Translation of peptidoglycan metabolites into immunotherapeutics (original) (raw)
Abstract
The discovery of defined peptidoglycan metabolites that activate host immunity and their specific receptors has revealed fundamental insights into host–microbe recognition and afforded new opportunities for therapeutic development against infection and cancer. In this review, we summarise the discovery of two key peptidoglycan metabolites, γ‐d‐glutamyl‐_meso_‐diaminopimelic acid (iE‐DAP) and muramyl dipeptide and their respective receptors, Nod1 and Nod2, and review progress towards translating these findings into therapeutic agents. Notably, synthetic derivatives of peptidoglycan metabolites have already yielded approved drugs for chemotherapy‐induced leukopenia and paediatric osteosarcoma; however, the broad effects of peptidoglycan metabolites on host immunity suggest additional translational opportunities for new therapeutics towards other cancers, microbial infections and inflammatory diseases.
Keywords: adjuvant, cancer, infection, microbiota, pattern recognition receptor, peptidoglycan
Peptidoglycan metabolites are key microbe‐associated molecular patterns that sensed by pattern recognition receptors. Modification of these microbiota‐derived metabolites is providing new therapeutic leads towards infection, cancer and inflammatory diseases.
Introduction
The immune system provides crucial defences against pathogens to protect the host from disease.1 At a molecular level, immune signalling is mediated through direct receptor binding to a variety of microbial factors, known collectively as microbial‐ or pathogen‐associated molecular patterns (MAMPs or PAMPs, respectively). In turn, recognition of MAMPs by cell‐surface or intracellular pattern recognition receptors (PRRs) stimulates transcriptional and cellular programmes to produce an immune response. The activation of these immune programmes mediates the rapid and direct clearance of microbes and primes adaptive responses to prevent subsequent infections.1 Conversely, hyperactivation of PRRs can lead to chronic inflammation.2 As such, MAMPs present attractive therapeutic leads to modulate host immunity during microbial infections, autoimmune diseases and even failures of normal immunosurveillance such as cancer.
One major source of MAMPs is peptidoglycan (PG), a rigid, mesh‐like glycopeptide polymer found in nearly all species of bacteria as protection from osmotic shock.3, 4 PG is composed of a polysaccharide of repeating _N_‐acetylglucosamine (GlcNAc) and _N_‐acetylmuramic acid (MurNAc) residues with a variable peptide stem attached to the 3‐_O_‐lactoyl group of MurNAc (Figure 1).5 The peptide chains of the PG monomers are then crosslinked during cell wall biosynthesis through bridging amino acid residues to provide further structural rigidity. Gram‐positive and Gram‐negative bacteria differ both in the amount and in chemical composition of their PG. Gram‐positive bacteria contain a thick PG layer up to 80 nm in diameter on their outer surface and generally possess a lysine residue at the third position of the peptide stem (Figure 1a). Conversely, Gram‐negative cells carry a much thinner layer (5–10 nm) between their inner and outer phospholipid membranes and utilise _meso_‐diaminopimelic acid (DAP) within their peptide stems (Figure 1b). Species‐specific differences are also observed in the composition and length of the crosslinking peptide bridge as well as chemical modifications to the polysaccharide backbone.5 Importantly, the degradation of PG by bacterial and host hydrolytic enzymes gives rise to numerous PG metabolites that can then be recognised by the host.6, 7
Figure 1.
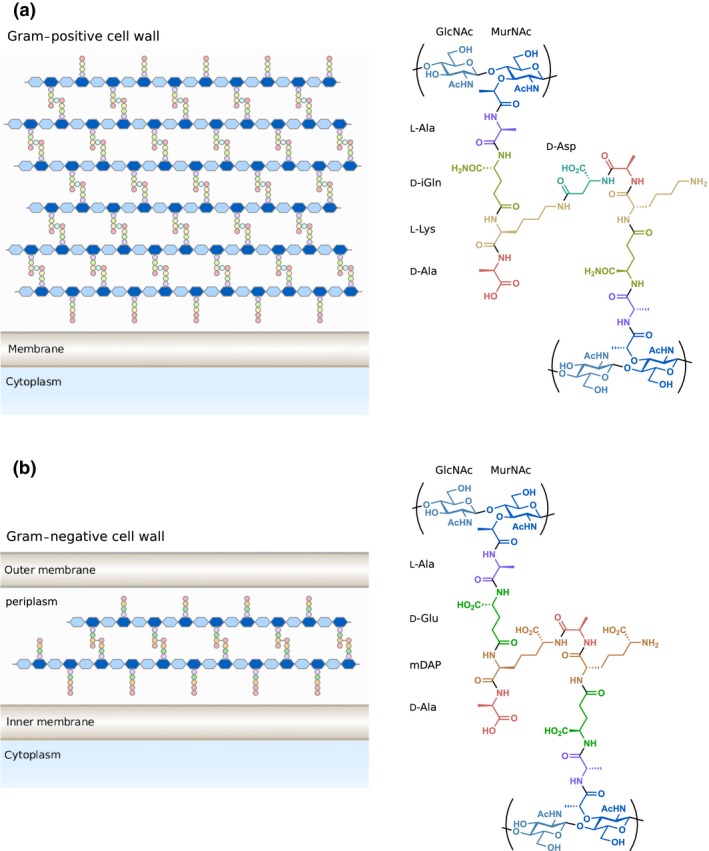
Structure of Gram‐positive and Gram‐negative peptidoglycan. (a) Gram‐positive bacteria like Enterococcus faecium contain a thick layer of peptidoglycan outside of their single membrane. Gram‐positive bacteria usually contain d‐isoglutamine (d‐iGln) and L‐lysine at the second and third positions of the peptide stem. (b) Gram‐negative bacteria such as Escherichia coli contain a thin layer of peptidoglycan within their periplasm. Gram‐negative bacteria generally utilise d‐glutamate (d‐Glu) and _meso_‐diaminopimelic acid (mDAP) at the second and third positions of the peptide stem. The location and composition of the crosslink between peptide stems vary between species. Ac, acetyl.
Host recognition of PG metabolites
The immunomodulatory activity of bacterial PG was first described at the molecular level in 19748 through fractionation studies of complete Freund’s adjuvant (CFA), an emulsion of heat‐inactivated mycobacterial cells, a surfactant and mineral oil first published in 1937.9 Here, isolated water‐soluble, low molecular weight PG fragments were demonstrated as the minimal components of CFA necessary for its adjuvant effects.8 However, the first evidence of host receptors for these metabolites came much later in the early 2000s with the discovery of the cytosolic nucleotide‐binding oligomerisation domain (NOD) proteins Nod1 and Nod2 (Figure 2a).10, 11, 12, 13 These PRRs share a similar domain architecture consisting of 1 or 2 caspase activation and recruitment domains (CARD), a NOD, and 10 or 11 leucine‐rich repeat (LRR) domains in Nod1 and Nod2, respectively.14, 15, 16 Nod1 is expressed in a variety of different cell types and tissues and is activated by PG fragments containing DAP mostly from Gram‐negative bacteria.10, 11 The fragment γ‐d‐glutamyl‐_meso_‐diaminopimelic acid (iE‐DAP, 1, Figure 2b) is often cited as the minimal motif necessary for binding to Nod110, 17; however, murine Nod1 requires the presence of an additional d‐Ala residue for activation.18 The activity of DAP alone is controversial as conflicting reports regarding its activity have been documented.17, 19, 20 Conversely, Nod2 expression is restricted to hematopoietic cells and the epithelium of barrier tissues such as the skin, lungs and gastrointestinal tract, including Lgr5+ stem cells21 and Paneth cells22 of the intestinal crypts. Nod2 recognises MurNAc‐containing PG fragments with _N_‐acetylmuramyl‐L‐alanyl‐d‐isoglutamine or muramyl dipeptide (MDP, 2, Figure 2b) as the minimal unit for Nod2 activation.12, 13 In addition to indirect, loss‐of‐function studies using genetic knockout, subsequent pulldown, surface plasmon resonance and photo‐crosslinking experiments established that the Nod receptors directly interact with these PG metabolites.23, 24, 25, 26 More recently, members of the membrane‐bound ADP ribosylation factor (Arf) family of proteins were also shown to directly interact with MDP.26 Arf6 and to a lesser extent Arf1 and Arf4 were found to associate with MDP in a Nod2‐dependent manner, suggesting that these Arf‐family GTPase may directly modulate MDP‐Nod2 signalling. The importance of membrane association for Nod signalling is further supported by the discovery of Nod1 and Nod2 S‐palmitoylation, which was critical for their downstream signalling.27
Figure 2.
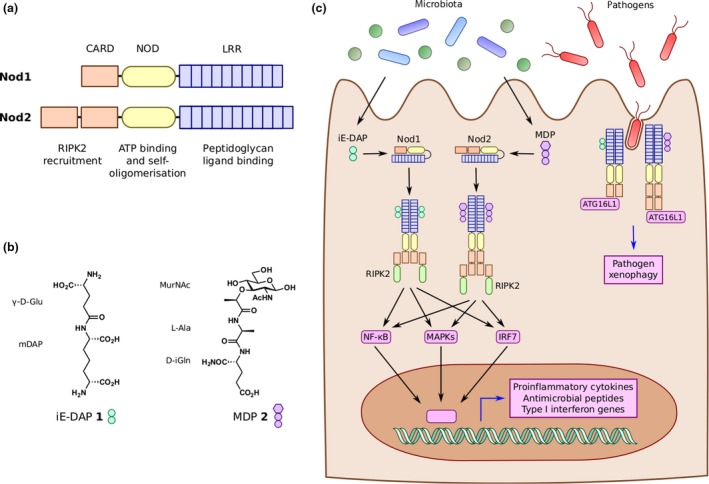
Peptidoglycan pattern recognition receptors. (a) Nucleotide‐binding oligomerisation domains 1 and 2 (Nod1 and Nod2) are conserved pattern recognition receptors of peptidoglycan fragments. Both proteins contain 1 or 2 caspase activation and recruitment domains (CARD), a NOD, and 10 or 11 leucine‐rich repeat (LRR) domains. (b) Nod1 recognises peptidoglycan fragments containing mDAP, the smallest of which is iE‐DAP 1. Conversely, Nod2 recognises muropeptides, with MDP 2 as the minimal active unit. (c) iE‐DAP and MDP derived from local bacteria or pathogens bind to Nod1 and Nod2, respectively. Activated Nod receptors oligomerise and recruit RIPK2. Through downstream adapter proteins and signalling cascades, Nod receptors activate NF‐κB, MAPK and IRF7 pathways to elicit expression of proinflammatory cytokines, antimicrobial peptides and type I interferon genes. Nod receptors also localise to sites of bacterial invasion to recruit ATG16L1 and induce autophagy pathways against the invading bacteria, known as xenophagy.
In addition to the Nods, other proteins have been implicated in PG metabolite sensing. Toll‐like receptor 2 (TLR2) had previously been shown to respond to PG stimulation with a preference for DAP‐containing fragments.28, 29 However, this finding has been controversial as other reports suggested that TLR2 activation was due instead to contaminating lipoproteins and lipoteichoic acids (LTAs) within the PG preparations.30 Hexokinase (HK) has also been demonstrated to sense PG through binding to GlcNAc derived from PG after phagosomal degradation.31 GlcNAc inhibited HK activity and led to its dissociation from the mitochondrial outer membrane, which in turn was sufficient to activate cytokine processing and secretion via the NOD‐like receptor family, pyrin domain‐containing 3 (NLRP3) inflammasome.
Upon binding to their respective PG metabolites, both Nod1 and Nod2 are believed to shift from an auto‐inhibited form to an active conformation that then can self‐oligomerise (Figure 2c).14, 15, 16 The oligomeric Nod complexes then recruit receptor interacting serine/threonine kinase 2 (RIPK2) via CARD‐CARD interactions.32, 33 In turn, the Nod/RIPK2 complex activates mitogen‐activated protein kinase (MAPK), nuclear factor kappa‐light‐chain enhancer of activated B cells (NF‐κB) and interferon transcription factor (IRF) pathways, which promotes the transcription of proinflammatory cytokines, antimicrobial peptides (AMPs) and type I interferon genes.14, 15, 16 Complex formation, stability and signal transduction are tightly controlled through numerous other proteins reviewed elsewhere.16 These regulatory mechanisms include the newly discovered role of heat‐shock protein B8 (HspB8), which directly binds to both Nod1 and Nod2, prevents their aggregation and may enhance the assembly of active Nod1/2 oligomers.34 In epithelial cells, Nod1/2 signalling leads to the cellular and tissue restriction of pathogens through induction of pathogen‐directed autophagy, or xenophagy,35 and the production of barrier‐promoting proteins and AMPs, respectively (Figures 2 and 3).36, 37 Moreover, activation of macrophages and other phagocytic myeloid cells elicits the direct killing of microbes and the clearance of host cells that present non‐self signals from either internal pathogens or cancer‐related mutations (Figures 2 and 3).1
Figure 3.
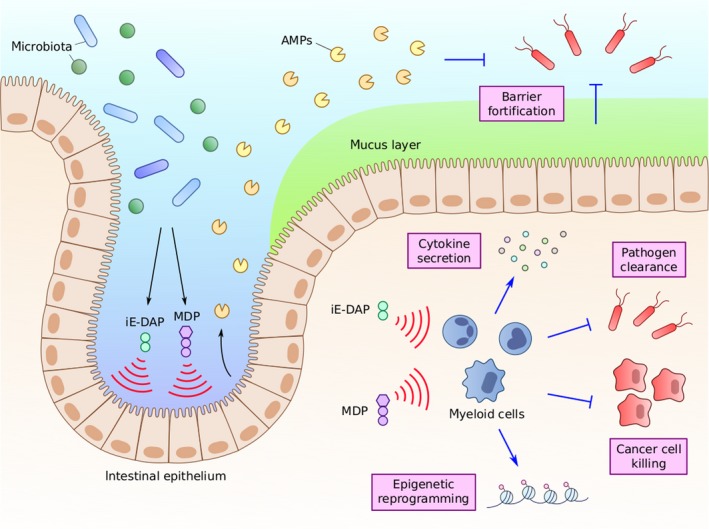
Peptidoglycan activation leads to diverse immune responses**.** Peptidoglycan fragments iE‐DAP and MDP exhibit a wide array of activities through the activation of numerous cell types. Activation of intestinal epithelial cells leads to the increased production of mucins and antimicrobial peptides (AMPs) to improve the intestinal epithelial barrier and prevent infection. Activation of different myeloid cell populations can lead to increased cytokine secretion, epigenetic reprogramming, and the direct killing of pathogens and other foreign‐presenting cells such as cancer cells.
In addition to rapid clearance of microbes, PG fragments have been implicated in priming longer term immune responses. For example, Nod2 activation trains monocytes via epigenetic reprogramming to better respond to subsequent microbial challenges.38, 39 Similarly, sensing of Nod1‐activating PG fragments by circulating monocytes is necessary to enhance systemic immune priming prior to infection.40, 41 PG‐mediated activation of Nods also promotes adaptive immune responses. Stimulation of either Nod1 or Nod2 leads to Th2 responses and the production of antigen‐specific antibodies.42, 43 Moreover, co‐stimulation with Nod1 or Nod2 agonists and other MAMPs that engage TLRs produces a combination of Th1, Th2 and Th17 responses.42, 43, 44
Through these small molecule–protein interactions, PG metabolites coordinate both the priming and activation of host immune responses to maintain health and combat infection. Therefore, iE‐DAP, MDP and their derivatives may function as possible drug candidates to improve immune activation before and during disease. In this review, we summarise efforts towards improved synthetic analogs of Nod1 and Nod2 agonists. We focus on chemical trends that have led to more potent derivatives and how these molecules have been employed to modulate a variety of host immune processes against infection and cancer.
Chemical derivatives of PG metabolites
Due to their wide‐ranging biological activities, the therapeutic application of PG metabolites has garnered significant attention since their initial discoveries. However, these molecules have many properties that complicate if not completely preclude them from direct clinical use. Both iE‐DAP and MDP are hydrophilic molecules that can easily be excreted from the body. MDP is more than 50% cleared by the kidneys in 30 min following intravenous or subcutaneous injection in mice and more than 90% by 2 h.45 Although both molecules contain chemical motifs that are uncommon or not present in animals such as the ɣ‐linkage and d‐isomer of Glu/Gln, PG derivatives may be subject to host‐mediated hydrolysis in circulation. For instance, rat serum was found to degrade MDP into its monomeric components, although the timescale of this process was much slower than excretion.46 Finally, the broad immune modulatory activity of these molecules may lead to undesired effects. MDP in particular has been demonstrated to be both pyrogenic (fever‐inducing)47, 48 and somnogenic (sleep‐inducing).49, 50 Therefore, chemical optimisation of the iE‐DAP and MDP scaffolds has focused not only on improved potency in vitro but also enhanced bioavailability and reduced side effects in vivo.
Although iE‐DAP has been described as the minimal active structure to stimulate Nod1, other naturally occurring, DAP‐containing compounds based on larger PG fragments retain activity. One of the earliest discovered immune active PG metabolites containing DAP was FK‐156 (3, Figure 4), the lactoyl‐conjugated tetrapeptide d‐Lac‐L‐Ala‐ɣ‐d‐Glu‐mDAP‐L‐Gly.51 Isolated in 1982 from Gram‐positive Streptomyces olivaceogriseus and S. violaceus strains,52 FK‐156 was found to induce proliferation of murine splenocytes, protect against lethal challenge with Escherichia coli and improve carbon clearance from the blood, an early assay for phagocytic activity in vivo.53 Further synthetic studies based on the FK‐156 scaffold yielded a number of other immune active analogs, the most widely used being the heptanoyl tripeptide FK‐565 (Hep‐ɣ‐d‐Glu‐mDAP‐L‐Gly, 4).54 Testing of these compounds including FK‐565 uncovered both that the d‐Lac‐L‐Ala residues of FK‐156 were not required for activity and that long‐chain fatty acid acylation improved overall activity, mirroring contemporary synthetic studies with MDP (see below). Using a reductivist approach, it was also found that ɣ‐d‐Glu‐mDAP or iE‐DAP was the smallest derivative of FK‐156 that could elicit activity,55 revealing the minimal Nod1 ligand over 20 years before the discovery of its receptor.
Figure 4.
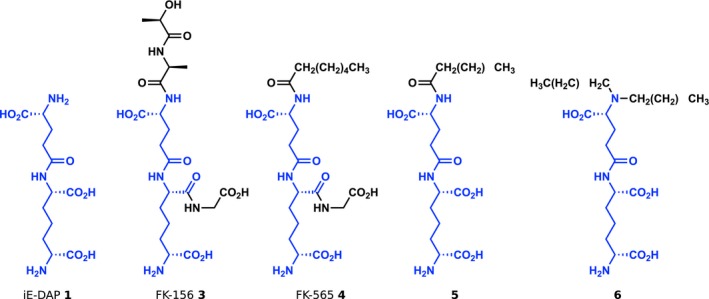
Representative iE‐DAP‐derived Nod1 agonists. The chemical structures of iE‐DAP and representative Nod1 agonists are shown. In all structures, the iE‐DAP scaffold is highlighted in blue. In general, a trend towards higher lipophilicity has resulted in increased biological activity.
The elucidation of Nod1 as the iE‐DAP receptor in 200310, 11 allowed for synthetic studies to focus more directly on receptor activation as a primary readout (Figure 2c). Using an in vitro NF‐κB reporter assay, Wolfert et al. 56 tested a panel of six synthetic iE‐DAP analogs, showing that amidation of ɣ‐d‐Glu (referred to as isoglutamine, iGln) abrogated Nod1 activation. As iGln is commonly found across different species, these findings revealed a Nod1‐dependent mechanism for immune evasion through endogenous PG modification. Similar to _N_‐acylated FK‐565, Hasegawa et al. 57 showed that _N_‐acyl derivatisation of the iE‐DAP dipeptide was well tolerated by Nod1 using NF‐κB reporter and monocyte chemokine secretion assays, with _N_‐myristoyl (C14) iE‐DAP (5) exhibiting more than 100‐fold higher potency than unmodified iE‐DAP. Subsequently, Agnihotri et al. 58 reported a thorough structure–activity relationship study examining modification of all terminal heteroatoms on iE‐DAP against commercial _N_‐lauroyl (C12) iE‐DAP as a standard. As expected, loss of either the terminal carboxylic acid or amine of DAP abolished Nod1 activation. No large changes in EC50 values were observed after esterification of any carboxylic acid of iE‐DAP; however, amidation of ɣ‐d‐Glu or the terminal amine of DAP with caused a significant drop in activity. Interestingly, dealkylation of the ɣ‐d‐Glu amine with lauryl groups provided an agonist over 10x more potent than C12‐iE‐DAP (6, EC50 = 0.0015 nm using the NF‐κB reporter assay), but this increase in activity did not extend to other N,_N_‐dialkyl compounds. Jakopin et al. 3 also examined constraining the orientation of the flexible sp carbon backbone of DAP through the introduction of an alkene.59 Although no differences in activity were observed for the four derivatives at a single concentration via an NF‐κB reporter assay, one compound based on the N,_N_‐dialkyl modification of Agnihotri et al. with an alkene between the ɣ and δ carbons showed higher activity than its counterpart with a β‐ɣ alkene in a cytokine secretion assay. However, this compound was not directly tested against the alkane derivative, so it is unclear whether conformational rigidity of the DAP moiety improves Nod1 activation or is simply tolerated.
Because MDP was found to be biologically active over 35 years ago, it is unsurprising that extensive efforts have been made to produce variants that improve upon its potency. As with iE‐DAP, these efforts have focused broadly on elucidating the necessary structural components, examining natural variants and improving bioavailability. Many early studies focused on the structural flexibility of three monomeric residues of MDP. For example, screening of L‐amino acids to replace L‐Ala found that the position was somewhat tolerant of other side groups, with L‐Val, L‐Ser and L‐ɑ‐aminobutyric acid able to slightly improve biological activity.60 The d‐iGln residue was less amenable to substitution, with L‐iGln or d‐Asp causing loss of activity.47, 61 Interestingly, d‐iGln could be substituted for d‐Glu to maintain adjuvant and anti‐infective activity, while causing a loss of pyrogenicity.62 Methyl esterification of d‐Glu showed similar activity as the diacid form of d‐Glu, whereas _N_‐methyl amides of d‐iGln caused inactivation.62 Most early modifications to MurNAc led to a decrease or loss of activity. For example, reduction to the sugar alcohol yielded an inactive molecule, whereas removal of the methyl group from the lactyl group of MurNAc showed a decrease in adjuvant activity as well.61
Analogs that mirror natural PG structures have also been thoroughly studied. Both GlcNAc‐MDP structural isomers containing the disaccharide repeating unit of PG were produced either chemoenzymatically or synthetically and demonstrated similar or higher adjuvant activity as MDP.63, 64 More recently, Wang et al. 65 synthesised longer tetrasaccharide fragments and found 10‐fold higher activation of Nod2 when treated with MurNAc(L‐Ala‐d‐iGln)‐GlcNAc‐MurNAc(L‐Ala‐d‐iGln)‐GlcNAc compared to the isomer with GlcNAc at the nonreducing end of the tetrasaccharide. Extension of the peptide stem showed residue‐specific effects. For example, inclusion of DAP to mimic most Gram‐negative PG fragments showed no Nod2 activation; however, the addition of L‐Lys or L‐ornithine to MDP had little detrimental effect on potency using an NF‐κB reporter assay.17 Further elongation of the peptide stem to include one or two naturally occurring d‐Ala residues led to a concomitant decrease in activity via cytokine secretion assay.66 Substitution of the _N_‐acetyl group of MurNAc with an _N_‐glycolyl group, a modification found in some Actinomycetes species including mycobacteria, showed higher Nod2 activation, supporting previous observations of the highly immunogenic potential of mycobacterial PG.67
As with iE‐DAP, the addition of acyl groups to MDP has been explored as a method to improve bioavailability presumably through both longer half‐life in vivo and better cellular uptake. Acylation of the MDP scaffold has been profiled both at the 6‐O position of MurNAc and at the C terminus of the peptide stem. For example, a series of 6‐_O_‐acyl derivatives with a range of lengths from acetyl (C2) to triacontanoyl (C30) was screened for protection of mice against lethal challenge with E. coli, where the 6‐_O_‐stearoyl (C18) modification 7 (Figure 5) showed the highest overall survival.68 Further extension of the 6‐_O_‐acyl group with linear or branched fatty acids up to 48 carbon atoms in total produced the 6‐_O_‐(2‐(tetradecyl)hexadecanoyl) variant B30‐MDP 8, which elicited heightened immunoadjuvant activity and increased serum antibody levels.69 Structural optimisation of these acyl MDP compounds led to the discovery of MDP‐Lys(L18) 9 also known as romurtide or muroctasin, in which ε‐_N_‐stearoyl‐L‐Lys was attached to the MDP scaffold.70 Romurtide has been shown to stimulate macrophages to release cytokines including colony‐stimulating factors that in turn can increase white blood cell and platelet counts,71, 72 and the molecule is currently in use in Japan to treat leukopenia after chemotherapy. Other acylpeptide MDP derivatives have also been reported including butyl ester‐containing murabutide, MurNAc‐L‐Ala‐d‐Gln(_O_‐_n_Bu) 10.73 Similar to the MDP analog containing the dimethyl ester of d‐Glu, murabutide retains its adjuvant and anti‐infective properties without any pyrogenic side effects even at high doses.73, 74
Figure 5.
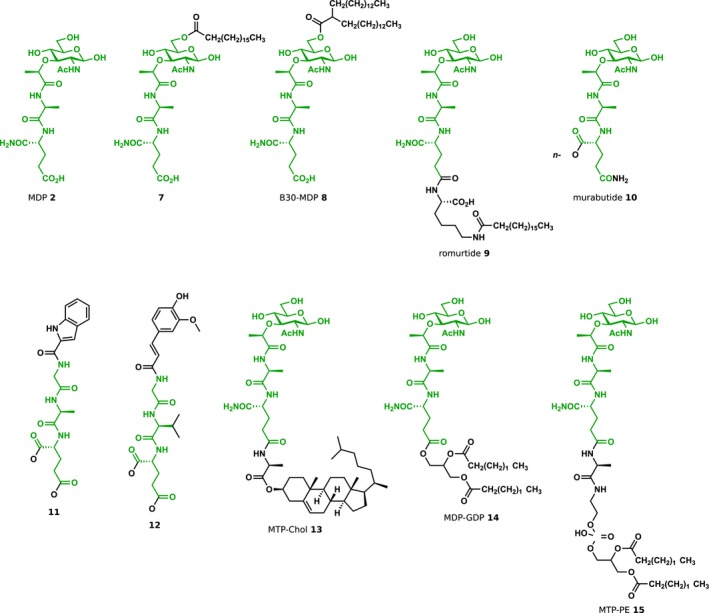
Representative MDP‐derived Nod2 agonists. The chemical structures of MDP and representative Nod2 agonists are shown. In all structures, the MDP scaffold is highlighted in green. As seen with Nod1‐targeted molecules, an increase in lipophilicity has resulted in more potent agonists. Modifications such as esterification also have decreased side effects such as pyrogenicity observed with the parental MDP molecule. Moreover, departure from the MurNAc monosaccharide has yielded compounds with similar efficacy. _n_‐Bu, _n_‐butyl; Et, ethyl.
Although removal of MurNAc from the dipeptide stem prevents Nod2 stimulation,17 the MurNAc moiety can be replaced with noncarbohydrate structures while retaining Nod2 activity. Termed ‘desmuramyl’ peptides, these aglycon structures generally utilise hydrophobic groups to replace MurNAc. For example, some early studies described the immune activity of a number of dipeptide structures conjugated to alkyl groups such as octadecanoic acid,75 7‐oxooctanoic acid,76 glyceryl mycolate77 and adamantane.78 More recently, substituted aromatic rings have also been explored to produce potent Nod2 agonists. The MDP derivative 11 in which MurNAc was replaced with an indole‐2‐ylcarboxamido group showed similar activity as murabutide in both NF‐κB reporter and cytokine secretion assays.79 The related _trans_‐feruloyl analog 12 exhibited low nanomolar activation (EC50 = 46 nm), providing functional MDP analogs with equivalent or even more potent activity without the hydrolysable reducing sugar.80
Peptidoglycan metabolites and infection response
Nod activation by PG fragments is an important determinant of host defences against many microbial infections. Indeed, Nod signalling via NF‐κB has been observed upon in vitro infection by a variety of Gram‐negative bacteria such as Campylobacter jejuni, Pseudomonas aeruginosa, and Helicobacter pylori and Gram‐positive bacteria including Clostridium difficile and Staphylococcus aureus as well as mycobacteria, as reviewed elsewhere.16, 81 In nonhematopoietic cells such as epithelial cells, PG metabolite sensing is thought to aid in the establishment and maintenance of the barrier between the host and environment. In the gut, this occurs through cell‐intrinsic xenophagy‐mediated clearance, the physical exclusion of pathogens by the production of mucins and direct killing via AMP secretion (Figures 2 and 3).36, 37 For example, Nod1 has been correlated with the expression of the mucin protein Muc2 and the AMPs ɑ‐ and β‐defensins.82, 83 Early studies indicated that Nod2 also altered the expression of ɑ‐defensins produced by Paneth cells in vivo 84; however, subsequent work did not corroborate this observation.85
Although the metabolites sensed by Nod1 and Nod2 may be derived from invading pathogens, associated commensal species may also contribute active PG fragments. Recently, the Gram‐positive species Enterococcus faecium was found to increase host tolerance to infection by both Salmonella enterica serovar Typhimurium and C. difficile.86, 87 Resistance to infection after E. faecium colonisation was lost in Nod2–/– animals, and the protective activity was associated with the production of Muc2 and AMPs as well as normalisation of the intestinal epithelium. The protective phenotype was linked to the secreted PG endopeptidase, SagA, which was sufficient to promote protection when heterologously expressed in the nonprotective species Enterococcus faecalis. Structural and biochemical characterisation of its catalytic domain revealed that SagA cleaves PG fragments to release the disaccharide‐dipeptide GlcNAc‐MDP (GMDP), which was shown to activate Nod2 in vitro.88 Probiotic Lactobacillus plantarum engineered to express wild‐type SagA recapitulated its protective phenotype against C. difficile, whereas no protective effect was observed upon colonisation with bacteria expressing mutant SagA variants in which the catalytic residues or secretion signal was ablated. Therefore, the delivery of active PG metabolites to the gut may be achieved not only through small molecules but also by genetically engineered probiotics.
Beyond the epithelial line of defence, myeloid cells initiate diverse immune pathways upon sensing PG metabolites (Figure 3). Phagocytosis along with the secretion of reactive oxygen species and proteases provides a direct and rapid means for certain myeloid cells to kill and clear invading pathogens. The activation of cytotoxic pathways particularly in macrophages after stimulation with synthetic Nod agonists has been examined extensively in terms of cancer cell killing (see next section). As an additional acute response, PG sensing can elicit the secretion of multiple cytokines. For example, stimulation of human monocyte‐derived macrophages with murabutide 10 led to an increase in expression and secretion of numerous cytokines including IL‐1β, IL‐6, TNFɑ, RANTES, IL‐8 and MIP‐1β.89 Additionally, stimulation of macrophages with romurtide 9 led to increased secretion of colony‐stimulating factors to promote hematopoiesis.71, 72
Peptidoglycan metabolites and adjuvanticity
In addition to native host sensing of PG metabolites during acute infections, noninfectious PG sources have been applied as adjuvants to prevent or combat disease for nearly 100 years. Through iterative passaging of the bacterium Mycobacterium bovis, Albert Calmette and Camille Guerin produced the weakened mycobacterial strain now known as Bacillus Calmette‐Guérin (BCG).90 First used in humans in 1921, BCG found success as a vaccine against tuberculosis,91 and derivatives of the BCG vaccine are still in use today in countries with prevalent tuberculosis infection rates. In 1937, Jules Freund utilised a dried, inactivated form of the related strain Mycobacterium tuberculosis to produce CFA,9 which was found to elicit a Th1 response and delayed‐type hypersensitivity.92 As described above, studies in the 1970s found that MDP 2 was the minimal bioactive component of CFA,8 paving the way for synthetic PG metabolites to be used as adjuvants.
Muramyl dipeptide derivatives have proven useful in numerous preclinical models of vaccination. Owl monkeys were successfully vaccinated against Plasmodium falciparum, the parasite responsible for malaria in humans, by co‐injection with 6‐_O_‐stearoyl‐MDP 7 in liposomes.93 Co‐administration of romurtide 9 with a temperature‐sensitive, live Salmonella enteritidis vaccine (Ts‐O) in mice led to an increase in response as demonstrated by footpad delayed‐type hypersensitivity.94 Interestingly, administration of romurtide either 48 h prior to or immediately after vaccination both augmented its efficacy. Combination of the inactivated B‐1 vaccine of hantavirus with either romurtide or B30‐MDP 8 improved delayed‐type hypersensitivity upon challenge seven days postinjection.95 B30‐MDP also increased immunisation in mice using X‐irradiated L5178Y‐ML25 lymphoma cells and prevented subsequent metastases better than vaccination with the irradiated cells alone.96 More recently, liposomal‐encapsulated desmuramyl compound 12 showed an increase in serum levels of antigen‐specific IgG upon OVA peptide vaccination; yet, its effects were less pronounced than MDP.80 Unfortunately, these preclinical observations have not directly translated to the clinic. The muramyl tripeptide‐phosphatidylethanolamine conjugate MTP‐PE 15 had been explored in Phase I clinical trials as a vaccine adjuvant for both influenza and human immunodeficiency virus type 1 (HIV‐1), but its addition to the vaccines demonstrated significantly increased adverse side effects with little to no improvement of immunogenicity.97, 98, 99
Along with vaccine adjuvant activity, MDP‐based molecules demonstrate nonspecific, therapeutic adjuvanticity. This general anti‐infective effect was first observed in children treated with the BCG vaccine during the early 20th century, where the mortality rate was notably reduced compared to unvaccinated children.100 The molecular basis of this epidemiological observation was discovered in 2012, when BCG vaccination was found to induce epigenetic reprogramming of monocytes in a Nod2‐dependent manner.38 This activity enhanced the response of innate immune cells against subsequent challenges in a memory‐like but nonspecific manner, a process now known as trained immunity.39 This phenotype was demonstrated in peripheral blood mononuclear cells isolated from healthy adults treated with the BCG vaccine, where cells exhibited increased cytokine production upon stimulation with not only the vaccine‐specific pathogen M. tuberculosis but also the unrelated bacterium S. aureus and even the yeast Candida albicans.38 Nevertheless, synthetic studies to identify more potent MDP analogs had previously demonstrated that these molecules exhibited nonspecific, anti‐infective activity when administered prior to pathogen challenge. For example, both 6‐_O_‐stearoyl‐MDP 7 and romurtide 9 were developed using a protection assay in a murine model of _E. coli_‐mediated sepsis.68, 70 The two molecules were also found to effectively limit infection of the opportunistic pathogen Cornyebacterium kutscheri in a cortisone‐mediated model of immunosuppression.101 Similarly, 6‐_O_‐stearoyl‐MDP enhanced host resistance against both E. coli and C. albicans infection in immunocompromised mice treated with either X‐ray irradiation or cyclophosphamide102 and limited _P. aeruginosa_‐associated pneumonia in immunocompromised guinea pigs.103 Nonspecific protection was observed as well during viral infections. Romurtide and B30‐MDP 8 were found to limit infection by herpes simplex virus type 1,104 herpes simplex virus type 2105 and vaccinia virus.105 Moreover, murabutide 10 has been shown to limit HIV‐1 viral loads in a humanised murine model.106 However, it remains uncertain whether these effects are truly via long‐term epigenetic reprogramming as many of the assays were conducted soon after adjuvant administration.
In addition to Nod2 agonists, Nod1 stimulation has yielded nonspecific adjuvants to combat infection. For instance, oral or subcutaneous dosing with Nod1 agonists FK‐156 3 and FK‐565 4 was shown to enhance host tolerance of subcutaneous S. aureus infection as well as systemic infection by a variety of Gram‐positive and Gram‐negative bacteria including E. coli, P. aeruginosa and Listeria monocytogenes.107 Nod1 agonists from endogenous sources have also been correlated with systemic, nonspecific immune priming. Neutrophils harvested from either germ‐free or antibiotic‐treated mice showed deficient killing of Streptococcus pneumoniae and S. aureus.40 Using knockout animal models, this deficiency was traced to Nod1 expression. Interestingly, neutrophils from animals prestimulated with heat‐killed, Gram‐negative Haemophilus influenzae, which produces mDAP‐containing PG, showed enhanced cytotoxic activity against both pathogens compared to unstimulated animals. The results were recapitulated by stimulation with a MurNAc‐tripeptide fragment containing mDAP. Although distinct from canonical trained immunity, these findings suggest that PG metabolites from persistent, nonpathogenic sources such as the gut microbiota can also mediate the systemic priming and increased activity of immune cells.
Peptidoglycan metabolites and cancer treatment
Peptidoglycan metabolites, and specifically muropeptides, have been shown for nearly 40 years to improve clearance and survival in animal models of cancer. In fact, evidence for the antitumoral activity of PG fragments was indirectly demonstrated even prior to the discovery of MDP as the minimal active component of CFA. In these experiments, direct, intratumoral injection of live BCG was shown to decrease tumor size and prevent metastases in a guinea pig model of cancer. However, BCG immunotherapy in humans led to numerous detrimental side effects, including persistent infection, fever and liver disease.108, 109 To alleviate these issues, considerable efforts were made towards identifying nonviable replacements for BCG. Among the studies, it was identified that MDP acylated at the 6‐O position with mycolic acid, a lipid found in Mycobacterium species, could suppress fibrosarcoma growth in mice when co‐injected intradermally.110 Notably, tumor growth was only limited when the acyl MDP was pre‐incubated with oil droplets, whereas buffer suspension of the molecule showed no effect. MDP derivatives that were acylated or that had replaced L‐Ala with certain bulkier amino acids also showed tumoricidal activity after intralesional injection into a tumor model in guinea pigs.111 Similarly, this activity depended on the formation of mineral oil emulsions containing trehalose dimycolate, with either component alone showing no effect.
Although these studies suggested that MDP derivatives in combination with other mycobacterial‐derived compounds could effectively treat tumors, it remained unclear whether MDP alone would prove an effective antitumor agent. At the same time, MDP was demonstrated to potently activate macrophages and lead to tumor cell killing in vitro,112, 113 and the effects were potentiated by encapsulation of MDP into multilamellar vesicles or liposomes.114 Accordingly, repeated intravenous injection of liposome‐encapsulated MDP in mice treated with a metastatic model of melanoma led to an overall decrease in pulmonary metastases and partial survival of the cohort.115 Here, free MDP at 40x the concentration of liposomal MDP did not prove effective, suggesting that soluble MDP may be cleared too rapidly from the animals and that liposome encapsulation improved endocytic uptake of the small molecule. Moreover, this process did not depend on indirect, T cell‐mediated activation of macrophages as tumoricidal activity was still observed in vivo using UV‐irradiated, thymectomised and X‐ray irradiated, or athymic nude mice.116
Based on these observations, new MDP derivatives were produced in which a muropeptide was conjugated to a variety of lipophilic molecules to improve liposome incorporation or produce micelles as a single agent. For example, liposomes containing MDP‐L‐alanyl‐cholesterol (MTP‐Chol, 13) developed in 1985 were shown to be eightfold more effective than liposome‐encapsulated MDP in inducing macrophage cytotoxic activity, putatively due in part to leakage of water‐soluble MDP from the liposome.117 Subsequent in vivo studies in which multiple doses of MTP‐Chol‐containing liposomes were administered intravenously showed that early MTP‐Chol treatment could prevent liver metastases after tail vein injection with the M5076 histiocytosarcoma cell line.118 Moreover, intragastric administrations of MTP‐Chol liposomes along with a prophylactic dose two days prior tumor cell injection also moderately limited hepatic metastases. Numerous other lipophilic derivatives of MDP were analysed as well, with similar antitumoral activities reported in vivo.119, 120
Of the many lipophilic MDP compounds, two formulations reached clinical trial stage. One molecule, which was developed in 1985, was produced by conjugating the muropeptide moiety to an analog of the dilipid tail of phospholipids to produce MDP‐glyceryldipalmitate (MDP‐GDP, 14).117 Similar to MTP‐Chol, liposomes containing MDP‐GDP were shown to be 10‐fold more effective than MDP‐containing liposomes in activating macrophage‐mediated cell killing of tumor cells. Starting three days after tail vein injection of the B16‐BL6 melanoma cell line, intravenous injections of MDP‐GDP‐containing liposomes led to a decrease in the number of pulmonary metastases. Interestingly, this treatment regimen led to the eradication of smaller metastases but appeared to have little effect on larger nodules. In a separate study using the B16‐F1 model of liver metastasis, it was seen that either a single prophylactic injection or multiple therapeutic injections of liposomal MDP‐GDP could effectively decrease the number of hepatic tumors.121 Similarly, using the H‐59 model of liver metastases, therapeutic activity of liposomal MDP‐GDP against hepatic tumor formation inversely correlated with the number of administered tumor cells, and pretreatment of the animals showed a similar enhancement of drug efficacy.122 Driven by preclinical results, MDP‐GDP was developed into the disaccharide‐tripeptide drug candidate ImmTher, which showed low toxicity in a Phase I clinical trial.123 Two Phase II clinical trials were established in 1997 and 1998, respectively, to examine the usefulness of ImmTher administration after chemotherapy and surgical resection in patients with high‐risk Ewing’s sarcoma,124, 125 a rare bone cancer found in children and young adults that most commonly metastasises to the lungs. The first trial (NCT00038142) was officially terminated in March 2016 due to low accrual.124 The second trial (NCT00003667) completed recruitment in August 2000,125 but no public results on the trial outcomes are available on clinicaltrials.gov as of November 2019.
Another structurally similar MDP derivative MTP‐PE 15 was developed in 1982 via conjugation of MDP‐L‐alanine to the phospholipid dipalmitoylphosphatidylethanolamine.126 MTP‐PE administration via liposome encapsulation showed increased survival in both a UV‐induced skin cancer model127 and spontaneous and resection‐based B16‐BL6 metastasis models in mice,128 with the best responses observed with more frequent and earlier therapeutic intervention. MTP‐PE could also be formulated as a micellar single agent, and oral administration of MTP‐PE micelles showed body‐wide distribution via radiotracing experiments and decreased the number of pulmonary and lymph node metastases in the B16/BL6 amputation model.129 However, oral MTP‐PE treatment did not show efficacy in the treatment of larger, established metastases, suggesting that MDP‐based activation of macrophages was only effective to inhibit rather than treat metastases. In further experiments, treatment of spontaneous canine osteosarcoma after limb amputation with liposomal MTP‐PE led to an overall increase in disease‐free and overall survival.130 Liposomal MTP‐PE, renamed mifamurtide and later Junovan (IDM Pharma) and now Mepact (Takeda), began Phase I and Phase II clinical trial enrolment in the mid‐1980s, where it showed no major toxicity in adults with refractory metastatic cancer and led to increased circulating TNFα and IL‐6 in patients with relapsed osteosarcoma, respectively.131, 132 These successes led to a large‐scale Phase III clinical trial started by the Children’s Cancer Group and Pediatric Oncology Group, later merged as the Children’s Oncology Group, in 1993.133, 134, 135 Enrolling patients under the age of 30 with newly diagnosed osteosarcoma, the INT 0133 study was conducted with a 2x2 factorial design to determine whether the addition of the alkylating agent ifosfamide and/or liposomal MTP‐PE to standard chemotherapy would improve event‐free and overall survival. Although results from the trial showed an overall increase in event‐free and overall survival upon administration of liposomal MTP‐PE from pooled groups with and without ifosfamide treatment,134 the trial received criticism due to the factorial study design and the possible interaction between ifosfamide and liposomal MTP‐PE.136 These concerns ultimately led to the rejection of liposomal MTP‐PE as a new osteosarcoma treatment by the US Food and Drug Administration. However, the drug is currently approved in the European Union for the treatment of nonmetastatic osteosarcoma after resection along with multidrug chemotherapy.
In addition to its well‐studied effects on macrophages, MDP and its derivatives have also been implicated to limit tumor growth through the activation of other myeloid populations including dendritic cells (DCs). For example, various 6‐_O_‐acylated MDP derivatives were found to improve expression of mature cell‐surface markers such as CD80, CD83 and CD86 and increased TNFα production in monocyte‐derived DCs (moDCs).137 However, these results were ablated using TLR2/4 blocking antibodies or genetic knockout, which suggested that this process occurred independent of Nod2. Later studies showed that treatment of moDCs with romurtide 9 in combination with IFNβ, a cytokine that leads to DC‐mediated cross‐presentation to CD8+ T cells in vitro,138 also led to increased phenotypic maturation as shown by flow cytometry and cytokine production.139 In vitro treatment of purified, allogeneic T cells with moDCs activated with IFNβ and romurtide showed a significant increase in IFNγ production. Furthermore, adjacent, intradermal co‐injection of IFNβ and varying concentrations of romurtide led to a significant decrease in primary tumor size and improved lymphocytic infiltration using a subcutaneous B16‐F10 melanoma model in mice. Similar activation results upon MDP treatment were also observed in human DCs differentiated in vitro using mononuclear bone marrow cells purified from acute leukaemia patients.140 More broadly, MDP was shown to elicit the migration of Ly6Chigh monocytes from the bone marrow to the lungs141 and to convert inflammatory, Ly6Chigh monocytes into Ly6Clow monocytes that exhibit patrolling properties,142 highlighting the complex and likely simultaneous activities of MDP in systemic circulation on myeloid cell populations.
Nod1 agonists FK‐156 3 and FK‐565 4 have demonstrated antitumoral activity in preclinical models as well. Direct intratumoral injection of either compound into established P388 leukaemia subcutaneous tumors led to a significant decrease in tumor growth.143 Distal, nuchal administration also modestly slowed tumor development. In addition, FK‐565 was shown to enhance cytotoxic activity of macrophages against B16 melanoma cells in vitro and in vivo.144 Although a clinical trial for FK‐565 was initiated based on these promising results, the trial was ended in 1995 without further progression. More recently, FK‐565 was shown to act on CD8ɑ+ DCs to promote cross‐presentation.145 Treatment of DC and OT‐I T‐cell co‐cultures with the OVA antigen and FK‐565 led to increased T‐cell proliferation. Treatment of intradermal, OVA‐expressing E.G7 lymphoma tumors with FK‐565 alone showed no difference in tumor growth, but co‐administration of FK‐565 and the OVA peptide significantly impeded tumor growth and increased OVA‐specific CD8+ T cells.
Conclusions and future perspectives
Since the elucidation of their chemical structures over 30 years ago, hundreds of MDP and iE‐DAP derivatives have been synthesised. However, only two of these molecules, romurtide 9 and mifamurtide 10, have successfully entered the clinic, highlighting the disconnect between preclinical activity and clinical implementation. Nevertheless, synthetic studies have yielded a number of molecules that have greatly improved activity over the natural precursors. Molecules such as _N,N_‐dilauryl‐iE‐DAP 6 and murabutide not only provide useful tools to interrogate the outcomes of Nod agonism in vitro but also allow for the direct testing of Nod activity in diverse in vivo contexts. Moreover, the development of desmuramyl compounds 11 and 12 underscores the essentially untapped chemical potential for Nod agonism found by moving away from the natural PG architecture.
A clear synthetic theme among MDP and iE‐DAP analogs is the marked increase in lipophilicity. This is somewhat unsurprising as both MDP and iE‐DAP are relatively hydrophilic molecules, and the addition of lipophilic tails as well as the formation of liposomes likely improve cellular uptake. Yet, these advances suggest that the enhanced activity from many current, ‘best‐in‐class’ molecules results largely from improved bioavailability rather than increased binding affinity towards the Nod proteins. The desmuramyl compounds 11 and 12 demonstrate that Nod2 agonism can be achieved without the complete glycopeptide scaffold, opening up the possibility for chemically distinct, small molecule Nod agonists. Therefore, approaches such as traditional, high‐throughput screening methods may help to identify Nod agonists with higher ‘on‐target’ binding affinity. The development of more ‘synthetic’ Nod agonists may also subvert other PG processing enzymes expressed in the host and microbiota that could interfere with naturally occurring peptide‐based PG ligands. Bacteria have evolved numerous modifications to their PG structure including glycan _O_‐acetylation and _N_‐deacetylation to avoid immune surveillance.146 Thus, defining the chemical diversity of bacterial PG may offer an alternative, native screen for structure–activity relationships to augment the development of Nod agonists. Unfortunately, no structure of either Nod protein in its active conformation has been solved, limiting in silico screening for Nod agonists. Although the recently solved apoprotein structure of rabbit Nod2147 has allowed for preliminary docking studies of the possible MDP binding site, it remains unclear whether Nods undergo significant structural changes upon binding to reveal altered or possibly new sites to chemically target. Therefore, the structural elucidation of active Nod1 and Nod2 with bound PG ligands could greatly facilitate progress towards next‐generation Nod agonists.
Finally, the broader success of Nod targeting in the clinic remains unclear. Complementary efforts to define the chemical determinants and biological activity of Nod agonists have yielded enticing preclinical results for the treatment of infection, cancer and even obesity‐induced insulin resistance.148 Through this iterative process, current Nod agonists have overcome a number of in vivo challenges faced by MDP and iE‐DAP. The addition of lipophilic tails has provided molecules that remain in the body much longer than the quickly excreted native structures. Moreover, murabutide and other analogs have demonstrated similar efficacy as MDP without pyrogenic side effects. Nevertheless, very few adjuvants overall have received clinical approval, underscoring the difficulty in selectively controlling immune responses from molecules with such multifaceted activities. For cancer treatment in particular, the use of Nod agonists must overcome lingering questions. Liposomal MTP‐PE 15 has received approval for use against paediatric osteosarcoma after both resection and multidrug chemotherapy. This indication reflects preclinical data in which MDP derivatives were successful only when administered prior to or at the onset of metastases. Thus, the strategic implementation of Nod agonists in combination with other therapies may provide the most effective path forward to improve patient outcomes.
Conflict of Interest
MEG and HCH are inventors on a patent filed by The Rockefeller University for the use of SagA towards the treatment of cancer and infection. Rise Therapeutics has licensed the patent on SagA for probiotic development.
Acknowledgments
MEG is a Hope Funds for Cancer Research Fellow supported by the Hope Funds for Cancer Research (HFCR‐19‐03‐02) and is supported in part by a research grant from the Melanoma Research Foundation. CWH is a Ruth L. Kirschstein National Research Service Award Postdoctoral Fellow supported by the National Institutes of Health (NIH‐NICCH F32 AT010087‐01A1). YCW was a Cancer Research Institute Irvington Fellow supported by the Cancer Research Institute. HCH acknowledges support from the National Institutes of Health (NIH‐NIGMS R01 GM103593) and the Kenneth Rainin Foundation (Synergy Award).
References
- 1.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature 2007; 449: 819–826. [DOI] [PubMed] [Google Scholar]
- 2.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140: 805–820. [DOI] [PubMed] [Google Scholar]
- 3.Wolf AJ, Underhill DM. Peptidoglycan recognition by the innate immune system. Nat Rev Immunol 2018; 18: 243–254. [DOI] [PubMed] [Google Scholar]
- 4.Typas A, Banzhaf M, Gross CA_et al_From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol 2011; 10: 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vollmer W, Blanot D, de Pedro MA. Peptidoglycan structure and architecture. FEMS Microbiol Rev 2008; 32: 149–167. [DOI] [PubMed] [Google Scholar]
- 6.Vollmer W, Joris B, Charlier P_et al_Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev 2008; 32: 259–286. [DOI] [PubMed] [Google Scholar]
- 7.Humann J, Lenz LL. Bacterial peptidoglycan degrading enzymes and their impact on host muropeptide detection. J Innate Immun 2009; 1: 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellouz F, Adam A, Ciorbaru R_et al_Minimal structural requirements for adjuvant activity of bacterial peptidoglycan derivatives. Biochem Biophys Res Comm 1974; 59: 1317–1325. [DOI] [PubMed] [Google Scholar]
- 9.Freund J, Casals J, Hosmer EP. Sensitization and antibody formation after injection of tubercle bacilli and paraffin oil. Proc Soc Exp Biol Med 1937; 37: 509–513. [Google Scholar]
- 10.Chamaillard M, Hashimoto M, Horie Y_et al_An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol 2003; 4: 702–707. [DOI] [PubMed] [Google Scholar]
- 11.Girardin SE, Boneca IG, Carneiro LAM_et al_Nod1 detects a unique muropeptide from gram‐negative bacterial peptidoglycan. Science 2003; 300: 1584–1587. [DOI] [PubMed] [Google Scholar]
- 12.Girardin SE, Boneca IG, Viala J_et al_Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 2003; 278: 8869–8872. [DOI] [PubMed] [Google Scholar]
- 13.Inohara N, Ogura Y, Fontalba A_et al_Host recognition of bacterial muramyl dipeptide mediated through NOD2: implications for Crohn’s disease. J Biol Chem 2003; 278: 5509–5512. [DOI] [PubMed] [Google Scholar]
- 14.Caruso R, Warner N, Inohara N_et al_NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 2014; 41: 898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Philpott DJ, Sorbara MT, Robertson SJ_et al_NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol 2014; 14: 9–23. [DOI] [PubMed] [Google Scholar]
- 16.Mukherjee T, Hovingh ES, Foerster EG_et al_NOD1 and NOD2 in inflammation, immunity and disease. Arch Biochem Biophys 2019; 670: 69–81. [DOI] [PubMed] [Google Scholar]
- 17.Girardin SE, Travassos LH, Hervé M_et al_Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem 2003; 278: 41702–41708. [DOI] [PubMed] [Google Scholar]
- 18.Magalhaes JG, Philpott DJ, Nahori M‐A_et al_Murine Nod1 but not its human orthologue mediates innate immune detection of tracheal cytotoxin. EMBO Rep 2005; 6: 1201–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uehara A, Fujimoto Y, Kawasaki A_et al_Meso‐diaminopimelic acid and meso‐lanthionine, amino acids specific to bacterial peptidoglycans, activate human epithelial cells through NOD1. J Immunol 2006; 177: 1796–1804. [DOI] [PubMed] [Google Scholar]
- 20.Hasegawa M, Kawasaki A, Yang K_et al_A role of lipophilic peptidoglycan‐related molecules in induction of Nod1‐mediated immune responses. J Biol Chem 2007; 282: 11757–11764. [DOI] [PubMed] [Google Scholar]
- 21.Nigro G, Rossi R, Commere P‐H_et al_The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe 2014; 15: 792–798. [DOI] [PubMed] [Google Scholar]
- 22.Ogura Y, Lala S, Xin W_et al_Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut 2003; 52: 1591–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laroui H, Yan Y, Narui Y_et al_L-Ala-γ-D-Glu-meso-diaminopimelic acid (DAP) interacts directly with leucine‐rich region domain of nucleotide‐binding oligomerization domain 1, increasing phosphorylation activity of receptor‐interacting serine/threonine‐protein kinase 2 and its interaction with nucleotide‐binding oligomerization domain 1. J Biol Chem 2011; 286: 31003–31013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mo J, Boyle JP, Howard CB_et al_Pathogen sensing by nucleotide‐binding oligomerization domain‐containing protein 2 (NOD2) is mediated by direct binding to muramyl dipeptide and ATP. J Biol Chem 2012; 287: 23057–23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grimes CL, Ariyananda LDZ, Melnyk JE_et al_The innate immune protein Nod2 binds directly to MDP, a bacterial cell wall fragment. J Am Chem Soc 2012; 134: 13535–13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y‐C, Westcott NP, Griffin ME_et al_Peptidoglycan metabolite photoaffinity reporters reveal direct binding to intracellular pattern recognition receptors and Arf GTPases. ACS Chem Biol 2019; 14: 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu Y, Zheng Y, Coyaud E_et al_Palmitoylation of NOD1 and NOD2 is required for bacterial sensing. Science 2019; 366: 460–467. [DOI] [PubMed] [Google Scholar]
- 28.Yoshimura A, Lien E, Ingalls RR_et al_Cutting edge: recognition of Gram‐positive bacterial cell wall components by the innate immune system occurs via Toll‐like receptor 2. J Immunol 1999; 163: 1–5. [PubMed] [Google Scholar]
- 29.Takeuchi O, Hoshino K, Kawai T_et al_Differential roles of TLR2 and TLR4 in recognition of gram‐negative and gram‐positive bacterial cell wall components. Immunity 1999; 11: 443–451. [DOI] [PubMed] [Google Scholar]
- 30.Travassos LH, Girardin SE, Philpott DJ_et al_Toll‐like receptor 2‐dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep 2004; 5: 1000–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolf AJ, Reyes CN, Liang W_et al_Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 2016; 166: 624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inohara N, Koseki T, del Peso L_et al_Nod1, an Apaf‐1‐like activator of caspase‐9 and nuclear factor‐kappaB. J Biol Chem 1999; 274: 14560–14567. [DOI] [PubMed] [Google Scholar]
- 33.Pellegrini E, Desfosses A, Wallmann A_et al_RIP2 filament formation is required for NOD2 dependent NF‐κB signalling. Nat Commun 2018; 9: 4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abdel‐Nour M, Carneiro LAM, Downey J_et al_The heme‐regulated inhibitor is a cytosolic sensor of protein misfolding that controls innate immune signaling. Science 2019; 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Travassos LH, Carneiro LAM, Ramjeet M_et al_Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 2010; 11: 55–62. [DOI] [PubMed] [Google Scholar]
- 36.Hansson GC. Role of mucus layers in gut infection and inflammation. Curr Opin Microbiol 2012; 15: 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mukherjee S, Hooper LV. Antimicrobial defense of the intestine. Immunity 2015; 42: 28–39. [DOI] [PubMed] [Google Scholar]
- 38.Kleinnijenhuis J, Quintin J, Preijers F_et al_Bacille Calmette‐Guerin induces NOD2‐dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA 2012; 109: 17537–17542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Netea MG, Quintin J, van der Meer JWM. Trained immunity: a memory for innate host defense. Cell Host Microbe 2011; 9: 355–361. [DOI] [PubMed] [Google Scholar]
- 40.Clarke TB, Davis KM, Lysenko ES_et al_Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med 2010; 16: 228–U137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hergott CB, Roche AM, Tamashiro E_et al_Peptidoglycan from the gut microbiota governs the lifespan of circulating phagocytes at homeostasis. Blood 2016; 127: 2460–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fritz JH, Le Bourhis L, Sellge G_et al_Nod1‐mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity 2007; 26: 445–459. [DOI] [PubMed] [Google Scholar]
- 43.Magalhaes JG, Fritz JH, Le Bourhis L_et al_Nod2‐dependent Th2 polarization of antigen‐specific immunity. J Immunol 2008; 181: 7925–7935. [DOI] [PubMed] [Google Scholar]
- 44.van Beelen AJ, Zelinkova Z, Taanman‐Kueter EW_et al_Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin‐17 production in human memory T cells. Immunity 2007; 27: 660–669. [DOI] [PubMed] [Google Scholar]
- 45.Parant M, Parant F, Chedid L_et al_Fate of the synthetic immunoadjuvant, muramyl dipeptide (14C‐labelled) in the mouse. Int J Immunopharmacol 1979; 1: 35–41. [DOI] [PubMed] [Google Scholar]
- 46.Harrison J, Fox A. Degradation of muramyl dipeptide by mammalian serum. Infect Immun 1985; 50: 320–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kotani S, Watanabe Y, Shimono T_et al_Correlation between the immunoadjuvant activities and pyrogenicities of synthetic N‐acetylmuramyl‐peptides or ‐amino acids. Biken J 1976; 19: 9–13. [PubMed] [Google Scholar]
- 48.Dinarello CA, Elin RJ, Chedid L_et al_The pyrogenicity of the synthetic adjuvant muramyl dipeptide and two structural analogues. J Infect Dis 1978; 138: 760–767. [DOI] [PubMed] [Google Scholar]
- 49.Krueger JM, Pappenheimer JR, Karnovsky ML. Sleep‐promoting effects of muramyl peptides. Proc Natl Acad Sci USA 1982; 79: 6102–6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krueger JM, Walter J, Karnovsky ML_et al_Muramyl peptides: variation of somnogenic activity with structure. J Exp Med 1984; 159: 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawai Y, Nakahara K, Gotoh T_et al_Studies on a new immunoactive peptide, FK‐ 156: III. Structure elucidation. J Antibiot (Tokyo) 1982; 35: 1293–1299. [DOI] [PubMed] [Google Scholar]
- 52.Gotoh T, Nakahara K, Iwami M_et al_Studies on a new immunoactive peptide, FK‐ 156: I. taxonomy of the producing strains. J Antibiot (Tokyo) 1982; 35: 1280–1285. [DOI] [PubMed] [Google Scholar]
- 53.Gotoh T, Nakahara K, Nishiura T_et al_Studies on a new immunoactive peptide, FK‐ 156: II. Fermentation, extraction and chemical and biological characterization. J Antibiot (Tokyo) 1982; 35: 1286–1292. [DOI] [PubMed] [Google Scholar]
- 54.Kitaura Y, Takeno H, Aratani M_et al_Synthesis and RES‐stimulating activity of bacterial cell‐wall peptidoglycan peptides related to FK‐156. Experientia 1982; 38: 1101–1103. [DOI] [PubMed] [Google Scholar]
- 55.Kitaura Y, Nakaguchi O, Takeno H_et al_N2‐(gamma‐D‐Glutamyl)‐meso‐2(L),2’(D)‐diaminopimelic acid as the minimal prerequisite structure of FK‐156: its acyl derivatives with potent immunostimulating activity. J Med Chem 1982; 25: 335–337. [DOI] [PubMed] [Google Scholar]
- 56.Wolfert MA, Roychowdhury A, Boons G‐J. Modification of the structure of peptidoglycan is a strategy to avoid detection by nucleotide‐binding oligomerization domain protein 1. Infect Immun 2007; 75: 706–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hasegawa M, Yamazaki T, Kamada N_et al_Nucleotide‐binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol 2011; 186: 4872–4880. [DOI] [PubMed] [Google Scholar]
- 58.Agnihotri G, Ukani R, Malladi SS_et al_Structure‐activity relationships in nucleotide oligomerization domain 1 (Nod1) agonistic γ‐glutamyldiaminopimelic acid derivatives. J Med Chem 2011; 54: 1490–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jakopin Z, Gobec M, Kodela J_et al_Synthesis of conformationally constrained γ‐D‐glutamyl‐meso‐diaminopimelic acid derivatives as ligands of nucleotide‐binding oligomerization domain protein 1 (Nod1). Eur J Med Chem 2013; 69: 232–243. [DOI] [PubMed] [Google Scholar]
- 60.Dukor P, Tarcsay L, Baschang G. Chapter 15: immunostimulants. Annu Rep Med Chem 1979; 14: 146–167. [Google Scholar]
- 61.Adam A, Devys M, Souvannavong V_et al_Correlation of structure and adjuvant activity of N‐acetyl‐muramyl‐L‐alanyl‐D‐isoglutamine (MDP), its derivatives and analogues: anti‐adjuvant and competition properties of stereoisomers. Biochem Biophys Res Comm 1976; 72: 339–346. [DOI] [PubMed] [Google Scholar]
- 62.Lederer E. Synthetic immunostimulants derived from the bacterial cell wall. J Med Chem 1980; 23: 819–825. [DOI] [PubMed] [Google Scholar]
- 63.Tsujimoto M, Kinoshita F, Okunaga T_et al_Higher immunoadjuvant activities of N‐acetyl‐β‐D‐glucosaminyl‐(1–4)‐N‐acetylmuramyl‐L‐alanyl‐D‐isoglutamine in comparison with N‐acetylmuramyl‐L‐alanyl‐D‐isoglutamine. Microbiol Immunol 1979; 23: 933–936. [DOI] [PubMed] [Google Scholar]
- 64.Durette PL, Meitzner EP, Shen TY. Bacterial cell wall constituents: II. synthesis of O‐(N‐acetyl‐β‐muramyl‐L‐alanyl‐D‐isoglutamine)‐(1→4)‐N‐acetyl‐D‐glucosamine. Tet Lett 1979; 20: 4013–4016. [Google Scholar]
- 65.Wang N, Huang C, Hasegawa M_et al_Glycan sequence‐dependent Nod2 activation investigated by using a chemically synthesized bacterial peptidoglycan fragment library. ChemBioChem 2013; 14: 482–488. [DOI] [PubMed] [Google Scholar]
- 66.Inamura S, Fujimoto Y, Kawasaki A_et al_Synthesis of peptidoglycan fragments and evaluation of their biological activity. Org Biomol Chem 2006; 4: 232–242. [DOI] [PubMed] [Google Scholar]
- 67.Coulombe F, Divangahi M, Veyrier F_et al_Increased NOD2‐mediated recognition of N‐glycolyl muramyl dipeptide. J Exp Med 2009; 206: 1709–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Matsumoto K, Ogawa H, Kusama T_et al_Stimulation of nonspecific resistance to infection induced by 6‐O‐acyl muramyl dipeptide analogs in mice. Infect Immun 1981; 32: 748–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tsujimoto M, Kotani S, Kinoshita F_et al_Adjuvant activity of 6‐O‐acyl‐muramyldipeptides to enhance primary cellular and humoral immune responses in guinea pigs: adaptability to various vehicles and pyrogenicity. Infect Immun 1986; 53: 511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matsumoto K, Otani T, Une T_et al_Stimulation of nonspecific resistance to infection induced by muramyl dipeptide analogs substituted in the gamma‐carboxyl group and evaluation of Nɑ‐muramyl dipeptide‐Nε‐stearoyllysine. Infect Immun 1983; 39: 1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ichiro A. Development of the cytokine inducer romurtide: experimental studies and clinical application. Trends Pharmacol Sci 1992; 13: 425–428. [DOI] [PubMed] [Google Scholar]
- 72.Ichiro A. Review: Inducer of cytokines in vivo: overview of field and romurtide experience. Int J Immunopharmacol 1992; 14: 487–496. [DOI] [PubMed] [Google Scholar]
- 73.Chedid LA, Parant MA, Audibert FM_et al_Biological activity of a new synthetic muramyl peptide adjuvant devoid of pyrogenicity. Infect Immun 1982; 35: 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jakopin Z. Murabutide revisited: a review of its pleiotropic biological effects. Curr Med Chem 2013; 20: 2068–2079. [DOI] [PubMed] [Google Scholar]
- 75.Sidwell RW, Smee DF, Huffman JH_et al_Antiviral activity of an immunomodulatory lipophilic desmuramyl dipeptide analog. Antiviral Res 1995; 26: 145–159. [DOI] [PubMed] [Google Scholar]
- 76.Sollner M, Kotnik V, Pecar S_et al_Apyrogenic synthetic desmuramyldipeptide, LK‐409, with immunomodulatory properties. Agents Actions 1993; 38: 273–280. [DOI] [PubMed] [Google Scholar]
- 77.Leclerc CD, Audibert FM, Chedid LA_et al_Comparison of immunomodulatory activities in mice and guinea pigs of a synthetic desmuramyl peptidolipid triglymyc. Infect Immun 1984; 43: 870–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zidek Z, Masek K, Frankova D_et al_T‐cell‐dependent immunobiological activity of a desmuramyl analog of muramyl dipeptide, adamantylamide dipeptide (AdDP), and D‐isoglutamine. Int J Immunopharmacol 1993; 15: 631–637. [DOI] [PubMed] [Google Scholar]
- 79.Jakopin Ž, Gobec M, Mlinarič‐Raščan I_et al_Immunomodulatory properties of novel nucleotide oligomerization domain 2 (Nod2) agonistic desmuramyldipeptides. J Med Chem 2012; 55: 6478–6488. [DOI] [PubMed] [Google Scholar]
- 80.Gobec M, Tomašič T, Štimac A_et al_Discovery of nanomolar desmuramylpeptide agonists of the innate immune receptor nucleotide‐binding oligomerization domain‐containing protein 2 (NOD2) possessing immunostimulatory properties. J Med Chem 2018; 61: 2707–2724. [DOI] [PubMed] [Google Scholar]
- 81.Moreira LO, Zamboni DS. NOD1 and NOD2 signaling in infection and inflammation. Front Immunol 2012; 3: 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boughan PK, Argent RH, Body‐Malapel M_et al_Nucleotide‐binding oligomerization domain‐1 and epidermal growth factor receptor: critical regulators of beta‐defensins during Helicobacter pylori infection. J Biol Chem 2006; 281: 11637–11648. [DOI] [PubMed] [Google Scholar]
- 83.Robertson SJ, Zhou JY, Geddes K_et al_Nod1 and Nod2 signaling does not alter the composition of intestinal bacterial communities at homeostasis. Gut Microbes 2013; 4: 222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kobayashi KS, Chamaillard M, Ogura Y_et al_Nod2‐dependent regulation of innate and adaptive immunity in the intestinal tract. Science 2005; 307: 731–734. [DOI] [PubMed] [Google Scholar]
- 85.Shanahan MT, Carroll IM, Grossniklaus E_et al_Mouse Paneth cell antimicrobial function is independent of Nod2. Gut 2014; 63: 903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rangan KJ, Pedicord VA, Wang Y‐C_et al_A secreted bacterial peptidoglycan hydrolase enhances tolerance to enteric pathogens. Science 2016; 353: 1434–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pedicord VA, Lockhart AAK, Rangan KJ_et al_Exploiting a host‐commensal interaction to promote intestinal barrier function and enteric pathogen tolerance. Sci Immunol 2016; 1: eaai7732–eaai7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim B, Wang Y‐C, Hespen CW_et al_ Enterococcus faecium secreted antigen A generates muropeptides to enhance host immunity and limit bacterial pathogenesis. eLife 2019; 8: e45343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Goasduff T, Darcissac ECA, Vidal V_et al_The transcriptional response of human macrophages to murabutide reflects a spectrum of biological effects for the synthetic immunomodulator. Clin Exp Immunol 2002; 128: 474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Calmette A. L’Infection bacillaire et la tuberculose chez l’homme et chez les animaux. Paris, France: Libraires de l"Académie de Médecine; 1920. [Google Scholar]
- 91.Calmette A. Preventive vaccination against tuberculosis with BCG. Proc R Soc Med 1931; 24: 1481–1490. [PMC free article] [PubMed] [Google Scholar]
- 92.Billiau A, Matthys P. Modes of action of Freund’s adjuvants in experimental models of autoimmune diseases. J Leukoc Biol 2001; 70: 849–860. [PubMed] [Google Scholar]
- 93.Siddiqui WA, Taylor DW, Kan SC_et al_Vaccination of experimental monkeys against Plasmodium falciparum: a possible safe adjuvant. Science 1978; 201: 1237–1239. [DOI] [PubMed] [Google Scholar]
- 94.Onozuka K, Shinomiya H, Cho N_et al_The adjuvant effect of a muramyl dipeptide (MDP) analog on temperature‐sensitive Salmonella mutant vaccine. Int J Immunopharmacol 1989; 11: 781–787. [DOI] [PubMed] [Google Scholar]
- 95.Yoo YC, Yoshimatsu K, Koike Y_et al_Adjuvant activity of muramyl dipeptide derivatives to enhance immunogenicity of a hantavirus‐inactivated vaccine. Vaccine 1998; 16: 216–224. [DOI] [PubMed] [Google Scholar]
- 96.Yoo YC, Saiki I, Sato K_et al_B30‐MDP, a synthetic muramyl dipeptide derivative for tumour vaccination to enhance antitumour immunity and antimetastatic effect in mice. Vaccine 1992; 10: 792–797. [DOI] [PubMed] [Google Scholar]
- 97.Keitel W, Couch R, Bond N_et al_Pilot evaluation of influenza virus vaccine (IVV) combined with adjuvant. Vaccine 1993; 11: 909–913. [DOI] [PubMed] [Google Scholar]
- 98.Keefer MC, Graham BS, McElrath MJ_et al_Safety and immunogenicity of Env 2–3, a human immunodeficiency virus type 1 candidate vaccine, in combination with a novel adjuvant, MTP‐PE/MF59. NIAID AIDS Vaccine Evaluation Group. AIDS Res Hum Retroviruses 1996; 12: 683–693. [DOI] [PubMed] [Google Scholar]
- 99.Graham BS, Keefer MC, McElrath MJ_et al_Safety and immunogenicity of a candidate HIV‐1 vaccine in healthy adults: recombinant glycoprotein (rgp) 120: a randomized, double‐blind trial. Ann Int Med 1996; 125: 270–279. [DOI] [PubMed] [Google Scholar]
- 100.Aaby P, Benn CS. Saving lives by training innate immunity with bacille Calmette‐Guerin vaccine. Proc Natl Acad Sci USA 2012; 109: 17317–17318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ishihara C, Yamamoto K, Hamada N_et al_Effect of stearoyl‐N‐acetylmuramyl‐L‐alanyl‐D‐isoglutamine on host resistance to Corynebacterium kutscheri infection in cortisone‐treated mice. Vaccine 1984; 2: 261–264. [DOI] [PubMed] [Google Scholar]
- 102.Osada Y, Mitsuyama M, Matsumoto K_et al_Stimulation of resistance of immunocompromised mice by a muramyl dipeptide analog. Infect Immun 1982; 37: 1285–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Osada Y, Ohtani T, Une T_et al_Enhancement of non‐specific resistance to Pseudomonas pneumonia by a synthetic derivative of muramoyl dipeptide in immunosuppressed guinea pigs. J Gen Microbiol 1982; 128: 2361–2370. [DOI] [PubMed] [Google Scholar]
- 104.Ishihara C, Iida J, Mizukoshi N_et al_Effect of Nɑ‐acetylmuramyl‐L‐alanyl‐D‐isoglutaminyl‐Nε‐stearoyl‐L‐lysine on resistance to herpes simplex virus type‐1 infection in cyclophosphamide‐treated mice. Vaccine 1989; 7: 309–313. [DOI] [PubMed] [Google Scholar]
- 105.Ikeda S, Negishi T, Nishimura C. Enhancement of non‐specific resistance to viral infection by muramyldipeptide and its analogs. Antiviral Res. 1985; 5: 207–215. [DOI] [PubMed] [Google Scholar]
- 106.Bahr GM, Darcissac EC, Casteran N_et al_Selective regulation of human immunodeficiency virus‐infected CD4+ lymphocytes by a synthetic immunomodulator leads to potent virus suppression in vitro and in hu‐PBL‐SCID mice. J Virol 2001; 75: 6941–6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mine Y, Yokota Y, Wakai Y_et al_Immunoactive peptides, FK‐156 and FK‐ 565: I. Enhancement of host resistance to microbial infection in mice. J Antibiot (Tokyo) 1983; 36: 1045–1050. [DOI] [PubMed] [Google Scholar]
- 108.Sparks FC, Silverstein MJ, Hunt JS_et al_Complications of BCG immunotherapy in patients with cancer. N Engl J Med 1973; 289: 827–830. [DOI] [PubMed] [Google Scholar]
- 109.Aungst CW, Sokal JE, Jager BV. Complications of BCG vaccination in neoplastic disease. Ann Intern Med 1975; 82: 666–669. [DOI] [PubMed] [Google Scholar]
- 110.Azuma I, Sugimura K, Yamawaki M_et al_Adjuvant activity of synthetic 6‐O‐"mycoloyl"‐N‐acetylmuramyl‐L‐alanyl‐D‐isoglutamine and related compounds. Infect Immun 1978; 20: 600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McLaughlin CA, Schwartzman SM, Horner BL_et al_Regression of tumors in guinea pigs after treatment with synthetic muramyl dipeptides and trehalose dimycolate. Science 1980; 208: 415–416. [DOI] [PubMed] [Google Scholar]
- 112.Taniyama T, Holden HT. Direct augmentation of cytolytic activity of tumor‐derived macrophages and macrophage cell lines by muramyl dipeptide. Cell Immunol 1979; 48: 369–374. [DOI] [PubMed] [Google Scholar]
- 113.Sone S, Fidler IJ. Synergistic activation by lymphokines and muramyl dipeptide of tumoricidal properties in rat alveolar macrophages. J Immunol 1980; 125: 2454–2460. [PubMed] [Google Scholar]
- 114.Sone S, Mutsuura S, Ogawara M_et al_Potentiating effect of muramyl dipeptide and its lipophilic analog encapsulated in liposomes on tumor cell killing by human monocytes. J Immunol 1984; 132: 2105–2110. [PubMed] [Google Scholar]
- 115.Fidler IJ, Sone S, Fogler WE_et al_Eradication of spontaneous metastases and activation of alveolar macrophages by intravenous injection of liposomes containing muramyl dipeptide. Proc Natl Acad Sci USA 1981; 78: 1680–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fidler IJ. The in situ induction of tumoricidal activity in alveolar macrophages by liposomes containing muramyl dipeptide is a thymus‐independent process. J Immunol 1981; 127: 1719–1720. [PubMed] [Google Scholar]
- 117.Phillips NC, Moras ML, Chedid L_et al_Activation of macrophage cytostatic and cytotoxic activity in vitro by liposomes containing a new lipophilic muramyl peptide derivative, MDP‐L‐alanyl‐cholesterol (MTP‐CHOL). J Biol Response Mod 1985; 4: 464–474. [PubMed] [Google Scholar]
- 118.Yu WP, Barratt GM, Devissaguet JP_et al_Anti‐metastatic activity in vivo of MDP‐L‐alanyl‐cholesterol (MTP‐Chol) entrapped in nanocapsules. Int J Immunopharmacol 1991; 13: 167–173. [DOI] [PubMed] [Google Scholar]
- 119.Lopez‐Berestein G, Milas L, Hunter N_et al_Prophylaxis and treatment of experimental lung metastases in mice after treatment with liposome‐encapsulated. Clin Exp Metastasis 1984; 2: 127–137. [DOI] [PubMed] [Google Scholar]
- 120.Yoo YC, Saiki I, Sato K_et al_MDP‐Lys(L18), a lipophilic derivative of muramyl dipeptide, inhibits the metastasis of haematogenous and non‐haematogenous tumours in mice. Vaccine 1994; 12: 175–160. [DOI] [PubMed] [Google Scholar]
- 121.Phillips NC, Tsao MS. Inhibition of experimental liver tumor growth in mice by liposomes containing a lipophilic muramyl dipeptide derivative. Cancer Res 1989; 49: 936–939. [PubMed] [Google Scholar]
- 122.Brodt P, Blore J, Phillips NC_et al_Inhibition of murine hepatic tumor growth by liposomes containing a lipophilic muramyl dipeptide. Cancer Immunol Immunother 1989; 28: 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vosika GJ, Cornelius DA, Gilbert CW_et al_Phase I trial of ImmTher, a new liposome‐incorporated lipophilic disaccharide tripeptide. J Immunother 1991; 10: 256–266. [DOI] [PubMed] [Google Scholar]
- 124.Vincristine, doxorubicin, cyclophosphamide and dexrazoxane (VACdxr) in high risk Ewing’s sarcoma patients. ClinicalTrials.gov. US National Library of Medicine; https://clinicaltrials.gov/ct2/show/NCT00038142. Accessed November 1, 2019. [Google Scholar]
- 125.Combination chemotherapy and biological therapy in treating patients with high‐risk Ewing’s sarcoma. ClinicalTrials.gov. US National Library of Medicine; https://clinicaltrials.gov/ct2/show/NCT00003667. Accessed November 1, 2019. [Google Scholar]
- 126.Gisler R, Schumann G, Sackmann W. _et al._A novel muramyl peptide, MTP‐PE: profile of biological activities In: Yamamura Y, & Kotani S, eds. Immunomodulation by microbial products and related synthetic compounds. 1982, pp. 167–170. [Google Scholar]
- 127.Talmadge JE, Lenz BF, Klabansky R_et al_Therapy of autochthonous skin cancers in mice with intravenously injected liposomes containing muramyltripeptide. Cancer Res 1986; 46: 1160–1163. [PubMed] [Google Scholar]
- 128.Fidler IJ. Optimization and limitations of systemic treatment of murine melanoma metastases with liposomes containing muramyl tripeptide phosphatidylethanolamine. Cancer Immunol Immunother 1986; 21: 169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fidler IJ, Fogler WE, Brownbill AF_et al_Systemic activation of tumoricidal properties in mouse macrophages and inhibition of melanoma metastases by the oral administration of MTP‐PE, a lipophilic muramyl dipeptide. J Immunol 1987; 138: 4509–4514. [PubMed] [Google Scholar]
- 130.MacEwen EG, Kurzman ID, Rosenthal RC_et al_Therapy for osteosarcoma in dogs with intravenous injection of liposome‐encapsulated muramyl tripeptide. J Natl Cancer Inst 1989; 81: 935–938. [DOI] [PubMed] [Google Scholar]
- 131.Murray JL, Kleinerman ES, Cunningham JE_et al_Phase I trial of liposomal muramyl tripeptide phosphatidylethanolamine in cancer patients. J Clin Oncol 1989; 7: 1915–1925. [DOI] [PubMed] [Google Scholar]
- 132.Kleinerman ES, Jia SF, Griffin J_et al_Phase II study of liposomal muramyl tripeptide in osteosarcoma: the cytokine cascade and monocyte activation following administration. J Clin Oncol 1992; 10: 1310–1316. [DOI] [PubMed] [Google Scholar]
- 133.Meyers PA, Schwartz CL, Krailo M_et al_Osteosarcoma: A randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high‐dose methotrexate. J Clin Oncol 2005; 23: 2004–2011. [DOI] [PubMed] [Google Scholar]
- 134.Meyers PA, Schwartz CL, Krailo MD_et al_Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival–a report from the Children’s Oncology Group. J Clin Oncol 2008; 26: 633–638. [DOI] [PubMed] [Google Scholar]
- 135.Chou AJ, Kleinerman ES, Krailo MD_et al_Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma: a report from the Children’s Oncology Group. Cancer 2009; 115: 5339–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hunsberger S, Freidlin B, Smith MA. Complexities in interpretation of osteosarcoma clinical trial results. J Clin Oncol 2008; 26: 3103–3104; author reply 3104–3105. [DOI] [PubMed] [Google Scholar]
- 137.Uehori J, Fukase K, Akazawa T_et al_Dendritic cell maturation induced by muramyl dipeptide (MDP) derivatives: monoacylated MDP confers TLR2/TLR4 activation. J Immunol 2005; 174: 7096–7103. [DOI] [PubMed] [Google Scholar]
- 138.Le Bon A, Etchart N, Rossmann C_et al_Cross‐priming of CD8+ T cells stimulated by virus‐induced type I interferon. Nat Immunol 2003; 4: 1009–1015. [DOI] [PubMed] [Google Scholar]
- 139.Fujimura T, Yamasaki K, Hidaka T_et al_A synthetic NOD2 agonist, muramyl dipeptide (MDP)‐Lys (L18) and IFNβ synergistically induce dendritic cell maturation with augmented IL‐12 production and suppress melanoma growth. J Dermatol Sci 2011; 62: 107–115. [DOI] [PubMed] [Google Scholar]
- 140.Wang L‐Z, Li X‐L, Pang X‐Y_et al_Muramyl dipeptide modulates differentiation, maturity of dendritic cells and anti‐tumor effect of DC‐mediated T cell in acute leukemia children. Hum Vaccin 2011; 7: 618–624. [DOI] [PubMed] [Google Scholar]
- 141.Coulombe F, Fiola S, Akira S_et al_Muramyl dipeptide induces NOD2‐dependent Ly6Chigh monocyte recruitment to the lungs and protects against influenza virus infection. PLoS One 2012; 7: e36734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lessard A‐J, LeBel M, Egarnes B_et al_Triggering of NOD2 receptor converts inflammatory Ly6Chigh into Ly6Clow monocytes with patrolling properties. Cell Rep 2017; 20: 1830–1843. [DOI] [PubMed] [Google Scholar]
- 143.Izumi S, Nakahara K, Gotoh T_et al_Antitumor effects of novel immunoactive peptides, FK‐156 and its synthetic derivatives. J Antibiot (Tokyo) 1983; 36: 566–574. [DOI] [PubMed] [Google Scholar]
- 144.Inamura N, Nakahara K, Kino T_et al_Activation of tumoricidal properties in macrophages and inhibition of experimentally‐induced murine metastases by a new synthetic acyltripeptide. J Biol Response Mod 1985; 4: 408–417. [PubMed] [Google Scholar]
- 145.Asano J, Tada H, Onai N_et al_Nucleotide oligomerization binding domain‐like receptor signaling enhances dendritic cell‐mediated cross‐priming in vivo. J Immunol 2010; 184: 736–745. [DOI] [PubMed] [Google Scholar]
- 146.Davis KM, Weiser JN. Modifications to the peptidoglycan backbone help bacteria to establish infection. Infect Immun 2011; 79: 562–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Maekawa S, Ohto U, Shibata T_et al_Crystal structure of NOD2 and its implications in human disease. Nat Commun 2016; 7: 11813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cavallari JF, Fullerton MD, Duggan BM_et al_Muramyl dipeptide‐based postbiotics mitigate obesity‐induced insulin resistance via IRF4. Cell Metab 2017; 25: 1063–1074.e3. [DOI] [PubMed] [Google Scholar]