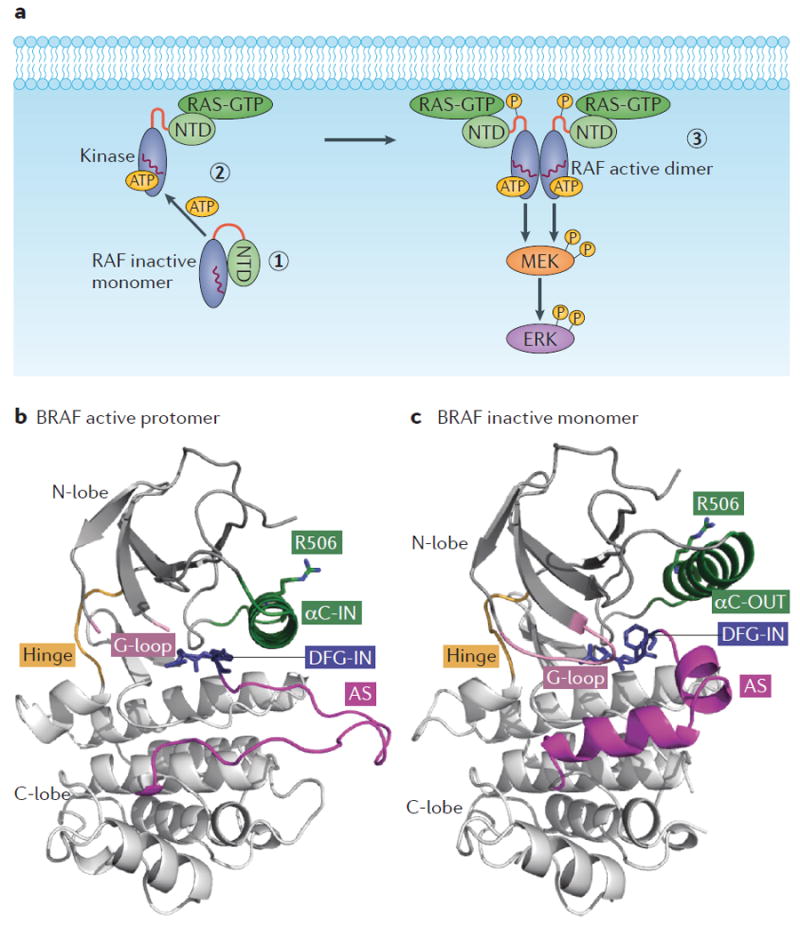
a ∣ Schematic showing canonical RAF activation. In conditions of low RAS-GTP, RAF is monomeric and inactive in the cytosol due to intramolecular interaction between the N-terminal domain (NTD; regulatory) and the C-terminal (kinase) domain (1). Upregulation of RAS-GTP promotes the formation of the RAF–RAS-GTP complex in the membrane due to the high affinity of RAS-GTP for the RAS-binding domain (RBD) present in the NTD of RAF (2). This step is followed by activating phosphorylation and dimerization for full RAF activation (3). αC-helix indicated by the red wavy line in the kinase domain. b ∣ Ribbon representation of one protomer of the active BRAF dimer (Protein Data Bank (PDB) ID: 4MNE). The kinase domain consists of N and C terminal lobes linked through a short flexible hinge. c ∣ Ribbon representation of the inactive and monomeric BRAF kinase domain (PDB ID: 4RZV). In parts b and c, note the different positions of the αC-helix (in green) and of the activation segment (AS, magenta), which highlight the movement of the αC-helix from the OUT to the IN position and the unfolding of the AS upon RAF activation. The R506 residue side chain in the IN (active) and the OUT (inactive) positions and the conformation of the glycine-rich loop (G-loop) and DFG motif in the active and inactive form are also depicted.
RAF has a typical kinase domain architecture, with the N-terminal lobe (N-lobe) and C-terminal lobe (C-lobe) linked through a short flexible hinge. At the interface of the two lobes lies the active site of the kinase that includes the nucleotide (ADP or ATP) binding site, the magnesium binding site (DFG motif), and the phospho-acceptor site (activation segment). The motions between the lobes are controlled by the flexible hinge and enable recruitment of the substrate and release of the product46. The catalytically active conformation of the kinase domain requires a closed conformation between the two lobes, an unfolded activation segment and the αC-helix of the N-terminal lobe fixed into the IN position to bring the catalytic residues into a productive distance and orientation (FIG. 1b). The active conformation in RAF is facilitated by dimerization. Dimerized RAF has been visualized by several crystal structures determined with BRAF or CRAF bound to inhibitors, providing insights into the dimerization interface10,30,47-50. The dimerization interface of each BRAF protomer includes the αC-helix of each kinase and specifically the interaction between the C-terminal R509 residue of each αC-helix. Mutagenesis studies have established the critical role of R509 and adjacent residues in mediating BRAF dimerization50-53.
Recently, two crystal structures of monomeric BRAF kinase bound to structurally related compounds30,54 and the crystal structure of the BRAF–MEK complex55 have been determined, thereby providing important insight on the conformational transitions and the structural basis of dimerization-dependent RAF activation. The structure of monomeric BRAF shows the kinase in an inactive conformation characterized by an open configuration between the N-lobe and C-lobe of the kinase domain30,54. The αC-helix is positioned in the OUT position, where it is stabilized by interactions with residues of the folded activation segment, which has also been determined30,54 (FIG. 1c). The structure of dimeric BRAF kinase bound to MEK enabled the visualization of an active conformation of the BRAF structure and the closed configuration between the N-lobe and C-lobe of the kinase55. In this conformation, the position of the αC-helix is shifted to the full IN position, and the activation segment is extended as an unstructured loop that enables the shift of the αC-helix to facilitate catalysis and the interaction with the substrate55. The crystal structures show the DFG motif to adopt an IN conformation in both the active and the inactive kinase conformations30,55 (FIG. 1b,c). These structural studies suggest that BRAF undergoes dynamic conformational transitions within the N-lobe, C-lobe, αC-helix and activation segment. These transitions are structurally interconnected and reveal how dimerization promotes activation of the kinase domain.
Despite a number of available crystal structures of either wild-type (WT) BRAF or BRAF-V600E, the detailed structural mechanism of BRAF activation by the most common BRAF mutation (V600 substitution) is still not well understood. Co-crystal structures of BRAF-V600E with RAF inhibitors suggested the potential for dimerization-induced activation by conformational changes promoted by the V600E substitution10,54. Recent studies suggested the importance of a salt bridge between V600E in the activation segment and K507 of the αC-helix in stabilizing the active conformation of BRAF-V600E as evidenced by crystal structures of the BRAF-V600E dimer54,55. However, cell-based and biochemical data established that BRAF-V600 proteins in the presence of low levels of RAS-GTP can be catalytically active monomers30,31,51-53,56. The reason for this discrepancy is presumably the absence of the N-terminal regulatory domain of BRAF in available crystal structures; this domain may be involved in the stabilization of an active monomeric conformation of V600-mutant BRAF kinase. Further studies are needed to elucidate the structural details of BRAF kinase activation. Structural understanding of the interaction of the N-terminal regulatory domain with the RAF kinase domain, in either the inactive conformation or the active conformation of the monomer, would be a major step towards elucidating the regulation of normal and oncogenic BRAF.
Beyond the physiological signalling mechanism of BRAF dimerization-induced activation through activated RAS, cell-based studies showed that certain oncogenic BRAF mutations promote spontaneous BRAF dimerization and activation, either by forming homodimers in the absence of RAS-GTP, such as BRAF-L597V or BRAF-G466E11,30,31,51, or by further enhancing RAF dimerization promoted by RAS-GTP, such as the catalytically inactive BRAF-D594N mutant or ‘impaired activity’ mutants57. As in the case of BRAF-V600 mutants, the exact mechanism by which these mutations induce dimerization of the kinase is not well understood, due to the lack of structural information that includes the N-terminal domain of BRAF, or at least the part that regulates dimerization of the catalytic domain. Furthermore, several mutations are found in residues in the activation segment or in the glycine-rich loop, which interacts with the activation segment9. In most co-crystal structures of BRAF kinase, several residues of the activation segment are not resolved, precluding information of the position of these residues and their interactions in the activated BRAF structure. The recently reported structures of BRAF monomers in the inactive conformation suggest that mutation in the activation segment such as V600E may disrupt the hydrophobic packing between the inactive state of the activation segment and the αC-helix through steric clashes or unfavourable interactions; in turn, this can promote allosteric movement of the αC-helix towards the IN position, which eventually favours dimerization of the RAF kinase domain30,54. Additional crystal structures of BRAF mutants other than the V600 substitution are required for more definitive and detailed understanding of the mechanisms involved.
Models of RAF inhibitor action
RAF inhibitors have unusual biochemical properties. Unlike most kinase inhibitors, which suppress their targets in all cells, current clinical RAF inhibitors suppress RAF activity and ERK signalling selectively in cells expressing mutant BRAF. In tumour and normal cells expressing WT BRAF, these RAF inhibitors do not inhibit but instead paradoxically activate RAF activity and downstream ERK signalling (the so-called ‘RAF inhibitor paradox’)1,47,49,56-63. The complex biochemical mechanism of action of RAF inhibitors has been the topic of intense investigation, and it was only recently, with the integration of structural, biochemical and cell-based analysis of a large number of structurally diverse RAF inhibitors, that it has been understood in more detail.
Structural insight into RAF inhibitor activity
Dependent on their chemical structure, RAF inhibitors stabilize the αC-helix in a position between the IN and OUT conformations, forming imperfect dimers due to differences in the position of the αC-helix within each protomer (FIG. 2a). A notable example is vemurafenib, which binds the first protomer and stabilizes the αC- helix towards the OUT position. In the second protomer, the IN position of the αC-helix creates negative allostery for binding a second vemurafenib molecule, because movement of the αC-helix towards the OUT position in that protomer would cause disruption of the dimer (Supplementary information S1 (movie)). By contrast, other RAF inhibitors, such as AZ628, that stabilize the αC-helix of the first protomer towards the IN position, sterically allow occupancy of the second protomer by a second drug molecule (Supplementary information S2 (movie)). Thus, RAF kinase inhibitors can be broadly classified according to the conformation in which they stabilize their target kinase — ‘αC-IN’ (either αC-helix-IN/DFG-IN (type I) or αC-helix-IN/DFG-OUT (type II)) or ‘αC-OUT’, the latter most commonly occurring as αC-helix-OUT/DFG-IN (type I1/2)64, which is the class to which the current clinical RAF inhibitors vemurafenib and dabrafenib belong (FIG. 2a and TABLE 1).
Figure 2. Mechanism of action of RAF inhibitors.
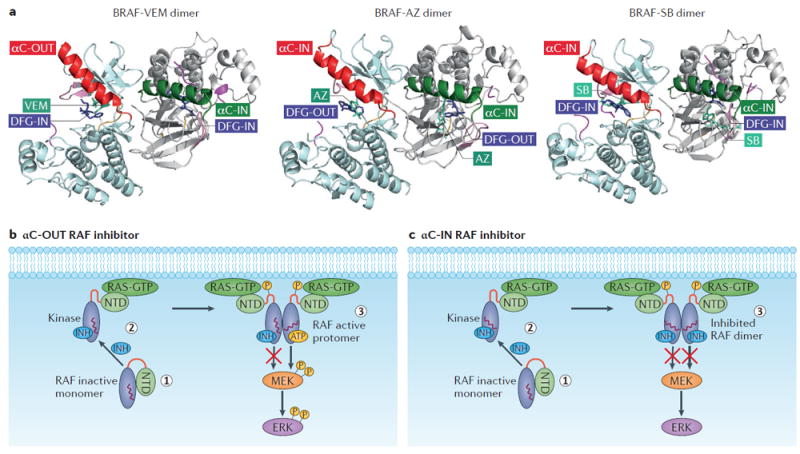
a ∣ Structural features of representative RAF inhibitors bound to a BRAF dimer. The three structural types of RAF inhibitor bound to BRAF are shown as ribbon representations: Vemurafenib (VEM), a type I1/2 inhibitor (αC-OUT/DFG-IN), AZ628 (AZ), a type II inhibitor (αC-IN/DFG-OUT), and SB590885 (SB), a type I inhibitor (αC-IN/DFG-IN). Vemurafenib and other type I1/2 inhibitors stabilize the αC-helix (red) of the first BRAF protomer to the OUT position and the αC-helix (green) of the second BRAF protomer to the IN position (Protein Data Bank (PDB) ID: 3OG7). AZ628 and other type II inhibitors stabilize the αC-helix of the first BRAF protomer (red) and the αC-helix of the second BRAF protomer (green) to the IN position (PDB ID: 4RZW). SB590885 and other type I inhibitors stabilize both the αC-helix of the first BRAF protomer (red) and the αC-helix of the second BRAF protomer (green) to the IN position (PDB ID: 2FB8). AZ628 and other type II inhibitors induce the DFG motif to an OUT conformation, while other inhibitors of the type I and I1/2 groups maintain the DFG in an IN conformation. b ∣ Schematic showing the αC-OUT RAF inhibitor-induced paradoxical RAF pathway activation and negative allostery. Most αC-OUT RAF inhibitors stabilize the αC-helix (indicated by the red wavy line in the kinase domain) in the overall OUT position (1). However, the C-terminal αC-helix residue R506 is displaced into the IN position, which promotes the interaction of RAF with RAS-GTP (2). Subsequently, RAF is primed through phosphorylation and dimerization. RAF inhibitor binding to one protomer and stabilization of the αC-helix in the OUT position sterically prevents inhibitor binding to the second monomer (negative allostery), leading to paradoxical pathway activity (3). c ∣ Schematic showing αC-IN RAF inhibitor-induced RAF activation and effective dimer inhibition. By stabilizing the αC-helix in the IN position (1), αC-IN RAF inhibitors promote the formation of the RAF–RAS–GTP complex (2). This is followed by RAF phosphorylation and dimerization. Due to low negative allostery, RAF inhibitors belonging to this group effectively bind and inhibit both protomers in the RAF dimer (3). INH, inhibitor; NTD, N-terminal domain.
Table 1.
Select RAF inhibitors and their stage of development
Linking structure to biochemical effects of RAF inhibitors in cells
The first studies focusing on the mechanisms underlying the unique biochemical properties of RAF inhibitors were published during the preclinical and clinical development of inhibitors targeting mutated BRAF47,56,57. Although not all studies concurred in their proposed models, they all agreed that paradoxical activation of ERK signalling by RAF inhibitors in WT BRAF-expressing cells required active RAS. However, the details of this mechanism remained elusive. Furthermore, in those and subsequent studies, RAF dimerization was shown to confer resistance to RAF inhibitors31,52, but the structural basis of the phenomenon remained unknown. More recently, detailed analysis of crystallographic data in combination with cell-based studies enabled the structural properties of RAF inhibitors to be linked to their biochemical effects in cells30,49 (FIG. 2b,c). Specifically, the biochemical effect of RAF inhibitors is the outcome of distinct allosteric mechanisms, determined by the combination of the specific conformation in which the RAF kinase is stabilized by the inhibitor and by the cellular levels of RAS-GTP.
First, resistance of dimeric RAF to inhibitors is the consequence of stabilization of the αC-helix in the OUT (inactive) position by the inhibitor, because this position of the αC-helix is not sterically allowed for both protomers in a RAF dimer. Thus, binding of an αC-OUT inhibitor to the first protomer stabilizes the position of the αC-helix of the second protomer towards the IN position, reducing the affinity of a second αC-OUT inhibitor for the second protomer (negative allostery). Such inhibitors are predicted to be ineffective inhibitors of dimeric RAF30 (FIG. 2b). Certain αC-OUT RAF inhibitors, such as dabrafenib, stabilize the αC-helix in a less OUT position compared to vemurafenib in both protomers, a position that allows the inhibitor to bind both protomers, as seen in the crystal structure with BRAF kinase. However, this might be an effect of crystallization forcing the two protomers into a ‘distorted’ dimer. In contrast, αC-IN RAF inhibitors bind both protomers of dimeric RAF with similar affinity and are thus predicted to be equipotent inhibitors of monomeric and dimeric RAF30 (FIG. 2c). Because CRAF signals as an obligatory dimer50,65, αC-OUT inhibitors fail to suppress its activity. Thus, αC-IN RAF inhibitors, by suppressing both BRAF and CRAF dimers can also be considered ‘pan-RAF inhibitors’66-68 and may be effective when used in combination in contexts such as RAS-mutant tumour cells, in which CRAF is known to be a major activator of ERK signalling downstream of RAS. However, a potentially narrow therapeutic window and allosteric priming (see below) are predicted to limit the effectiveness of αC-IN RAF inhibitor monotherapy in RAS-mutant tumours30. Although the model of negative allostery due to the OUT position of the αC-helix is mostly inferred based on structural and cell-based data and remains to be shown directly by biophysical methods, it helps explain why αC-OUT inhibitors are selectively effective in cellular contexts in which RAF signals as a monomer (that is, BRAF-V600 tumour cells with low RAS-GTP) and why increased RAF dimerization is a common mechanism of resistance to these drugs30.
Second, the basis for priming and RAS-dependent dimerization of RAF promoted by RAF inhibitors is the stabilization of the αC-helix towards the IN (active) position. αC-IN RAF inhibitor binding to RAF promotes the interaction of RAF with RAS-GTP, increasing activating phosphorylation and RAF dimerization, and essentially ‘hijacking’ the process of canonical growth factor-induced RAF activation30. The mechanism is reminiscent of allosteric priming by inhibitor, which has been observed with other kinase inhibitors (such as those targeting AKT69, protein kinase C (PKC)70, ribosomal 6-kinase 1 (S6K1)71 and Janus kinase 2 (JAK2)72), although in these cases virtually all the primed and phosphorylated kinase molecules are drug-bound and inhibited. Analysis of RAF priming by various inhibitors, led to the hypothesis that it is the area around a specific amino acid of the αC-helix (R506) and not the overall position of the αC-helix that determines the extent of the RAF–RAS-GTP interaction induced by the inhibitor30. According to this model, αC-IN inhibitors, such as GDC0879 or SB590885, displace R506 from the OUT position and thus promote RAF–RAS-GTP interaction30. Importantly, dabrafenib and to a lesser extent vemurafenib, although they stabilize the αC-helix in an overall OUT position, also promote RAF–RAS-GTP interaction and RAF dimerization due to displacement of the R506 residue towards the IN position30. Members of a certain class of next-generation inhibitors (such as PLX7904 and PLX8394; see below for more details) stabilize the entire αC-helix, including the R506 residue, in the OUT position and thus do not promote paradoxical activation of ERK signalling in cells30,73. Although further experimental data are required to substantiate this hypothesis, it may explain the requirement of RAS-GTP for paradoxical activation of ERK signalling by inhibitors and the concurrent finding of mutant-RAS expression in the squamous-cell carcinomas and keratoacanthomas, commonly induced as second-site tumours in patients with melanoma treated with second-generation RAF inhibitors74.
Based on this model, paradoxical activation of ERK signalling by RAF inhibitors is the consequence of inhibitor binding to RAF, which in turn promotes RAF binding to RAS-GTP and results in an increase in active RAF dimers. Overall, RAF dimers are ineffectively inhibited by RAF inhibitors due to negative allostery for binding to the second protomer (FIG. 2b). αC-IN RAF inhibitors promote the RAS–RAF interaction at lower concentrations and effectively suppress ERK signalling at moderate and high concentrations, due to lower negative allostery (FIG. 2c). In contrast, αC-OUT inhibitors promote the RAS–RAF interaction at moderate and high concentrations and fail to suppress ERK signalling at these concentrations due to higher negative allostery30.
Finally, there are data indicating that the position of the αC-helix and the DFG motif affect the interaction of RAF with MEK. αC-OUT RAF inhibitors (for example, vemurafenib or dabrafenib) tend to disrupt the RAF–MEK complex. Among αC-IN RAF inhibitors, those stabilizing a conformation closer to the native active kinase (αC-IN/DFG-IN, for example, GDC0879 or SB590885) allosterically promote the formation of the RAF–MEK complex and thus display more pronounced paradoxical ERK signalling activation than do αC-IN/DFG-OUT inhibitors30,55.
It should be emphasized that despite the major contributions by a number of groups to our understanding of the complex mechanism of action of RAF inhibitors, more work is required to further elucidate the highly dynamic structural rearrangement that RAF proteins undergo upon inhibitor binding. Adding to this complexity is the variation in the details of the mechanism of action, even among inhibitors of the same class, and for example, as a result of the mutational status of the RAF proteins, the cell-specific stoichiometry of the various RAF interacting proteins and the levels of RAS activity.
Nonetheless, the mechanism of action of RAF inhibitors has important clinical implications. First, αC-OUT RAF inhibitors are predicted to be effective selectively against monomeric forms of RAF, but any mechanism of RAF dimerization will cause drug resistance, a notion that has been confirmed both in the laboratory and in the clinic30,31,62,75. Second, αC-IN RAF inhibitors, by inhibiting both monomeric and dimeric RAF to a similar extent, will overcome resistance due to RAF dimerization. However, they are predicted to have a fairly narrow therapeutic window, given that they will suppress ERK signalling in normal, WT BRAF-expressing cells at concentrations similar to those used to suppress ERK signalling in tumour cells, as suggested by preclinical data30.
Activation of mutant BRAF
Linking BRAF mutational alterations to predicted RAF inhibitor response
A critical breakthrough in elucidating the mechanism of action of RAF inhibitors was the discovery that, unlike WT BRAF that requires dimerization for its catalytic activation, mutant BRAF-V600E is able to signal as an active monomer in the absence of RAS-GTP30,31,51,52,56. These studies were carried out using ectopically expressed proteins and mutational analysis; thus, the cellular status and the composition of endogenous BRAF-V600E containing complexes have yet to be unequivocally determined53,76. Nonetheless, the basis for the wide clinical therapeutic window of the current clinical αC-OUT RAF inhibitors vemurafenib and dabrafenib is believed to be their relative inability to inhibit dimeric RAF in normal cells, while potently inhibiting mutant BRAF in tumour cells.
Recent work has revealed important differences in the mechanism by which BRAF mutations other than BRAF-V600E promote catalytic activation of BRAF (FIG. 3). First, similar to BRAF-V600E, other BRAF-V600 point mutant forms, such as V600K, V600D and V600R, are also catalytically active monomers in the absence of RAS-GTP and are sensitive to inhibition by current clinical αC-OUT RAF inhibitors30,31. By contrast, the catalytic activity of non-V600 BRAF point mutants depends either partially or fully on dimerization30,31,51. Thus, non-V600 BRAF mutants, including BRAF-L597V, BRAF-K601E and BRAF-G466E, signal as RAS-GTP-independent RAF dimers by spontaneously dimerizing, and they are thus resistant to vemurafenib and other αC-OUT inhibitors11,30,31,51. However, this has been shown only in cell-based experiments so far30,31,51. The structural basis for the difference between RAF inhibitor activity upon V600 and non-V600 BRAF point mutants will presumably require biochemical and crystallographic studies including the N-terminal domain of BRAF or at least the part of the N-terminal domain that regulates RAF dimerization. Nonetheless, as with other forms of dimeric RAF, αC-OUT RAF inhibitors are not predicted to be effective in tumours with non-V600 mutant BRAF. Equipotent inhibition of monomeric and dimeric RAF with αC-IN RAF inhibitors may be a promising alternative for this class of tumours30,31.
Figure 3. Categories of RAF alterations found in tumours and the types of RAF inhibitor predicted to be effective against them.
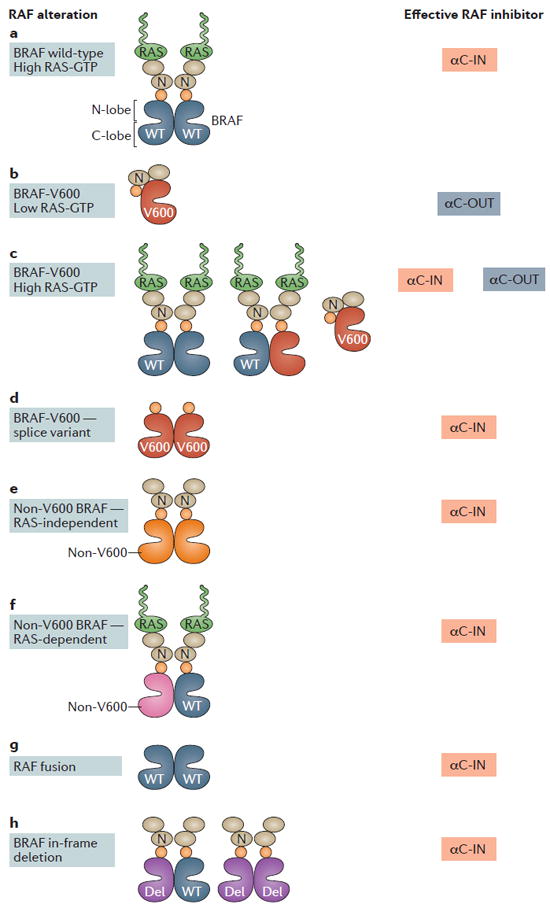
a ∣ In the cellular context of RAS-activated wild-type (WT) BRAF, BRAF, CRAF and ARAF form active homodimers and heterodimers that are resistant to αC-OUT RAF inhibitors. αC-IN RAF inhibitors are predicted to potently inhibit RAF dimers, although their effectiveness will be limited due to concurrent formation of active RAF dimers by the inhibitor. b ∣ Mutant BRAF-V600 proteins in the absence of RAS-GTP signal as a monomer and are potently inhibited by either αC-OUT or αC-IN inhibitors. In this context, αC-OUT inhibitors are predicted to be more effective treatments due to their wide therapeutic window. c ∣ In the context of tumours expressing BRAF-V600 in the presence of active RAS, ERK is activated by both BRAF-V600E monomers and RAF dimers (homodimers and heterodimers of BRAF-V600E, WT BRAF, CRAF and ARAF). A combination of a αC-OUT inhibitor with a αC-IN RAF inhibitor is predicted to result in effective RAF inhibition in the tumour while retaining a wide therapeutic window. d ∣ A BRAF-V600E splice variant that lacks part of the N-terminal domain and constitutively dimerizes has been identified in tumours that have developed clinical resistance to αC-OUT RAF inhibitors. In this context, ERK is activated by BRAF-V600E homodimers, and αC-IN RAF inhibitors may be an effective therapeutic option. e ∣ For tumours expressing mutant BRAF proteins other than BRAF-V600 that signal as RAS-independent dimers, αC-IN RAF inhibitors may be an effective therapeutic option. f ∣ For tumours expressing mutant BRAF proteins other than V600 that require RAS activity, αC-IN RAF inhibitors may be an effective therapeutic option. g ∣ Fusion transcripts of various genes with the catalytic domain of either WT BRAF or, in some cases, CRAF, lack the N-terminal domain that prevents RAF dimerization, and their protein products thus signal as RAS-independent RAF dimers. For patients whose tumours depend on such fusion proteins, αC-IN RAF inhibitors may be an effective therapeutic option. h ∣ In-frame deletions in BRAF that stabilize the αC-helix in the active (IN) position have been found in tumours. For patients with such tumours, αC-IN RAF inhibitors may be an effective therapeutic option. C-lobe, C-terminal lobe; N, N-terminal domain; N-lobe, N-terminal lobe; Del, in-frame deletion; αC-IN, αC-helix-IN RAF inhibitor; αC-OUT, αC-helix-OUT RAF inhibitor.
In addition to point mutations, BRAF (and more rarely CRAF) is found as part of fusion transcripts and proteins caused by translocations77. Such fusions are commonly found in paediatric gliomas78 but also in other solid tumours, including lung cancers, advanced prostate cancers, gastric cancers, pancreatic cancers and melanoma77,79. Typically, translation of the fusion transcript results in the expression of RAF proteins either as the intact kinase domain only or as the kinase domain fused with a partner protein that has replaced the N-terminal domain of RAF77. Deletion of the N-terminal domain of RAF has been shown to promote RAF activation by constitutive dimerization44,52; thus, RAF fusion proteins are predicted to be resistant to αC-OUT RAF inhibitors. As αC-IN RAF inhibitors potently suppress dimeric RAF, they may be a therapeutic option in this context.
In-frame deletions in BRAF, such as L485–P490-deleted BRAF and N486–P490-deleted BRAF, have also been found in a small percentage of certain tumours, with the highest prevalence in pancreatic carcinomas (4.21%), as well as in lung, ovarian and thyroid tumours80. BRAF in-frame deletions do not overlap with KRAS or other BRAF mutations80, suggesting that they are sufficient to drive ERK signalling hyperactivation in these tumours. The deletions are mapped within the conserved β3/αC-helix loop of the kinase domain of BRAF, and they are predicted to activate the kinase by shortening the β3/αC-helix loop in a fashion analogous to that of in-frame deletions in epidermal growth factor receptor (EGFR) and HER2 (also known as ERBB2), also found in tumours12. This shortening of the β3/αC-helix loop results in a unique conformation that locks the αC-helix in the IN position through the formation of an intact K483-to-E501 salt bridge. Two recent studies focused on the mechanism of activation of in-frame BRAF deletions and their response to RAF inhibitors12,80. Chen et al.80 proposed that the increased activity of these genetic alterations is caused by increased BRAF homodimerization. Therefore, BRAF proteins carrying in-frame deletions are maintained in the active state that favours BRAF homodimerization and were shown to be resistant to vemurafenib but sensitive to the next-generation αC-IN RAF dimer inhibitor LY3009120 (REF. 80) (see below for more detail). By contrast, Foster et al.12 suggested that the activity of BRAF deletions is independent of RAF dimerization and that these in-frame-deletion mutants can signal as monomers, similar to BRAF-V600E. Structural analysis revealed that the αC-helix-IN active conformation of the kinase domain of BRAF harbouring a deletion, renders binding of vemurafenib incompatible due to steric effects12. Although the two studies differ on the proposed mechanism of BRAF activation by in-frame deletions, they both conclude that tumours expressing BRAF deletions will be resistant to current clinical RAF inhibitors and likely to respond to equipotent inhibitors of monomeric and dimeric RAF12,80.
Finally, there are a class of point mutations found in either the activation segment or the glycine-rich loop of BRAF that are not activating but that instead result in a protein that either is catalytically inactive or shows severely impaired activity compared to WT BRAF10,11,57. These forms of mutant BRAF with impaired activity have been found mainly in colorectal, melanoma and lung tumours81, require RAS activity and dimerization to hyperactivate ERK signalling and transform cells11,31,57 and are commonly co-expressed with activated receptor tyrosine kinases (RTKs) or upstream alterations of RAF signalling, such as RAS mutations or neurofibromin 1 (NF1) loss82,83. As these mutant BRAF proteins form dimers, tumours harbouring them are predicted to be insensitive to current clinical αC-OUT RAF inhibitors, but αC-IN RAF inhibitors may be an effective option.
Mechanisms of resistance to RAF inhibitors
The notion that second-generation RAF inhibitors do not effectively suppress dimeric RAF was emphatically confirmed with the discovery of increased RAF dimerization as a common cause of clinical acquired resistance to RAF inhibitors30,31,52,75,84. Various molecular mechanisms have been identified that confer this clinical resistance to second-generation RAF inhibitors. RAS activation75, either by RAS mutation or by overexpression of upstream RTKs75,85, promotes RAF dimerization as well as activation of parallel survival pathways, such as PI3K–AKT signalling. BRAF-V600E amplification has been found in a subset of resistant melanoma tumours28,84, with the resulting overexpression of the BRAF-V600E protein predicted to promote resistance to RAF inhibitors by both increasing abundance of the target (that is, BRAF-V600E) and spontaneous BRAF-V600E dimerization31. Another common mechanism of clinical resistance due to increased BRAF-V600E dimerization is the expression of splice variants of BRAF-V600E that lack exons encoding part of the N terminus of the protein, specifically those including the RBD, which prevents dimerization in the context of full-length BRAF52,86. Finally, mutational activation of MEK has also been reported in a subset of BRAF-V600E tumours that developed resistance to RAF inhibitors87,88.
In addition to acquired resistance, increased RAF dimerization may confer an adaptive response to RAF inhibitor therapy. In BRAF-V600E-expressing tumour cells, any increase in RAS activity due to upstream activation will cause formation of RAF dimers, including homodimers and heterodimers of WT and BRAF-V600E, CRAF and BRAF-V600E and ARAF and BRAF-V600E, thus hindering the effect of RAF inhibitors30,89. A number of studies have proposed mechanisms by which tumours may adapt to RAF inhibitor therapy via upstream RTK and RAS activation, either as a result of relief of negative feedback37-39,89 or due to cytokine and growth factor release from the tumour microenvironment90-92 (FIG. 4).
Figure 4. The interplay of mechanisms of adaptive response to RAF inhibitors.
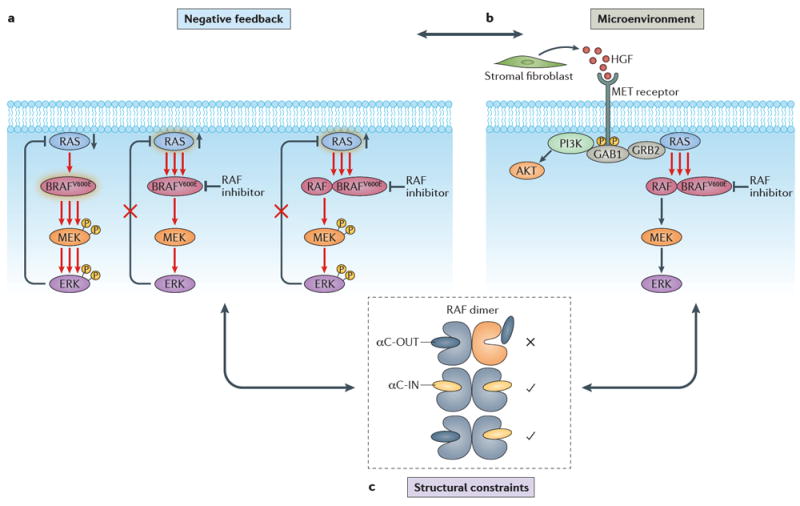
a ∣ Relief of negative feedback. At steady state, hyperactivated ERK downstream of mutant BRAF suppresses RAS due to negative feedback. Addition of a RAF inhibitor results in relief of negative feedback and RAS activation, which in turn promotes increased flux through ERK signalling and RAF dimerization41,73,74. Activation indicated by a green halo around the protein. b ∣ Pathway reactivation due to paracrine signalling by components of the tumour microenvironment (TME). Stromal cells in the TME release growth factors (exemplified here by hepatocyte growth factor (HGF)) and cytokines, which promote resistance to RAF inhibitors by activating parallel survival pathways, such as PI3K signalling, or by activating RAS75-77. c ∣ Structural constraints. RAF dimerization promotes an adaptive response to αC-OUT RAF inhibitors due to negative allostery for inhibitor binding to the second protomer of a RAF dimer when the first is occupied by inhibitor. αC-IN RAF inhibitors are predicted to overcome this resistance mechanism by binding with similar affinity to both protomers of the RAF dimer41. All of these mechanisms of adaptive response are interconnected and converge upon structural constraints for inhibitor binding. Relief of negative feedback promotes upstream receptor tyrosine kinase activation, which may be further enhanced by growth factors and cytokines released in the TME. Both mechanisms promote increased flux in the pathway and levels of active RAF molecules. They also promote RAF dimerization, which prevents effective RAF inhibition by current clinical αC-OUT RAF inhibitors due to negative allostery. GAB1, growth factor receptor-bound protein 2-associated binding protein 1; GRB2, growth factor receptor-bound protein 2.
In tumour types other than melanomas harbouring BRAF-V600E mutations, such as in colorectal and thyroid cancers, the response rates to second-generation RAF inhibitors are low93,94. In these tumours, increased upstream RTK expression and activity at baseline compared to melanoma (mostly through EGFR in colorectal cancers and HER2–HER3 (also known as ERBB3) signalling in thyroid cancers) has been reported to account for the unexpected unresponsiveness37-39. Consequently, feedback reactivation of upstream RTKs and RAS upon RAF inhibitor treatment is more pronounced in these tumours, profoundly enhancing RAF dimerization and thus limiting the effect of αC-OUT inhibitors30.
Development of RAF inhibitors
First-generation (αC-IN) RAF inhibitors
The first small-molecule ATP-competitive RAF inhibitors were reported before the discovery of BRAF mutations and were developed with the goal of targeting CRAF in cancer95. The compound ZM336372 (likely to be αC-IN/DFG-IN) was identified from a chemical screen as both a CRAF and BRAF inhibitor in vitro and was associated with the first report of the phenomenon of paradoxical ERK activation induced by a RAF inhibitor in cells95. Sorafenib (Nexavar, BAY43-9006; Bayer/Onyx Pharmaceuticals) (αC-IN/DFG-OUT) is the only first-generation RAF inhibitor that advanced to the clinic96 (and received FDA approval for the treatment of advanced renal cell carcinoma and hepatocellular carcinoma97,98). Sorafenib shows weak activity against BRAF-V600E in cells, and its clinical activity in the aforementioned tumours with WT BRAF is likely due to its multi-kinase profile. Other first-generation RAF inhibitors that did not advance to the clinic include SB590885 (REF. 99) and GDC0879 (REF. 100) (both αC-IN/DFG-IN) and GW5074 (REFS 101,102) and L779450 (REF. 87) (there are currently no crystal structures for GW5074 and L779450, but it is likely they are αC-IN/DFG-IN).
Second-generation, BRAF-V600E-selective (αC-OUT) RAF inhibitors
The discovery of BRAF mutations in 2002 (REF. 9) renewed interest in developing RAF inhibitors, leading to second-generation compounds, which were identified after screening for inhibitors of BRAF-V600E48. The first such RAF inhibitor to enter clinical trials was vemurafenib (Zelboraf, PLX4032; Plexxikon/Genentech), developed by a structure-guided discovery approach23,48. Vemurafenib was approved by the FDA in 2011 for the treatment of patients with BRAF-V600E metastatic melanoma22,33, followed by dabrafenib (Tafinlar, GSK2118436; GlaxoSmithKline), the second RAF inhibitor to receive approval by the FDA in 2013 for the treatment of melanoma patients with BRAF-V600E/K mutations21,103. The two inhibitors showed similar clinical efficacy and induced similar on-target effects related to paradoxical activation of ERK signalling in normal cells. Approximately 14–26% of patients with melanoma treated with second-generation RAF inhibitors were diagnosed with the secondary cancers keratoacanthomas and squamous-cell carcinomas, which can be treated by simple excision74. These second- site cancers are commonly associated with expression of a RAS mutation and have been almost eliminated with the combination of a RAF and a MEK inhibi tor, which is the current standard of care (see below). Other side effects, such as photosensitivity caused by vemurafenib22 and fever caused by dabrafenib21, appear to be more drug-specific, presumably related to different off-target effects of the two drugs.
LGX818 (Encorafenib, Array BioPharma) is another RAF inhibitor selective for mutant BRAF that has shown promising clinical activity and is currently in phase III clinical trials104. Preclinically, the cellular profile of the compound is similar to other αC-OUT inhibitors. It has been reported to show longer residence time (lower offrate) compared to vemurafenib or dabrafenib31, a property that may positively affect its clinical profile by prolonging target inhibition.
Third-generation RAF inhibitors
Third-generation RAF inhibitors, which are currently under preclinical and clinical development, fall structurally and biochemically into two main classes (TABLE 1). One group includes compounds that equipotently inhibit the monomeric and dimeric forms of RAF and are thus expected to overcome resistance due to RAF dimerization. Among them are TAK632 (REF. 105) and MLN2480 (TAK580)106 (Takeda/Millennium), LY3009120 (REF. 68) (Eli Lilly) and CCT3833 (BAL3833) (REF. 107) (ICR/Royal Marsden NHS Foundation Trust). Crystallographic data obtained with TAK632 or LY3009120 bound to BRAF show a type II conformation (αC-IN/DFG-OUT), which explains the similar affinity for both protomers in the RAF dimer, as well as the generally slow off-rate of these compounds67,68,105.
In preclinical studies, TAK632 demonstrated potent antiproliferative activities in multiple cancer cell lines harbouring BRAF, NRAS or KRAS mutations with the potential to delay the emergence of resistance30,67. The co-crystal structure of TAK632 bound to WT BRAF revealed compound occupancy and similar conformations of both protomers of the BRAF dimer105. TAK632 exhibited equipotent inhibition of monomeric and dimeric RAF in cells30,67 and is expected to be effective in vemurafenib-resistant melanomas harbouring BRAF or NRAS mutations and BRAF-V600E colorectal or thyroid tumours67. MLN2480, which has characteristics similar to TAK632, is currently in a phase I clinical trial for patients with melanomas and other solid tumours108. MLN2480 will also be evaluated in a phase I clinical trial in combination with the programmed cell death 1 (PD1) monoclonal antibody (nivolumab) for patients with melanoma109. Finally, a phase I clinical trial of MLN2480 in combination with inhibitors targeting other families of kinases (MLN0128, an mTOR complex 1 (mTORC1) and mTORC2 inhibitor, and alisertib, an Aurora kinase A inhibitor), chemotherapy regimens (paclitaxel and irinotecan) or the EGFR monoclonal antibody (cetuximab) will be conducted for patients with advanced solid malignancies110.
LY3009120 is another αC-IN/DFG-OUT RAF inhibitor that showed antitumour activity in preclinical studies against NRAS or KRAS mutant tumours and vemurafenib-resistant melanomas68,111. LY3009120 binds to both RAF protomers within the BRAF dimer, stabilizing them in similar conformations, and, like other αC-IN RAF inhibitors, it inhibits monomeric and dimeric BRAF with similar potency30,41,93. LY3009120 was also effective against BRAF deletions, including BRAF-N486–P490 and other in-frame BRAF deletions that have been identified in pancreatic and thyroid tumours80. Currently, LY3009120 is in a phase I clinical trial for patients with advanced cancers112.
A compound from BeiGene, BGB659, showed properties of equipotent inhibitors of monomeric and dimeric RAF31. In preclinical studies, BGB659 was effective in melanomas with V600E and non-V600 BRAF mutations, in vemurafenib-resistant BRAF-V600E melanomas, as well as against ectopically expressed BRAF fusions31. A related compound, BGB283, acts as a dual RAF and EGFR inhibitor113. In preclinical studies, it suppressed the proliferation of cells harbouring BRAF-V600E or EGFR mutations and inhibited EGFR reactivation in BRAF-V600E colorectal tumours. A phase I clinical trial of BGB283 for patients with solid tumours is ongoing114.
CCT196969 and CCT241161 have been reported as dual pan-RAF and SRC kinase inhibitors66. Although crystallographic data are not available, these compounds behave biochemically in a similar manner to αC-IN RAF inhibitors. In preclinical studies, both CCT196969 and CCT241161 inhibited the growth of BRAF-V600E melanomas as well as RAS-mutant melanomas and colorectal tumours66. CCT196969 and CCT241161 were also effective in patient-derived xenograft (PDX) models that included melanomas with intrinsic or acquired resistance to second-generation RAF and MEK inhibitors66. The study of the safety and tolerability of the lead compound of this class of inhibitors, CCT3833 (BAL3833), is ongoing in a phase I clinical trial for patients with advanced solid tumours107. Melanoma patients harbouring BRAF-V600E (either treatment-naive or after progression on RAF inhibitor therapy) or RAS mutations are also included in the trial. Finally, LXH254 and RAF709, both currently under development by Novartis are also equipotent inhibitors of monomeric and dimeric RAF. An ongoing phase I clinical trial is recruiting patients with tumours with ERK signalling alterations to be treated with LXH254 (REF. 115).
The other group of third-generation RAF inhibitors includes the ‘paradox breakers’, which are αC-OUT RAF-inhibitor derivatives with variable terminal sulfonamide and sulfamide substitutions. Both PLX7904 and its analogue PLX8394 (Plexxikon) are more potent inhibitors of BRAF-V600E than vemurafenib, and unlike vemurafenib, they do not promote paradoxical ERK activation in WT BRAF cells or squamous-cell carcinomas in preclinical models73. These compounds stabilize the αC-helix of BRAF in the OUT position, and structural analysis showed that the position of the R506 residue of the αC-helix with PLX7904 has the furthest OUT position compared to that with non-paradox breakers30,73. Thus, the ‘paradox breakers’ are predicted to have fewer on-target toxicities than second-generation RAF inhibitors and may be an effective option for chronic administration in more indolent neoplasias, such as in LCH patients with tumours expressing BRAF-V600E19. PLX8394 is in a phase I/II clinical trial for the treatment of patients with advanced unresectable solid tumours116. Preclinical studies using ‘paradox breakers’ have also suggested that they will be effective against vemurafenib-resistant tumours and BRAF fusions that are common in paediatric astrocytomas, presumably due to their higher potency against RAF when compared with vemurafenib117-119. However, as they are αC-OUT inhibitors, RAF dimerization is predicted to limit the effectiveness of these compounds.
Finally, BI882370 is an αC-OUT RAF inhibitor under development by Boehringer Ingelheim that showed promising preclinical activity against BRAF-mutant melanoma and colorectal tumours, both in cell line models and in vivo120. This compound differs from other αC-OUT RAF inhibitors by stabilizing a conformation in which it interacts with the Phe595 from the DFG motif and keeps the DFG motif in an inactive (OUT) position120. However, further studies are required to determine potential functional consequences of these differences.
Currently in clinical trials are additional RAF inhibitors that cannot yet be classified due to a lack of structural data, including HM95573 (Hanmi Pharmaceutical/Genentech) and CEP32496 (RXDX-105, Ambit Biosciences/Ignyta). HM95573 is currently in an expansion clinical trial for solid tumours harbouring mutations in either BRAF, KRAS or NRAS genes121,122. CEP32496 (REF. 123) is a quinazoline BRAF inhibitor that has demonstrated multikinase activity against BRAF-V600E, BCR-ABL, RET and ephrin type A receptor 2 (EPHA2) and is reported to be effective in preclinical studies of melanomas and colorectal tumours harbouring BRAF-V600E123. CEP32496 will be evaluated in a phase I clinical trial for the treatment of patients with solid tumours124.
Based on preclinical data, clinical efficacy of next-generation RAF inhibitors may also be limited by either on-target toxicities or development of drug resistance. Third-generation αC-OUT RAF inhibitors (such as the ‘paradox breakers’) are expected to retain the wide therapeutic window of second-generation αC-OUT RAF inhibitors, without the on-target toxicities of second-site tumours due to paradoxical ERK activation in normal cells. However, RAF dimerization is predicted to limit the effectiveness of these inhibitors. In contrast, αC-IN RAF inhibitors are predicted to overcome resistance due to RAF dimerization, but resistance to these compounds is anticipated to be conferred by other mechanisms, such as other mutations in BRAF or alterations that would enable cell proliferation and survival in the absence of ERK signalling. However, allosteric priming and a fairly narrow therapeutic window due to inhibition of ERK signalling in normal cells at lower drug concentrations is expected to limit the effectiveness of αC-IN RAF inhibitors. Therapeutic strategies combining structurally complementary RAF inhibitors with inhibitors of downstream components of ERK signalling (MEK and ERK inhibitors) may enable sufficient inhibition of ERK signalling in the tumour and thus provide durable responses and perhaps even cures for patients with BRAF-mutant tumours.
Combinatorial therapeutic strategies
As in both preclinical models and in patients, potency of ERK inhibition in the tumour was found to correlate with response to RAF inhibitor treatment23, RAF inhibitors were combined with MEK inhibitors in melanoma patients with BRAF-V600E tumours in order to achieve more potent and durable inhibition of ERK signalling in the tumour24,25. RAF–MEK inhibitor combinations demonstrated increased progression-free survival (PFS) and objective responses compared to RAF inhibitor monotherapy24,25. Moreover, the cutaneous toxicities induced by RAF inhibitors were dramatically decreased in the combination, further confirming that they were the result of paradoxical activation of ERK signalling in normal tissue. The dabrafenib and trametinib combination24 (Novartis) increased the response rate to 76% and the median duration of response to 10.5 months as compared with a 54% response rate and 5.8 months duration of response with dabrafenib alone, and was approved by the FDA in 2014. The combination of vemurafenib and cobimetinib25 (Roche/Genentech) demonstrated similar responses (response rate 68% and duration of response 9.9 months as compared with response rate of 45% and median duration of response of 6.2 months in patients treated with vemurafenib alone) and was also approved by the FDA in 2015. Given their superior efficacy over RAF inhibitor monotherapies, these RAF–MEK inhibitor combinations are currently the standard of practice for the treatment of melanoma patients with BRAF-V600E/K mutations.
Clinical trials of drug combinations including the second-generation RAF inhibitor LGX818 are currently underway: one with the MEK inhibitor MEK162 (binimetinib, Array BioPharma) in patients with BRAF-V600E melanomas (COLUMBUS)104, and another in combination with MEK162 and the EGFR antibody cetuximab in patients with colorectal BRAF-V600E tumours (BEACON CRC)125. Preliminary results from the COLUMBUS trial presented on the Array BioPharma website indicate significant responses, including increased median PFS and improvement in overall response rate compared with vemurafenib monotherapy.
Despite the improvement of clinical outcome, responses to RAF–MEK inhibitor combinations are variable, and resistance develops for the majority of patients treated with these regimens. Most mechanisms of resistance to RAF–MEK inhibitor combination are similar to the ones identified with RAF inhibitor monotherapy and are associated with recovery of ERK signalling in the presence of inhibitors27,126. This suggests that strategies that would more potently and durably suppress RAF–ERK signalling in the tumour, while retaining a wide therapeutic window, may yield even better responses and further forestall drug resistance. The wide therapeutic window of αC-OUT RAF inhibitors suggests that they will most likely remain components of any future combinatorial strategy for BRAF-V600E tumours.
A number of next-generation αC-IN RAF inhibitors are currently in clinical trials (TABLE 1). Such compounds are predicted to suppress dimeric RAF and thus be more effective in a broad range of RAF-dependent tumours, including tumours expressing non-V600 BRAF mutations, or perhaps RAS-mutant tumours. However, the narrow therapeutic window of these inhibitors may be a critical factor limiting their efficacy as single agents. As aforementioned, adding a modest dose of an αC-IN RAF inhibitor to the current standard αC-OUT–MEK inhibitor combination may be an effective strategy for patients with BRAF-V600E tumours30. Preclinical work supports the idea of including αC-IN RAF inhibitors in combination with αC-OUT RAF inhibitors (and also MEK or ERK inhibitors) to overcome feedback recovery and thus achieve durable suppression of ERK signalling30,127.
RAF inhibitors are also included in a number of trials that are undertaking a combinatorial approach to target parallel pathways (for example, RTKs, cyclin-dependent kinase 4 (CDK4) and CDK6 and PI3K) in addition to ERK signalling128-131. Although in principle, these strategies are usually based on strong preclinical rationale, additive on-target and off-target toxicities tend to result in lower dosing and thus limit the efficacy of the individual compounds in the clinic. Again, it is critical that the need for a wide therapeutic window be taken into account in the design of clinical trials assessing combinations of targeted agents.
Finally, a great deal of interest is being directed towards elucidating the combinatorial interaction of targeted therapy with immunotherapy, especially in melanoma treatment. Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and PD1 inhibitors have shown remarkable clinical efficacy and prolonged survival of patients with melanoma132-134 and are now FDA-approved for the treatment of metastatic melanoma135. Trials with concomitant administration of immune checkpoint inhibitors and RAF and MEK inhibitors have so far been discontinued due to unacceptable adverse effects, although it is not inconceivable that certain combinations and scheduling may be better tolerated than others. If simultaneous treatment is not an option, whether immunotherapy or targeted therapy should be the first-line treatment for patients with BRAF-V600E melanomas remains an open question, as there is reported evidence for cross-resistance136. In current clinical practice, targeted therapy is the preferred first-line therapy for rapidly growing tumours because it tends to yield faster remissions than immunotherapy, but a number of ongoing clinical trials are aimed at exploring this question more broadly137.
Challenges and future perspectives
Since the validation of BRAF-V600E as a drug target, as well as outcomes from sequencing projects highlighting the role of _BRAF_V600E and other BRAF mutations in several cancers, drug discovery and development of BRAF inhibitors has progressed at a fast pace. However, there is a pressing need for more effective RAF signalling inhibition that would overcome resistance due to mechanisms of BRAF dimerization. As our understanding of the structural and biochemical regulation of BRAF progresses, novel and more specific targeting strategies should be developed. Future efforts should explore the possibility for improving current ATP-competitive αC-OUT inhibitors to prevent negative allostery and bypass paradoxical activation. In addition, third-generation αC-IN inhibitors are more effective inhibitors of RAF dimers than are αC-OUT inhibitors, although their selectivity for mutant or WT BRAF dimers versus BRAF monomers is not optimal. Finally, one could envision the development of allosteric inhibitors that effectively inhibit the mutant or WT BRAF dimer and/or BRAF monomer but that do not promote the RAF priming that leads to paradoxical activation.
Key biochemical data suggest the importance of the BRAF–CRAF heterodimer in RAF oncogenic signalling56,57,65,138, although the structure of this heterodimer has not yet been determined. Nevertheless, peptides generated from the dimerization interface of BRAF have been shown to prevent formation of homodimers and heterodimers of BRAF–CRAF in a BRAF-mutant or RAS-mutant cellular context51, suggesting the possibility of an alternative strategy for developing inhibitors of RAF dimerization. Lastly, we mentioned that several BRAF mutations other than V600 can spontaneously induce BRAF dimerization through the kinase domain30,31,51. Structures of such BRAF mutants favouring dimer formation are not available and should provide further understanding about their mechanism of activation, as well as serve as templates for tailored drug design to these mutations.
Importantly, the structural basis of the interaction of the N-terminal regulatory domain and the kinase domain in the inactive conformation and the active conformation remains elusive. Such information would enable our understanding of the architectural organization of BRAF and the conformational transitions involved in the context of the full-length BRAF from its inactive monomer conformation to the active dimer and active monomer, particularly in the case of the BRAF-V600 mutant. The effects of the current inhibitors upon these inter-domain interactions in the active and inactive BRAF conformations are not well understood. Given the conformational changes in the kinase domain and dimerization process observed with the current BRAF inhibitors, it is likely that these same inhibitors will have an impact on inter-domain interactions. Such information will enable further improvement of the current inhibitors but will also provide unique opportunities for BRAF allosteric inhibitors outside of the catalytic pocket.
The oncoprotein BRAF is a validated therapeutic target in a large number of human tumours. Current clinical RAF inhibitors have improved survival of patients with melanoma whose tumours harbour BRAF mutations, but resistance limits their effectiveness in these patients, as well as in patients with BRAF-V600 tumours other than melanomas, or harbouring BRAF mutations other than V600. Our deeper understanding of the molecular and biochemical mechanisms of resistance to current clinical RAF inhibitors, combined with the development of next-generation RAF inhibitors with different structural and biochemical properties, will enable the design of innovative, potentially more effective, RAF-inhibitor-based therapeutic strategies tailored to the clinical context.
Supplementary Material
Video1
Video2
Acknowledgments
The authors are grateful to Bogos Agianian for generating the movies. E.G. is supported by NIH grant R01CA178394 and awards from the Melanoma Research Alliance, the Pershing Square Sohn Cancer Research Alliance, the Irma T. Hirschl Trust, the Gabrielle’s Angels Foundation for Cancer Research, and the Sidney Kimmel Foundation for Cancer Research. P.I.P. is supported by NIH grant R01CA204314, the Sidney Kimmel Foundation for Cancer Research, the Melanoma Research Foundation, the Dermatology Foundation and the Melanoma Research Alliance.
Glossary
Langerhans cell histiocytosis (LCH)
A myeloid neoplasia characterized by inflammatory lesions containing pathological dendritic cells, frequently harbouring the BRAF-V600E mutation.
Protomer
The structural unit of an oligomeric protein; in the case of wild-type BRAF, a functional BRAF dimer is composed of two protomers.
Steric clashes
When atoms from different residues come into close proximity, the resultant repulsion between the atoms leads to a change in conformation.
Negative allostery
Also known as allosteric inhibition, occurs when binding of one ligand to a substrate (in this case a BRAF protomer) decreases the affinity of another ligand at a different site (that is, the other protomer).
Therapeutic window
The range of concentrations of a drug in the patient that provides safe effective therapy. The therapeutic window is wide when the minimum toxic concentration is much higher than the minimum effective concentration.
Allosteric priming
A phenomenon recently observed with several small-molecule inhibitors, by which binding of the inhibitor induces the active conformation of the target kinase, which results in its increased interaction with upstream regulators and consequent kinase priming and activating phosphorylation.
Steric effects
A phenomenon arising as a result of the fact that each residue and its atoms occupy a certain amount of space in a defined conformation.
Residence time
The period for which the target kinase is occupied by the small-molecule inhibitor.
Off-rate
The rate of dissociation of the small-molecule inhibitor from the kinase.
Cross-resistance
The development of tumour resistance to a potential therapy after treatment of a patient with a different therapeutic agent.
Footnotes
Author contributions
Z.K., E.G. and P.I.P. contributed equally to researching the literature, to writing the article and to reviewing and editing the manuscript before submission.
Competing interests statement
The authors declare no competing interests.
SUPPLEMENTARY INFORMATION
See online article: S1 (movie) ∣ S2 (movie)
References
- 1.Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16:281–298. doi: 10.1038/nrm3979. [DOI] [PubMed] [Google Scholar]
- 2.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 3.Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 4.Matallanas D, et al. Raf family kinases: old dogs have learned new tricks. Genes Cancer. 2011;2:232–260. doi: 10.1177/1947601911407323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 6.Young A, et al. Ras signaling and therapies. Adv Cancer Res. 2009;102:1–17. doi: 10.1016/S0065-230X(09)02001-6. [DOI] [PubMed] [Google Scholar]
- 7.Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011;3:1787–1808. doi: 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer. 2015;15:577–592. doi: 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]
- 9.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 10.Wan PT, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. This paper provides the first crystal structure of BRAF-V600E bound to inhibitor, along with biochemical insight into the mechanism of BRAF activation by mutations. [DOI] [PubMed] [Google Scholar]
- 11.Garnett MJ, Rana S, Paterson H, Barford D, Marais R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell. 2005;20:963–969. doi: 10.1016/j.molcel.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 12.Foster SA, et al. Activation mechanism of oncogenic deletion mutations in BRAF, EGFR, and HER2. Cancer Cell. 2016;29:477–493. doi: 10.1016/j.ccell.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 13.Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14:455–467. doi: 10.1038/nrc3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimura ET, et al. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63:1454–1457. [PubMed] [Google Scholar]
- 16.Rajagopalan H, et al. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tiacci E, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364:2305–2315. doi: 10.1056/NEJMoa1014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Badalian-Very G, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919–1923. doi: 10.1182/blood-2010-04-279083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Solit DB, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–362. doi: 10.1038/nature04304. This paper provides preclinical evidence supporting the targeting of BRAF–ERK signalling in tumours harbouring the mutation BRAF-V600E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hauschild A, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 22.Chapman PB, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bollag G, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. This paper reports on the discovery and early clinical results of the first FDA-approved RAF inhibitor, vemurafenib. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long GV, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877–1888. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 25.Larkin J, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 26.Carlino MS, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther. 2013;12:1332–1342. doi: 10.1158/1535-7163.MCT-13-0011. [DOI] [PubMed] [Google Scholar]
- 27.Moriceau G, et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell. 2015;27:240–256. doi: 10.1016/j.ccell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Villanueva J, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013;4:1090–1099. doi: 10.1016/j.celrep.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang H, et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res. 2010;70:5518–5527. doi: 10.1158/0008-5472.CAN-10-0646. [DOI] [PubMed] [Google Scholar]
- 30.Karoulia Z, et al. An integrated model of RAF inhibitor action predicts inhibitor activity against oncogenic BRAF signaling. Cancer Cell. 2016;30:485–498. doi: 10.1016/j.ccell.2016.06.024. This study provides a unified mechanistic model of RAF inhibitor action and a roadmap for the clinical use of next-generation RAF inhibitors based on their structural properties. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yao Z, et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell. 2015;28:370–383. doi: 10.1016/j.ccell.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gentilcore G, et al. Effect of dabrafenib on melanoma cell lines harbouring the BRAF(V600D/R) mutations. BMC Cancer. 2013;13:17. doi: 10.1186/1471-2407-13-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flaherty KT, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McArthur GA, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323–332. doi: 10.1016/S1470-2045(14)70012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klein O, et al. BRAF inhibitor activity in V600R metastatic melanoma. Eur J Cancer. 2013;49:1073–1079. doi: 10.1016/j.ejca.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 36.Dahlman KB, et al. BRAF(L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov. 2012;2:791–797. doi: 10.1158/2159-8290.CD-12-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prahallad A, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 38.Corcoran RB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. This paper shows that insensitivity of colorectal BRAF-V600E tumours to RAF inhibitors is caused by relief of negative feedback and upstream activation of RTK signalling, most commonly through EGFR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montero-Conde C, et al. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013;3:520–533. doi: 10.1158/2159-8290.CD-12-0531. This paper shows that insensitivity of thyroid BRAF-V600E tumours to RAF inhibitors is caused by relief of negative feedback and upstream activation of RTK signalling, most commonly through HER3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warne PH, Viciana PR, Downward J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature. 1993;364:352–355. doi: 10.1038/364352a0. [DOI] [PubMed] [Google Scholar]
- 41.Cutler RE, Jr, Stephens RM, Saracino MR, Morrison DK. Autoregulation of the Raf-1 serine/threonine kinase. Proc Natl Acad Sci USA. 1998;95:9214–9219. doi: 10.1073/pnas.95.16.9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993;74:205–214. doi: 10.1016/0092-8674(93)90307-c. [DOI] [PubMed] [Google Scholar]
- 43.Moodie SA, Willumsen BM, Weber MJ, Wolfman A. Complexes of Ras. GTP with Raf-1 and mitogen-activated protein kinase kinase. Science. 1993;260:1658–1661. doi: 10.1126/science.8503013. [DOI] [PubMed] [Google Scholar]
- 44.Weber CK, Slupsky JR, Kalmes HA, Rapp UR. Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res. 2001;61:3595–3598. [PubMed] [Google Scholar]
- 45.Avruch J, et al. Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog Horm Res. 2001;56:127–155. doi: 10.1210/rp.56.1.127. [DOI] [PubMed] [Google Scholar]
- 46.Kornev AP, Taylor SS. Dynamics-driven allostery in protein kinases. Trends Biochem Sci. 2015;40:628–647. doi: 10.1016/j.tibs.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hatzivassiliou G, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. This study provides insight into the unique biochemical mechanism of RAF inhibitor action. [DOI] [PubMed] [Google Scholar]
- 48.Tsai J, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lavoie H, et al. Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat Chem Biol. 2013;9:428–436. doi: 10.1038/nchembio.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rajakulendran T, Sahmi M, Lefrancois M, Sicheri F, Therrien M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature. 2009;461:542–545. doi: 10.1038/nature08314. This paper provides mechanistic insight into the mechanism of RAF activation by dimerization. [DOI] [PubMed] [Google Scholar]
- 51.Freeman AK, Ritt DA, Morrison DK. Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol Cell. 2013;49:751–758. doi: 10.1016/j.molcel.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poulikakos PI, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. This paper provides information identifying the expression of splice variants of BRAF-V600E, which cause clinical resistance to RAF inhibitors by enhanced homodimerization, and confirming that RAF dimerization is a common mechanism of resistance to RAF inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roring M, et al. Distinct requirement for an intact dimer interface in wild-type, V600E and kinase-dead B-Raf signalling. EMBO J. 2012;31:2629–2647. doi: 10.1038/emboj.2012.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thevakumaran N, et al. Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat Struct Mol Biol. 2015;22:37–43. doi: 10.1038/nsmb.2924. [DOI] [PubMed] [Google Scholar]
- 55.Haling JR, et al. Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell. 2014;26:402–413. doi: 10.1016/j.ccr.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 56.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. This paper provides insight into the unique biochemical mechanism of RAF inhibitor action. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heidorn SJ, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. This paper shows that mutant BRAF with impaired kinase activity can cooperate with active RAS to promote tumour progression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Holderfield M, et al. RAF inhibitors activate the MAPK pathway by relieving inhibitory autophosphorylation. Cancer Cell. 2013;23:594–602. doi: 10.1016/j.ccr.2013.03.033. [DOI] [PubMed] [Google Scholar]
- 59.Halaban R, et al. PLX4032, a selective BRAF(V600E) kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF melanoma cells. Pigment Cell Melanoma Res. 2010;23:190–200. doi: 10.1111/j.1755-148X.2010.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Joseph EW, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci USA. 2010;107:14903–14908. doi: 10.1073/pnas.1008990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holderfield M, Nagel TE, Stuart DD. Mechanism and consequences of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640–645. doi: 10.1038/bjc.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poulikakos PI, Rosen N. Mutant BRAF melanomas — dependence and resistance. Cancer Cell. 2011;19:11–15. doi: 10.1016/j.ccr.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 63.Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13:928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 64.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 65.Hu J, et al. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell. 2013;154:1036–1046. doi: 10.1016/j.cell.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Girotti MR, et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell. 2015;27:85–96. doi: 10.1016/j.ccell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nakamura A, et al. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res. 2013;73:7043–7055. doi: 10.1158/0008-5472.CAN-13-1825. [DOI] [PubMed] [Google Scholar]
- 68.Peng SB, et al. Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell. 2015;28:384–398. doi: 10.1016/j.ccell.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 69.Okuzumi T, et al. Inhibitor hijacking of Akt activation. Nat Chem Biol. 2009;5:484–493. doi: 10.1038/nchembio.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cameron AJ, Escribano C, Saurin AT, Kostelecky B, Parker PJ. PKC maturation is promoted by nucleotide pocket occupation independently of intrinsic kinase activity. Nat Struct Mol Biol. 2009;16:624–630. doi: 10.1038/nsmb.1606. [DOI] [PubMed] [Google Scholar]
- 71.Pearce LR, et al. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1) Biochem J. 2010;431:245–255. doi: 10.1042/BJ20101024. [DOI] [PubMed] [Google Scholar]
- 72.Andraos R, et al. Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov. 2012;2:512–523. doi: 10.1158/2159-8290.CD-11-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang C, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526:583–586. doi: 10.1038/nature14982. [DOI] [PubMed] [Google Scholar]
- 74.Su F, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–215. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nazarian R, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. This paper provides evidence identifying RAS mutation and RTK upregulation as major mechanisms of acquired clinical resistance of BRAF-V600E melanomas to RAF inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Diedrich B, et al. Discrete cytosolic macromolecular BRAF complexes exhibit distinct activities and composition. EMBO J. 2017;36:646–663. doi: 10.15252/embj.201694732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ross JS, et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer. 2016;138:881–890. doi: 10.1002/ijc.29825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jones DT, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673–8677. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Palanisamy N, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med. 2010;16:793–798. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen SH, et al. Oncogenic BRAF deletions that function as homodimers and are sensitive to inhibition by RAF dimer inhibitor LY3009120. Cancer Discov. 2016;6:300–315. doi: 10.1158/2159-8290.CD-15-0896. [DOI] [PubMed] [Google Scholar]
- 81.Zheng G, et al. Clinical detection and categorization of uncommon and concomitant mutations involving BRAF. BMC Cancer. 2015;15:779. doi: 10.1186/s12885-015-1811-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nieto P, et al. A Braf kinase-inactive mutant induces lung adenocarcinoma. Nature. 2017;548:239–243. doi: 10.1038/nature23297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yao Z, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature. 2017;548:234–238. doi: 10.1038/nature23291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shi H, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Villanueva J, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shi H, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Emery CM, et al. BRAF-inhibitor associated MEK mutations increase RAF-dependent and-independent enzymatic activity. Mol Cancer Res. 2017 doi: 10.1158/1541-7786.MCR-17-0211. http://dx.doi.org/10.1158/1541-7786.MCR-17-0211. [DOI] [PubMed]
- 88.Wagle N, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–3096. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lito P, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–682. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Obenauf AC, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015;520:368–372. doi: 10.1038/nature14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Straussman R, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi: 10.1038/nature11183. This paper shows that adaptive response to RAF inhibitors is promoted by growth factors in the tumour microenvironment and upstream RTK activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wilson TR, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kopetz S, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33:4032–4038. doi: 10.1200/JCO.2015.63.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hyman DM, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373:726–736. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hall-Jackson CA, et al. Paradoxical activation of Raf by a novel Raf inhibitor. Chem Biol. 1999;6:559–568. doi: 10.1016/s1074-5521(99)80088-x. [DOI] [PubMed] [Google Scholar]
- 96.Lyons JF, Wilhelm S, Hibner B, Bollag G. Discovery of a novel Raf kinase inhibitor. Endocr Relat Cancer. 2001;8:219–225. doi: 10.1677/erc.0.0080219. [DOI] [PubMed] [Google Scholar]
- 97.Escudier B, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 98.Llovet JM, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 99.King AJ, et al. Demonstration of a genetic therapeutic index for tumors expressing oncogenic BRAF by the kinase inhibitor SB-590885. Cancer Res. 2006;66:11100–11105. doi: 10.1158/0008-5472.CAN-06-2554. [DOI] [PubMed] [Google Scholar]
- 100.Hoeflich KP, et al. Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res. 2009;69:3042–3051. doi: 10.1158/0008-5472.CAN-08-3563. [DOI] [PubMed] [Google Scholar]
- 101.Shelton JG, et al. Differential effects of kinase cascade inhibitors on neoplastic and cytokine-mediated cell proliferation. Leukemia. 2003;17:1765–1782. doi: 10.1038/sj.leu.2403052. [DOI] [PubMed] [Google Scholar]
- 102.Chin PC, et al. The c-Raf inhibitor GW5074 provides neuroprotection in vitro and in an animal model of neurodegeneration through a MEK-ERK and Akt-independent mechanism. J Neurochem. 2004;90:595–608. doi: 10.1111/j.1471-4159.2004.02530.x. [DOI] [PubMed] [Google Scholar]
- 103.Falchook GS, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379:1893–1901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT01909453.
- 105.Okaniwa M, et al. Discovery of a selective kinase inhibitor (TAK-632) targeting pan-RAF inhibition: design, synthesis, and biological evaluation of C-7-substituted 1,3-benzothiazole derivatives. J Med Chem. 2013;56:6478–6494. doi: 10.1021/jm400778d. [DOI] [PubMed] [Google Scholar]
- 106.Sun Y, et al. A brain-penetrant RAF dimer antagonist for the noncanonical BRAF oncoprotein of pediatric low-grade astrocytomas. Neuro Oncol. 2017;19:774–785. doi: 10.1093/neuonc/now261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT02437227.
- 108.US National Library of Medicine. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/ct2/show/NCT01425008.
- 109.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT02723006.
- 110.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT02327169.
- 111.Henry JR, et al. Discovery of 1-(3,3-dimethylbutyl)-3-(2-fluoro-4-methyl-5-(7-methyl-2-(methylamino) pyrido[2,3-d]pyrimidin-6-yl)phenyl)urea (LY3009120) as a pan-RAF inhibitor with minimal paradoxical activation and activity against BRAF or RAS mutant tumor cells. J Med Chem. 2015;58:4165–4179. doi: 10.1021/acs.jmedchem.5b00067. [DOI] [PubMed] [Google Scholar]
- 112.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT02014116.
- 113.Tang Z, et al. BGB-283, a novel RAF kinase and EGFR inhibitor, displays potent antitumor activity in BRAF-mutated colorectal cancers. Mol Cancer Ther. 2015;14:2187–2197. doi: 10.1158/1535-7163.MCT-15-0262. [DOI] [PubMed] [Google Scholar]
- 114.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT02610361.
- 115.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT02607813.
- 116.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT02428712.
- 117.Sievert AJ, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci USA. 2013;110:5957–5962. doi: 10.1073/pnas.1219232110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Le K, Blomain ES, Rodeck U, Aplin AE. Selective RAF inhibitor impairs ERK1/2 phosphorylation and growth in mutant NRAS, vemurafenib-resistant melanoma cells. Pigment Cell Melanoma Res. 2013;26:509–517. doi: 10.1111/pcmr.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Basile KJ, Le K, Hartsough EJ, Aplin AE. Inhibition of mutant BRAF splice variant signaling by next-generation, selective RAF inhibitors. Pigment Cell Melanoma Res. 2014;27:479–484. doi: 10.1111/pcmr.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Waizenegger IC, et al. A novel RAF kinase inhibitor with DFG-out-binding mode: high efficacy in BRAF-mutant tumor xenograft models in the absence of normal tissue hyperproliferation. Mol Cancer Ther. 2016;15:354–365. doi: 10.1158/1535-7163.MCT-15-0617. [DOI] [PubMed] [Google Scholar]
- 121.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT03118817.
- 122.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT02405065.
- 123.James J, et al. CEP-32496: a novel orally active BRAF(V600E) inhibitor with selective cellular and in vivo antitumor activity. Mol Cancer Ther. 2012;11:930–941. doi: 10.1158/1535-7163.MCT-11-0645. [DOI] [PubMed] [Google Scholar]
- 124.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT01877811.
- 125.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT02928224.
- 126.Long GV, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5:5694. doi: 10.1038/ncomms6694. [DOI] [PubMed] [Google Scholar]
- 127.Carlino MS, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol. 2014;8:544–554. doi: 10.1016/j.molonc.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT02872259.
- 129.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT01719380.
- 130.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT01543698.
- 131.US National Library of Medicine. ClinicalTrials.gov. 2017 https://clinicaltrials.gov/ct2/show/NCT02159066.
- 132.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Robert C, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 135.Hoos A. Development of immuno-oncology drugs— from CTLA4 to PD1 to the next generations. Nat Rev Drug Discov. 2016;15:235–247. doi: 10.1038/nrd.2015.35. [DOI] [PubMed] [Google Scholar]
- 136.Hugo W, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mandalà M, Massi D. Immunotolerance as a mechanism of resistance to targeted therapies in melanoma. Handb Exp Pharmacol. 2017 doi: 10.1007/164_2017_5. http://dx.doi.org/10.1007/164_2017_5. [DOI] [PubMed]
- 138.Rushworth LK, Hindley AD, O’Neill E, Kolch W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol Cell Biol. 2006;26:2262–2272. doi: 10.1128/MCB.26.6.2262-2272.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video1
Video2