The Kinetics of Synaptic Vesicle Reacidification at Hippocampal Nerve Terminals (original) (raw)
Abstract
After exocytosis, synaptic vesicles are recycled locally in the synaptic terminal and are refilled with neurotransmitter via vesicular transporters. The biophysical mechanisms of refilling are poorly understood, but it is clear that the generation of a proton gradient across the vesicle membrane is crucial. To better understand the determinants of vesicle refilling, we developed a novel method to measure unambiguously the kinetics of synaptic vesicle reacidification at individual synaptic terminals. Hippocampal neurons transfected with synapto-pHluorin (SpH), a synaptic vesicle-targeted lumenal GFP (green fluorescent protein), whose fluorescence is quenched when protonated (pKa ∼ 7.1), were rapidly surface-quenched immediately after trains of repetitive electrical stimulation. The recently endocytosed alkaline pool of SpH is protected from such surface quenching, and its fluorescence decay reflects reacidification kinetics. These measurements indicate that, after compensatory endocytosis, synaptic vesicles reacidify with first-order kinetics (τ ∼ 4–5 s) and that their rate of reacidification is subject to slowing by increased external buffer.
Keywords: hippocampus, fluorescence microscopy, synaptic vesicle release, optical imaging, synaptic transmission, synaptic
Introduction
In newly endocytosed synaptic vesicles, a proton electrochemical gradient (Grabe and Oster, 2001) produced by the V-type H+-ATPase2 (Nishi and Forgac, 2002) powers transport of neurotransmitter molecules into the vesicle lumen (Liu and Edwards, 1997; Fremeau et al., 2004). Previous studies indicate that quantal size and release probability may be related to steady-state vesicle pH (Wolosker et al., 1996; Pothos et al., 2002). In principle, postendocytic reacidification kinetics could influence short-term synaptic plasticity by modulation of the function of vesicular proteins involved in exocytosis or by limiting vesicle refilling speed.
Despite the potential importance of reacidification kinetics, technical limitations have hampered direct measurements in intact synapses. Experiments using synapto-pHluorin (SpH) (Miesenbock et al., 1998) in our laboratory suggested that, after trains of stimulation, vesicles reacidify with a time constant at most on the order of seconds (Sankaranarayanan and Ryan, 2000). SpH was used recently with single action potential (AP) stimulation to measure rates of endocytosis/reacidification (Gandhi and Stevens, 2003). The authors found that SpH fluorescence transients, thought to represent individual vesicle fusion, uptake, and acidification, can decay very rapidly (<500 ms). However, whether such rapid reacidification occurs after trains of AP remains unknown.
Here, we use repetitive electrical stimulation of SpH-transfected, cultured hippocampal neurons to measure precisely postendocytic reacidification kinetics. The poststimulus decay of SpH fluorescence ordinarily reflects a combination of the kinetics of both endocytosis and subsequent reacidification. To overcome this problem, we used pulse-pressure application of impermeant acid to isolate the kinetics of reacidification from that of endocytosis. The amplitudes of surface-quenched SpH fluorescence transients allowed us to estimate the dynamics of the quench-protected, “alkaline pool” of vesicles. Deconvolution was then used to estimate the time course of endocytosis after brief stimulus trains such as those that lead to exocytosis of the readily releasable pool (RRP) of vesicles.
Materials and Methods
Preparation and solutions.
On postnatal day 2, Sprague Dawley rat hippocampi (Ca3–Ca1 region) were dissected, dissociated, and plated on poly-ornithine-coated glass coverslips, as described previously (Ryan, 1999). On postnatal day 6 or 7, neurons were transfected with superecliptic SpH as described previously (Sankaranarayanan and Ryan, 2000). Experiments were performed 5–20 d after transfection, and coverslips were mounted in a laminar-flow perfusion and stimulation chamber. Cells were perfused continuously at room temperature (25°C). Control Tyrode’s solution contained (in mm) 119 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 25 HEPES, and 30 glucose; osmolarity was adjusted to 300 mOsm with sucrose, pH 7.40, with NaOH. In addition, all solutions contained 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione (Research Biochemicals, Natick, MA), and 50 μm d,l-2-amino-5-phosphonovaleric acid (Research Biochemicals). In bafilomycin A1 (Baf; Calbiochem, San Diego, CA) experiments, Baf (stored in frozen aliquots at 200 μm in DMSO) was diluted to 1 μm in 500 μl of control Tyrode’s and added dropwise to the preparation for 30 s before washout with Baf-free Tyrode’s perfusion. Because Baf causes a slow alkalinization of internal vesicles (Sankaranarayanan and Ryan, 2001), it was important to start the trial soon (<90 s) after Baf application. In buffer experiments, glucose was lowered to 10 mm. HEPES was used at 5, 25, or 40 mm. In Tris experiments, NaCl was reduced to 91 mm; this led to a reduction in size of SpH responses; thus, longer trains (300 AP, 10 Hz) were used to compensate. Tris base was used at 10, 20, or 50 mm, pH 7.40, with HCl. Attempts to use Tris-base concentration of 100 mm were unsuccessful, because they led to solutions with osmolarity of >360 mOsm after pH titration. SpH signals were diminished in this high-osmolarity solution, and cells did not survive for long.
Line-scan fluorescence measurements.
SpH fluorescence from individual boutons was measured using a custom-built laser confocal-imaging system in line-scan mode, through a 40× 1.3 numerical aperture Zeiss (Oberkochen, Germany) Fluar objective. An argon ion laser was used to illuminate the sample at 488 nm; emitted light was passed through a 498–538 nm bandpass filter and detected with a photomultiplier tube. Custom software was used to acquire and store fluorescence measurements. Cells were stimulated through platinum-iridium field electrodes located at the inlet and outlet of the perfusion chamber, using 1 ms pulses and a field of 10 V/cm. Boutons from SpH-transfected cells that responded (>20% increase in fluorescence) to stimuli of 60 AP at 20 Hz were selected for study. In quenching experiments, only responding boutons that were readily (i.e., rapidly, deeply, and reliably) quenched by 300 ms test pulses of acidic solution were studied. Sampling intervals ranged from 21 ms (to optimize puffer position) to 346 ms (to minimize bleaching).
Measurements of reacidification kinetics.
To isolate SpH fluorescence from recently endocytosed synaptic vesicles, surface SpH was rapidly quenched by pulse-pressure application (PicoSpritzer II; General Valve, Fairfield, NJ) of impermeant acid. The puffer pipette had a tip diameter of 5–10 μm and was generally positioned 30–100 μm (in the horizontal plane) from the target bouton and 30–50 μm above. The puffer pipette was aimed directly at the target bouton, but high perfusion velocity (2–3 ml/min; 150–300 μl chamber volume) limited exposure of the target cell to acidic solution leaking from the puffer pipette. In most experiments, the puffer pipette contained MES-buffered (pKa 6.1, 45 mm) Tyrode’s solution, pH 5.25. We found that neurons survived only a few repeated MES puffs after Baf application. HEPES-buffered (25 mm) Tyrode’s, pH 5.25, was better tolerated and was used in the puffer pipette for Baf experiments. HEPES-buffered quenching gave reacidification kinetics identical to that obtained in MES. These buffers appear adequate for both quenching the surface and maintaining the low pH after endocytosis, because quenching in bafilomycin showed almost constant fluorescence in the poststimulus quench period (see Fig. 2A).
Figure 2.
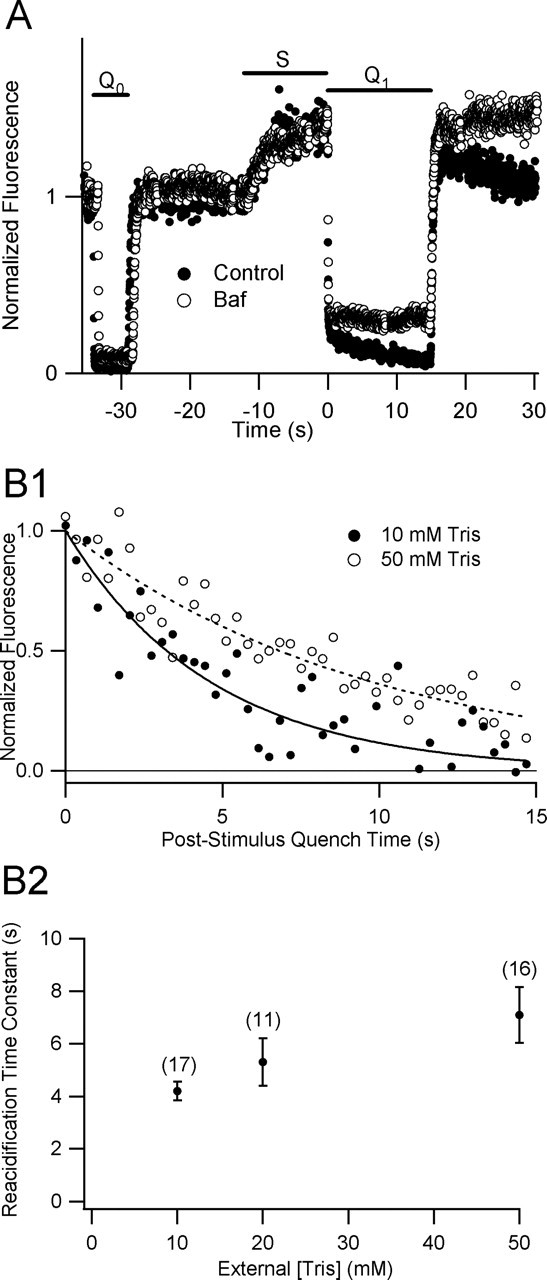
A, SpH fluorescence decay during poststimulus acid quench is blocked by a specific inhibitor of V-ATPase. SpH fluorescence sampled every 21 ms from a presynaptic bouton in control solution (closed circles) and after 30 s treatment with 1 μm Baf (open circles). Pressure-pulse application of impermeant acid before (_Q_0, 5 s) and immediately after (_Q_1, 15 s) a stimulus train (S, 12 s, 20 Hz) reveals a stimulation-dependent quench-resistant fluorescence transient whose decay is blocked by inhibition of the V-ATPase. Similar results were observed in a series of 13 paired trials on 8 separate preparations using a stimulus of 30 s at 10 Hz and a sampling interval of 342 ms. B, Sensitivity of reacidification kinetics to extracellular proton buffer. B1, Two fluorescence transients from the poststimulus quench periods of successive trials at the same bouton, normalized to unitary amplitude B2, Summary plot of reacidification time constants (mean ± SEM) versus external [Tris]. Parentheses indicate number of trials for each point. All Tris experiments were performed in 91 mm NaCl (see Materials and Methods) with stimulus trains of 30 s at 10 Hz. Unpaired two-tailed Student’s t tests indicate a statistically significant difference (p < 0.013) between mean time constants in 10 and 50 mm Tris but no significant difference between 10 and 20 mm Tris (_p_ > 0.19) or between 20 and 50 mm Tris (p > 0.23).
Figure 1.
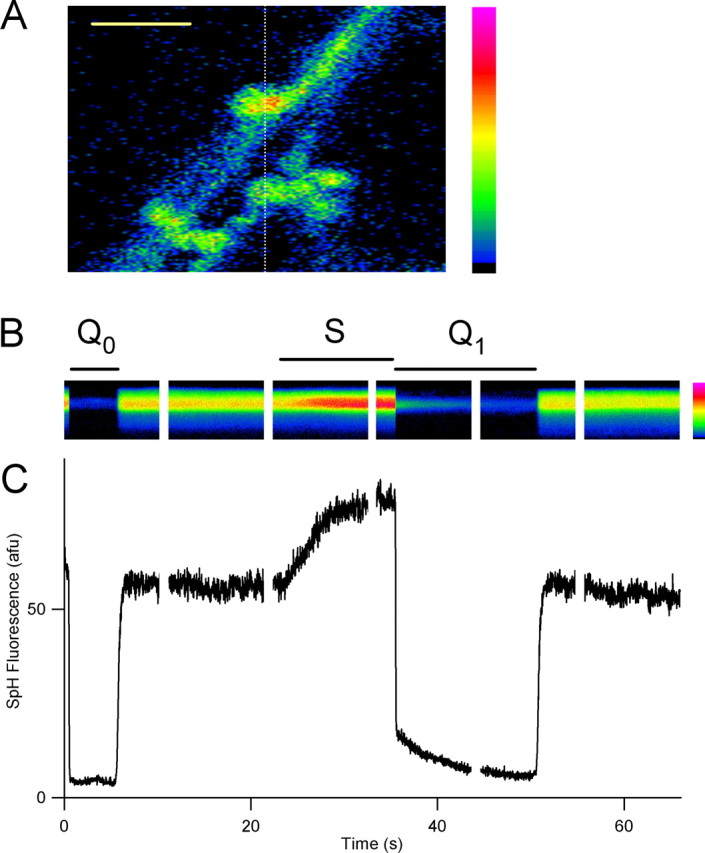
Rapid acid quench of surface SpH fluorescence allows measurement of internal alkaline vesicle pool and postendocytic reacidification time course. A, Confocal image of resting fluorescence of axons and presynaptic terminals transfected with synapto-pHluorin, with a dashed line at the position of the scan line for B. Scale bar, 3.4 μm. B, Line-scan time series of fluorescence from a single bouton, with rapid surface quenching by pulsed-pressure application of impermeant acid (45 mm MES, pH 5.25) before (_Q_0, 5 s duration) and immediately after (_Q_1, 15 s) field electrical stimulation (S; 12 s, 20 Hz). A, B, Pseudocolor scale bars (right) are linear, with black indicating zero fluorescence. C, Plot of fluorescence time course, computed by averaging those rows from B with stimulus-evoked fluorescence increase. Time scale is the same as that in B.
If the quench is adequate, the time course of fluorescence decay during the quench period should not be sensitive to vesicles endocytosed during the quench period, because these vesicles should be already acidic and should not contribute to the time-varying signal. However, turbulent mixing of the acidic quench solution with the alkaline bath perfusion might lead to contamination of the reacidification signal by fluorescence from ongoing endocytosis of partially quenched vesicles during the quench period. Thus, it was important to optimize the acid quench. Before most trials, the acid quench depth and rapidity of onset were probed with brief (300 ms) pulses and optimized by slightly repositioning the puffer pipette or adjusting the amplitude of the pressure pulse. Control experiments (data not shown) indicated that, after optimization, the depth of quench achieved during the pressure pulse was equivalent to that achieved by bath application of Tyrode’s solution, pH 5.5. Some of the boutons examined could not be adequately quenched, perhaps because of glial barriers to diffusion and convection, which may interfere with rapid access of the acid solution to the bouton.
Alkaline pool dynamics.
We define the “alkaline pool” as recently endocytosed vesicles alkaline with respect to the pH of resting vesicles (i.e., pH ∼5.5). The “size” of the alkaline pool, as reported by SpH fluorescence during the quench period, is a sum over recently endocytosed vesicles weighted by the instantaneous fluorescence of each vesicle.
We studied alkaline pool dynamics by varying Δ_t_, the time between onset of the 1 s, 40 Hz stimulus and the beginning of a 15 s acid quench. Five to ten trials were performed on each bouton. To minimize acid exposure and prolong tissue viability, we omitted the 5 s prequench from these experiments. However, the quench was optimized before each trial (as explained above). For each bouton, the alkaline pool amplitude for a particular Δ_t_ was calculated as the difference between the average fluorescence of five points (sampling interval, 346 ms) at the beginning and end of the quench.
Because the alkaline pool plots from different boutons have different peak times, it was not clear how best to summarize their time courses. Figure 3B (bottom) shows a simple average of the alkaline pool amplitudes (see above) without normalization. We elected to fit the plots of alkaline pool dynamics for each bouton to an α function with a delay term (see Fig. 3B, top). The fit parameters for each bouton studied were averaged and used to arrive at a “mean” α function (i.e., one with mean parameters). This method of averaging the alkaline pool time courses gave a mean α function (see Figs. 4, 5) with parameters similar to those from a fit to the simple average of the data (see Fig. 3B, bottom). The choice of the α function for curve fitting was somewhat arbitrary, but convenient. We do not imply that there are not many other ways to parameterize these curves.
Figure 3.
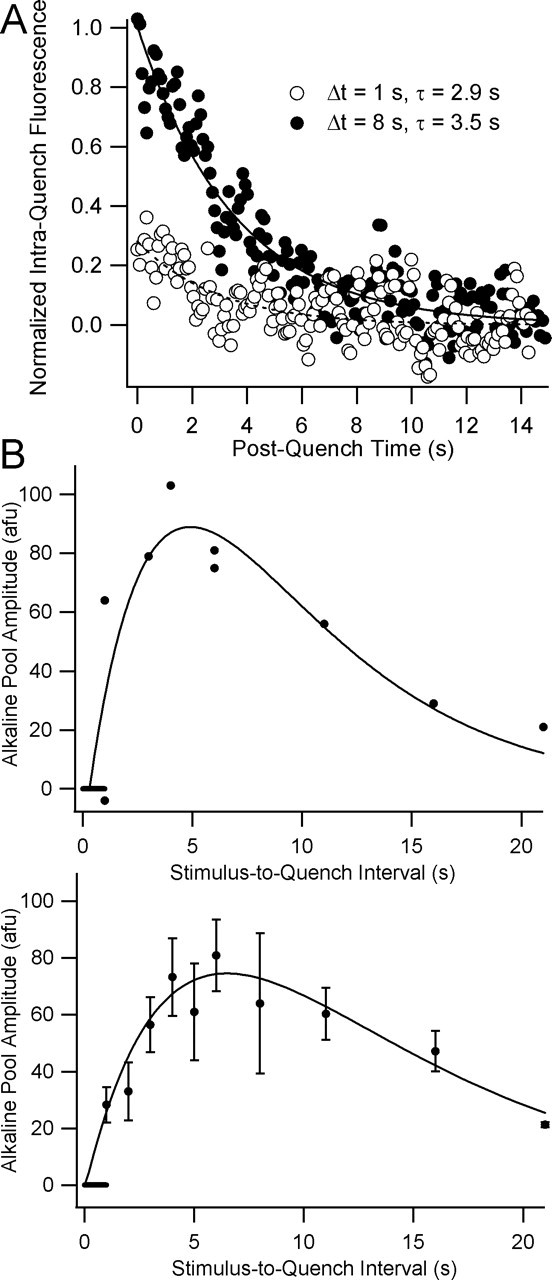
Time course of alkaline pool growth and decay after a 1 s, 40 Hz train. A, Normalized fluorescence transients measured during 15 s quenches begun 1 s and 8 s after stimulus onset. The alkaline pool amplitude at time Δ_t_ is estimated by taking the difference of the means of the first five and the last five points of the quench period. Sampling interval for these experiments is 85.4 ms. B, Plots of alkaline pool amplitudes from a representative experiment (top) and from seven such experiments (bottom; mean ± SEM) versus the interval between stimulus and quench onsets. Curves are fits to an α function of the following form: y = k*(Δ_t_ − delay)*exp(−(Δ_t_ − delay)/τ), where k is a scaling factor, and Δ_t_ is the stimulus–quench interval, and delay is a time offset term. The fit parameters {k, delay, τ} (± SD) were {53 ± 16, 0.3 ± 0.5 s, 4.6 ± 1.0 s} for the curve in the top panel and {31 ± 6, 0.1 ± 0.4 s, 6.4 ± 1.0 s} for the curve in the bottom panel (see Materials and Methods). Horizontal bars at origins indicate the stimulus period.
Figure 4.

Deconvolution of reacidification time course (thin solid line) from alkaline pool dynamics (thick solid line) gives the estimated time course of endocytosis (dotted line) after a 1 s, 40 Hz train. Each curve was normalized to its peak value.
Figure 5.
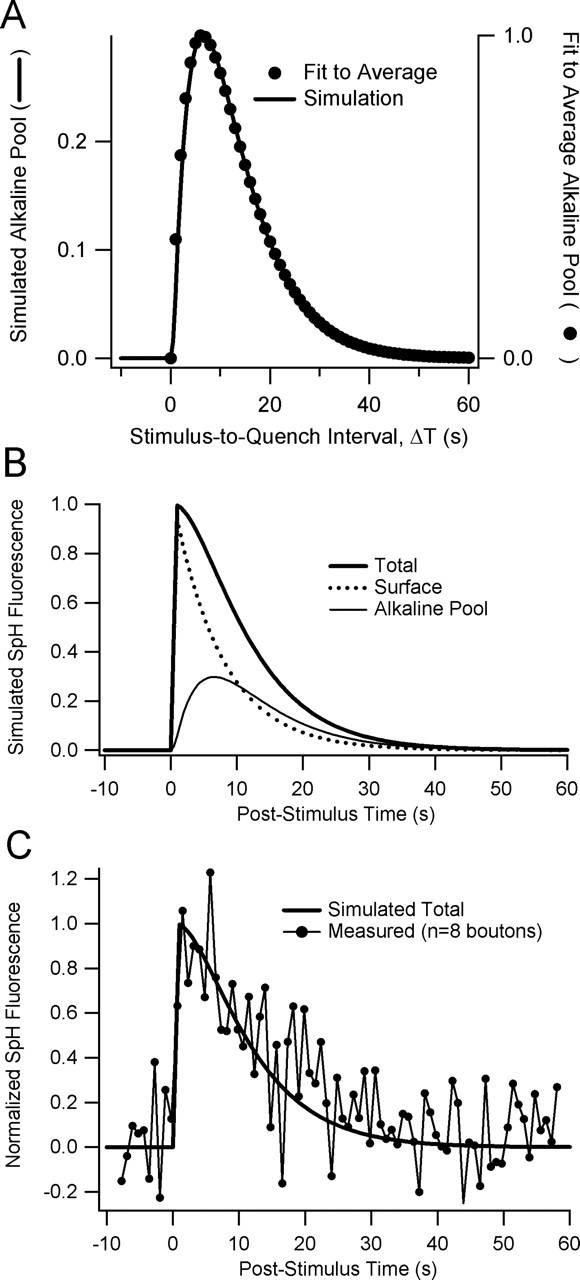
Simple simulation of alkaline pool dynamics. A, Superposition of average alkaline pool time course (closed circles) and simulated alkaline pool time course (solid line). Numerical simulation used a zero-order rate constant of 1 s−1 for exocytosis and first-order rate constants of 0.135 and 0.2 s−1 for endocytosis and reacidification, respectively. B, Time courses of surface (dotted line) and alkaline pool (thin solid line) SpH fluorescence; their sum is the expected SpH fluorescence transient (thick solid line) with no surface quenching. The time course of reacidification significantly influences the time course of SpH fluorescence transient after brief trains. C, The simulated SpH curve (smooth line) from (B) with superimposed data from an experiment with 1 s, 40 Hz stimulation and field scanning of eight boutons with sampling interval ∼700 ms. Error bars were omitted for clarity.
Deconvolution.
If endocytosis and reacidification are the only contributants to alkaline pool amplitude, and if they are linear time-invariant processes, we can recover the endocytosis time course by deconvolution of reacidification from the alkaline pool time course. A linear time-invariant process is one in which (1) the amplitude of the response (intraquench fluorescence transient) varies linearly with the amplitude of the input (endocytosis); and (2) the response to a particular input does not depend on the time of that input. Deconvolution was calculated by dividing the fast Fourier transform (FFT) of the alkaline pool time course by the FFT of the reacidification time course and then performing an inverse FFT on the result.
After a 1 s, 40 Hz stimulus, there was a slight increase in the time constant of reacidification with increasing stimulus to quench intervals (data not shown). One interpretation is that the impulse response is not strictly time invariant; instead, it slightly slows with longer poststimulus times. Another possibility is that there is a small, slower second component to reacidification, which we do not resolve because of our limited (15 s) quench window. Because endocytosis rates decline with poststimulus time, the mean delay between vesicle entry and the quench will increase for larger stimulus–quench intervals. The greater mean delay would be expected to accentuate the contribution of a second, slower component of reacidification.
Simulation.
We numerically simulated alkaline pool dynamics using a fourth-order Runge-Kutta algorithm implemented in IGOR Pro 4 (WaveMetrics, Lake Oswego, OR) and assumed that exocytosis proceeded at a constant velocity during the stimulus and was zero otherwise. Similarly, we assumed that, before exocytosis, endocytosis was zero. We also assumed that endocytosis and reacidification during the poststimulus period were first-order processes whose velocities were proportional to the amount of SpH in the surface and alkaline pools, respectively.
Results
We used laser-confocal microscopy of cultured hippocampal neurons transfected with SpH to monitor the lumenal pH of synaptic vesicles in individual presynaptic terminals during and after brief trains of electrical stimuli. SpH is a pH-sensitive (pKa ∼ 7.1) green fluorescent protein (GFP) conjugated to the lumenal domain of VAMP2 (vesicle-associated membrane protein)/synaptobrevin, a v-SNARE (soluble _N_-ethylmaleimide-sensitive factor attached protein receptor) located in synaptic vesicle membranes (Miesenbock et al., 1998). In fully acidified (pH ∼ 5.5) synaptic vesicles of a resting bouton, SpH molecules spend ∼97% of the time in the quenched, proton-bound state (Sankaranarayanan and Ryan, 2000). Electrical stimulation triggers synaptic vesicles to fuse with the plasma membrane, in which SpH accumulates (Fernandez-Alfonso and Ryan, 2004), rapidly equilibrating with the alkaline (pH ∼ 7.4) extracellular medium. This relief from proton quench leads to a fluorescence increase of ∼20-fold. SpH thereafter is removed from the plasma membrane by internalization of synaptic vesicles whose fluorescence relaxes to baseline levels as they reacidify.
Previous work in our laboratory suggested that the slow decay (τ ∼ 60 s) of SpH fluorescence after a long stimulus train reflects the kinetics of endocytosis (Sankaranarayanan and Ryan, 2000). This work rested on the assumption that synaptic vesicle reacidification kinetics is sufficiently rapid with respect to internalization kinetics that reacidification may be safely ignored in interpreting SpH fluorescence transients. Although this was a reasonable assumption with the long stimulus trains that were used in previous work, here we are interested in short stimulus trains that preferentially mobilize the RRP. Previous work in our laboratory showed that internalization kinetics accelerates as we shorten the stimulus, leading to our concern that, after brief trains, it would be no longer safe simply to assume that reacidification kinetics could be safely discounted.
In principle, instantaneous presynaptic terminal SpH fluorescence can be separated into three components. The first derives from internal SpH quenched in acidic synaptic vesicles: the “acidic pool.” The second derives from unquenched SpH exposed to the alkaline extracellular medium: the “surface pool.” The third derives from SpH partially quenched in recently internalized synaptic vesicles that have not yet fully reacidified: the “alkaline pool.” Exocytosis (whether classical or via a transient fusion pore) causes a rapid rise in SpH fluorescence, as the vesicles move from the acidic to the surface pool. Internalization (whether by classical endocytosis or by other mechanisms) is the movement of vesicles from the surface to the alkaline pool. SpH fluorescence is not diminished by internalization per se; fluorescence decays by proton quench as internalized vesicles in the alkaline pool reacidify, returning to the acidic pool.
The decay of SpH fluorescence after a short train is thus controlled by two factors: reacidification kinetics and internalization kinetics. We reasoned that, if we measured both reacidification kinetics and the time course of expansion and contraction of the alkaline pool, we could deconvolve the reacidification time course to estimate the internalization time course.
Our goal, then, was to devise ways to measure both reacidification kinetics and alkaline pool dynamics. If successful, we would be in a position to estimate internalization kinetics after short trains that selectively mobilize the RRP. This would directly test the hypothesis that rapid internalization is characteristic of RRP mobilization in small CNS terminals.
Measurement of reacidification kinetics
To measure reacidification kinetics, our strategy was to monitor fluorescence in single presynaptic boutons stimulated with brief trains, and then to quench surface SpH for 15 s by local pressure-pulse application of acid. SpH in the alkaline pool (i.e., in recently endocytosed, partially reacidified vesicles) will be protected from this 15 s surface quench. SpH in fully acidified (pH 5.5) vesicles will contribute a component of fluorescence detected during this period, but this component should be nearly constant. The decaying component of fluorescence during the quench should be a measure of reacidification of the alkaline pool.
As shown in Figure 1, we were able to detect fluorescence in line-scan mode from single boutons at a high temporal resolution (50 Hz sampling). A 5-s-long prestimulus acid quench (_Q_0) showed that the majority (typically 85–95%) of resting fluorescence was attributable to surface SpH, but a small, constant quench-resistant fraction remained, corresponding to quench-protected pools of SpH. The quenching of surface fluorescence was very rapid, with a first-order time constant τq of 32 ms; in all such experiments accepted for additional analysis, τq ranged in the tens of milliseconds (see Materials and Methods). After termination of the prequench, surface SpH rapidly re-equilibrates with the alkaline medium, and fluorescence recovers. During the stimulation period (S), fluorescence increased as vesicular SpH was added to the surface. The poststimulus quench (_Q_1) revealed a much larger quench-resistant fluorescence signal than that during _Q_0. This transient had a time course well approximated by a monoexponential decay with a time constant of 5.0 s and a baseline of 5.1 arbitrary fluorescence units (afu), slightly higher than the prequench mean fluorescence of 4.5 afu.
If the gradually declining fluorescence signal observed during the poststimulus quench (_Q_1) represents the reacidification kinetics of the alkaline pool, its decay should be blocked by Baf (Sankaranarayanan and Ryan, 2001). In Figure 2A, we show a representative experiment in which brief exposure to 1 μm Baf completely blocked the decay of fluorescence seen previously during the quench period. In similar experiments on eight different preparations stimulated for 30 s at 10 Hz, the time courses of quench period fluorescence in control conditions were well fit by monoexponential decays with a mean time constant (mean ± SEM) of 4.4 ± 0.4 s (n = 12) and a normalized amplitude of 1 ± 0.13 U (n = 12). After Baf treatment, fluorescence during the quench period did not decay. Instead, quench period fluorescence gradually increased in intensity, with a mean slope of 1.4E-2 ± 1.1E-2 U/s (n = 8). In addition, the baseline fluorescence both before and during the quench is slightly elevated in the presence of bafilomycin. Both of these features are most likely attributable to slow alkalinization of synaptic vesicles, arising from either spontaneous exocytosis or a small proton leak from the vesicles. The effectiveness of Baf in blocking the fluorescence transient strongly supports the idea that the quench period fluorescence transient is a valid measure of reacidification kinetics.
To test whether the external pH buffer distorts reacidification kinetics, we varied external HEPES concentration. For 5 s 20 Hz stimuli, the poststimulus quench-resistant fluorescence decay time course was indifferent to external HEPES concentration between 5 and 40 mm (data not shown). For these experiments, time constants of decay were 3.8 ± 0.4 s (5 mm HEPES; n = 9), 4.3 ± 0.3 s (25 mm; n = 5), and 4.0 ± 0.4 s (40 mm; n = 9). In several of these experiments, solution changes were followed by a long train of electrical stimulation (90 s, 10 Hz), to test whether extensive endo-exo cycling favors access of HEPES to the vesicle lumina, as reported previously (Gandhi and Stevens, 2003). However, we found no effect of vesicle turnover on reacidification kinetics; it appears likely that either HEPES is excluded from vesicles undergoing “compensatory” endocytosis or that it is ineffective at proton buffering within the microenvironment of the synaptic vesicle lumen.
We also tested the effect of external Tris on reacidification kinetics (Fig. 2B1,B2). To facilitate comparison with previous work (Gandhi and Stevens, 2003), we used a low Na+ Tyrode’s solution for these experiments (see Materials and Methods). After a 30 s, 10 Hz stimuli we found that high Tris slows the decay of the reacidification transient (Fig. 2B1). In a series of such experiments on nine individual boutons from six separate preparations, reacidification time constants (mean ± SEM) of 4.2 ± 0.35 (n = 17), 5.3 ± 0.90 (n = 11), and 7.1 ± 1.1 s (n = 16) were observed with 10, 20, and 50 mm Tris, respectively (Fig. 2B2). Unpaired two-tailed Student’s t tests indicate a statistically significant difference (p < 0.013) between mean time constants in 10 and 50 mm Tris. Thus, as reported previously, external Tris slows reacidification, most likely by entering nascent vesicles and augmenting their buffering capacity.
To test whether photobleaching of the alkaline pool distorts the fluorescence time course, we performed surface-quenching experiments while varying the line-scan sampling rate from 1 to 50 Hz. We saw no change in the time course of fluorescence decay (data not shown).
The observed sensitivity of SpH fluorescence transients during the quench period to Baf and to external Tris strongly supports the idea that these signals represent postinternalization reacidification kinetics.
Subsequently, we wished to test whether there was a difference between reacidification kinetics of vesicles in the RRP versus the recycling pool (RP). In our system, a 40 ap, 1 s stimulus mobilizes only 9.1 ± 0.7% of the total recycling pool (102 boutons in four separate experiments; data not shown), whereas a 300 ap, 10 s stimulus mobilizes ∼75% (Ryan et al., 1996). Thus, we expect short trains selectively to mobilize the RRP, whereas longer trains will mobilize both RRP and the entire RP. We found that for both short and long trains, reacidification time course during the 15 s quench period was well approximated by a monoexponential decay. Similar time constants were observed for experiments with 40 APs at 40 Hz (4.5 ± 1.2; n = 4), 100 at 20 Hz (4.3 ± 0.3; n = 5), 120 at 20 Hz (4.8 ± 0.1; n = 2), 240 at 20 Hz (4.8 ± 0.5; n = 14), and 300 at 10 Hz (4.4 ± 0.4; n = 12). These experiments suggest that vesicles in the RRP and the RP share similar reacidification kinetics.
Measurement of alkaline pool dynamics
Having measured the time course of reacidification after RRP mobilization, we next sought to measure alkaline pool dynamics, with a view toward eventually estimating internalization kinetics. We noted that alkaline pool amplitude at time Δ_t_ after stimulus onset is proportional to the initial amplitude of the quench-resistant SpH fluorescence transient. We performed experiments in which the interval between stimulus and quench onsets, Δ_t_, was varied between 1 and 20 s, and the size of the alkaline pool was measured at each Δ_t_ (for details, see Materials and Methods).
In two successive trials from a representative experiment (Fig. 3A), quenching immediately after a 1 s, 40 Hz train (Δ_t_ = 1 s) revealed a small alkaline pool that decayed exponentially. Delaying the quench (Δ_t_ = 8 s) revealed a larger alkaline pool that also decayed exponentially. In a representative experiment (Fig. 3B, top) with nine such trials in the same bouton, the plot of alkaline pool size versus Δ_t_ resembled an α function (see Materials and Methods) with time constant (mean ± SD) of 4.6 ± 1 s and time intercept of 0.31 ± 0.49 s. Data from 10 such experiments (Fig. 3B, bottom), with 5–10 trials in each bouton, shows that, after a 1 s, 40 Hz train, alkaline pool dynamics may be approximated by an α function with a time constant of 6.4 ± 0.95 s (mean ± SD) and a time intercept at 0.05 ± 0.42 s. We note that the appearance of a small but measurable alkaline pool in trials with Δ_t_ = 1 s argues against any significant delay in the onset of internalization after these stimuli.
In summary, after brief stimulus trains, the fluorescence of the alkaline pool peaks ∼6 s after the stimulus and then slowly decays over tens of seconds. With these measurements of alkaline pool dynamics and with our measurements of reacidification kinetics, we were now in a position to use deconvolution to estimate the time course of internalization of synaptic vesicles after brief trains.
Alkaline pool deconvolution and simulation
If we assume that the processes governing alkaline pool dynamics are linear and time invariant (see Materials and Methods), we can deconvolve the output process (reacidification time course) from alkaline pool dynamics to estimate the input process (endocytosis time course). Deconvolution (Fig. 4) allowed us to estimate the time course of endocytosis after a 1 s, 40 Hz train as a curve whose shape is closely approximated by an exponential decay with a time constant of 7.6 s. Notably, there is no indication of a very rapid component of endocytosis, as would be expected if a significant fraction of vesicles were undergoing rapid reuse.
To estimate the relative contributions of surface and alkaline pools to the fluorescence transients observed after a brief train, we made a very simple numerical simulation (Fig. 5) that implemented the following scheme:

The rate constants _k_exo and _k_endo describe the kinetics of exocytosis and endocytosis; _k_reacid is the rate constant of vesicle reacidification. We modeled exocytosis as a zero-order reaction with rate constant _k_exo = 1 s−1. Our estimate of _k_reacid was 0.2 s−1, the reciprocal of the time constant of reacidification. We then searched for a value of _k_endo such that the simulated time course of the alkaline pool matched the average alkaline pool time course we measured after a 1 s, 40 Hz stimulus. We found that a rate constant of 0.135 s−1 gave a good match to the time course of the alkaline pool (Fig. 5A). These rate constants for endocytosis and reacidification imply a relative amplitude of ∼0.3 for the peak of the alkaline pool, where 1 corresponds to the amount of SpH exocytosed during a 1 s, 40 Hz train.
This simulation helps to illustrate the importance of considering alkaline pool dynamics in interpreting rapidly decaying SpH transients. In Figure 5B, the predicted total SpH fluorescence decays with a time constant of ∼11 s and closely matches a measured SpH transient after a 1 s, 40 Hz stimulus (Fig. 5C). SpH internalization occurs with a time constant of ∼7.4 s. Ignoring alkaline pool dynamics would have led to a 50% overestimate of the time constant of internalization.
Discussion
To understand vesicle refilling and quantal size determinants during ongoing activity, we set out to study synaptic vesicle reacidification kinetics by monitoring the fluorescence of SpH in CNS boutons after trains of APs. By rapidly quenching surface SpH with pressure-pulse applications of acid, we isolated a quench-protected fluorescence signal that reflects reacidification kinetics isolated from distortion by ongoing endocytosis. Surface quenching also eliminates alternate fluorescence decay pathways, such as that which might be caused by lateral diffusion of vesicular SpH from sites of exocytosis into neighboring axon (Sankaranarayanan and Ryan, 2000). A possible artifact of quench experiments could arise from slow access of the acid to surface SpH in tortuous membrane invaginations, posited by some (Wilkinson and Cole, 2001) to arise in the poststimulus period. The complete block of the decay of fluorescence during the quench period by exposure to the V-ATPase inhibitor Baf (Fig. 2) demonstrates that these fluorescence transients reflect reacidification kinetics rather than lateral diffusion of SpH or slow quench access. To our knowledge, these are the first direct measurements in intact terminals of postendocytic reacidification kinetics, unambiguously isolated from the kinetics of endocytosis and that of lateral diffusion of SpH in the plasma membrane.
Our principle findings are as follows: (1) rapid acid quench of surface SpH may be used to isolate fluorescence from the alkaline pool of reacidifying vesicles and to measure directly the kinetics of reacidification; (2) after compensatory endocytosis, reacidification of synaptic vesicles occurs with apparent first-order kinetics and a time constant of 4–5 s; (3) similar reacidification kinetics are observed after a variety of stimulus pulse protocols; (4) external pH buffers (Tris but not HEPES) can slow such reacidification kinetics; (5) by varying the stimulus-to-quench interval, the growth and decay of the alkaline pool may be estimated after brief stimuli; and (6) the time course of endocytosis after brief trains, otherwise very difficult to measure in small presynaptic terminals, may be estimated by deconvolution of reacidification kinetics from alkaline pool dynamics.
Our most important finding is that postendocytic reacidification, after trains of 40–300 AP, occurs quite slowly, with a time constant of 4–5 s. Reacidification on this time scale is not expected to contribute significantly to the time course of SpH fluorescence decay (time constants, 20–80 s) after trains longer than ∼60 AP at 10 Hz (Sankaranarayanan and Ryan, 2000). For such trains, SpH fluorescence decay may be safely interpreted as reflecting the time course of endocytosis. However, after brief trains, SpH fluorescence transients decay more rapidly, on a time scale similar to reacidification; their decay times result from a combination of endocytosis and reacidification kinetics. We found that, by varying the stimulus-to-quench interval, we could measure alkaline pool dynamics (Fig. 3), from which we deconvolved reacidification kinetics to estimate endocytosis kinetics (Fig. 4). After a 1 s, 40 Hz stimulus, surface SpH is internalized with a time constant of ∼7.6 s. It is important to note that it would be invalid to try to estimate endocytosis rates by deconvolving reacidification from the unquenched SpH fluorescence transient, which includes fluorescence both from internalized, reacidifying compartments and from surface SpH not subject to reacidification.
Our approach has allowed us to test the hypothesis that, in small CNS presynaptic terminals, mobilization of the vesicles of the RRP by short trains of electrical stimulation leads to “kiss-and-run” cycling with rapid internalization and presumably rapid reuse of vesicles, enabling the synapse to avoid exhaustion of the RRP and to maintain signaling capacity. Evidence for kiss-and-run cycling at central synapses has emerged from a variety of experiments based on destaining of amphipathic probes that differ significantly in their conclusions about both the fusion-to-fission time scale as well as the conditions under which kiss-and-run will dominate (Ryan et al., 1996; Klingauf et al., 1998; Pyle et al., 2000; Stevens and Williams, 2000; Zenisek et al., 2002; Aravanis et al., 2003; Richards et al., 2005) However, definitive conclusions based on FM destaining characteristics are difficult to achieve because the detailed physical behavior of the dye within the confines of the vesicle, such as whether it dissociates from the vesicular membrane with the same dissociation constant as that from the plasma membrane, are not known.
Alternative approaches to examining the details of fusion-to-fission part of the vesicle cycle have become possible with the use of SpH, a modified GFP tethered to a synaptic vesicle protein. These experiments exploit the fact that SpH has a proton-binding site whose occupancy reversibly quenches SpH fluorescence by ∼20-fold; this site is in rapid equilibrium with protons in the vesicle lumen or at the cell surface, allowing SpH to rapidly signal synaptic vesicle fusion with the plasma membrane or changes in synaptic vesicle pH. Gandhi and Stevens (2003) measured small fluorescence transients in SpH-transfected hippocampal neurons after a single AP. These transients were interpreted as representing the fusion, uptake, and reacidification of individual synaptic vesicles. The authors propose a scheme in which there are three modes of internalization after single APs: fast (or kiss-and-run), slow (or compensatory), and triggered (or “stranded”). Reacidification of vesicles internalized by the fast or triggered modes was found to be very rapid, with a time constant of ∼500 ms. In this study, the ratio of kiss-and-run events to compensatory endocytosis was highly dependent on the release probability, was skewed toward kiss-and-run only for very low release probability conditions, and was only tested for a small number of stimuli at very long interpulse intervals. Furthermore, the interpretation of the data in that work, in particular the fluorescence decreases after exocytosis, implicitly assumed that synaptic vesicle proteins do not escape the active zone by lateral diffusion. Postexocytosis lateral diffusion of SpH, which would also lead to fluorescence decreases have been shown to occur in these synapses (Sankaranarayanan and Ryan, 2000) and would be even more prevalent under conditions in which the surface molecules were photobleached before stimulation. All of these difficulties underscore the need to use means that discriminate against surface versus internal fluorescence when interpreting SpH signals.
Using such means, we found that, after a 1 s, 40 Hz stimulus that preferentially mobilizes the RRP, surface SpH is internalized with a time constant of ∼7.6 s. There was no sign of rapid internalization, arguing against rapid reuse. This time scale is consistent with the saturation model that we originally proposed for these nerve terminals (Sankaranarayanan and Ryan, 2000), whereby the time constant for vesicle retrieval after the stimulus is proportional to the amount of accumulated membrane during stimulation and agrees well with the recent findings that vesicles that comprise the RRP are not preferentially reused after exocytosis (Li et al., 2005). The continuous progression of endocytosis over a several second time period, even after brief stimuli expected to mobilize the RRP, but not the RP, is not consistent with the recent suggestion that, although RP vesicles may use the “slow” classical cycling pathway, RRP vesicle cycling occurs predominantly by the “fast” kiss-and-run pathway. It might be argued that kiss-and-run cycling with rapid internalization is occurring in our system but that we are insensitive to these events because the “kiss” is so ephemeral that protons do not have a chance to equilibrate across the fusion pore. This seems unlikely for three reasons. First, if the fusion pore were so short-lived that protons did not equilibrate, it would be unable to release a significant quantity of neurotransmitter. Second, Gandhi and Stevens (2003) report quantal SpH signals, which suggest but do not imply full pH equilibration. Third, Aravanis et al. (2003) argue for a mean fusion pore lifetime on the order of 1 s after short trains.
Thus, our data impose significant restrictions on the degree to which a rapid kiss-and-run type mechanism of internalization occurs at these nerve terminals and argue strongly against the predominance of this pathway after rapid mobilization of the RRP. These results agree well with recent measurements of membrane retrieval at the calyx of Held, which also argue against a kiss-and-run mechanism during repetitive stimulation at that synapse (Yamashita et al., 2005). Our measurements do not lend themselves well to measurements at very-low-frequency stimulation, and thus it is possible that, only under those conditions, kiss-and-run would dominate, which would be consistent with recent measurements of subquantal dye destaining using low-frequency stimulation (Aravanis et al., 2003; Richards et al., 2005).
It is important to note that, although accurate measurement of the kinetics of synaptic vesicle reacidification will be crucial for understanding how vesicles are readied for reuse, the precise relationship between vesicle reacidification and refilling kinetics remains to be determined (Tabb et al., 1992; Wolosker et al., 1996; Huang et al., 2001; Stobrawa et al., 2001; Sonawane et al., 2002; Vanoye and George, 2002; Robinson et al., 2004).
Footnotes
P. P. Atluri’s present address: Department of Psychiatry, NewYork-Presbyterian Hospital, Weill Cornell Medical Center, 525 East 68th Street, NY, New York 10021.
This work was supported by the National Institutes of Health (T.A.R.), the Hirschl Trust (T.A.R.), and the Charles H. Revson Foundation (P.P.A.). We thank James Rothman for kindly providing the superecliptic synapto-pHluorin construct, Tomás Fernández-Alfonso for careful reading of this manuscript, members of the Ryan laboratory for useful discussions, and Wayne Yan for technical assistance.
References
- Aravanis AM, Pyle JL, Tsien RW (2003). Single synaptic vesicles fusing transiently and successively without loss of identity. Nature 423:643–647. [DOI] [PubMed] [Google Scholar]
- Fernandez-Alfonso T, Ryan TA (2004). The kinetics of synaptic vesicle pool depletion at CNS synaptic terminals. Neuron 41:943–953. [DOI] [PubMed] [Google Scholar]
- Fremeau RT Jr, Voglmaier S, Seal RP, Edwards RH (2004). VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci 27:98–103. [DOI] [PubMed] [Google Scholar]
- Gandhi SP, Stevens CF (2003). Three modes of synaptic vesicular recycling revealed by single-vesicle imaging. Nature 423:607–613. [DOI] [PubMed] [Google Scholar]
- Grabe M, Oster G (2001). Regulation of organelle acidity. J Gen Physiol 117:329–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Liu J, Di A, Robinson NC, Musch MW, Kaetzel MA, Nelson DJ (2001). Regulation of human CLC-3 channels by multifunctional Ca2+/calmodulin-dependent protein kinase. J Biol Chem 276:20093–20100. [DOI] [PubMed] [Google Scholar]
- Klingauf J, Kavalali ET, Tsien RW (1998). Kinetics and regulation of fast endocytosis at hippocampal synapses. Nature 394:581–585. [DOI] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE (1998). Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394:192–195. [DOI] [PubMed] [Google Scholar]
- Li Z, Li Z, Burrone J, Tyler WJ, Hartman KN, Albeanu DF, Murthy VN (2005). Synaptic vesicle recycling studied in transgenic mice expressing synaptopHluorin. Proc Natl Acad Sci USA 102:6131–6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Edwards RH (1997). The role of vesicular transport proteins in synaptic transmission and neural degeneration. Annu Rev Neurosci 20:125–156. [DOI] [PubMed] [Google Scholar]
- Nishi T, Forgac M (2002). The vacuolar (H+)-ATPases—nature’s most versatile proton pumps. Nat Rev Mol Cell Biol 3:94–103. [DOI] [PubMed] [Google Scholar]
- Pothos EN, Mosharov E, Liu KP, Setlik W, Haburcak M, Baldini G, Gershon MD, Tamir H, Sulzer D (2002). Stimulation-dependent regulation of the pH, volume and quantal size of bovine and rodent secretory vesicles. J Physiol 542:453–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Piedras-Renteria ES, Tsien RW (2000). Rapid reuse of readily releasable pool vesicles at hippocampal synapses. Neuron 28:221–231. [DOI] [PubMed] [Google Scholar]
- Richards DA, Bai J, Chapman ER (2005). Two modes of exocytosis at hippocampal synapses revealed by rate of FM1–43 efflux from individual vesicles. J Cell Biol 168:929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson NC, Huang P, Kaetzel MA, Lamb FS, Nelson DJ (2004). Identification of an N-terminal amino acid of the CLC-3 chloride channel critical in phosphorylation-dependent activation of a CaMKII-activated chloride current. J Physiol 556:353–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan TA (1999). Inhibitors of myosin light chain kinase block synaptic vesicle pool mobilization during action potential firing. J Neurosci 19:1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan TA, Smith SJ, Reuter H (1996). The timing of synaptic vesicle endocytosis. Proc Natl Acad Sci USA 93:5567–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA (2000). Real-time measurements of vesicle-SNARE recycling in synapses of the central nervous system. Nat Cell Biol 2:197–204. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA (2001). Calcium accelerates endocytosis of vSNAREs at hippocampal synapses. Nat Neurosci 4:129–136. [DOI] [PubMed] [Google Scholar]
- Sonawane ND, Thiagarajah JR, Verkman AS (2002). Chloride concentration in endosomes measured using a ratioable fluorescent Cl− indicator: evidence for chloride accumulation during acidification. J Biol Chem 277:5506–5513. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Williams JH (2000). “Kiss and run” exocytosis at hippocampal synapses. Proc Natl Acad Sci USA 97:12828–12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, Bosl MR, Ruether K, Jahn H, Draguhn A, Jahn R, Jentsch TJ (2001). Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 29:185–196. [DOI] [PubMed] [Google Scholar]
- Tabb JS, Kish PE, Van Dyke R, Ueda T (1992). Glutamate transport into synaptic vesicles. Roles of membrane potential, pH gradient, and intravesicular pH. J Biol Chem 267:15412–15418. [PubMed] [Google Scholar]
- Vanoye CG, George AL Jr (2002). Functional characterization of recombinant human ClC-4 chloride channels in cultured mammalian cells. J Physiol (Lond) 539:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson RS, Cole JC (2001). Resolving the Heuser-Ceccarelli debate. Trends Neurosci 24:195–197. [DOI] [PubMed] [Google Scholar]
- Wolosker H, de Souza DO, de Meis L (1996). Regulation of glutamate transport into synaptic vesicles by chloride and proton gradient. J Biol Chem 271:11726–11731. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Hige T, Takahashi T (2005). Vesicle endocytosis requires dynamin-dependent GTP hydrolysis at a fast CNS synapse. Science 307:124–127. [DOI] [PubMed] [Google Scholar]
- Zenisek D, Steyer JA, Feldman ME, Almers W (2002). A membrane marker leaves synaptic vesicles in milliseconds after exocytosis in retinal bipolar cells. Neuron 35:1085–1097. [DOI] [PubMed] [Google Scholar]