Extracellular Cations Sensitize and Gate Capsaicin Receptor TRPV1 Modulating Pain Signaling (original) (raw)
Abstract
Transient receptor potential (TRP) channels detect diverse sensory stimuli, including alterations in osmolarity. However, a molecular detector of noxious hypertonic stimuli has not yet been identified. We show here that acute pain-related behavior evoked by elevated ionic strength is abolished in TRP vanilloid subtype 1 (TRPV1)-null mice and inhibited by iodoresiniferatoxin, a potent TRPV1 antagonist. Electrophysiological recordings demonstrate a novel form of ion channel modulation by which extracellular Na+, Mg2+, and Ca2+ ions sensitize and activate the capsaicin receptor, TRPV1. At room temperature, increasing extracellular Mg2+ (from 1 to 5 mm) or Na+ (+50 mm) increased ligand-activated currents up to fourfold, and 10 mm Mg2+ reduced the EC50 for activation by capsaicin from 890 to 450 nm. Moreover, concentrations of divalent cations >10 mm directly gate the receptor. These effects occur via electrostatic interactions with two glutamates (E600 and E648) formerly identified as proton-binding residues. Furthermore, phospholipase C-mediated signaling enhances the effects of cations, and physiological concentrations of cations contribute to the bradykinin-evoked activation of TRPV1 and the sensitization of the receptor to heat. Thus, the modulation of TRPV1 by cationic strength may contribute to inflammatory pain signaling.
Keywords: TRPV1, VR1, nociception, cations, ionic strength, pain
Introduction
Mammals vigorously defend ionic homeostasis, and alterations in ionic strength and osmolarity are often perceived as painful (Steinbrocker et al., 1953; Alessandri-Haber et al., 2003). Furthermore, elevated ionic strength (for example, MgSO4 or NaCl) is used in models of muscle and visceral “pain” (Fukawa et al., 1980; Gyires and Torma, 1984; Graven-Nielsen and Mense, 2001). Indeed, injections of MgSO4 in the clinic are reported as being intensely painful (Agarwal et al., 2004), and NaCl produces a very familiar “burning” pain when applied to injured tissue, hence the phrase “rubbing salt into wounds.” These disturbances in extracellular ion concentrations are perceived as painful because they activate nociceptive sensory neurons. However, little is known about the molecular mechanisms or identity of receptors that detect these stimuli. Understanding this process is significant not only in the context of hyperosmotic stimuli, but also because it may reveal fundamental information about general nociceptive signaling.
Elevations in extracellular ions such as NaCl may stimulate sensory neurons either directly by regulating membrane ion channels or indirectly by osmotic deformation of the cell membrane. The burning sensation evoked by salt is similar to that evoked by capsaicin, heat, extracellular protons, and ethanol, all of which are known to act through the capsaicin receptor [transient receptor potential vanilloid subtype 1 (TRPV1)] (Caterina et al., 1997; Tominaga et al., 1998; Trevisani et al., 2002). We therefore hypothesized that ionic strength may regulate the function of TRPV1. TRPV1 is a nonselective “cation” channel expressed in a subset of sensory C-fibers. Targeted deletion of the TRPV1 gene in mice has demonstrated an essential role for this ion channel in inflammatory thermal hyperalgesia (Caterina et al., 2000; Davis et al., 2000). We demonstrate here that cations gate TRPV1 directly and that this mechanism may contribute to the nociceptive response to elevated ionic strength. Moreover, we show that under inflammatory conditions, physiological levels of cations contribute to the activation of TRPV1.
Materials and Methods
Behavioral analysis. All experimental procedures were approved by the Georgetown University Animal Care and Use Committee and conformed to National Institutes of Health guidelines. Animals from the following strains were obtained from The Jackson Laboratory (Bar Harbor, ME): CBA/J, C57BL/6J, and mice lacking the TRPV1 receptor (B6.129S4-Trpv1tm1Jul/J). Mice were either bred in house or allowed at least 10 d to acclimate before behavioral analysis. Male mice 4-6 weeks of age were allowed to habituate to the testing room for 1 h and to the observation chamber for 10 min before testing. Wild-type and TRPV1 null mice received an intraperitoneal injection (10 ml/kg) of MgSO4 heptahydrate (120 mg/kg) (Mogil et al., 1999), MgCl2, CaCl2, or SrCl2 (50 mm) in distilled water. Writhing (lengthwise stretch of the torso with a concomitant concave arching of the back) was monitored over a 40 min period. To test pharmacological inhibition of TRPV1 in the MgSO4 writhing test, we used CBA mice, because this strain performs better in this assay (Mogil et al., 1999). CBA mice writhed for only ∼10 min and did not demonstrate a secondary phase, unlike C57BL/6 mice (see Fig 1 A, B). CBA/J mice received a subcutaneous injection (10 ml/kg) of either 1 μmol/kg iodoresiniferatoxin (I-RTX) or vehicle (1% ethanol in 0.9% saline) 1 h before an intraperitoneal injection (10 ml/kg) of anhydrous MgSO4. Writhing was monitored over 10 min.
Figure 1.
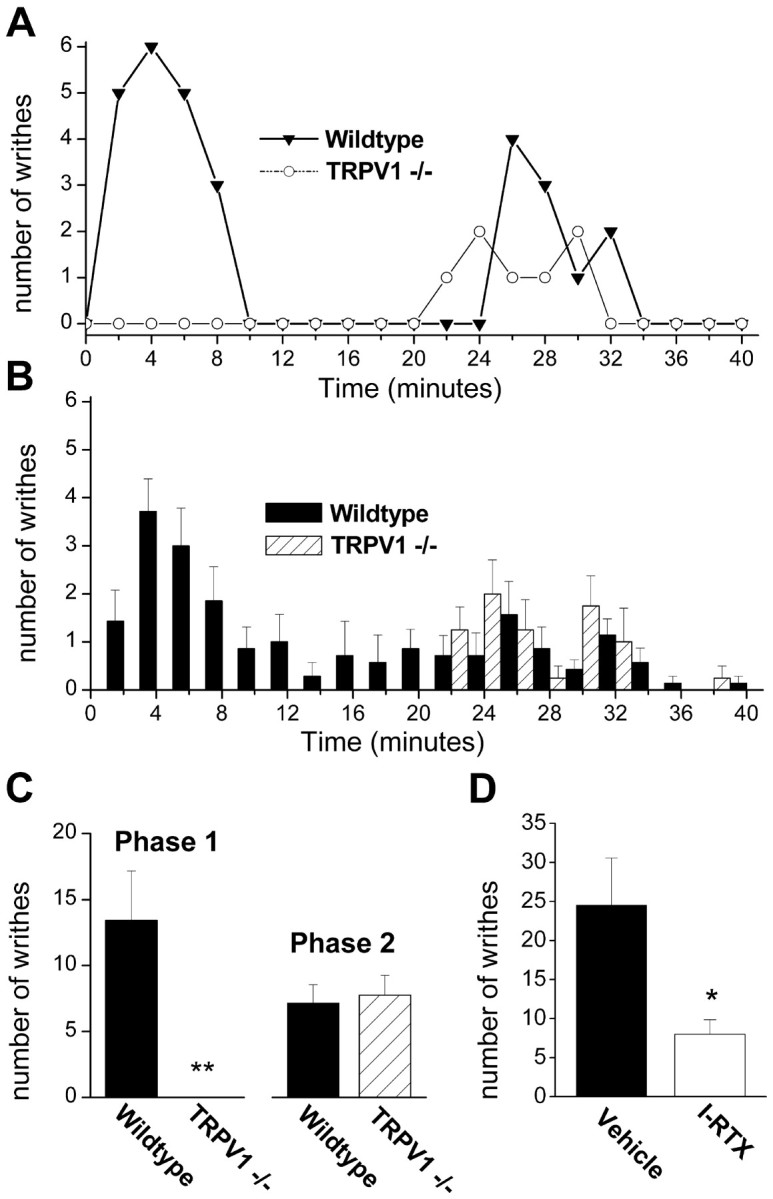
Mg2+ evokes visceral pain in mice via the activation of TRPV1. A, B, Time distribution of writhes (2 min bins) for 40 min after intraperitoneal injection of MgSO4 heptahydrate (12 mg/ml, 10 ml/kg). Responses of individual wild-type and TRPV1-/- mice (C57BL/6J) are shown in A, and mean responses of seven wild-type and four TRPV1-/- mice are shown in B. C, Mean number of writhes for wild-type and TRPV1-/- mice for the first phase (0-18 min) and second phase (18-40 min) of writhing; **p < 0.01. D, Mean number of writhes over 10 min for CBA mice after injection with anhydrous MgSO4 (12 mg/ml, 10 ml/kg; n = 3-4); *p < 0.05. Mice were preinjected (10 ml/kg, s.c.) with either 1 μmol/kg I-RTX or vehicle (1% EtOH in 0.9% saline) 1 h before MgSO4 injection. Error bars represent SEM.
Oocyte electrophysiology. Defolliculated Xenopus laevis oocytes (harvested from adult females anesthetized with 0.5 g/L tricaine methanesulfonate) were injected with ∼10 ng of wild-type, E600Q, or E648A TRPV1 cRNA (a gift from David Julius, University of California, San Francisco, San Francisco, CA). Oocytes were placed in a Perspex chamber and continuously superfused (5 ml/min) with Ca2+-free solution containing the following (in mm): 100 NaCl, 2.5 KCl, 5 HEPES, and 1 MgCl2, titrated to pH 7.3 with ∼5 mm NaOH. Iso-osmotic MgCl2 and CaCl2 solutions (200 ± 5 mOsm) contained the following (in mm): 10-70 MgCl2/CaCl2 and 0-87 NaCl, with 5 HEPES and 2.5 KCl. For solutions of pH <6.0, HEPES was replaced with 5 mm MES. Oocytes were routinely voltage clamped at -60 mV at 22-23°C. For comparison of MgCl2 and CaCl2 responses, oocytes were voltage clamped at -20 mV (the chloride reversal potential) and injected with 100 mm BAPTA (Sigma, St. Louis, MO) prepared in 10 mm HEPES (brought to pH 7.3 with KOH). BAPTA was pressure injected (20 psi) 15-60 min before the experiment with 10 pulses of 5 s duration.
For heat activation, bath temperature was raised from ∼22 to 50°C over ∼100 s using an in-line solution heater (Warner Instruments, Hamden, CT). The temperature was continuously monitored with a probe placed within 2 mm of the oocyte. The temperature-activation threshold was defined as a 10% deviation from baseline.
Human embryonic kidney cell and sensory neuron electrophysiology. Human embryonic kidney 293F (HEK293F) cells (Invitrogen, Carlsbad, CA) were cultured in DMEM supplemented with 1% nonessential amino acids and 10% fetal calf serum. Cell cultures were maintained at 37°C with 5% CO2. Cells were transfected with rat TRPV1 (a gift from David Julius) and green fluorescent protein (GFP) cDNA using Lipofectamine transfection reagent (Invitrogen) according to the instructions of the manufacturer and used 24-48 h after transfection.
Nodose ganglia were obtained from adult mice (C57BL/6J and TRPV1-null), cut, digested with collagenase, and cultured in Neurobasal plus 2% B-27 medium (Invitrogen), 0.1% l-glutamine, and 1% penicillin/streptomycin on poly-d-lysine-coated glass coverslips at 37°C in 5% CO2. Neurons were used within 24-36 h of culture.
Whole-cell and single-channel patch-clamp recordings were performed using an EPC 8 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany). The current signal was low-pass filtered at 1-3 kHz and sampled at 4 kHz. Currents were further filtered for display purposes. For whole-cell and excised patch recordings, the bath solution contained the following (in mm): 140 NaCl, 4 KCl, 1 MgCl2, 1 EGTA, 10 HEPES, and 10 glucose, pH 7.3 (290 mOsm). Divalent solutions (295 ± 5 mOsm) contained the following (in mm): 10-100 MgCl2/CaCl2/SrCl2, 0-140 NaCl, and 10 HEPES, pH 7.3. The pipette solution contained the following (in mm): 140 CsCl, 10 NaCl, 10 HEPES, 5 EGTA, 2 MgATP, and 0.03 GTP, pH 7.3. For measurements of divalent cation permeability, the bath contained the following (in mm): 110 MgCl2/CaCl2, 5 EGTA, and 10 HEPES, pH 7.3, buffered with 2 Mg(OH)2 or Ca(OH)2; the pipette solution contained the following (in mm): 150 NaCl, 5 EGTA, and 10 HEPES, pH 7.3, buffered with NaOH. Measured liquid junction potentials were <1 mV. Permeability ratios were calculated as follows: P_Y/P_Na = [Na+]i exp(Δ_V_rev_F/RT) (1 + exp(Δ_V_rev_F/RT))/4[_Y_2+]O, where Y represents a divalent cation. Ion activity coefficients were 0.75 for monovalents and 0.25 for divalents.
Chemicals. Capsazepine, phorbol-12,13-dibutyrate (PDBu), and phenylarsine oxide (PAO) were obtained from Sigma. Anandamide (AEA), capsaicin, and I-RTX were obtained from Tocris Cookson (Ellisville, MO). _N_-arachidonoyl dopamine (NADA) was obtained from Cayman Chemicals (Ann Arbor, MI), and 1-oleoyl-2-acetyl-_sn_-glycerol was from Calbiochem (La Jolla, CA). Drugs were prepared as stock solutions in DMSO or ethanol and diluted into a physiological solution before experiments.
Statistical analysis. Data are given as mean ± SEM, and statistical significance was evaluated using unpaired Student's t test.
Results
TRPV1 is essential for acute Mg2+-induced pain
Intraperitoneal infusion of MgSO4 elicits writhing in mice (Gyires and Torma, 1984), and we investigated whether TRPV1 is necessary for this response. As reported previously (Mogil et al., 1999), intraperitoneal injection of MgSO4 (120 mg/kg) evoked writhing responses in wild-type mice (C57BL/6J) that occurred shortly after injection with a duration of ∼5-10 min (Fig. 1_A-D_). This effect was independent of osmolarity, because we observed similar effects with the heptahydrate and anhydrous solutions of MgSO4 in water (∼75-110 mOsm) or 0.9% saline (345 mOsm). In addition to this acute writhing response, we identified a slower secondary phase of writhing with an onset ∼20 min after injection that may represent a non-neurogenic response. Strikingly, we found that the acute phase of writhing was completely absent in TRPV1-/- mice, whereas the latter phase was unaffected (Fig. 1_A-C_). Moreover, this acute writhing was inhibited by the specific TRPV1 antagonist iodoresiniferatoxin (Wahl et al., 2001) (Fig. 1_D_). Thus, these data show that TRPV1 is essential for the acute noxious effects of Mg2+ and are consistent with a noninflammatory mechanism; writhing after MgSO4 injection is not inhibited by nonsteroidal anti-inflammatory drugs and is not accompanied by an increase in prostaglandins in the peritoneum (Gyires and Torma, 1984).
Extracellular Mg2+ and Ca2+ ions directly gate TRPV1 expressed in HEK293 cells and oocytes
Elevated extracellular Mg2+ may directly stimulate TRPV1 or, alternatively, indirectly activate the channel by releasing a TRPV1 ligand. To examine the first possibility, we tested the effects of raised [Mg2+] in TRPV1-expressing HEK293 cells. Figure 2_A_ shows that iso-osmotic Mg2+ (30 and 100 mm) activated inward currents in a dose-dependent manner. Similarly, elevated Ca2+ (30 and 100 mm) evoked inward currents; however, in contrast to Mg2+, these currents often decayed rapidly during the course of Ca2+ application (Fig. 2_B_). This fast inactivation occurred in all cells treated with 100 mm Ca2+ (11 of 11 cells) and in a subset of cells challenged with 30 mm Ca2+ (3 of 12 cells) and is consistent with Ca2+-mediated desensitization of TRPV1 (Caterina et al., 1997; Koplas et al., 1997). The TRPV1 inhibitor capsazepine (5 μm) blocked these cation-activated currents by ∼70-80% (Fig. 2_A,B_) (n = 6), to an extent similar to that of “proton”-gated currents (pH 5.5; 64 ± 5%; n = 4). This latter finding is in agreement with the electrophysiological data of Tominaga et al. (1998) but at variance with McIntyre et al. (2001), who reported that capsazepine minimally blocked proton-evoked calcium responses with rat TRPV1. Furthermore, little to no cation-gated current was observed in cells transfected with GFP alone (<1 pA/pF; n = 6). Together, these data confirm that TRPV1 is required for responses to cations. Figure 2_C_ shows the current-voltage relationships of cation-activated currents under bi-ionic conditions compared with 100 nm capsaicin in standard buffer; mean reversal potentials of +18 ± 2 and +31 ± 2 mV (n = 4) for 110 Mg2+ and 110 Ca2+, respectively, yield a _P_Mg/_P_Na of 6.1 and a _P_Ca/_P_Na of 14.8. These values are slightly greater than those reported previously for capsaicin-activated currents (Caterina et al., 1997).
Figure 2.
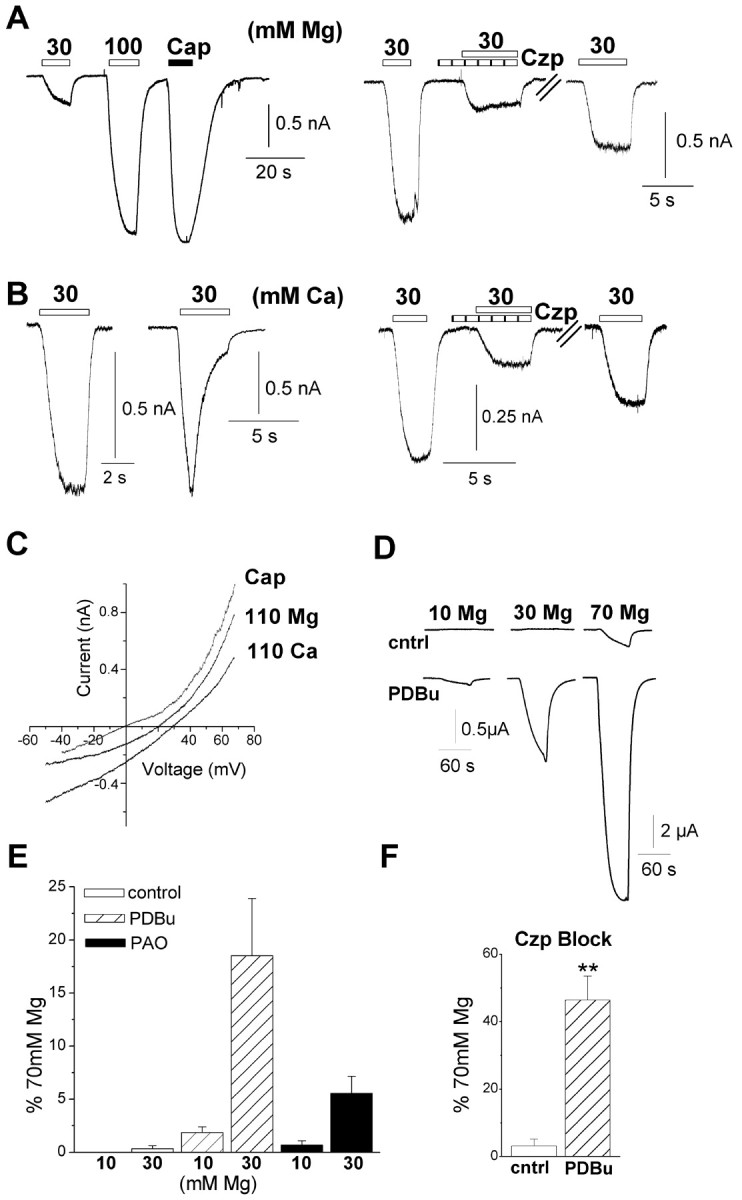
Extracellular cations activate TRPV1 currents in HEK293 cells and oocytes. A, HEK293 cells were voltage clamped at -50 mV in a nominally Ca2+-free medium with a CsCl pipette solution. Representative currents evoked by MgCl2 (30 and 100 mm) and capsaicin (Cap; 100 nm) in TRPV1-expressing HEK293 cells (left) and reversible inhibition with capsazepine (Czp; 5 μm; right). B, Representative currents evoked by 30 mm CaCl2 (left) that exhibit noninactivating (9 of 12 cells) and inactivating (3 of 12 cells) responses and reversible inhibition by capsazepine (5 μm; right). C, Current-voltage relationship for responses evoked by MgCl2 and CaCl2 (110 mm) in a single HEK293 cell compared with the response evoked by 100 nm capsaicin in control solution from a different cell (the baseline current under control conditions is subtracted from each trace). The mean reversal potentials for MgCl2 and CaCl2 from four cells were +18 ± 2 and +31 ± 2 mV, respectively. D, Currents in TRPV1-expressing oocytes evoked by 10, 30, and 70 mm MgCl2 before and after 3 min treatment with 100 nm PDBu. Note the different scale for the bottom right trace. E, Mean current evoked by 10 and 30 mm Mg2+ normalized to the current evoked by 70 mm concentrations, under control conditions (open bar) or after pretreatment with 100 nm PDBu (striped bar) and 30 μm PAO (filled bar). Data are mean of three to five oocytes. F, Currents evoked by 70 mm MgCl2 are inhibited by capsazepine (5 μm) under control conditions (cntrl, n = 4) and, to a lesser extent, after PDBu treatment (n = 5; **p < 0.001 PDBu compared with control). Error bars represent SEM.
A similar, if somewhat less potent, activation of TRPV1 was observed in the oocyte expression system in response to iso-osmotic (∼200 mOsm) MgCl2 solutions ranging from 10 to 70 mm. Under resting conditions, 70 mm Mg2+ alone evoked large inward currents (Fig. 2_D_). However, currents were seen at all Mg2+ concentrations after stimulation of protein kinase C (PKC) with PDBu, which is known to sensitize TRPV1 to agonists (Crandall et al., 2002; Ahern, 2003) (Fig. 2_D,E_). We obtained equivalent results using the cell-permeable diacylglycerol analog 1-oleoyl-2-acetyl-_sn_-glycerol to stimulate PKC (data not shown). These Mg2+-evoked currents were blocked by capsazepine (5 μm), although the block was less effective after PKC stimulation (Fig. 2_F_) (p < 0.001). Furthermore, no currents were seen in water-injected oocytes, confirming a specific activation of TRPV1. Another pathway known to augment TRPV1 responses is the hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2) (Chuang et al., 2001). We observed that treating oocytes with PAO to deplete PIP2 (Prescott and Julius, 2003) sensitized TRPV1 to Mg2+ in a manner similar to that of PKC (Fig. 2_E_). Thus, these data show that two signaling pathways downstream of phospholipase C (PLC) activation (PIP2 depletion and PKC stimulation) sensitize TRPV1 to direct activation by cations.
Finally, we tested whether high concentrations of monovalent or trivalent cations could directly gate TRPV1. LiCl (100 mm) activated outwardly rectifying currents (1 ± 0.7% of 10 μm capsaicin; n = 5) that were enhanced after PKC stimulation (11 ± 5% of 10 μm capsaicin; n = 5), but NaCl at concentrations up to 400 mm was without effect in both oocytes and HEK293 cells (data not shown). This lack of effect with hypertonic NaCl is consistent with a previous report (Caterina et al., 1997). Submillimolar concentrations of trivalent cations Gd3+ and La3+ completely blocked capsaicin-evoked currents (n = 3-4), suggesting that these cations produce an open-channel block. Thus, cation activation of TRPV1 requires cation permeation and appears to correlate with charge and charge density (Ca2+ = Mg2+ ≫ Li+ ≫ Na+).
Mg2+ and Ca2+ ions directly gate TRPV1 in sensory neurons
To test whether cations regulated native TRPV1 channels, we turned to voltage-clamped sensory neurons. We found that 10-100 mm Mg2+ and Ca2+ evoked large inward currents in capsaicin-sensitive neurons (Fig. 3_B-D_) (n = 23) but not in capsaicin-insensitive neurons (Fig. 3_A_, top) (<0.1 nA; n = 13). Similarly, no cation-evoked currents were seen in neurons cultured from TRPV1-null mice (Fig. 3_A_, bottom) (n = 6). All of these neurons exhibited ATP-evoked currents reflecting expression of P2X channels and confirming their status as nociceptors. Figure 3_D_ shows that Mg2+ (10-100 mm) acted in a dose-dependent manner, with 100 mm concentrations eliciting a slightly greater response than 100 nm capsaicin. Capsazepine (5 μm) inhibited these Mg2+ evoked currents by ∼90% (Fig. 3_D_). These responses are in agreement with those observed in HEK293 cells and oocytes. This indicates that, in sensory neurons, Mg2+ and Ca2+ primarily act at TRPV1 with little contribution from other cation-sensing receptors.
Figure 3.

Mg2+ and Ca2+ directly activate TRPV1 in sensory neurons. Neurons were voltage clamped at -60 mV in a nominally Ca2+-free medium with a CsCl pipette solution. A, Response of a capsaicin (Cap)-insensitive neuron (top) to 100 mm Mg2+, 100 mm Ca2+, and 100 nm capsaicin and response of a neuron cultured from a TRPV1-null mouse (bottom) to 100 mm Mg2+ and 100 μm ATP. B, Mg2+ (30 and 100 mm)-evoked inward currents in a neuron that responded to capsaicin (100 nm). C, Ca2+ (100 mm)-evoked currents that exhibited desensitization. D, Dose-dependent activation of currents by 30-100 mm Mg2+ (n = 4-9 for each point) and comparison with 100 nm capsaicin (n = 14; left). Block of 30 mm Mg2+ with 5 μm capsazepine (Czp; n = 3; right); **p < 0.01. E, Mg2+ (10 mm)-activated TRPV1 channels in an outside-out patch (holding potential, +50 mV). Error bars represent SEM.
Direct cation activation of TRPV1 was observed at the single-channel level in cell-free patches (n = 6). Figure 3_E_ shows that 10 mm Mg2+ activated single channels at +50 mV that were identified as TRPV1 by subsequent sensitivity to capsaicin (data not shown). High [Mg2+] also caused a small reduction in single-channel conductance at positive potentials (from ∼90 to 80 pS). Thus, cations can activate TRPV1 in a membrane-delimited manner, and this is consistent with cations interacting directly with extracellular glutamate residues (see Fig. 7).
Figure 7.
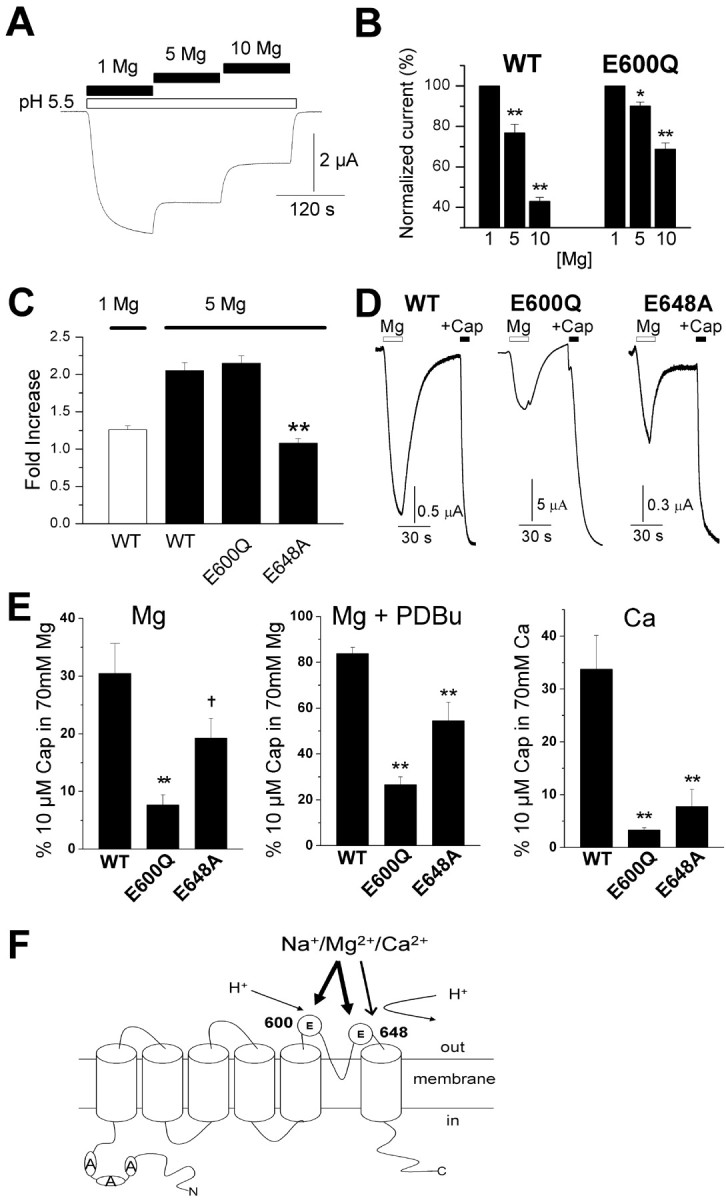
Cations regulate TRPV1 via extracellular proton-binding sites. A, B, Increasing [Mg2+]o from 1 to 10 mm dose-dependently inhibited the current evoked by protons (pH 5.5) in wild-type (WT) receptors (n = 6-7). A smaller reduction was observed in E600Q receptors (n = 3; *p < 0.05 and **p < 0.01 compared with 1 mm Mg control). E648A is very insensitive to proton activation and therefore was not tested. C, Mg2+ potentiation of currents evoked by capsaicin in oocytes expressing wild-type, E600Q, and E648A TRPV1. The capsaicin concentrations used were 500 nm for WT and E648 and 25 nm for E600Q and were selected based on the respective EC50 values of these receptors (Jordt et al., 2000). The mean fold potentiation is shown for three to four oocytes (**p < 0.01 compared with WT). D, Currents evoked by 70 mm MgCl2 and 10 μm capsaicin (Cap) in 70 mm MgCl2 solution in oocytes expressing wild-type, E600Q, and E648A receptors after phosphorylation of PKC with 100 nm PDBu. E, Currents from three to nine oocytes were normalized to the response evoked by 10 μm capsaicin in 70 mm Mg2+ or 70 mm Ca2+ solution (used to control for changes in Mg2+ or Ca2+ permeability; †p < 0.06 and **p < 0.01 compared with WT). Oocytes in D and E were held at -20 mV (Cl- reversal potential found for these oocytes) and injected with 100 mm BAPTA 15-60 min before recording. F, Model of cation-mediated modulation of TRPV1 at glutamate (E) residues. Sensitization (arrow) occurs selectively via interactions at E648, whereas activation (thick arrow) requires interaction with both E600 and E648. This model also accounts for cation inhibition of H+ activation (thin arrow). Error bars represent SEM.
Cation-induced writhing correlates with activation of TRPV1
Our data show that divalent cations (>10 mm) directly activate TRPV1 and suggest a mechanism for Mg2+-induced writhing. However, it remains possible that other cation-sensitive pathways contribute to writhing. For example, millimolar concentrations of divalent cations are known to activate the Ca2+-sensing receptor (CaR), a G-protein-coupled receptor that regulates Ca2+ homeostasis (Hofer and Brown, 2003). In turn, activation of CaR or another cation-sensitive pathway could indirectly lead to activation of TRPV1. Therefore, to examine whether cation writhing is directly related to the activation of TRPV1, we tested whether the magnitude of acute writhing correlated with the activation of TRPV1 currents. Figure 4_A_ shows representative responses of voltage-clamped sensory neurons to 100 mm concentrations of MgCl2, CaCl2, or SrCl2. As described above, Mg2+ produced a sustained inward current, whereas Ca2+ evoked a rapidly desensitizing response. In addition, Sr2+ evoked somewhat smaller desensitizing currents (similar responses were seen in TRPV1-expressing HEK293 cells). Thus, high concentrations of Sr2+ can activate TRPV1, but, like Ca2+, also promote TRPV1 desensitization. Next, we compared the acute writhing responses induced by intraperitoneal injections of these cations. Figure 4_B_ shows that Ca2+ and Sr2+ elicited significantly less writhing than Mg2+, consistent with the reduced ability of these cations to activate sustained TRPV1 current. In contrast, all of these cations activated the second phase of writhing in a statistically indistinguishable manner (data not shown). These concentrations of Ca2+ and Sr2+ are sufficient to robustly activate CaR (Hofer and Brown, 2003; Coulombe et al., 2004). Thus, the acute phase of writhing evoked by cations corresponds with the direct activation of TRPV1 and not via another cation sensing pathway.
Figure 4.

Cation-induced writhing behavior correlates with activation of TRPV1. A, Representative currents evoked by 100 mm MgCl2, CaCl2, and SrCl2. Mg2+ produces sustained inward currents; in contrast, both Ca2+ and Sr2+ elicit transient, desensitizing responses. B, Mean number of writhes within 16 min (first phase) after intraperitoneal injection (10 ml/kg) of 50 mm MgCl2, CaCl2, or SrCl2 (*p < 0.05 and **p < 0.001 compared with MgCl2). Error bars represent SEM.
Cations sensitize TRPV1 currents in oocytes and neurons
We next asked whether monovalent or divalent cations at concentrations below the threshold for direct activation (<10 mm) could modulate agonist-evoked TRPV1 responses. Elevated extracellular MgCl2 (1-10 mm) enhanced the capsaicin-activated current by up to 4.4-fold (Fig. 5_A,C,E_) (p < 0.05; n = 17), whereas NaCl (+50 mm) increased the current 2.3-fold (Fig. 5_A,B_) (n = 9; p < 0.05). The effects of raised NaCl were not merely attributable to an increase in driving force (∼10 mV), because the current was increased at all potentials (Fig. 5_B_). It was also not attributable to changes in osmolarity; supplementing the extracellular solution with 100 mm mannitol did not alter the capsaicin-evoked response (data not shown). Similarly, in sensory neurons, increasing Mg2+ from 1 to 5 mm enhanced the current activated by capsaicin (100 nm) by 4.1 ± 0.3-fold (n = 5). We also found that the addition of 50 mm concentrations of the alkali metal cations Li+ (Fig. 5_A_) and Cs+ enhanced the capsaicin-activated current by 3.1 ± 0.3-fold (n = 3; p < 0.05) and 2.1 ± 0.2-fold (n = 4; p < 0.05), respectively, with both inducing an ∼10-11 mV shift in reversal potential similar to Na+. These experiments demonstrate that capsaicin-activated TRPV1 currents are enhanced by cationic strength.
Figure 5.

Extracellular cations enhance TRPV1 currents in oocytes. A, Currents evoked by successive 60 s applications of 500 nm capsaicin (Cap) in normal buffer (100 mm NaCl/1 mm MgCl2) or buffer supplemented with 50 mm NaCl, 4 mm MgCl2, or 50 mm LiCl. Traces shown are from four separate oocytes. Calibration, 2 μA. B, Current-voltage relationships for TRPV1 in oocytes activated by 250 nm capsaicin before and after the addition of 50 mm NaCl. C, Current-voltage relationship for TRPV1 activated by 100 nm capsaicin with 1, 5, and 10 mm Mg2+ in the extracellular solutions. D, Dose-response relationship for capsaicin-activated currents in 1 or 10 mm Mg2+. Each point is the mean of four to eight oocytes, and the smooth lines are best fits to a Hill function. EC50 values were 889 ± 45 and 454 ± 63 μm, respectively. E, Mean potentiation of TRPV1 currents by 5 mm Mg2+ in oocytes activated with capsaicin (100 nm; n = 4), anandamide (15 μm; n = 5), NADA (5 μm; n = 4), and pH 5.5 (n = 6; * p < 0.05 and ** p < 0.01 compared with control). Error bars represent SEM.
Subsequently, we asked whether extracellular cations might also modulate the action of endogenous TRPV1 agonists, including AEA (Zygmunt et al., 1999), NADA (Huang et al., 2002), and protons (Bevan and Geppetti, 1994; Tominaga et al., 1998). As with capsaicin, elevated [Mg2+] elicited robust twofold to threefold increases in currents evoked by AEA and NADA (Fig. 5_E_). In contrast, elevated Mg2+, pH 5.5, blocked the current activated by protons (Fig. 5_E_). Thus, ionic strength appears to selectively increase the sensitivity of TRPV1 to ligands acting at the intracellular capsaicin binding site(s) (Jordt and Julius, 2002). Indeed, dose-response curves show that raising [Mg2+] from 1 to 10 mm increased TRPV1 sensitivity to capsaicin, shifting the EC50 from 890 to 450 nm (Fig. 5_D_).
Physiological concentrations of cations contribute to bradykinin and heat-evoked TRPV1 activation
Our data show that a modest increase in [Mg2+](4 mm) is capable of sensitizing TRPV1 to a variety of agonists. In addition, cation regulation of TRPV1 is enhanced by PLC signaling (Fig. 2_D,E_). We therefore asked whether normal concentrations of cations can contribute to the activation of TRPV1 and, in particular, under conditions that simulate inflammatory pain signaling. The inflammatory mediator bradykinin (BK) plays a key role in signaling during tissue injury. BK sensitizes and activates TRPV1 currents via PLC, PKC, and lipoxygenase-derived products (Premkumar and Ahern, 2000; Chuang et al., 2001; Shin et al., 2002; Sugiura et al., 2002). We tested the response of sensory neurons to brief applications of BK (100 nm) in either a 1 or 3 mm MgCl solution, which approximates the total physiological concentration of divalent cations (i.e., ∼2 mm Ca2+ and 1 mm Mg2+). Figure 6, A and B, shows that BK evoked substantially greater responses in 3 mm compared with 1 mm Mg2+ solutions, and this was apparent in both whole-cell (Fig. 6_A_) and single-channel (Fig. 6_B_) recordings from three separate neurons.
Figure 6.

Physiological cation concentrations enhance TRPV1 activity during inflammatory conditions. A, B, MgCl2 (3 mm) enhances BK (100 nm)-evoked activation of TRPV1 in a whole-cell recording (-60 mV) and in an outside-out patch (+50 mV). C-E, Mg2+ lowers the TRPV1 temperature threshold in oocytes. Current versus temperature relationships are plotted for TRPV1-expressing oocytes in solutions containing 1-10 mm MgCl2 before (C) and after PDBu treatment (D). The mean temperature thresholds of three to seven oocytes are shown under stated conditions (**p < 0.001 compared with 1 mm Mg control).
TRPV1 is a heat-gated channel (Cesare and McNaughton, 1996; Caterina et al., 1997), and a reduction in the temperature threshold for activation by PLC signaling is considered a hall-mark of inflammatory hyperalgesia. We therefore explored the effects of cations on heat activation of TRPV1 expressed in ooctyes (Fig. 6_C-E_). Under control conditions, 10 mm Mg2+ did not significantly alter the temperature threshold of ∼47°C (Fig. 6_C,E_). However, after the stimulation of PKC, the threshold decreased in an Mg2+-dependent manner (Fig. 6_D,E_). Significantly, a marked sensitization occurred with 3 mm Mg2+ (∼40°C). Importantly, these experiments with bradykinin and heat suggest that physiological concentrations of divalent cations can contribute to the sensitization and activation of TRPV1 during inflammatory pain signaling.
Cations act at TRPV1 proton-binding residues E600 and E648
We next sought to determine the molecular mechanism(s) underlying cation modulation of TRPV1. Elevations in cation concentrations (in particular divalents) will change the membrane surface potential, and it is possible that this might activate/sensitize TRPV1 via its voltage-sensitive properties (Ahern and Premkumar, 2002; Voets et al., 2004). However, the reduction in membrane surface potential produced by cations would oppose TRPV1 voltage activation.
Because elevated Mg2+ failed to augment currents evoked by protons (pH 5.5) (Fig. 5_E_), we asked whether cations modulate TRPV1 by interacting at proton-binding sites. To address this possibility, we tested whether Mg2+ could effectively compete with protons for channel activation. Indeed, increasing [Mg2+] from 1 to 10 mm produced a dose-dependent block of proton-evoked current (Fig. 7_A,B_), whereas capsaicin-evoked currents were enhanced over the same range of [Mg2+] (Fig. 5). To confirm an action at proton-binding sites, we used mutant receptors lacking acidic extracellular amino acids identified as a key for proton activation and modulation. E600 is believed to be important for proton sensitization of agonist-evoked responses, whereas E648 is important for direct proton activation (Jordt et al., 2000).
First, we tested the responses of these mutants to sensitization by cations. Mutation of E600 (E600Q) did not significantly affect the Mg2+ enhancement of capsaicin-evoked currents (Fig. 7_C_). Furthermore, these receptors still exhibited Mg2+ blockade of proton currents (Fig. 7_B_), although the effect was smaller compared with wild-type receptors. In contrast, potentiation by Mg2+ was completely inhibited by mutation of E648 (E648A) (Fig. 7_C_). (Note that these receptors are not activated by protons, and we therefore could not test Mg2+ blockade of proton currents.) Second, we tested whether mutants retained the ability to be directly activated by Ca2+ and Mg2+. Mutation of E600 inhibited Ca2+- and Mg2+-evoked currents (Fig. 7_D,E_) without affecting responses to 10 μm capsaicin (applied with 70 mm MgCl2 or CaCl2). Mutation of E648 substantially inhibited Ca2+-evoked currents. This mutation also reduced the current activated by Mg2+, this being significant after PKC stimulation (note that PDBu treatment was used here to reduce variability in the control data). Thus, these data support a model (Fig. 7_F_) whereby E600 and, to a lesser degree, E648 mediate cation activation of TRPV1, whereas E648 selectively supports cation sensitization of agonist-evoked responses. Interestingly, this is the converse of the response seen with protons, in which E600 mediates proton sensitization of agonist-mediated currents (Jordt et al., 2000). Like protons, activation by cations may occur via charge neutralization of these residues. Divalent cations are presumably more potent agonists, because they impart a greater net positive charge at these sites. This is consistent with observations that substitution of acidic residues with basic residues can produce constitutively open channels (Jordt et al., 2000).
Discussion
Cations directly gate TRPV1
Our results demonstrate that extracellular cations can directly gate and sensitize the TRPV1 channel. This represents a new mechanism for modulating TRPV1 function and extends the list of TRPV1 activators that includes heat, protons, capsaicin (Caterina et al., 1997; Tominaga et al., 1998), and several lipid metabolites: eicosanoids (Hwang et al., 2000), AEA (Zygmunt et al., 1999), NADA (Huang et al., 2002), and oleoylethanolamide (Ahern, 2003). Cation gating of TRPV1 appears to be unique. Although the brain NaX channel is gated by monovalent sodium ions (Hiyama et al., 2002), we are not aware of a previous report that describes a channel being gated by extracellular divalent cations. Several ion channels are instead known to be sensitized by cations, including P2X3 receptors (Cook et al., 1998), nicotinic acetylcholine receptors (Mulle et al., 1992), and acid-sensitive ion channels (Babini et al., 2002; Immke and McCleskey, 2003). Interestingly, TRPV1 shares functional homology not with an ion channel but with a G-protein-coupled receptor, the Ca2+-sensing receptor, in that it is directly activated by Ca2+ and Mg2+ ions (Hofer and Brown, 2003). This form of activation expands the repertoire of sensory modalities detected by the TRP channel family. The TRPV4 channel is gated by hypotonic stimuli and may transduce hypotonic nociception (Alessandri-Haber et al., 2003). Conversely, TRPV1 may transduce a subset of hypertonic nociception.
Cations interact with extracellular glutamate residues
We showed that cations modulate TRPV1 by interacting with two glutamates (E600 and E648) located between transmembrane domains 5 and 6 near the channel pore. Sensitization with Mg2+ was occluded in the E648A mutant, whereas activation by Mg2+ and Ca2+ was inhibited in both E600Q and E648A mutants. However, the responses to Ca2+ were more markedly inhibited compared with Mg2+, suggesting that additional acidic residues may also contribute to Mg2+ activation. E600 and E648 have been implicated previously in the activation and sensitization of TRPV1 by protons (Jordt et al., 2000). Acidosis to pH 6 sensitizes TRPV1 to agonists including heat and capsaicin, whereas acidosis <pH 6 directly gates the receptor. Similarly, extracellular cations sensitize and directly gate TRPV1 in a concentration- and charge-dependent manner. At room temperature, 1-10 mm divalent cations sensitize TRPV1, whereas divalent cations >10 mm directly gate the receptor. Monovalent cations require concentration increases of ∼50 mm to exert significant effects on activity. Nevertheless, all of these processes are likely to be enhanced at 37°C, given the temperature dependence of TRPV1 gating. Although proton regulation of TRPV1 is likely to be manifest only during tissue acidosis, sensitization of TRPV1 by cations can occur under normal physiological conditions. Thus, extracellular glutamates might serve interchangeably as both proton- and cation-binding sites. Indeed, we found that cations could inhibit proton-gated responses presumably because of competitive binding at these sites. However, a significant block of proton responses only occurred at 5-10 mm Mg2+, suggesting that this effect will not play a general role under normal physiological conditions. Interestingly, competitive cation-proton interactions also occur in acid-sensitive ion channels (Immke and McCleskey, 2003) and may subserve gating of these channels.
TRPV1 may transduce nociceptive responses to elevated ionic strength
Elevations in ionic strength are often perceived as painful and are used in models of visceral and muscle pain (Fukawa et al., 1980; Gyires and Torma, 1984; Graven-Nielsen and Mense, 2001). Cation regulation of TRPV1 may explain why high concentrations of exogenous Mg2+, salt, and seawater (∼11 mm Ca2+, 55 mm Mg2+, and 460 mm NaCl) cause pain and why this response is more marked in the setting of tissue injury. Moreover, concentrations of cations sufficient to directly gate TRPV1 are found in various tissues and extracellular microenvironments. For example, sensory neurons innervate the epithelial tissues of skin and airways that commonly encounter elevated ionic strength (Anderson, 1984), as well as bone, in which the [Ca2+] surrounding resorbing osteoclasts approaches 20 mm (Silver et al., 1988). TRPV1 activation in these settings may contribute to the pain of exercise-induced asthma (Anderson, 1984; Wiens et al., 1992) and bone cancer (Walls et al., 1995). In addition, cation regulation of TRPV1 may participate in non-nociceptive functions. In the urinary bladder, TRPV1 channels are present in both sensory nerves and the bladder epithelium, in which they contribute to voiding behavior (Birder et al., 2001, 2002). Interestingly, in the tongue, TRPV1-positive fibers innervate the taste papillae (Ishida et al., 2002; Kido et al., 2003), and amiloride-insensitive salt responses in the chorda tympani are abolished in TRPV1-null animals (Lyall et al., 2004). Thus, TRPV1 or a splice variant may transduce some forms of salt taste perception.
Cation sensitization of TRPV1 may contribute to pain signaling
Although direct activation of TRPV1 may occur under extreme or pathophysiological cation concentrations, a more significant effect of cations may occur under normal physiological conditions. Our data show that millimolar increases in cation concentations (1-5 mm) are sufficient to sensitize TRPV1 to various ligands including capsaicin, AEA, and NADA. This sensitization is quite marked; in sensory neurons, Mg2+ produced an approximately fourfold increase in capsaicin-evoked current, whereas in oocytes, increasing Mg2+ from 1 to 10 mm halved the capsaicin EC50, equivalent to the shift produced by acidosis (pH 6.4) (Tominaga et al., 1998). In addition, we demonstrate that physiological concentrations of cations are capable of regulating TRPV1 in the setting of tissue injury and inflammation. Tissue damage leads to the generation of chemical mediators, such as bradykinin, which activate and sensitize TRPV1 (Premkumar and Ahern, 2000; Chuang et al., 2001). In turn, this manifests as a marked reduction in the threshold for heat activation (Cesare and McNaughton, 1996; Tominaga et al., 2001; Vellani et al., 2001) and accounts for inflammatory thermal hyperalgesia in animals (Caterina et al., 2000; Davis et al., 2000). Importantly, our data show that normal levels of divalent cations contribute to both of these processes. Thus, cations may act in a constitutive manner to regulate TRPV1 sensitivity. Even small changes in cation concentrations may therefore influence nociception by modulating the magnitude of TRPV1 responses.
Footnotes
We thank David Julius for kindly providing us with cDNA for wild-type and mutant (E600Q and E648A) rat TRPV1.
Correspondence should be addressed to Gerard Ahern at the above address. E-mail: gpa3@georgetown.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255109-08$15.00/0
References
- Agarwal A, Dhiraj S, Raza M, Pandey R, Pandey CK, Singh PK, Singh U, Gupta D (2004) Vein pretreatment with magnesium sulfate to prevent pain on injection of propofol is not justified. Can J Anaesth 51: 130-133. [DOI] [PubMed] [Google Scholar]
- Ahern GP (2003) Activation of TRPV1 by the satiety factor oleoylethanolamide. J Biol Chem 278: 30429-30434. [DOI] [PubMed] [Google Scholar]
- Ahern GP, Premkumar LS (2002) Voltage-dependent priming of rat vanilloid receptor: effects of agonist and protein kinase C activation. J Physiol (Lond) 545: 441-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessandri-Haber N, Yeh JJ, Boyd AE, Parada CA, Chen X, Reichling DB, Levine JD (2003) Hypotonicity induces TRPV4-mediated nociception in rat. Neuron 39: 497-511. [DOI] [PubMed] [Google Scholar]
- Anderson SD (1984) Is there a unifying hypothesis for exercise-induced asthma? J Allergy Clin Immunol 73: 660-665. [DOI] [PubMed] [Google Scholar]
- Babini E, Paukert M, Geisler HS, Grunder S (2002) Alternative splicing and interaction with di- and polyvalent cations control the dynamic range of acid-sensing ion channel 1 (ASIC1). J Biol Chem 277: 41597-41603. [DOI] [PubMed] [Google Scholar]
- Bevan S, Geppetti P (1994) Protons: small stimulants of capsaicin-sensitive sensory nerves. Trends Neurosci 17: 509-512. [DOI] [PubMed] [Google Scholar]
- Birder LA, Kanai AJ, de Groat WC, Kiss S, Nealen ML, Burke NE, Dineley KE, Watkins S, Reynolds IJ, Caterina MJ (2001) Vanilloid receptor expression suggests a sensory role for urinary bladder epithelial cells. Proc Natl Acad Sci USA 98: 13396-13401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birder LA, Nakamura Y, Kiss S, Nealen ML, Barrick S, Kanai AJ, Wang E, Ruiz G, De Groat WC, Apodaca G, Watkins S, Caterina MJ (2002) Altered urinary bladder function in mice lacking the vanilloid receptor TRPV1. Nat Neurosci 5: 856-860. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389: 816-824. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D (2000) Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288: 306-313. [DOI] [PubMed] [Google Scholar]
- Cesare P, McNaughton P (1996) A novel heat-activated current in nociceptive neurons and its sensitization by bradykinin. Proc Natl Acad Sci USA 93: 15435-15439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D (2001) Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411: 957-962. [DOI] [PubMed] [Google Scholar]
- Cook SP, Rodland KD, McCleskey EW (1998) A memory for extracellular Ca2+ by speeding recovery of P2X receptors from desensitization. J Neurosci 18: 9238-9244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe J, Faure H, Robin B, Ruat M (2004) In vitro effects of strontium ranelate on the extracellular calcium-sensing receptor. Biochem Biophys Res Commun 323: 1184-1190. [DOI] [PubMed] [Google Scholar]
- Crandall M, Kwash J, Yu W, White G (2002) Activation of protein kinase C sensitizes human VR1 to capsaicin and to moderate decreases in pH at physiological temperatures in Xenopus oocytes. Pain 98: 109-117. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA (2000) Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405: 183-187. [DOI] [PubMed] [Google Scholar]
- Fukawa K, Kawano O, Hibi M, Misaki N, Ohba S, Hatanaka Y (1980) A method for evaluating analgesic agents in rats. J Pharmacol Methods 4: 251-259. [DOI] [PubMed] [Google Scholar]
- Graven-Nielsen T, Mense S (2001) The peripheral apparatus of muscle pain: evidence from animal and human studies. Clin J Pain 17: 2-10. [DOI] [PubMed] [Google Scholar]
- Gyires K, Torma Z (1984) The use of the writhing test in mice for screening different types of analgesics. Arch Int Pharmacodyn Ther 267: 131-140. [PubMed] [Google Scholar]
- Hiyama TY, Watanabe E, Ono K, Inenaga K, Tamkun MM, Yoshida S, Noda M (2002) Na(x) channel involved in CNS sodium-level sensing. Nat Neurosci 5: 511-512. [DOI] [PubMed] [Google Scholar]
- Hofer AM, Brown EM (2003) Extracellular calcium sensing and signalling. Nat Rev Mol Cell Biol 4: 530-538. [DOI] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, Tognetto M, Petros TJ, Krey JF, Chu CJ, Miller JD, Davies SN, Geppetti P, Walker JM, Di Marzo V (2002) An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci USA 99: 8400-8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, Cho S, Min KH, Suh YG, Kim D, Oh U (2000) Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc Natl Acad Sci USA 97: 6155-6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immke DC, McCleskey EW (2003) Protons open acid-sensing ion channels by catalyzing relief of Ca2+ blockade. Neuron 37: 75-84. [DOI] [PubMed] [Google Scholar]
- Ishida Y, Ugawa S, Ueda T, Murakami S, Shimada S (2002) Vanilloid receptor subtype-1 (VR1) is specifically localized to taste papillae. Brain Res Mol Brain Res 107: 17-22. [DOI] [PubMed] [Google Scholar]
- Jordt SE, Julius D (2002) Molecular basis for species-specific sensitivity to “hot” chili peppers. Cell 108: 421-430. [DOI] [PubMed] [Google Scholar]
- Jordt SE, Tominaga M, Julius D (2000) Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc Natl Acad Sci USA 97: 8134-8139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kido MA, Muroya H, Yamaza T, Terada Y, Tanaka T (2003) Vanilloid receptor expression in the rat tongue and palate. J Dent Res 82: 393-397. [DOI] [PubMed] [Google Scholar]
- Koplas PA, Rosenberg RL, Oxford GS (1997) The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. J Neurosci 17: 3525-3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyall V, Heck GL, Vinnikova AK, Ghosh S, Phan TH, Alam RI, Russell OF, Malik SA, Bigbee JW, DeSimone JA (2004) The mammalian amiloride-insensitive non-specific salt taste receptor is a vanilloid receptor-1 variant. J Physiol (Lond) 558: 147-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre P, McLatchie LM, Chambers A, Phillips E, Clarke M, Savidge J, Toms C, Peacock M, Shah K, Winter J, Weerasakera N, Webb M, Rang HP, Bevan S, James IF (2001) Pharmacological differences between the human and rat vanilloid receptor 1 (VR1). Br J Pharmacol 132: 1084-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS, Wilson SG, Bon K, Lee SE, Chung K, Raber P, Pieper JO, Hain HS, Belknap JK, Hubert L, Elmer GI, Chung JM, Devor M (1999) Heritability of nociception I: responses of 11 inbred mouse strains on 12 measures of nociception. Pain 80: 67-82. [DOI] [PubMed] [Google Scholar]
- Mulle C, Lena C, Changeux JP (1992) Potentiation of nicotinic receptor response by external calcium in rat central neurons. Neuron 8: 937-945. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Ahern GP (2000) Induction of vanilloid receptor channel activity by protein kinase C. Nature 408: 985-990. [DOI] [PubMed] [Google Scholar]
- Prescott ED, Julius D (2003) A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science 300: 1284-1288. [DOI] [PubMed] [Google Scholar]
- Shin J, Cho H, Hwang SW, Jung J, Shin CY, Lee SY, Kim SH, Lee MG, Choi YH, Kim J, Haber NA, Reichling DB, Khasar S, Levine JD, Oh U (2002) Bradykinin-12-lipoxygenase-VR1 signaling pathway for inflammatory hyperalgesia. Proc Natl Acad Sci USA 99: 10150-10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver IA, Murrills RJ, Etherington DJ (1988) Microelectrode studies on the acid microenvironment beneath adherent macrophages and osteoclasts. Exp Cell Res 175: 266-276. [DOI] [PubMed] [Google Scholar]
- Steinbrocker O, Isenberg SA, Silver M, Neustadt D, Kuhn P, Schittone M (1953) Observations on pain produced by injection of hypertonic saline into muscles and other supportive tissues. J Clin Invest 32: 1045-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T, Tominaga M, Katsuya H, Mizumura K (2002) Bradykinin lowers the threshold temperature for heat activation of vanilloid receptor 1. J Neurophysiol 88: 544-548. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D (1998) The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21: 531-543. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Wada M, Masu M (2001) Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proc Natl Acad Sci USA 98: 6951-6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisani M, Smart D, Gunthorpe MJ, Tognetto M, Barbieri M, Campi B, Amadesi S, Gray J, Jerman JC, Brough SJ, Owen D, Smith GD, Randall AD, Harrison S, Bianchi A, Davis JB, Geppetti P (2002) Ethanol elicits and potentiates nociceptor responses via the vanilloid receptor-1. Nat Neurosci 5: 546-551. [DOI] [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA (2001) Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol (Lond) 534: 813-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T, Droogmans G, Wissenbach U, Janssens A, Flockerzi V, Nilius B (2004) The principle of temperature-dependent gating in cold- and heat-sensitive TRP channels. Nature 430: 748-754. [DOI] [PubMed] [Google Scholar]
- Wahl P, Foged C, Tullin S, Thomsen C (2001) Iodo-resiniferatoxin, a new potent vanilloid receptor antagonist. Mol Pharmacol 59: 9-15. [DOI] [PubMed] [Google Scholar]
- Walls J, Bundred N, Howell A (1995) Hypercalcemia and bone resorption in malignancy. Clin Orthop Relat Res 51-63. [PubMed]
- Wiens L, Sabath R, Ewing L, Gowdamarajan R, Portnoy J, Scagliotti D (1992) Chest pain in otherwise healthy children and adolescents is frequently caused by exercise-induced asthma. Pediatrics 90: 350-353. [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D, Hogestatt ED (1999) Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400: 452-457. [DOI] [PubMed] [Google Scholar]