Cdh1/Hct1-APC Is Essential for the Survival of Postmitotic Neurons (original) (raw)
Abstract
Cell division at the end of mitosis and G1 is controlled by Cdh1/Hct1, an activator of the E3-ubiquitin ligase anaphase-promoting complex (APC) that promotes the ubiquitylation and degradation of mitotic cyclins and other substrates. Cdh1–APC is active in postmitotic neurons, where it regulates axonal growth and patterning in the developing brain. However, it remains unknown whether Cdh1–APC is involved in preventing cell-cycle progression in terminally differentiated neurons. To address this issue, we used the small hairpin RNA strategy to deplete Cdh1 in postmitotic neurons. We observed that Cdh1 silencing rapidly triggered apoptotic neuronal death. To investigate the underlying mechanism, we focused on cyclin B1, a major Cdh1–APC substrate. Our results demonstrate that Cdh1 is required to prevent the accumulation of cyclin B1 in terminally differentiated neurons. Moreover, by keeping cyclin B1 low, Cdh1 prevented these neurons from entering an aberrant S phase that led to apoptotic cell death. These results provide an explanation for the mechanism of cyclin B1 reactivation that occurs in the brain of patients suffering from neurodegenerative diseases, such as Alzheimer's disease.
Keywords: neurodegeneration, cyclin B1, cell cycle, Cdh1/Hct1, APC, cell death
Introduction
Differentiated cells, such as neurons, exit the cell cycle to remain resting in the G0 phase because of an active downregulation of mitotic proteins. Aberrant expression of cell-cycle proteins has been reported to be an important mechanism leading to developmentally regulated neuronal loss and apoptosis in the adult brain (Becker and Bonni, 2004). In this sense, there is an increasing body of evidence to suggest that a mitotic cyclin(s) would be involved in the mechanism of neuronal death in neurodegenerative diseases (Zhu et al., 2004). The strongest evidence for this notion comes from the finding that the Cdk1–cyclin B1 complex is reactivated in degenerating neurons of patients suffering from Alzheimer's disease (Vincent et al., 1997) and in experimental models of ischemia (Wen et al., 2004). Consistent with this idea, Konishi et al. (2002) have shown that overexpression of cyclin B1 in neurons triggers neuronal apoptosis via the phosphorylation and activation of BAD. However, the mechanism that promotes cyclin B1 accumulation in these postmitotic neurons remains elusive.
During the cell cycle, cyclin B1 stability is regulated by proteolysis mediated by the E3-ubiquitin ligase anaphase-promoting complex (APC) (Irniger et al., 1995; King et al., 1995; Sudakin et al., 1995). APC is activated in a cell-cycle-dependent manner by two factors, namely Cdc20 and Cdh1 (also known as Hct1, Fizzy-related, Srw1, or Ste9) (Schwab et al., 1997; Sigrist and Lehner, 1997; Visintin et al., 1997). Whereas Cdc20–APC activity is required early on in mitosis, Cdh1–APC functions at the end of mitosis and G1 (Schwab et al., 1997; Sigrist and Lehner, 1997; Visintin et al., 1997; Yamaguchi et al., 1997, 2000; Kitamura et al., 1998; Blanco et al., 2000; Bashir et al., 2004; Wei et al., 2004). Moreover, the finding that Cdh1–APC activity is elevated in postmitotic neurons (Gieffers et al., 1999) suggests that Cdh1–APC might also play novel functions in G0.
It has recently been found that Cdh1–APC modulates axonal growth during the differentiating process of neurons in the mammalian brain (Konishi et al., 2004) as well as neuronal synaptic plasticity in Drosophila and Caenorhabditis elegans (Juo and Kaplan, 2004; van Roessel et al., 2004). These reported functions for Cdh1–APC are independent of the modulation of cell-cycle-related factors, leaving open the question as to whether Cdh1–APC might be able to regulate mitotic cyclin instability in terminally differentiated neurons. We addressed this issue and found that Cdh1–APC is required to promote cyclin B1 degradation in fully differentiated cells, such as postmitotic cortical neurons. Moreover, we show that Cdh1 plays a key role in neuronal survival because Cdh1 depletion in these neurons triggered cyclin B1-mediated entry into S phase that led to apoptotic cell death. These results point to a possible mechanism through which cyclin B1 might be reactivated in degenerating neurons and are consistent with the notion (Zhu et al., 2004) that neurodegenerative diseases may involve processes related to an aberrant attempt of postmitotic neurons to reenter the cell cycle.
Materials and Methods
Neurons in primary culture. Cortical neurons in primary culture were prepared from fetal [embryonic day 16 (E16)] Wistar rats as described previously (Almeida et al., 2004). In brief, cells were seeded (2.5 × 105 cells/cm2) in DMEM (Sigma, Madrid, Spain) supplemented with 10% (v/v) fetal calf serum (FCS; Roche Diagnostics, Heidelberg, Germany) and incubated at 37°C under a humidified 5% CO2-containing atmosphere. Forty-eight hours after plating, the medium was replaced with DMEM supplemented with 5% horse serum (Sigma), 20 mm d-glucose, and 10 μm cytosine arabinoside (to prevent non-neuronal proliferation). All transfections in primary neurons were performed in fresh medium (without cytosine arabinoside) after 4 d in vitro (DIV) (i.e., when cells displayed a terminally differentiated neuronal phenotype), and the analyses were performed 3 d after transfections. Transfection efficiency was ∼7%.
SH-SY5Y cells. SH-SY5Y human neuroblastoma cells were generously supplied by Dr. C. Gallego (University of Lleida, Lleida, Spain) and grown in DMEM supplemented with 10% (v/v) FCS. To induce full differentiation, SH-SY5Y cells were seeded at 104 cells/cm2, and all-_trans_-retinoic acid (RA; 10 μm; Sigma) was added 24 h later. After 5 d, cells were washed three times with DMEM and further incubated with 50 ng/ml brain-derived neurotrophic factor (BDNF; PeproTrech, Rocky Hill, NJ) for 3 d. Transfections of SH-SY5Y cells were performed after 5 d in RA (i.e., when cells displayed a terminally differentiated neuronal-like phenotype) (efficiency, ∼7%). Exceptionally, some transfections were performed in dividing SH-SY5Y cells (efficiency, ∼60%). Unless specified otherwise, the analyses were performed 3 d after transfections.
293T cells. Human embryonic kidney 293T cells were maintained in DMEM supplemented with 10% (v/v) FCS. Cells were reseeded at 105 cells/cm2 1 d before transfections, after which FCS was reduced to 0.5% (v/v). The analyses were performed 3 d after transfections.
Design and expression of Cdh1 and cyclin B1 small hairpin RNAs. Cdh1 and cyclin B1 depletion was achieved by RNA interference using a vector-based small hairpin RNA (shRNA) approach (Brummelkamp et al., 2002). For Cdh1, we used the target sequence 5′-TGAGAAGTCTCCCAGTCAG-3′ (nucleotides 235–253; GenBank accession number NM_016263), as reported previously (Brummelkamp et al., 2002). For cyclin B1, we used the 5′-GATGGAGCTGATCCAAACC-3′ (nucleotides 478–496; GenBank accession number AY338491) target sequence. As controls, we used the firefly luciferase-targeted oligonucleotide 5′-CTGACGCGGAATACTTCGA-3′, as reported previously (Ohtsuka et al., 2004). These sequences were BLAST (Basic Local Alignment Search Tool) confirmed for specificity. The forward and reverse synthetic 64 nt oligonucleotides (Isogen Life Technologies, Maarsen, The Netherlands) were designed, annealed, and inserted into the _Bgl_II/_Hind_III sites of either pSuper or pSuper.GFP vectors, following the manufacturer's instructions (Oligoengine, Seattle, WA). These constructions express a 19 bp, 9 nt stem-loop shRNA specifically targeted against Cdh1 (Cdh1 shRNA), cyclin B1 (cyclin B1 shRNA), or luciferase (control shRNA) mRNAs.
Cell transfections. Transfections of cells were performed with these plasmid constructions using Lipofectamine 2000 (Invitrogen, Madrid, Spain) and following the manufacturer's instructions.
APC inhibitor hEmi1. hEmi1 was used to inhibit the APC complex by cotransfecting cortical primary neurons with pCS2-hEmi1 (generously donated by Dr. P. Jackson, Stanford University School of Medicine, Stanford, CA) plus pEGFP (Clontech, Palo Alto, CA), and control transfections were performed with a truncated form of hEmi1 carrying a deletion of the zinc-binding region that causes loss of function (pCS2-Myc5-hEmi1ΔZBR; generously donated by Dr. P. Jackson) plus pEGFP.
Site-directed mutagenesis. To assess whether the selected 19 nt sequence used for shRNA was specific, wild-type (pcDNA3-Cdh1 or pcDNA3-myc-cyclin B1-GFP) or mutant (mutCdh1 or mutCyclin B1) forms of Cdh1 or cyclin B1, respectively, were used. pcDNA3-Cdh1 and pcDNA3-myc-cyclin B1-GFP [the latter encoding cyclin B1 fused to green fluorescent protein (GFP)] were generously donated by Dr. J. Pines (Gurdon Institute, University of Cambridge, Cambridge, UK). mutCdh1 and mutCyclin B1 were obtained by PCR site-directed mutagenesis, using pcDNA3-Cdh1 or pcDNA3-myc-cyclin B1-GFP, respectively, as templates, followed by _Dpn_I digestion (QuikChange XL; Stratagene, La Jolla, CA). The selected PCR forward and reverse 50 nt oligonucleotides carried silent third-codon point mutations within the RNA interfering target sequence (5′-CGAGAAATCGCCTAGCCAA-3′ for Cdh1; 5′-GACGGTGCGGACCCGAATC-3′ for cyclin B1; mutated nucleotides are underlined).
Western blotting. Cells were washed with cold PBS, lysed (2% SDS, 2 mm EDTA, 2 mm EGTA, 5 mm Tris, 100 μm phenylmethylsulfonyl fluoride, 50 μg/ml anti-papain, 50 μg/ml pepstatin, 50 μg/ml amastatin, 50 μg/ml leupeptin, 50 μg/ml bestatin, and 50 μg/ml soybean trypsin inhibitor), and boiled for 5 min. Aliquots (40 μg of protein) of cell lysates were centrifuged (14,000 × g, 10 min) and electrophoresed in an SDS/8% polyacrylamide gel (MiniProtean; Bio-Rad, Hercules, CA) using a BenchMark prestained protein ladder (Isogen Life Technologies). The resolved proteins were transferred electrophoretically to nitrocellulose membranes, which were blocked in 5% (w/v) low-fat milk in 20 mm Tris, 500 mm NaCl, and 0.1% (w/v) Tween 20, pH 7.5, for 1 h, and further incubated with anti-Cdh1 (AR38; a generous gift from Dr. J. Gannon, Clare Hall Laboratories, Cancer Research UK, South Mimms, UK), anticyclin B1 (GNS-1; Becton Dickinson-PharMingen, Erembodegen, Belgium), anti-Cdc20 (H175; Santa Cruz Biotechnology, Santa Cruz, CA), anti-Cdk1 (SC54; Santa Cruz Biotechnology), or anti-Pfk1 (Almeida et al., 2004). HRP-conjugated anti-rabbit IgG (Santa Cruz Biotechnology)-treated membranes were developed by luminol-chemiluminescence.
Northern blotting. Purified (Sigma) total RNA samples were electrophoresed (15 μg of RNA per line) on a 1% (w/v) agarose-formaldehyde gel. After transfer to a GeneScreen Plus membrane (NEN, Boston, MA) and cross-linking by UV irradiation, membranes were hybridized for 18 h at 65°C in the presence of the appropriate random-primed [α-32P]dCTP-radiolabeled (Amersham Biosciences, Buckinghamshire, UK) cDNA probe and exposed to Kodak XAR-5 film. We used a cDNA probe either against mouse cyclin B1 or against rat cyclophilin.
Flow cytometric analysis of apoptotic cell death. APC-conjugated annexin-V and 7-amino-actinomycin D (7-AAD; Becton Dickinson) were used to quantitatively determine the percentage of apoptotic cells by flow cytometry. Cells were stained with annexin-V–APC and 7-AAD, following the manufacturer's instructions, and analyzed on a FACSCalibur flow cytometer (15 mW argon ion laser tuned at 488 nm; CellQuest software; Becton Dickinson). Annexin-V–APC-stained cells that were 7-AAD negative were considered apoptotic (Almeida et al., 2004). Each data point represents the acquisition of ∼15,000 cells.
Determination of apoptotic cell death by terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay. Apoptotic neurons were also quantified by the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay (Roche Diagnostics) following the manufacturer's instructions. Each data point was obtained by counting 500 cells from 10 microscopic fields.
Cell-cycle analysis. To analyze cell-cycle distribution, cells were fixed with 70% ethanol and stained with 50 μg/ml propidium iodide and 20 μg/ml RNase on ice for 30 min before flow-cytometric analysis. The proportion of neurons entering S phase was also assessed by flow-cytometric analysis of bromodeoxyuridine (BrdU) incorporation after 18 h of incubation with 10 μg/ml BrdU using the APC BrdU Flow kit (Becton Dickinson Biosciences), following the manufacturer's instructions.
Immunocytochemistry. Immunocytochemistry was performed on neurons grown on glass coverslips, which were fixed for 30 min in PBS containing 4% paraformaldehyde, rinsed with PBS, and permeabilized for 10 min with 0.1% Triton X-100. Cells were then incubated at room temperature in PBS containing blocking serum (10% normal goat serum) plus either anti-Map2 antibody (1:200 dilution; Sigma) for 2 h or anti-Cdh1 antibody (1:100 dilution; Zymed, San Francisco, CA) for 30 min. After washing with PBS, cells were incubated in PBS containing the secondary antibody (Molecular Probes, Eugene, OR) for 30 min. To visualize cyclin B1, we used anti-cyclin B1 phycoerythrin-conjugated antibody (Santa Cruz Biotechnology). Coverslips were washed and mounted in glycerol/PBS (90/10, by volume) on glass slides for phase contrast or fluorescence microphotographs at 10× or 20× magnifications. Analysis of cyclin B1 immunoreactivity in differentiated SH-SY5Y cells was performed by confocal microscopy.
Statistical analyses. Significances were assessed by ANOVA, followed by the least significant difference multiple range test. Results are the mean ± SEM values for at least three different culture preparations.
Results
Expression of APC activators in terminally differentiated neurons
We first investigated the expression of the APC activators Cdh1 and Cdc20 in postmitotic neurons. For this purpose, rat cortical neurons were subjected to full differentiation in culture (Fig. 1_A_), and protein extracts were analyzed at key time points. As shown in Figure 1_B_, these neurons were Map2 positive, whereas <1% were glial fibrillary acidic protein positive (data not shown). Moreover, all Map2-positive neurons expressed Cdh1 (Fig. 1_B_). As shown in Figure 1_C_, Cdh1 protein was expressed prominently throughout the terminal differentiation process of these neurons, whereas Cdc20 protein decreased dramatically to undetectable levels. Because Cdh1 was the only APC activator expressed in postmitotic neurons, we analyzed the expression of cyclin B1, which is a major substrate for Cdh1–APC-dependent proteolytic activity (Irniger et al., 1995; King et al., 1995; Sudakin et al., 1995). As shown in Figure 1_C_, cyclin B1 protein decreased dramatically along terminal differentiation, whereas its mRNA was present during this process (Fig. 1_D_). In contrast to cyclin B1, its counterpart kinase, Cdk1, was prominently expressed in differentiated cells (Fig. 1_C_).
Figure 1.

Cdh1, but not Cdc20, is present in postmitotic neurons. A, Phase-contrast microscopy of E16 rat cortical neurons along in vitro differentiation at 1, 4, and 7 DIV showing neurite processes as from 4 DIV. B, Identification of 7 DIV neuronal cultures by immunocytochemistry showing that all Map2-positive cells expressed Cdh1. C, Western blot analyses confirmed Cdh1 expression throughout terminal differentiation of cortical neurons in primary culture (from 1 to 7 DIV), whereas Cdc20 and cyclin B1 decreased progressively to undetectable levels after 7 DIV, and Cdk1 was still present in neurons after 7 DIV. D, Cyclin B1 mRNA was unchanged from 1 to 7 DIV. Pfk1 (6-phosphofructo-1-kinase) and cyclophilin were used as loading controls for protein and RNA, respectively.
Cell-death phenotype after Cdh1 inhibition in postmitotic neurons
To perform specific Cdh1–APC inhibition, an shRNA specifically targeted against Cdh1 was expressed from a GFP mammalian expression vector (pSuper) (Brummelkamp et al., 2002). To validate the specificity of the Cdh1 shRNA, Western blot analyses were performed after the expression of a Cdh1 cDNA refractory to the RNA interfering sequence [editorial (no authors listed), 2003]. For this purpose, 293T cells were transfected to express either wild-type (Cdh1) or mutant (mutCdh1) forms of Cdh1 cDNA. mutCdh1 carried silent third-codon point mutations within the RNA interfering target sequence. As control shRNA, we used luciferase shRNA. Expression of the control shRNA did not alter the protein levels of expressed Cdh1, regardless of the Cdh1 cDNA coexpressed (wild type or mutant) (Fig. 2_A_). However, Cdh1 shRNA treatment decreased Cdh1 protein levels expressed from wild-type but not from mutant Cdh1 cDNA (Fig. 2_A_).
Figure 2.
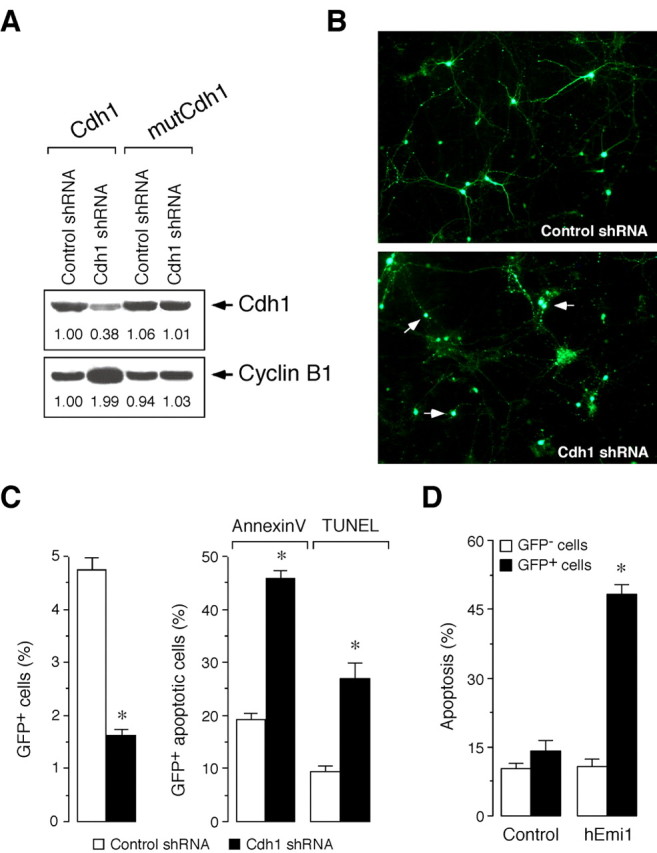
Cdh1 is essential for the survival of postmitotic neurons. A, Cdh1 shRNA is specific, as assessed by expression of target gene refractory to the RNA interfering sequence. Thus, transfection of 293T cells with control shRNA (pSuper.GFP.luciferase) did not alter the protein levels of expressed Cdh1, regardless of the Cdh1 cDNA coexpressed (wild type or mutant). However, transfection of cells with Cdh1 shRNA (pSuper.GFP.Cdh1) decreased Cdh1 protein levels expressed from wild-type (Cdh1) but not from mutant (mutCdh1) Cdh1. Moreover, endogenous cyclin B1 protein levels increased in those cells having lower Cdh1 protein levels. B, Fluorescence images show neurite disintegration and cell body condensation in cortical primary neurons transfected with Cdh1 shRNA compared with the control shRNA. C, Transfection of cortical primary neurons with pSuper.GFP.Cdh1 (Cdh1 shRNA) decreased the number of GFP+ cells (left) and increased apoptotic cells within the GFP+ subpopulation, as assessed both by annexin-V and TUNEL (right); neurons transfected with pSuper.GFP.luciferase were used as controls (control shRNA). D, Transfection of primary neurons with APC inhibitor hEmi1 increased the number of apoptotic cells within the GFP+ subpopulation but not within the GFP– subpopulation compared with control cells expressing a nonfunctional truncated form of hEmi1. *p < 0.05 versus the corresponding control shRNA.
We next investigated the function of Cdh1 in postmitotic neurons. To achieve this without affecting any potential effect on axonal growth (Konishi et al., 2004), transfections with Cdh1 shRNA were performed after 4 DIV (i.e., when cells displayed a neuronal phenotype). This was evident from the appearance of characteristic long axonal processes (Fig. 1_A_) and very low cyclin B1 protein levels (Fig. 1_C_). Neurons expressing Cdh1 shRNA, identified as GFP+ cells, were analyzed 3 d later. Examination by fluorescence microscopy revealed that GFP+ neurons displayed neurite disintegration and cell-body condensation (Fig. 2_B_), suggesting apoptotic cell death. To confirm this, we used flow cytometry and TUNEL assay to quantify cell death more accurately. Flow-cytometric analyses of postmitotic neurons showed a significant and dramatic decrease in the proportion of GFP+ cells transfected with the Cdh1 shRNA compared with those transfected with the control shRNA (Fig. 2_C_, left). Moreover, the loss of GFP+ cells was parallel to an increase in apoptotic GFP+ neurons, as assessed by immunostaining with either the specific apoptotic kit annexin-V/7-AAD or the TUNEL assay (Fig. 2_C_, right).
To inhibit the Cdh1–APC complex with a different approach, we used hEmi1, a well known inhibitor of Cdh1-dependent APC activity (Reimann et al., 2001; Hsu et al., 2002). Thus, transfection of cortical primary neurons with hEmi1, but not with an hEmi1 truncated form that carries a deletion of the zinc-binding region that causes loss of function, triggered apoptotic cell death (Fig. 2_D_). Together, these results suggest that Cdh1 function is required to prevent the apoptotic cell death of differentiated neurons.
Cdh1 inhibition promotes stabilization of cyclin B1 in differentiated cells
Cyclin B1 is a major substrate for Cdh1–APC-dependent proteolytic activity (Irniger et al., 1995; King et al., 1995; Sudakin et al., 1995). To elucidate whether Cdh1 inhibition promoted stabilization of cyclin B1, we used the human-derived SH-SY5Y neuroblastoma cell line because they can either be maintained in culture in a dividing state or be induced to full differentiation. To induce full differentiation, these cells were treated with RA and BDNF (Fig. 3_A_) (Encinas et al., 2000). We further characterized the transition from proliferation to differentiation by flow cytometry, observing that there was a progressive reduction of cells in the S and G2/M phases along the differentiation process (Fig. 3_B_). Consistent with our observations in primary neurons, full differentiation of SH-SY5Y cells was associated with a decline in cyclin B1 protein (Fig. 3_C_) but not mRNA levels (Fig. 3_D_). Furthermore, Cdh1 and Cdk1 levels (Fig. 3_C_) were maintained as cells progressed to terminal differentiation, whereas those of Cdc20 decreased dramatically (Fig. 3_C_). Accordingly, SH-SY5Y cells offer a suitable system to investigate the role of Cdh1 both in proliferating and in postmitotic cells.
Figure 3.

Characterization of SH-SY5Y neuroblastoma cells subjected to terminal differentiation and effect of Cdh1 depletion in the dividing SH-SY5Y cells. A, Phase-contrast microscopy of differentiating SH-SY5Y cells, showing the morphology of the postmitotic neuronal phenotype. B, Cell-cycle distribution of SH-SY5Y cells along the differentiation process, showing G0/G1 arrest after full differentiation. C, Western blot analysis revealed that Cdh1 levels were maintained and Cdk1 levels were present from the dividing to the terminally differentiated stages, whereas cyclin B1 and Cdc20 levels decreased. D, Cyclin B1 mRNA was maintained in terminally differentiated SH-SY5Y cells. E, Depletion of Cdh1 in dividing SH-SY5Y cells triggered cyclin B1 protein stabilization (left) but did not affect cell survival (right).
As shown in Figure 3_E_ (left), depletion of Cdh1 triggered the stabilization of cyclin B1 in dividing SH-SY5Y cells, as it did in 293T cells (Fig. 2_A_). However, Cdh1 inhibition did not affect cell survival as long as these cells were proliferating (Fig. 3_E_, right). Consistent with data published recently (Bashir et al., 2004; Wei et al., 2004), depletion of Cdh1 promoted the entry of these cells into S phase (G0/G1,58 ± 3 and 48 ± 3%; S, 32 ± 2 and 40 ± 2%; control and Cdh1 shRNA, respectively).
Cyclin B1 is responsible for postmitotic neuronal death after Cdh1 silencing
Next, we depleted Cdh1 in postmitotic SH-SY5Y cells. To accomplish this, cells were transfected with the Cdh1 shRNA after they displayed a differentiated neuronal-like phenotype, as evidenced by long axonal processes (Fig. 3_A_), cell-cycle parameters (Fig. 3_B_), and low cyclin B1 protein levels (Fig. 3_C_). Cyclin B1 protein levels were investigated in these cells by confocal microscopy, and we observed that only cells expressing Cdh1 shRNA (identified as GFP+ neurons) displayed cyclin B1 immunostaining (Fig. 4_A_). In light of the results shown in Figures 3_E_ and 4_A_, we may therefore conclude that inhibition of Cdh1 causes an increase in cyclin B1 levels in both proliferating and differentiating cells.
Figure 4.

Cdh1 promotes survival of terminally differentiated SH-SY5Y cells by preventing cyclin B1 accumulation. A, Representative confocal microscopy image (6 cells in this field) of terminally differentiated SH-SY5Y cells 3 d after Cdh1 shRNA treatment, indicating that only efficiently transfected cells (identified as GFP+ cells; 2 cells in this field) displayed cyclin B1 immunoreactivity (identified as red fluorescent cells). In contrast, nontransfected cells (identified as GFP– cells) showed no cyclin B1 immunoreactivity. The same pattern was observed in all fields examined. B, Cyclin B1 shRNA is specific, as assessed by expression of target gene refractory to the RNA interfering sequence. Thus, transfection of 293T cells with control shRNA (pSuper.GFP.luciferase) did not alter the protein levels of expressed cyclin B1, regardless of the cyclin B1 cDNA coexpressed (wild type or mutant). However, transfection of cells with cyclin B1 shRNA (pSuper.GFP.cyclin B1) decreased cyclin B1 protein levels expressed from wild-type (cyclin B1) but not from mutant (mutCyclin B1) cyclin B1. Endogenous Cdh1 protein levels were unaffected by cyclin B1 shRNA. C, Cyclin B1 shRNA prevented the increase in cyclin B1 triggered by Cdh1 shRNA in 293T cells. D, Cdh1 silencing in terminally differentiated SH-SY5Y cells triggered an increase in the proportion of GFP+ apoptotic cells. This effect was prevented by cotransfection with cyclin B1 shRNA. *p < 0.05 versus corresponding control shRNA. Control shRNA, pSuper.GFP.luciferase plus pSuper; Cdh1 shRNA, pSuper.GFP.Cdh1 plus pSuper; cyclin B1 shRNA, pSuper.GFP.Cdh1 plus pSuper.cyclin B1.
Then we investigated whether cyclin B1 protein accumulation attributable to Cdh1 silencing was involved in the apoptotic death of neurons. To do so, we first selected a shRNA sequence specifically targeted against cyclin B1 that was expressed from a GFP cDNA-carrying pSuper vector. The specificity of this cyclin B1 shRNA was confirmed by Western blot analyses after the expression of target genes refractory to the RNA interfering sequence using the same approach as that described above for Cdh1 shRNA. Thus, 293T cells were transfected to express either the wild-type (cyclin B1) or mutant (mutCyclin B1) forms of cyclin B1 carrying silent third-codon point mutations within the RNA interfering target sequence. Transfection of 293T cells with control shRNA (luciferase-targeted shRNA) did not alter the protein levels of expressed cyclin B1, regardless of the cyclin B1 cDNA coexpressed (wild type or mutant) (Fig. 4_B_). However, transfection of cells with cyclin B1 shRNA decreased cyclin B1 protein levels expressed from wild-type (cyclin B1) but not from mutant (mutCyclin B1) cyclin B1 (Fig. 4_B_). In contrast to Cdh1 silencing, which increased endogenous cyclin B1 (Fig. 2_A_), cyclin B1 knocking down did not affect Cdh1 protein levels (Fig. 4_B_). Moreover, cyclin B1 shRNA prevented the increase in cyclin B1 triggered by Cdh1 silencing (Fig. 4_C_).
To elucidate whether Cdh1 silencing promoted cell death in differentiated SH-SY5Y cells, as it did in postmitotic cortical primary neurons, we quantified cell death by flow cytometry. We observed a time-dependent increase in GFP+ apoptotic cells when transfected with the Cdh1 shRNA, but not when they were transfected with the control shRNA (Fig. 4_D_). Furthermore, the apoptotic cell death triggered by Cdh1 depletion was prevented by cyclin B1 silencing, as revealed by cotransfection with the shRNA expression vector specifically targeting cyclin B1 mRNA (Fig. 4_D_).
We further assessed whether the accumulation of cyclin B1 that followed Cdh1 silencing triggered apoptotic cell death in cortical primary neurons. Cyclin B1 was immunocytochemically detected only in those Map2-positive neurons displaying GFP fluorescence (Fig. 5_A_). Quantification of the apoptotic neurons by both flow cytometry and TUNEL assay confirmed that the apoptotic cell death triggered by Cdh1 depletion in primary neurons (Fig. 5_B_) was prevented by cyclin B1 silencing, as revealed by cotransfection with the shRNA expression vector specifically targeting cyclin B1 mRNA.
Figure 5.

Cdh1 promotes survival of primary cortical neurons by preventing cyclin B1 accumulation. A, Representative fluorescence microscopy image of 7 DIV neurons (identified as Map2-positive cells) 3 d after Cdh1 shRNA treatment, indicating that only efficiently transfected cells (identified as GFP+ cells; 2 cells in this field) displayed cyclin B1 immunoreactivity (identified as red fluorescent cells). The localization of cyclin B1 was displayed in a condensed manner, suggesting nuclear localization. In contrast, nontransfected cells (identified as GFP– cells) showed no cyclin B1 immunoreactivity. The same pattern was observed in all fields examined. B, Cdh1 silencing in primary cortical neurons triggered an increase in the proportion of GFP+ apoptotic cells, as assessed both by annexin-V and TUNEL. This effect was prevented by cotransfection with cyclin B1 shRNA. Control shRNA, pSuper.GFP.luciferase plus pSuper; Cdh1 shRNA, pSuper.GFP.Cdh1 plus pSuper; cyclin B1 shRNA, pSuper.GFP.Cdh1 plus pSuper.cyclin B1. C, Overexpression of cyclin B1 by transfection of cortical primary neurons with a plasmid construction encoding a nondegradable form of cyclin B1 fused to GFP (cyclin B1-R42) increased apoptotic cells within the GFP+ subpopulation but not within the GFP– subpopulation compared with control cells. D, Overexpression of Cdh1 in cortical primary neurons did not affect neuronal survival but significantly prevented apoptotic neuronal death triggered by β-amyloid. *p < 0.05 versus corresponding control shRNA.
To confirm that cyclin B1 accumulation would be responsible for neuronal death, cyclin B1 was overexpressed by transfection of primary neurons with a plasmid construction encoding a nondegradable form of cyclin B1 fused to GFP (cyclin B1-R42). As shown in Figure 5_C_, such treatment increased apoptotic cells within the GFP+ subpopulation but not within the GFP– subpopulation compared with control cells. Finally, because cyclin B1 accumulation has been associated with neuronal death in Alzheimer's disease (Vincent et al., 1997; Konishi et al., 2002), we next aimed to investigate whether Cdh1 protected against the neurotoxicity of β-amyloid. Thus, Cdh1 was overexpressed in cortical primary neurons, a treatment that did not affect neuronal survival but significantly prevented apoptotic neuronal death triggered by β-amyloid (Fig. 5_D_).
Cdh1-mediated cyclin B1 accumulation stimulates S phase entry of postmitotic neurons
It has recently been shown that Cdh1–APC activity is required to prevent the progression of dividing cells from G1 into S phase (Bashir et al., 2004; Wei et al., 2004). We reasoned that a possible explanation for the cell-death phenotype induced by Cdh1 depletion in postmitotic cells is that these would attempt to enter S phase before undergoing apoptosis. We analyzed the rate of BrdU incorporation in postmitotic SH-SY5Y cells and primary cortical neurons after Cdh1 depletion. The results, shown in Figure 6_A_ indicated that Cdh1 silencing in both systems elicited an increase in the proportion of cells in S phase. Furthermore, such an increase was counteracted by concomitant depletion of cyclin B1 (Fig. 6_B_), suggesting that cyclin B1 accumulation, after Cdh1 depletion, is strongly associated with the onset of cell death.
Figure 6.

Depletion of Cdh1 promotes cyclin B1-mediated re-entry of postmitotic neurons into S phase. A, Transfection of terminally differentiated SH-SY5Y or cortical neurons with Cdh1 shRNA increased the proportion of cells in S phase, as assessed by BrdU incorporation. B, Cyclin B1 shRNA counteracted the increase in the proportion of postmitotic SH-SY5Y cells entering S phase triggered by Cdh1 silencing. *p < 0.05 versus corresponding control. Control shRNA, pSuper.GFP.luciferase plus pSuper; Cdh1 shRNA, pSuper.GFP.Cdh1 plus pSuper; cyclin B1 shRNA, pSuper.GFP.Cdh1 plus pSuper.cyclin B1.
Discussion
In the present study, we show that the APC activator Cdh1 plays an important survival role in postmitotic neurons by preventing them from an attempt to re-enter into the cell cycle. In fact, it has been postulated that the possibility of the stability of mitotic cyclins outside the cell cycle would be regulated by APC-mediated protein degradation (Gieffers et al., 1999; Harper et al., 2002). Here, we show that Cdh1 protein is indeed prominently expressed throughout the terminal differentiation of rat cortical neurons in primary culture. Furthermore, we also show that the protein levels of the other APC activator, Cdc20, decreased dramatically along the differentiation process of these cells. Together, these results strongly suggest that Cdh1 would be the only APC activator that is present in postmitotic neurons. Accordingly, we were next prompted to investigate the possible role of Cdh1 in the degradation of a mitotic cyclin(s) in neurons.
We first considered cyclin A because this cyclin has been shown to be one target of Cdh1-mediated APC activity, at least in other cell types (Geley et al., 2001). However, it is unlikely that cyclin A would be a major Cdh1 substrate because cyclin A mRNA expression is repressed in differentiated neurons (Freeman et al., 1994). Instead, we focused on cyclin B1 because its mRNA was expressed while the protein was undetectable, which is consistent with the notion that cyclin B1 continues to be actively degraded in postmitotic neurons. In addition, this cyclin is a major substrate for Cdh1–APC-dependent proteolytic activity, at least during the G1 phase of the cell cycle (Irniger et al., 1995; King et al., 1995; Sudakin et al., 1995), and its counterpart kinase, Cdk1, was expressed prominently in postmitotic neurons (present study).
To investigate the relationship between Cdh1 and neuronal survival, we undertook two different approaches [i.e., direct inhibition of Cdh1 activity by ectopic expression of hEmi1 (Reimann et al., 2001; Hsu et al., 2002) or specific Cdh1 degradation by RNA interference]. Both strategies yielded consistent data indicating that Cdh1 loss of function triggered apoptotic neuronal death. Moreover, we found that this phenotype was only observed in differentiated but not in proliferating cells. These results led us to suggest that Cdh1 would be essential for the survival of postmitotic neurons. To investigate cyclin B1 stability after Cdh1 inhibition, we assessed by immunocytochemistry cyclin B1 protein in those neurons that were committed to apoptotic death. We showed that only those neurons expressing the Cdh1 shRNA, as revealed by GFP fluorescence, displayed cyclin B1 accumulation. Moreover, only such GFP+ subpopulations of neurons underwent subsequent cell death, strongly suggesting that cyclin B1 accumulation would be involved in the apoptotic process. In fact, cyclin B1 overexpression was itself neurotoxic, confirming a previous work (Konishi et al., 2002). Interestingly, the depletion of Cdh1 was not essential for the survival of proliferating cells, despite the fact that cyclin B1 was accumulated. In this context, it should be mentioned that others (Konishi et al., 2004) have shown that when Cdh1 is silenced before the terminal differentiation of hippocampal neurons, no cell death occurred; instead, it subsequently modulated axonal growth. Whether this apparent discrepancy would also be attributable to differences in the vulnerability between hippocampal (Konishi et al., 2004) and cortical (present study) neurons against Cdh1 silencing is an issue that remains to be elucidated. In any case, together these results suggest that Cdh1 depletion would only increase the vulnerability of cells against cyclin B1 after they have completely exited from the cell cycle.
To assess directly whether cyclin B1 was responsible for the apoptotic death of postmitotic neurons, we used an shRNA specifically targeted against cyclin B1. Thus, the inhibition of cyclin B1 accumulation significantly increased the survival of Cdh1-depleted neurons, indicating that Cdh1 promotes active degradation of cyclin B1 to ensure cell survival of postmitotic neurons. Cdh1 silencing promoted entry into S phase in dividing (SH-SY5Y) cells, as described recently (Bashir et al., 2004; Wei et al., 2004); however, cell death was not apparent as far as these cells were proliferating. In contrast, Cdh1 silencing in both (SH-SY5Y) cells previously subjected to in vitro differentiation or post-mitotic cortical neurons significantly promoted entry into S phase and triggered apoptotic death. Moreover, concomitant depletion of cyclin B1 prevented neurons from re-entering the cell cycle and rescued the cell-death phenotype. The molecular mechanism by which cyclin B1 triggers neurodegeneration after Cdh1 inactivation is intriguing at the moment. It has been shown that Cdk1/cyclin B1 activity could mediate the cell death by catalyzing BAD phosphorylation at Ser-128 in cerebellar granule neurons (Konishi et al., 2002). Another possibility is that cyclin B1 could directly trigger entry into S phase and mitosis that would be lethal in terminally differentitated cells. In this sense, our data suggest nuclear localization of cyclin B1 after Cdh1 silencing in neurons, a result that is consistent with previous findings showing that the Cdk1–cyclin B1 kinase complex has cryptic S-phase-promoting abilities that can be unmasked by forcing cyclin B1 nuclear localization (Moore et al., 2003).
In conclusion, we propose that Cdh1 would be a key survival protein for differentiated neurons by ensuring the degradation of cyclin B1 to prevent an aberrant re-entry into the cell cycle. This is reinforced by the observation that Cdh1 overexpression protected neurons against the neurotoxicity of β-amyloid. Hence, our results may provide an explanation for the aberrant reactivation of cyclin B1 that has been reported previously to precede cell-cycle re-entry and subsequent apoptosis of degenerating neurons in Alzheimer's disease and transient cerebral ischemia (Vincent et al., 1997; Konishi et al., 2002; Wen et al., 2004).
Footnotes
This work was supported by a Fundación La Caixa grant for the study of neurodegenerative diseases and by a biomedical research grant from Fondo de Investigacìones Sanitarias (FIS01/1215). A.A. was supported by Grant FIS03/1055, J.P.B. was supported by Grant SAF2004/2038, and S.M. was supported by Grant BMC2002-0283. We thank S. Pino, F. Garcia del Rey, and C. Gallego for help in setting up the conditions for the differentiation experiments with the SH-SY5Y cell line; K. Labib, I. Garcia-Higuera, B. Alvarez, J. Font de Mora, and D. Burks for comments on this manuscript; and M. Delgado-Esteban, M. Resch (Centro Nacional de Investigaciones Cardiovasculares, Salamanca, Spain), M. D. Algueró, and R. Valle for technical assistance.
Correspondence should be addressed to Dr. Sergio Moreno, Instituto de Biología Molecular y Celular del Cáncer, Consejo Superior de Investigacìones Científicas/Universidad de Salamanca, Campus Miguel de Unamuno, 37007 Salamanca, Spain. E-mail: smo@usal.es.
DOI:10.1523/JNEUROSCI.1143-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258115-07$15.00/0
References
- Almeida A, Moncada S, Bolaños JP (2004) Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol 6: 45–51. [DOI] [PubMed] [Google Scholar]
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M (2004) Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 428: 190–193. [DOI] [PubMed] [Google Scholar]
- Becker EBE, Bonni A (2004) Cell cycle regulation of neuronal apoptosis in development and disease. Prog Neurobiol 72: 1–25. [DOI] [PubMed] [Google Scholar]
- Blanco MA, Sanchez-Diaz A, de Prada JM, Moreno S (2000) APC(ste9/srw1) promotes degradation of mitotic cyclins in G(1) and is inhibited by cdc2 phosphorylation. EMBO J 19: 3945–3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550–553. [DOI] [PubMed] [Google Scholar]
- Encinas M, Iglesias M, Liu Y, Wang H, Muhaisen A, Cena V, Gallego C, Comella JX (2000) Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J Neurochem 75: 991–1003. [DOI] [PubMed] [Google Scholar]
- Freeman RS, Estus S, Johnson EM (1994) Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron 12: 343–355. [DOI] [PubMed] [Google Scholar]
- Geley S, Kramer ER, Gieffers C, Gannon J, Peters JM, Hunt T (2001) Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J Cell Biol 153: 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieffers C, Peters BH, Kramer ER, Dotti CG, Peters JM (1999) Expression of the CDH1-associated form of the anaphase-promoting complex in postmitotic neurons. Proc Natl Acad Sci USA 96: 11317–11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Burton JL, Solomon MJ (2002) The anaphase-promoting complex: it's not just for mitosis any more. Genes Dev 16: 2179–2206. [DOI] [PubMed] [Google Scholar]
- Hsu JY, Reimann JDR, Sørensen CS, Lukas J, Jackson PK (2002) EF2-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APC-Cdh1. Nat Cell Biol 4: 358–366. [DOI] [PubMed] [Google Scholar]
- Irniger S, Piatti S, Michaelis C, Nasmyth K (1995) Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell 81: 269–728. [DOI] [PubMed] [Google Scholar]
- Juo P, Kaplan JM (2004) The anaphase-promoting complex regulates the abundance of GLR-1 glutamate receptors in the ventral nerve cord of _C. elegans_Curr Biol 14: 2057–2062. [DOI] [PubMed] [Google Scholar]
- King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW (1995) A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 81: 279–288. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Maekawa H, Shimoda C (1998) Fission yeast Ste9, a homolog of Hct1/Cdh1 and Fizzy-related, is a novel negative regulator of cell cycle progression during G1-phase. Mol Biol Cell 9: 1065–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Lehtinen M, Donovan N, Bonni A (2002) Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell 9: 1005–1016. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Stegmuller J, Matsuda T, Bonni S, Bonni A (2004) Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 303: 1026–1030. [DOI] [PubMed] [Google Scholar]
- Moore JD, Kirk JA, Hunt T (2003) Unmasking the S-phase-promoting potential of cyclin B1. Science 300: 987–990. [DOI] [PubMed] [Google Scholar]
- [No authors listed] (2003) Whither RNAi? [editorial] Nat Cell Biol 5: 489–490. [DOI] [PubMed] [Google Scholar]
- Ohtsuka T, Ryu H, Minamishima YA, Macip S, Sagara J, Nakayama KI, Aaronson SA, Lee SW (2004) ASC is a Bax adaptor and regulates the p53-Bax mitochondrial apoptosis pathway. Nat Cell Biol 6: 121–128. [DOI] [PubMed] [Google Scholar]
- Reimann JD, Freed E, Hsu JY, Kramer ER, Peters JM, Jackson PK (2001) Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell 105: 645–655. [DOI] [PubMed] [Google Scholar]
- Schwab M, Lutum AS, Seufert W (1997) Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell 90: 683–693. [DOI] [PubMed] [Google Scholar]
- Sigrist SJ, Lehner CF (1997) Drosophila fizzy-related down-regulates mitotic cyclins and is required for cell proliferation arrest and entry into endocycles. Cell 90: 671–681. [DOI] [PubMed] [Google Scholar]
- Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, Luca FC, Ruderman JV, Hershko A (1995) The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell 6: 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Roessel DA, Elliott DA, Robinson IM, Prokop A, Brand AH (2004) Independent regulation of synaptic size and activity by the anaphase-promoting complex. Cell 119: 707–718. [DOI] [PubMed] [Google Scholar]
- Vincent I, Jicha G, Rosado M, Dickson DW (1997) Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci 17: 3588–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin R, Prinz S, Amon A (1997) CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science 278: 460–463. [DOI] [PubMed] [Google Scholar]
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG (2004) Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 428: 194–198. [DOI] [PubMed] [Google Scholar]
- Wen Y, Yang S, Liu R, Brun-Zinkernagels AM, Koulen P, Simpkins JW (2004) Transient cerebral ischemia induces aberrant neuronal cell cycle re-rentry and Alzheimer's disease-like tauopathy in female rats. J Biol Chem 279: 22684–22692. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Murakami H, Okayama H (1997) A WD repeat protein controls the cell cycle and differentiation by negatively regulating Cdc2/B-type cyclin complexes. Mol Biol Cell 8: 2475–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Okayama H, Nurse P (2000) Fission yeast Fizzy-related protein srw1p is a G(1)-specific promoter of mitotic cyclin B degradation. EMBO J 19: 3968–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Raina AK, Perry G, Smith MA (2004) Alzheimer's disease: the two-hit hypothesis. Lancet Neurol 3: 219–226. [DOI] [PubMed] [Google Scholar]