Second messengers and divergent HD-GYP phosphodiesterases regulate 3’,3’-cGAMP signaling (original) (raw)
. Author manuscript; available in PMC: 2021 Jan 1.
Published in final edited form as: Mol Microbiol. 2019 Nov 17;113(1):222–236. doi: 10.1111/mmi.14412
Abstract
3’,3’-cyclic GMP-AMP (cGAMP) is the third cyclic dinucleotide (CDN) to be discovered in bacteria. No activators of cGAMP signaling have yet been identified, and the signaling pathways for cGAMP have been inferred to display a narrow distribution based upon the characterized synthases, DncV and Hypr GGDEFs. Here we report that the ubiquitous second messenger cyclic AMP (cAMP) is an activator of the Hypr GGDEF enzyme GacB from Myxococcus xanthus. Furthermore, we show that GacB is inhibited directly by cyclic di-GMP, which provides evidence for cross-regulation between different CDN pathways. Finally, we reveal that the HD-GYP enzyme PmxA is a cGAMP-specific phosphodiesterase (GAP) that promotes resistance to osmotic stress in M. xanthus. A signature amino acid change in PmxA was found to reprogram substrate specificity and was applied to predict the presence of non-canonical HD-GYP phosphodiesterases in many bacterial species, including phyla previously not known to utilize cGAMP signaling.
ABBREVIATED SUMMARY
There are two distinct functions and pathways for cGAMP signalling in bacteria. We have discovered the first regulatory mechanisms for the Hypr GGDEF pathway. The synthase GacB from Myxococcus xanthus is activated by cAMP through its GAF domain and inhibited by cyclic di-GMP through the I-site. We also discovered that some HD-GYP enzymes with signature amino acid changes are cGAMP-specific phosphodiesterases (GAPs). Using bioinformatics, we predict GAP enzymes in bacteria not known to utilize cGAMP.
INTRODUCTION
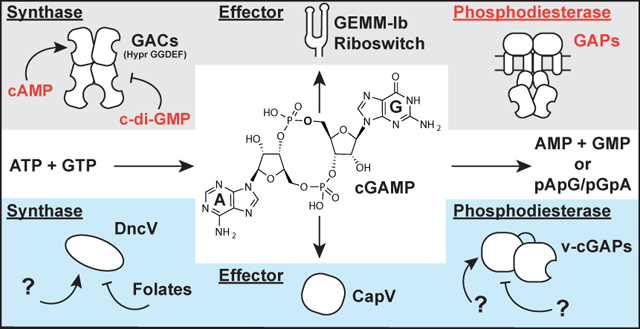
Diverse signaling pathways are comprised of specific enzyme classes that produce and degrade a signal, as well as biomolecules that respond to the signal. In cyclic dinucleotide signaling, synthases produce the cyclic dinucleotide (CDN) signal from nucleotide triphosphate (NTP) substrates, phosphodiesterases (PDEs) degrade the CDN via hydrolysis of one or both nucleotide linkages, and effectors, including transcription factors, enzymes, and riboswitches, sense the CDN signal (Römling et al., 2017; Römling et al., 2013). So far, three CDN second messengers have been found to control critical bacterial processes, including biofilm formation, cell wall homeostasis, intestinal colonization, and transient surface interactions (Römling et al., 2013; Corrigan and Gründling, 2013; Davies et al., 2012; Hallberg et al., 2019). Whereas representative examples of each of the three components required for signaling have been identified for both cyclic di-GMP and cyclic di-AMP, the recently discovered cyclic GMP-AMP (cGAMP) signaling pathways have remained incomplete (Fig. 1).
Figure 1. Two distinct pathways for cGAMP signaling in bacteria.
Current cGAMP signaling pathways are found in deltaproteobacteria (red) and in the gammaproteobacterium Vibrio cholerae (blue). While the central second messenger, cGAMP, is identical, these pathways appear to use different classes of synthases, effectors, and phosphodiesterases. Text in red indicate new components revealed in this paper.
A distinguishing feature of cGAMP signaling is that two distinct pathways apparently arose from independent evolution in different bacteria. The initial discovery of cGAMP as a second messenger resulted from characterization of the synthase DncV in V. cholerae El Tor (Davies et al., 2012). This enzyme is structurally related to the OAS-like family of enzymes that includes cGAS, a mammalian innate immune sensor that produces mixed-linkage cGAMP (Kranzusch et al., 2014). More recently, we discovered a different class of GMP-AMP cyclases (GACs) found in diverse deltaproteobacteria, including the environmental species Geobacter sulfurreducens, predatory species Bdellovibrio bacteriovorus (now within the class Oligoflexia), and social species Myxococcus xanthus. GACs are structurally related to the GGDEF family of diguanylate cyclases (DGCs), which are classically associated with cyclic di-GMP signaling, but instead have ‘Hypr’ GGDEF domains harboring specific variations in the active site that favor cGAMP production (Hallberg et al., 2016). We recently demonstrated that the Hypr GGDEF GSU1658 (_Gs_GacA) from Geobacter sulfurreducens regulates cGAMP but not cyclic di-GMP levels in vivo, resulting in a specific phenotype distinct from the biofilm-associated phenotype for the DGC EsnD (Hallberg et al., 2019).
While our focus to date has been on understanding the basis for cGAMP production by GACs with Hypr GGDEF domains (Hallberg et al., 2016; Hallberg et al., 2019), the regulation of GAC activity is relatively unexplored (Fig. 1). Specifically, it was not known how the conserved inhibitory site (I-site) often found in GGDEF domains acts to regulate Hypr GGDEF enzymes. Also, no activator of bacterial cGAMP signaling has yet been identified. _Gs_GacA and its homologs in other bacteria contain receiver (Rec) domains, but the partner histidine kinases and their signal inputs have not been identified. Based on structural and in vitro evidence, V. cholerae DncV is proposed to be inhibited by folates (Zhu et al., 2014), but this effect has not been tested in vivo. In any case, DncV does not appear to require activation by an exogenous factor.
In this study, we uncover three different regulatory mechanisms for the cGAMP signaling pathway: activation by cAMP, inhibition by cyclic di-GMP, and degradation by a newfound cGAMP-specific phosphodiesterase (GAP). A signature amino acid change in the GAP enzyme active site appears to give rise to cGAMP specificity. Using this signature to search bioinformatically, we show that GAP enzymes are found in many different bacteria, which expands the scope of cGAMP signaling to new organisms.
RESULTS
The second messenger cAMP binds and activates GacB, a Hypr GGDEF enzyme from M. xanthus
To elucidate the missing parts of the Hypr-cGAMP signaling pathway and to broaden its scope to other bacteria, we analyzed the system in the social bacterium Myxococcus xanthus. M. xanthus contains two GACs, MXAN_4463 (_Mx_GacA) and MXAN_2643 (_Mx_GacB), with Rec-GGDEF and GAF-GGDEF domain architectures, respectively (Fig. 2A). _Mx_GacA is homologous to _Gs_GacA (36.4% identity for the full-length protein) and has an orphan receiver domain that lacks an associated histidine kinase. Aravind and colleagues first identified the GAF domain by bioinformatics and coined the name for its presence in cGMP-specific phosphodiesterases, Anabaena adenylate cyclases, and Escherichia coli FhlA (Aravind and Ponting, 1997). Since then, GAF domains have been shown to respond to a diverse set of small molecule signals, including cyclic nucleotides, amino acids, and metabolites, as well as redox and light (Heikaus et al., 2009; Villapakkam et al., 2009; Little and Dixon, 2003; Sardiwal et al., 2005; Tang et al., 2015; Enomoto et al., 2014). Our initial in vitro experiments with _Mx_GacB included measuring enzyme activity in the presence of ATP, GTP, and related analogs. Serendipitously, high activity was observed in the presence of millimolar levels of ATP, which had not been observed with _Gs_GacA, a Rec-GGDEF. This led to the hypothesis that an adenine-containing compound may be a physiological activator of _Mx_GacB (herein referred to as GacB).
Figure 2. GacB, a Hypr GGDEF enzyme, is activated by cAMP.
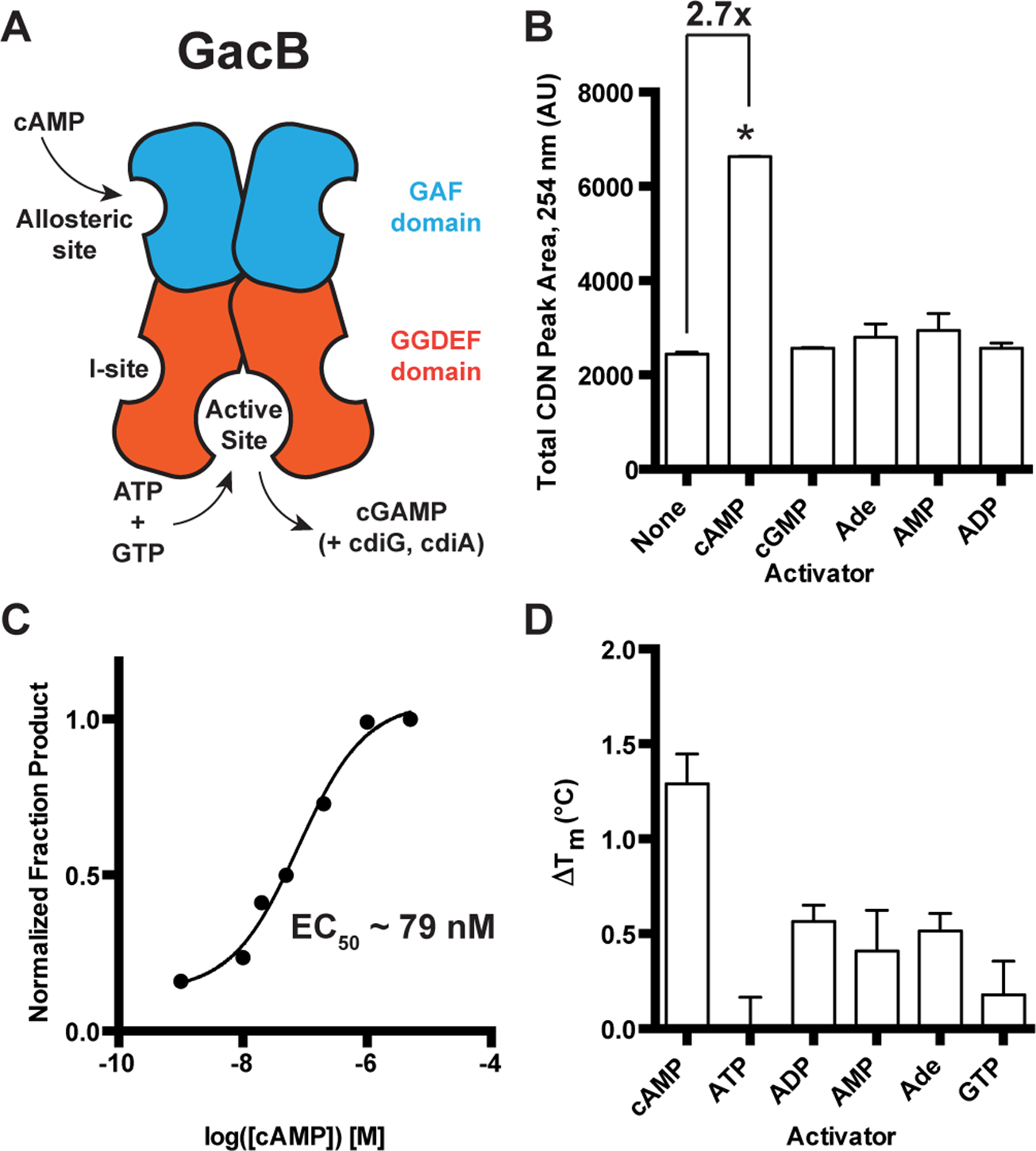
(A) Schematic of GacB showing the allosteric site on the N-terminal GAF domain (blue) and predicted inhibitory (I-site) in the GGDEF domain (red).
(B) Synthase activity of GacB (0.5 μM) with 1:1 ATP and GTP was measured by LC-MS in the absence or presence of candidate activators (1 μM). The sum of cGAMP and cdiG UV peak areas at 254 nm is reported, as the amount of cdiA produced was undetectable (p <0.05, n=2).
(C) Synthase activity of GacB (5 nM) with GTP doped with trace [α−32P] GTP was measured by TLC in the presence of increasing amounts of cAMP (0–5 μM). Production of cdiG was quantified by 32P signal density and plotted versus [cAMP] (n=1).
(D) Ligand binding to the GacB GAF domain was assessed by thermal shift assay using an MBP-GAF domain construct (1 μM). ΔTm was calculated as the difference between Tm with ligand (1 μM) and without ligand (n=3).
To identify the activator of GacB, an in vitro screen was conducted by incubating GacB with equimolar amounts of ATP and GTP substrates, with or without addition of candidate activators. A sum of all cyclic dinucleotide products was measured due to the promiscuous nature of GACs in vitro, an activity also seen with _Gs_GacA under these conditions (Hallberg et al., 2016). Only cAMP increased GacB activity (~2.7x) when added at a 10 μM concentration (Fig. 2B). Using a radiolabeled thin-layer chromatography (TLC) assay with GTP as the substrate to simplify product analysis, the effective concentration for GacB activation by cAMP (EC50) was found to be ~79 nM (Fig. 2C). A thermal shift assay conducted with the GAF domain of GacB (GacB1–180) shows that cAMP gives the greatest stabilization effect relative to other nucleotide ligands (Fig. 2D), supporting that the cyclic nucleotide binds in this domain. Taken together, these results demonstrate that cAMP is a selective activator that enhances GacB activity upon binding to the GAF domain.
GAF domains from bacteria and eukaryotes that bind cAMP or cGMP have been previously identified and structurally analyzed (Heikaus et al., 2009), including a recent structure of a cAMP-binding DGC enzyme, Lcd1 from Leptospira interrogans, which has high sequence homology (43.4% identity) to the GAF domain of GacB (da Costa Vasconcelos et al., 2017). However, to our knowledge, this is the first evidence linking cAMP signaling to cGAMP signaling, which expands the type of cAMP effectors in bacteria to include GAC enzymes. In addition, cAMP is the first identified small molecule activator of cGAMP synthesis. Since osmotic stress has been reported to increase cAMP levels in M. xanthus (Kimura et al., 2002; Kimura et al., 2005), the biochemical connection between these two pathways led us to examine the phenotypic effects of cGAMP signaling under osmotic stress conditions (see later section, Fig. 6). We expect the relationship between cAMP and cGAMP signaling to hold across Myxobacteria, as GacB is conserved within the genus and in the related fruiting Myxobacteria Corallococcus coralloides and Stigmatella aurantiaca (Skotnicka, Petters, et al., 2016).
Figure 6. PmxA has mild effects on type IV motility but promotes resistance to osmotic stress in M. xanthus.
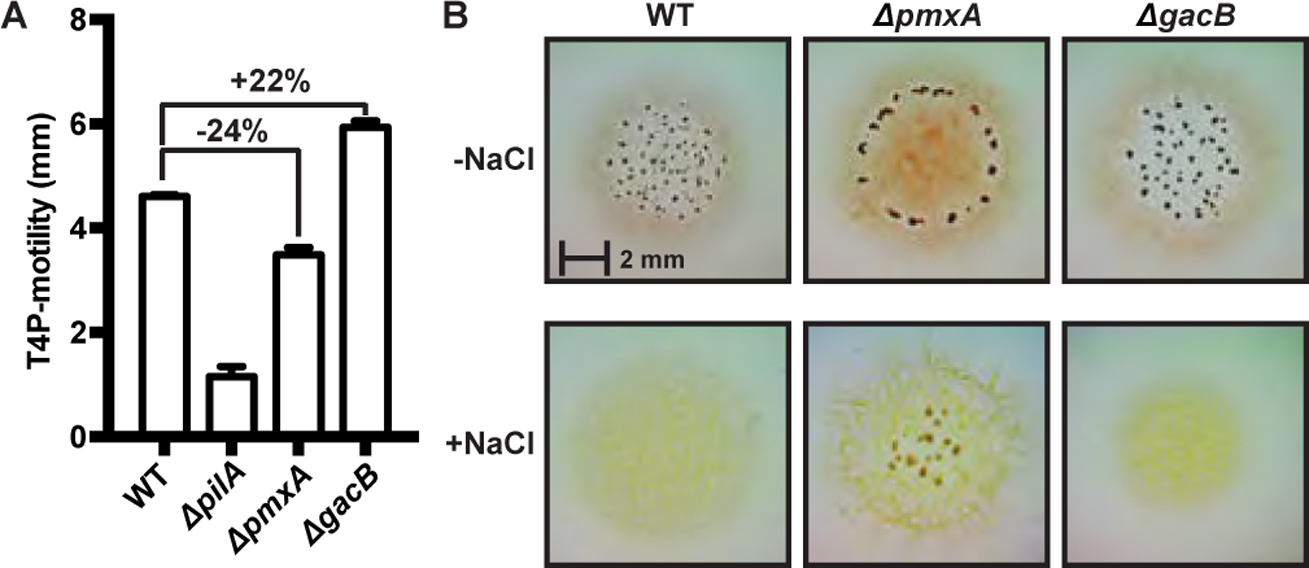
(A) Type IV pili (S) motility is mildly inhibited by cGAMP in M. xanthus, as shown by comparison of wild-type strain (DZ2) versus Δ_pmxA_ and Δ_gacB_ mutants.
(B) The Δ_pmxA_ mutant forms fruiting bodies in high (+NaCl) or low (-NaCl) salt conditions, whereas fruiting body formation is inhibited in both wild-type (DZ2) and Δ_gacB_ strains under osmotic stress conditions. Interestingly, the Δ_pmxA_ strain forms fruiting bodies at the periphery of the swarm at low osmolarity and towards the center under high osmolarity.
Analysis of the GacB allosteric inhibitory site reveals cross-regulation by cyclic di-GMP
Besides regulation by sensory domains like the GAF domain, canonical GGDEF domain-containing DGCs also contain an allosteric inhibitory site (I-site) that binds cyclic di-GMP and leads to allosteric feedback inhibition of DGC activity (Christen et al., 2006). However, since GacB is a GMP-AMP cyclase with a divergent Hypr GGDEF domain, we had to consider whether it was selectively regulated by a given cyclic dinucleotide. GacB could be subject to product auto-inhibition by cGAMP, or it could be cross-regulated by cyclic di-GMP or cyclic di-AMP. Recent studies of _Gs_GacA yielded phenotypic and transcriptomic evidence consistent with cross-inhibition by cyclic di-GMP, but did not show a direct mechanism (Hallberg et al., 2019).
In our initial inhibition assays, both ATP and GTP were used as substrates, which complicated the analysis as GacB produced all three bacterial cyclic dinucleotides in situ (SI Appendix, Fig. S1). We predicted that cyclic di-AMP was least likely to interact with GacB due to its structural dissimilarity to cyclic di-GMP, the canonical inhibitor of GGDEF enzymes. Therefore, the assays were repeated using ATP as the sole substrate, taking advantage of the in vitro promiscuity of GacB so that only cyclic di-AMP was produced in situ. Clear inhibition of enzyme activity was observed with 10 μM of cyclic di-GMP but with none of the other cyclic dinucleotides (Fig. 3A). We tested potential mutants to knock out the I-site and found that R286A was not inhibited by cyclic dinucleotides. The IC50 value was determined to be 17.80 +/− 1.06 μM for WT GacB inhibition by cyclic di-GMP (Fig. 3B), which is similar to the published IC50 value (5.1 +/− 1.4 μM) for the DGC PleD by cyclic di-GMP (Paul et al., 2007).
Figure 3. GacB is selectively inhibited by cdiG.
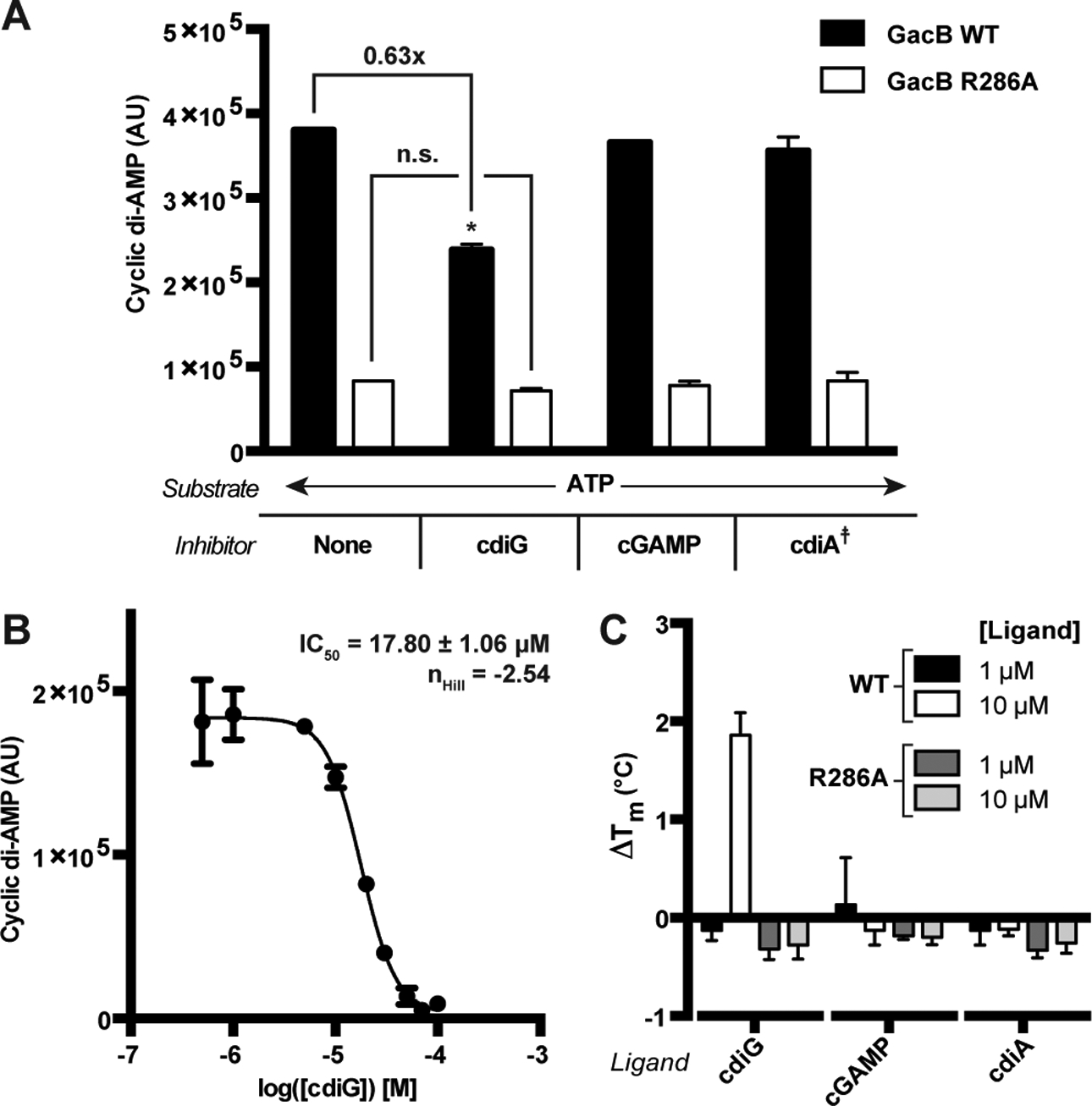
(A) Inhibition of GacB WT or I-site mutant (1 μM) by CDNs (10 μM) was measured by LC-MS (p<0.05, n=2). Due to potential in situ inhibition of GacB by other CDNs, cdiA production from ATP as sole substrate was used to quantify activity. The product cdiA (659 m/z) is reported as an ion-extracted signal from the mass spectra. For reactions testing cdiA as the candidate inhibitor, the background signal from added cdiA was subtracted (☨).
(B) Determination of IC50 value for cdiG inhibition of GacB using LC-MS (n=3). (C) CDN binding to full-length GacB WT or I-site mutant was assessed by thermal shift assay using MBP-GacB (1 μM) and 1 and 10 μM of each CDN. ΔTm was calculated as the difference between Tm with CDN and without (n=3).
We also conducted a thermal shift assay of full length GacB WT and R286A with various amounts of each CDN. Significant stabilization of GacB WT was only observed with 10 μM of cyclic di-GMP, which is consistent with the activity assays and measured IC50 value (Fig. 3C). GacB R286A was not stabilized by cyclic dinucleotides. Thus, cyclic di-GMP binds and inhibits both Hypr and canonical GGDEF enzymes via the conserved I-site. However, whereas I-site regulation leads to auto-inhibition for DGCs, our data strongly support that inhibition of GacB by cyclic di-GMP instead is a mechanism for cross-regulation of cGAMP signaling by cyclic di-GMP. Correspondingly, we observe that decreasing cyclic di-GMP levels in vivo activates cGAMP synthesis by a Hypr GGDEF enzyme (see later section, Fig. 7C).
Figure 7. An RNA-based fluorescent biosensor assay for cyclic dinucleotide phosphodiesterases.
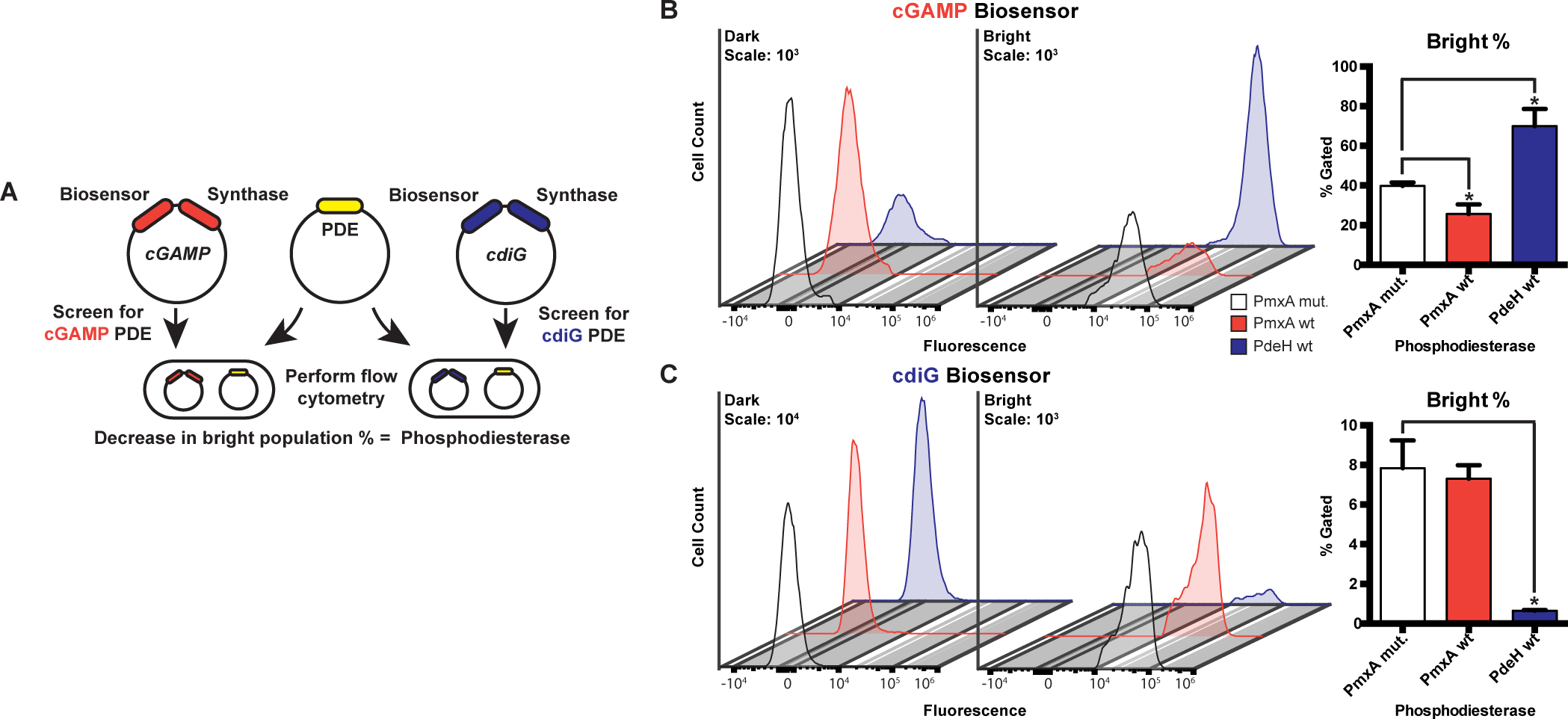
(A) Schematic of RBF biosensor assay showing that a PDE candidate (yellow) and cGAMP (red) or cdiG (blue) biosensor-synthase plasmids are co-transformed and overexpressed in E. coli BL21* (DE3) cells. Active PDEs lead to a decrease in the bright cell population as analyzed by flow cytometry in the presence of the DFHBI dye.
(B) Change in cellular cGAMP levels due to enzyme activity was measured by flow cytometry. Representative histograms show the dark (left) and bright (right) cell populations upon expression of WT PmxA (red), cdiG PDE PdeH (blue), and inactive PmxA mutant as a control (white). Y-axes are scaled by total cells (103), while the x-axes depict logarithmic fluorescence intensity. Bar graph depicts the change in % bright relative to the inactive control (p<0.05, n=3).
(C) Change in cellular cdiG levels due to enzyme activity was measured by flow cytometry. Same as in part B (p<0.05, n=3), except the histogram scale for the dark cell population is 104.
A divergent HD-GYP enzyme is a cGAMP-specific phosphodiesterase
Signal output can be decreased by inhibition of cGAMP synthesis or by degradation of cGAMP. So far, no cGAMP-specific PDE has been identified in bacteria. Three PDEs harboring HD-GYP domains from V. cholerae, including VCA0681, were shown to degrade 3’,3’-cGAMP in preference to the mammalian mixed-linkage 2’,3’-cGAMP. When 3’,3’-linkage cyclic dinucleotides are compared, however, VCA0681 clearly favors degradation of cyclic di-GMP over 3’,3’-cGAMP (Gao et al., 2015). VCA0681 previously was shown to control cyclic di-GMP levels in V. cholerae (Hammer and Bassler, 2009), so does not appear to act as a cGAMP-specific PDE in cells.
We had hypothesized that the PDEs involved in cGAMP signaling would be specific, but still may have evolved from components of cyclic di-GMP signaling, in line with how we discovered GACs and cGAMP-specific riboswitches as sub-classes of GGDEF enzymes and GEMM-I riboswitches, respectively (Hallberg et al., 2016; Kellenberger et al., 2015; Nelson et al., 2015). In both cases, it was possible to identify specific residues in the active site or ligand binding pocket that rationally reprogrammed the synthase or riboswitch to gain new specificities. The M. xanthus genome contains six HD-GYP and two EAL domain containing genes that are candidate phosphodiesterases. Based on structure-guided sequence analysis, both M. xanthus EAL proteins contain conserved residues that enforce selectivity for cyclic di-GMP (Minasov et al., 2009). However, when we compared to the HD-GYP domain from PmGH, a cyclic di-GMP PDE, we found that one M. xanthus HD-GYP protein does not harbor the two basic residues, R314 and K317, of the conserved Rxx(K/R) motif that recognizes a guanine nucleobase of cyclic di-GMP (Bellini et al., 2014). This exception was MXAN_2061, also called PmxA, which contains an R515 and Q518 pair instead. In vitro studies identified PmxA as a cyclic di-GMP PDE, but deletion of this gene did not show significant changes in cyclic di-GMP levels in vivo (Skotnicka, Smaldone, et al., 2016). The switch from a hydrogen-bond donor (K/R) to a potential hydrogen-bond acceptor (Q) was promising for altering specificity toward cGAMP (Fig. 4).
Figure 4. Sequence and structural analysis of PmxA reveals signature active site variation.
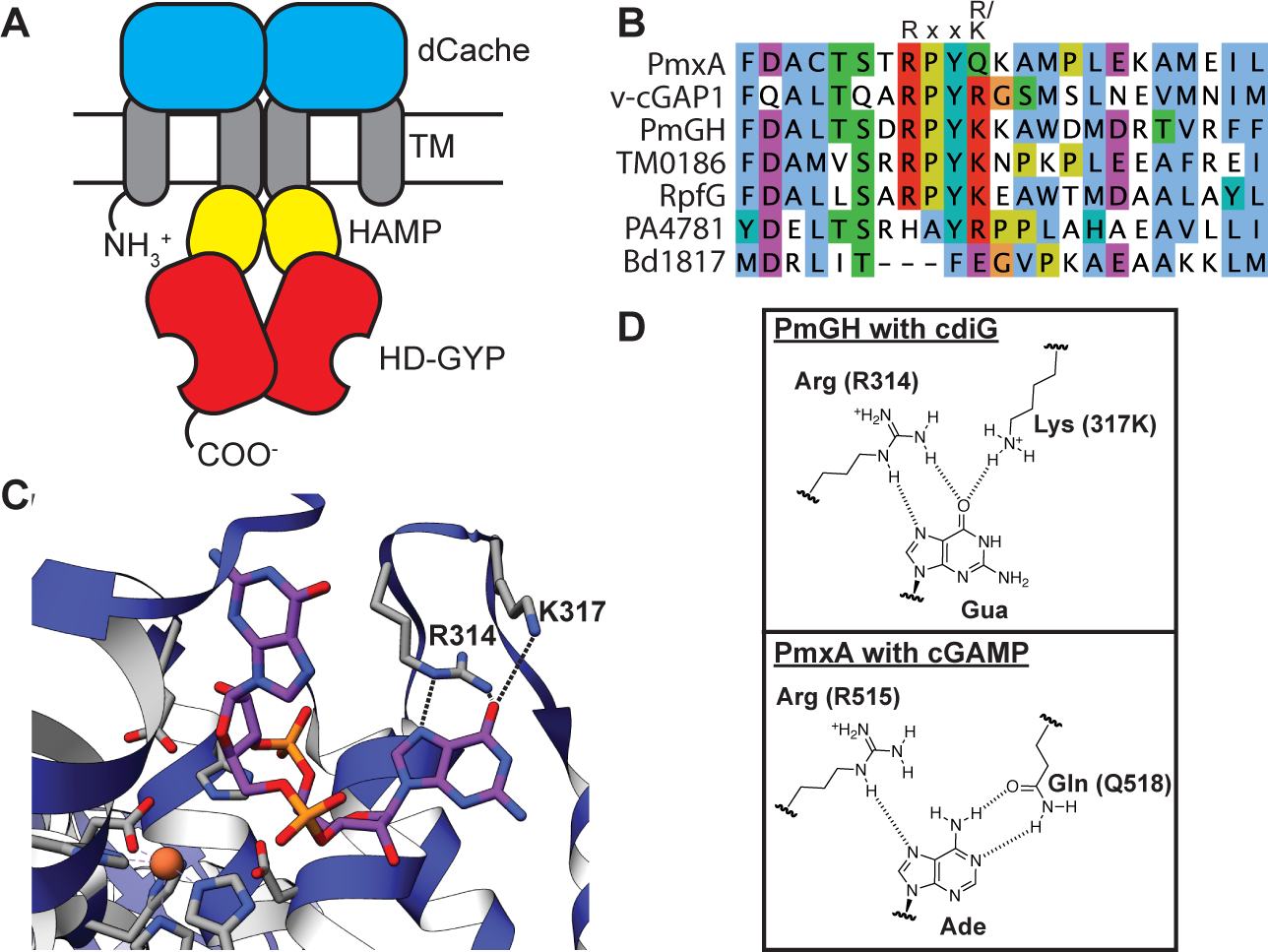
(A) Schematic of the PmxA (MXAN_2061) predicted homodimer. A single monomer contains an N-terminal sensory Cache domain, two TM helices, a coiled-coiled HAMP domain, and C-terminal HD-GYP domain with PDE activity.
(B) Sequence alignment of PmxA with previously reported HD-GYP domain PDEs using the MUSCLE alignment tool. The canonical Rxx(R/K) motif is shown. PmxA has a Gln (Q) at the fourth position of the motif.
(C) View of the active site of cdiG PDE PmGH (PDB 4MDZ) from Persephonella marina shows the two canonical motif residues, R314 and K317, forming hydrogen bond contacts with one of the two guanine nucleobases in cdiG.
(D) Molecular diagrams that show the structurally characterized interaction of the PmGH RxxK motif with cdiG and the proposed interaction of PmxA RxxQ motif with cGAMP.
PmxA is a multi-domain protein, composed of two transmembrane regions, an extracellular dCache domain, a coiled-coiled HAMP domain, and the catalytic C-terminal HD-GYP domain (Fig. 4A). The Søgaard-Andersen group previously identified a truncated PmxA construct that contains the HD-GYP domain that was soluble and active, as well as developed buffer conditions to assay its PDE activity (Skotnicka, Smaldone, et al., 2016), which were critical advances for in vitro studies (Fig. 5A). Using an MBP-tagged version of this construct, we detected robust degradation of cGAMP after 4 h, whereas little to no degradation was observed for cyclic di-GMP and cyclic di-AMP under the same conditions (Fig. 5B). The AA-GYP double mutant rendered the PDE inactive toward cGAMP, confirming that the HD-GYP domain was necessary for this activity (SI Appendix, Fig. S2).
Figure 5. PmxA is a selective cGAMP phosphodiesterase that can be reprogrammed via the signature residue.
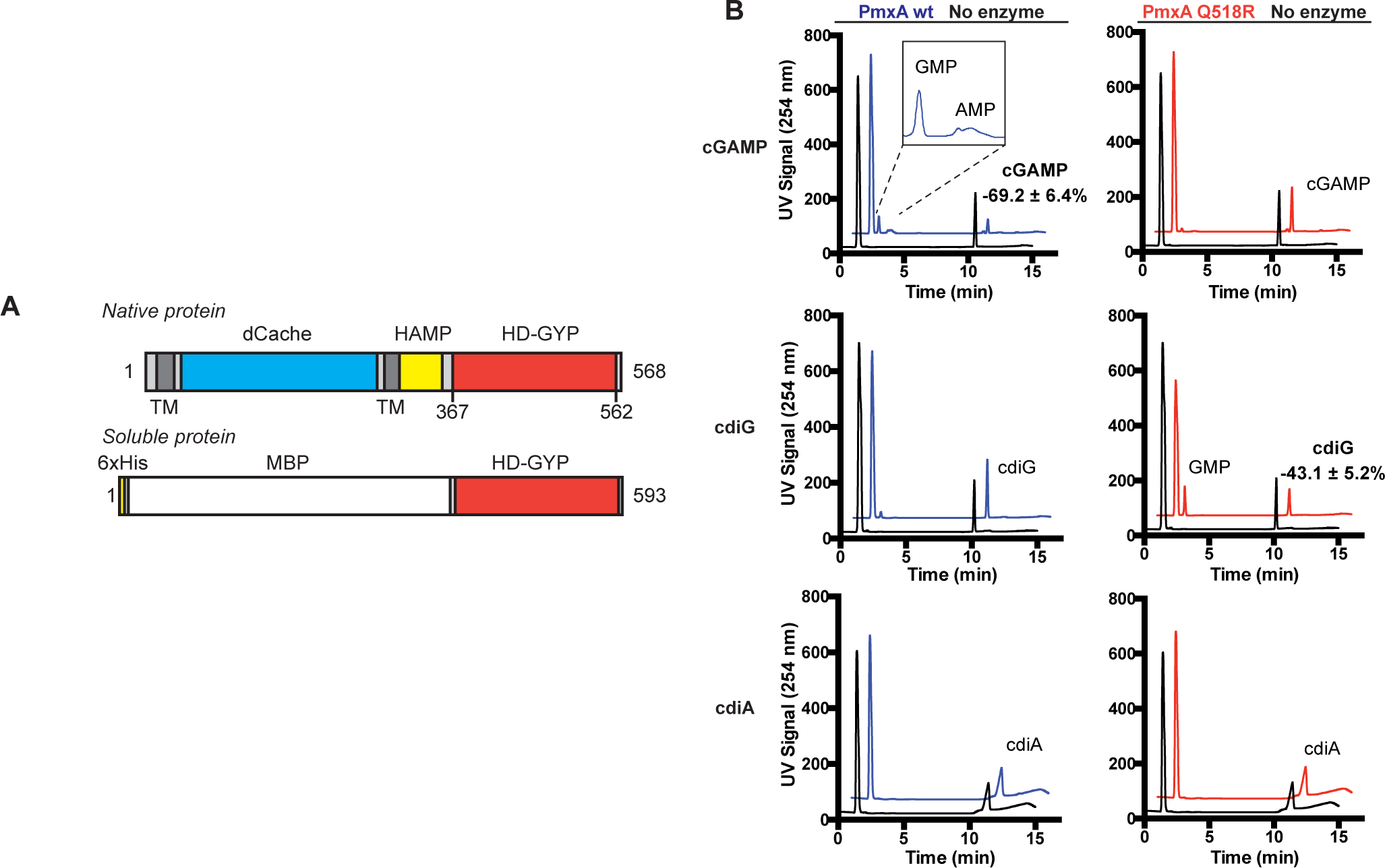
(A) Schematic of native PmxA and the MBP-tagged soluble PmxA construct used for in vitro activity assays, which was first reported by Skotnicka et al.
(B) In vitro PDE activity of MBP-PmxA wt (blue) and Q518R mutant (red) was analyzed by LC-MS. Enzyme (10 μM) or control (no enzyme, black) was incubated with 50 μM of cGAMP (top), cdiG (middle), or cdiA (bottom) for 4 h at 30 °C (n=2). Representative UV traces for each reaction are shown, with CDN peaks labeled based on MS analysis. Inset highlights the observation of mononucleotide products for PmxA wt with cGAMP.
To test whether the Q518 residue was responsible for the unique substrate selectivity of PmxA, the activity of the Q518R mutant PmxA was analyzed. This mutant exhibited little to no degradation of cGAMP after 4 h, and instead gained activity against cyclic di-GMP (Fig. 5B). The substrate selectivity of this point mutant is comparable to the published activity of v-cGAP1 (VCA0681), which harbors an R residue at this position (Gao et al., 2015). To our knowledge, PmxA is the first bacterial PDE shown to preferentially degrade 3’,3’-cGAMP over cyclic di-GMP. Moreover, our data reveal that Q518 may serve as a signature residue for the sub-class of HD-GYP domains that act specifically as cyclic GMP-AMP phosphodiesterases (GAPs). Homologs to PmxA that harbor the Q518 variation in their HD-GYP domain have the same conservation across Myxobacteria (Myxococcus, Corallococcus, Stigmatella) as GacB.
The Søgaard-Andersen group previously showed that 10 μM of PmxA degrades cyclic di-GMP using a radiolabeled substrate TLC assay, but did not test cGAMP as a substrate (Skotnicka, Smaldone, et al., 2016). This result actually is consistent with our results, as we also observe that PmxA is active against cyclic di-GMP, albeit to a much weaker extent than to cGAMP. Our selectivity assays were performed with 10 μM of PmxA and an excess (50 μM) of each cyclic dinucleotide, and thus we do not observe complete degradation in 4 h (Fig. 5B). The low in vitro activity of this enzyme construct may be due to the truncation, which removed the transmembrane regions to aid solubility but also removed the sensory domain that regulates enzyme activity. Also, while the enzyme is active with Mg2+, a later screen revealed higher activity with other divalent metals (SI Appendix, Fig. S4, S5).
Phenotypic analysis suggests a role for cGAMP signaling in osmotic stress
To investigate the regulatory function of cGAMP signaling in M. xanthus, the phenotype of a wild type strain (DZ2) was compared to the corresponding ΔgacB and ΔpmxA mutants. Type IV pilus (T4P)-dependent motility, a.k.a. twitching or social motility, was assayed on CYE plates that contained 0.5% agar, where gliding motility contributes very little to colony expansion. Consistent with a previous report that used DK1622 as the WT strain, neither gacB nor pmxA is essential for motility (Skotnicka, Petters, et al., 2016). However, after quantifying T4P-dependent motility using colony expansion as the readout, we found that ΔgacB slightly increased colony expansion (+22%) relative to WT, whereas ΔpmxA had the opposite effect (−24%) (Fig. 6A). These data indicate that cGAMP mildly inhibits T4P-dependent motility under these conditions.
M. xanthus form fruiting bodies to resist harsh environmental stresses and starvation. A previous report showed that deleting pmxA from the M. xanthus DK1622 background inhibited fruiting body formation in submerged culture (28). We followed up by investigating the role of pmxA in fruiting body formation in the DZ2 background. Specifically, we examined the effect of high salt (0.15 M NaCl) conditions, because earlier studies reported that osmotic stress increases the concentration of cAMP in M. xanthus (Kimura et al., 2002; Kimura et al., 2005) and our results show that cAMP activates the synthase activity of GacB (Fig. 2B). Consistent with the minor defect in fruiting in the DK1622 background (28), deleting pmxA from the DZ2 strain causes an altered development pattern. Compared to the DZ2 strain that forms scattered fruiting bodies throughout the colony, the ΔpmxA strain forms fruiting bodies only along the initial inoculum.
Furthermore, in the presence of osmotic stress, among the three strains tested (DZ2, ΔgacB, and ΔpmxA) only ΔpmxA is capable of forming fruiting bodies (Fig. 6B). This striking result indicates that while high cGAMP levels appears to inhibit development in regular laboratory conditions, it might provide resistance against osmotic stress during fruiting body formation in the natural habitats of M. xanthus. The fact that the DZ2 strain is unable to form fruiting bodies under high salt conditions suggests that in addition to increased activity of GacB, the PDE activity of PmxA also should be repressed to provide sufficient resistance against osmotic stress. However, the coordination between GacB and PmxA in vivo, especially during development, still remains to be understood. It is currently unclear whether PmxA is in the same pathway with GacB or with GacA, which harbors a receiver (Rec) domain. Nevertheless, our results provide initial characterization of cGAMP signaling phenotypes with a potential connection to stress responses.
An RNA-based fluorescent biosensor assay for cyclic dinucleotide phosphodiesterase activity
To further explore the function of PmxA and its homologs in cells, we developed a fluorescent biosensor assay for PDE activity based on RNA-based fluorescent (RBF) biosensors. RBF biosensors specific for each of the bacterial cyclic dinucleotides have been used previously to analyze synthase activities (Hallberg et al., 2017). This assay was adapted to screen PDE activities by encoding a candidate PDE in one plasmid and a matched pair of biosensor and synthase on a second plasmid (Fig. 7A). For example, the DGC WspR was co-expressed with the Dp biosensor, which responds to cyclic di-GMP (Wang et al., 2016), and the well-characterized GAC _Gs_GacA was co-expressed with the Gm biosensor, which responds to cGAMP (Kellenberger et al., 2015). These two matched synthase-sensor pairs result in highly fluorescent (bright) cell populations as measured by flow cytometry. Co-expression of an active PDE then should cause the bright cell population to decrease, due to degradation of the cyclic dinucleotide sensed by the RBF biosensor. Thus, PDE selectivity can be preliminarily assessed by screening against the two synthase-sensor pairs.
Since this assay requires balanced co-expression of three components (synthase, biosensor, and PDE), growth and induction conditions had to be optimized, especially for a high-throughput, 96-well format. Two factors that significantly improved assay results were the culture volume per well and % lactose in the autoinduction media. We also found that the addition of a third component, even an inactive protein (PmxA mut, SI Appendix, Fig. S2), leads to a mixed population of bright and dark cells rather than a single bright population as seen in prior studies co-expressing just synthase and biosensor (Fig. 7B, C). Most likely, this effect is due to stochastically reduced synthase activity from non-uniform expression levels. The synthases are particularly sensitive to this, because as GGDEF enzymes they are active only as dimers or higher order oligomers.
To assess PDE activity in the context of these bifurcated cell populations, we decided to analyze the percentage of the bright cells in the sample. Using this approach, it was verified that wild-type PmxA decreases the population of cGAMP-bright cells relative to the inactive PmxA mutant, but it has no effect on the population of cyclic di-GMP-bright cells (Fig. 7B, C). This result shows that PmxA is a cGAMP-specific PDE in the live cell context and corroborates our biochemical data. Notably, we expressed the full-length PmxA gene in these experiments and were able to observe activity of this membrane-bound PDE.
We had expected the opposite trend upon addition of PdeH, which is a well-studied cyclic di-GMP-specific PDE from E. coli previously called YhjH (Sarenko et al., 2017; Reinders et al., 2015). While PdeH did decrease the population of cyclic di-GMP-bright cells (Fig. 7C), it counterintuitively increased the population of cGAMP-bright cells. One reasonable explanation for this effect is that PdeH de-represses the _Gs_GacA synthase by lowering global cyclic di-GMP levels in E. coli. This result is in line with our demonstration that GAC activity is inhibited by cyclic di-GMP in vitro (Fig. 3), is consistent with reporter assay results in G. sulfurreducens (Hallberg et al., 2016), and further supports that this cross-regulation occurs in cells.
Both RxxQ and RxxN signature motifs give rise to cGAMP-specific phosphodiesterase activity
With this assay and analysis method validated using PmxA and PdeH, it was applied to screen HD-GYP domains from other bacteria for cGAMP-specific PDE (GAP) activity. We first performed a bioinformatic search outside the Myxococcus genus for HD-GYP domains homologous to PmxA that harbor the RxxQ motif, which was shown to be important for cGAMP specificity. HD-GYP domains with the RxxN motif were added to this search, as N contains an amide functional group similar to Q. The majority of homologs (87.97%) harbored the canonical Rxx(K/R) motif, but 539 sequences (4.69%) contained an Rxx(Q/N) sequence (Fig. 8A). Excitingly, while some candidates are from bacterial species previously predicted to have cGAMP signaling due to the presence of the synthase (Hallberg et al., 2016), the majority are from bacterial species not previously associated with cGAMP signaling (Fig. 8B). Candidate GAPs appear well represented in both Proteobacteria and Firmicutes, and are associated with a variety of sensory domains (SI Appendix, Table S1).
Figure 8. The variant signature Rxx(Q/N) motif is broadly distributed and most prevalent in Firmicutes and Proteobacteria.
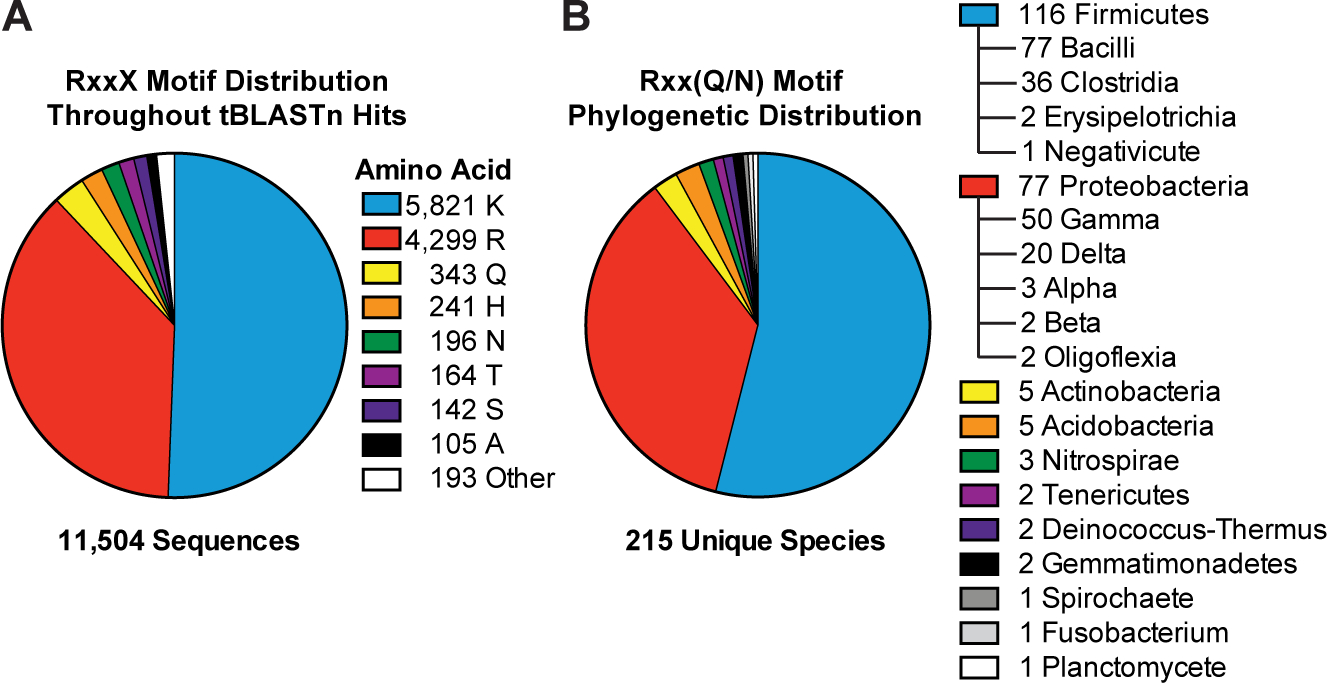
(A) The prevalence of the top 8 variants for the RxxX motif in bacterial HD-GYP domains is shown. The sequence analysis used 11,504 putative HD-GYP domains containing aligned RxxX motifs out of 14,326 total tBLASTn hits against the PmxA HD-GYP domain.
(B) The distribution of RxxQ and RxxN HD-GYP motifs in Bacteria. A total of 539 sequences containing RxxQ or RxxN motifs were sorted based on unique species, leading to the identification of unique 215 bacterial species containing HD-GYP domains with these variant motifs. The species were categorized using NCBI Taxonomy.
Out of 539 total candidate GAPs matching the Rxx(Q/N) motif (RxxQ, 343; RxxN, 196), six candidate genes were selected based on their phylogenetic diversity and prediction to be soluble, cytosolic proteins. The full length PDEs were cloned and screened for activity against cGAMP and cyclic di-GMP using the in vivo biosensor assay (Fig. 8A). Gratifyingly, five out of six candidates showed PDE activity against cGAMP, whereas the last candidate appears inactive or poorly expressed in the assay. Of the five active PDEs, three appear selective for cGAMP and two appear to degrade both cyclic dinucleotides. Thus, the candidates from Geobacter metallireducens, Bdellovibrio bacteriovorus, and Vibrio tapetis are putative GAP enzymes, whereas additional analysis is required to determine substrate preferences for the enzymes from Moorella thermoacetica and Phaeobacter piscinae.
B. bacteriovorus HD-GYP Bd2325 (Bd) demonstrated the strongest cGAMP-specific PDE activity in our screen and harbors the alternative RxxN motif. Interestingly, this gene was identified previously in a transposon screen for genes important for predatory activity in the B. bacteriovorus 109JA strain (Tudor et al., 2008). Bd2325 was cloned as a full-length, N-terminal MBP-tagged protein for testing in vitro. No PDE activity against either cGAMP or cyclic di-GMP was observed using in vitro assay conditions developed for PmxA, which included 10 mM Mg2+ as the added divalent metal (Fig. 9A, B). However, different divalent metals and pH have been shown to enhance HD-GYP activity (Gao et al., 2015; Deng et al., 2018), as HD-GYP active sites are known to contain redox-sensitive bi- and tri-nuclear cores with variable metal selectivity (Lovering et al., 2011; Rinaldo et al., 2015; Miner and Kurtz, 2016). We screened Bd2325 with different metals (Mg2+, Mn2+, Ca2+, Fe2+, and Ni2+, SI Appendix, Fig. S3), and indeed found that two types of divalent metals activate the enzyme in vitro.
Figure 9. Identification of selective cGAMP phosphodiesterases (GAPs) from other bacteria, including one with the RxxN motif.
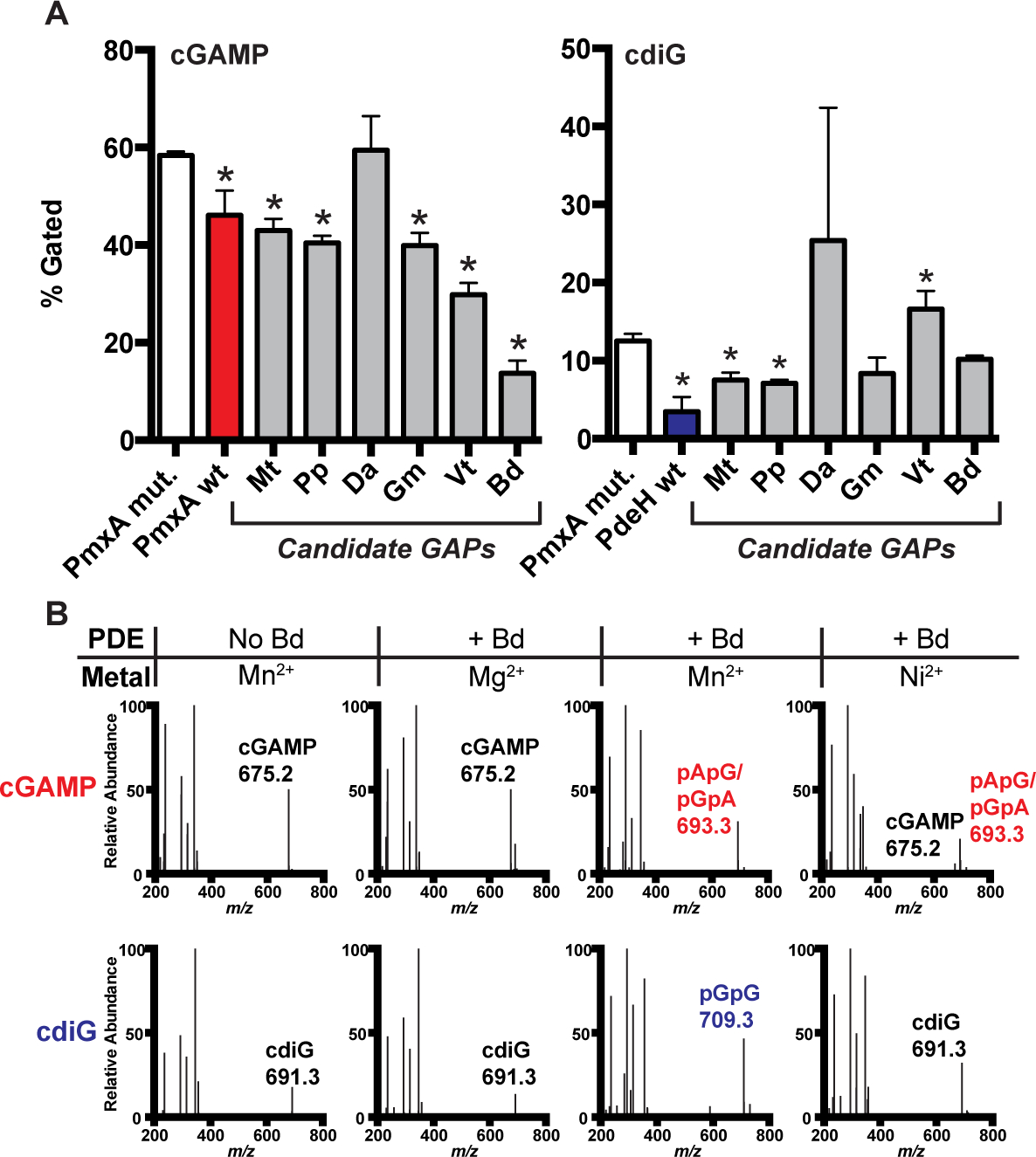
(A) PDE activity screen of six candidate GAPs using the biosensor-based flow cytometry assay depicted in Fig. 7A. PDEs contain a Q (Mt: Moorella thermoacetica, Pp: Phaeobacter piscinae, Da: Dechloromonas aromatica, Gm: Geobacter metallireducens, Vt: Vibrio tapetis) or N (Bd: Bdellovibrio bacteriovorus) at the selectivity position. Activity of PDE candidates were compared to an inactive control, PmxA mutant (p < 0.05, n=3).
(B) In vitro PDE activity of MBP-Bd2325 with different divalent metals was analyzed by LC-MS. Enzyme (10 μM) or control (no enzyme) was incubated with 50 μM of cGAMP or cdiG for 4 h at 30 °C in the presence of Mg2+, Mn2+, or Ni2+. A representative mass spectrum for each reaction is shown, with CDN or product peaks labeled based on MS analysis (n=2).
Bd2325 is most active in the presence of Mn2+, as it degraded both cGAMP and cyclic di-GMP to their respective linear dinucleotides within 4 h (Fig. 9A, B). In fact, under these conditions, this enzyme degraded a measurable amount of cGAMP within the ~30 seconds required to prepare the ‘0’ minute time point of a time course, but was less active against cyclic di-GMP (SI Appendix, Fig. S4, S5). In other words, pApG/pGpA already is detected at ‘0’ minute, but pGpG is not detected at this time point. After 60 min, the linear products appear to be further processed, leading to loss of the selective ion signal.
Observation of the linear dinucleotide product contrasts with the mononucleotides AMP and GMP detected with PmxA and Mg2+ in earlier preparations. HD-GYP protein TM0186 from Thermotoga maritima shifts from production of linear to mononucleotide product depending on active site metal occupancy, which may also be true for other HD-GYP PDEs (Miner and Kurtz, 2016). The preference for cGAMP was even clearer in the presence of Ni2+. Bd2325 in the presence of Ni2+ exhibited lower activity than with Mn2+, which meant that the difference between reactivities could be more easily observed in the chosen time course. After 4 h, cGAMP was fully degraded, but the majority of the cyclic di-GMP sample remained.
PmxA also is much more active in the presence of Mn2+ and Ni2+, now showing some activity against cyclic di-GMP but still clearly greater activity against cGAMP (SI Appendix, Fig. S4, S5). Perhaps due to the use of a truncated construct for PmxA, it is less active than Bd2325, which is a soluble protein. With these metals, PmxA generates linear dinucleotide products. In the presence of Mn2+ pApG/pGpA is detected at the 30 min time point, whereas pGpG is not detected until the 60 min time point. The same trend is observed for PmxA with Ni2+ but at later time points.
Thus, these in vitro experiments confirm that some variant HD-GYP domains harboring either the RxxQ or RxxN motifs are selective for cGAMP over cyclic di-GMP. Further work will be needed to probe whether this active site variation is necessary and sufficient to switch substrate selectivity. In the case of the Hypr GGDEFs, for example, we recently showed the mutational pathway involved additional residues (7).
DISCUSSION
According to our bioinformatics analysis, the newfound GAP enzymes may be more widespread in bacteria than the known set of Hypr GGDEF synthases and cGAMP riboswitches that have defined the Hypr-cGAMP signaling pathway to date. Very recently, it has been shown that the DncV-cGAMP signaling pathway is part of a microbial defense system against phages (Cohen et al., 2019). Thus, it is plausible that we may find instances where these GAP enzymes are co-opted by phages as an anti-defense mechanism. Another intriguing possibility we want to pursue is that some HD-GYP variants may have activity against newly discovered cyclic dinucleotide signals (Whiteley et al., 2019). For example, our bioinformatic analysis revealed several other amino acid substitutions at the signature position (Fig. 8A), and ongoing work is pursuing the specificities of these other variants. There is still high potential for novel enzyme classes associated with cyclic dinucleotide signaling, as well as novel effectors that bind cyclic dinucleotides.
When cellular signaling pathways are highly integrated, phenotype-based studies may become challenging to interpret or use for deconvoluting pathways and discovering novel components. To address this issue, we have worked on developing high-throughput fluorescence assays that read out specific cyclic dinucleotide levels in vivo (Hallberg et al., 2017). We recently showed that RBF biosensors can be applied to detect modulation of endogenous cyclic di-GMP signaling (Yeo et al., 2018; Yeo et al., 2017). Here we show for the first time that RBF biosensors can be used to discover and evaluate specific PDE activity.
Taken together, this study expands our knowledge of how cGAMP signaling is regulated, by identifying the first activator molecule, cAMP, and the founding members of a class of cGAMP specific phosphodiesterases (GAPs). In addition, we show that cGAMP signaling is integrated with other nucleotide signaling pathways via allosteric regulation of the synthase, GacB, by cAMP and cyclic di-GMP. This result reinforces the growing appreciation that nucleotide signaling pathways form an integrated, multi-layered network, allowing bacterial cells to respond in a coordinated, sophisticated manner to complex feedback from their environment and metabolic status.
EXPERIMENTAL PROCEDURES
General Reagents and Oligonucleotides
Oligonucleotide primers were purchased from either Elim Biopharmaceuticals (Hayward, CA) or Integrated DNA Technologies (Coralville, IA). M. xanthus genes were amplified from genomic DNA. GacB mutant R292A was previously reported in Hallberg et al. (Hallberg et al., 2016). Candidate cGAMP PDE genes were purchased as gBlocks (IDT). Nucleotide reagents for activator screening were obtained from Sigma-Aldrich (St. Louis, MO), Acros Organics (Belgium), and Biolog (Germany). Cyclic dinucleotide standards for inhibition assays and phosphodiesterase experiments were purchased from Biolog and Axxora (Farmingdale, NY). PEI-cellulose TLC plates and Sypro Orange (5,000x in DMSO) were bought from Sigma-Aldrich. DFHBI-1T used for flow cytometry screening was chemically synthesized from established protocols (Song et al., 2014).
Molecular Cloning
All gene constructs for in vitro assays were cloned into a pET16 vector containing an N-terminal purification tag that included both 6xHis tag and maltose binding protein (MBP) between NdeI and BamHI restriction sites. Gene constructs were cloned as fusions to 6xHis-MBP using BamHI and NotI restriction sites. PmxA was analyzed as the truncation PmxA384−568 containing the HD-GYP domain first described by Skotnicka et al. (Skotnicka, Smaldone, et al., 2016). Mutations were generated by the around-the-horn method. Untagged candidate PDE genes for flow cytometry assays were cloned into pCOLADuet-1 between restriction sites NdeI and XhoI in the second multi-cloning site. Alternative sites were used for sequences where these cut sites were found within the gene (BglII/XhoI for Pp, NdeI/KpnI for Gm). Gibson assembly was used to construct vectors containing a CDN synthase and biosensor in plasmid pETDuet-1. The synthase (WspR or _Gs_GacA) was placed 5’ to the biosensor (Dp17 or Gm790P1–4ΔA) under control of separate T7 promoters, with a short spacer sequence in between. Appropriate overhangs for Gibson assembly were added to the pETDuet-1 vector backbone, the synthase gene (WspR or _Gs_GacA), and the biosensors (Dp or Gm) by PCR with extended primers (Table S1). Templates for the biosensor have the design T7 promoter – tRNALys(5’ half)– Spinach2(5’ half) – biosensor – Spinach2(3’ half) – tRNALys(3’ half)– T7 terminator. These components were combined through a three piece Gibson assembly into the final plasmid construct.
Protein Overexpression and Purification
Proteins for in vitro analysis were overexpressed in BL21* (DE3) cells using either ZYP-5052 autoinduction media (Studier, 2005) or 2x YT media with IPTG induction with carbenicillin at 50 μg/mL final concentration. Autoinduction was used for production of GacB and PmxA constructs, whereas IPTG induction was used for Bd2325 due to poor expression observed in ZYP-5052 media. In ZYP-5052 media, cultures were inoculated and grown at 37 °C for 20–24 h. In 2x YT media, cells were grown at 37 °C until OD600 ~ 0.6–0.8 and induced with 1 mM IPTG, followed by 20 h expression at 18 °C. Cultures were grown at either 250 or 750 mL volumes. Subsequent purification steps were identical for all proteins. Cells were harvested by centrifugation and lysed by sonication in lysis buffer containing 25 mM Tris-HCl pH 8.2, 500 mM NaCl, 30 mM imidazole, 5 mM 2-mercaptoethanol, and 5% glycerol. Clarified lysates were generated by centrifugation at 9,200 rpm for 40 min then were incubated with 1.5–4 mL HisPur™ Ni-NTA Resin (Thermo Scientific). The resin was loaded onto a disposable protein chromatography column, the flow-through was drained, and the resin bed was washed with 60 mL lysis buffer. Protein was eluted with 10 mL lysis buffer containing 300 mM imidazole. Eluted proteins were buffer exchanged by dialysis or centrifugation into storage buffer containing 20 mM HEPES pH 7.5, 250 mM KCl, 5 mM 2-mercaptoethanol, and 5% glycerol, followed by concentration using centrifugal concentrator columns. Final protein concentrations were calculated by absorbance at 280 nm. Purified protein aliquots were flash frozen in liquid nitrogen and stored at −80 °C until further use.
In Vitro Activity Assay for GacB
For activation assays, 6xHis-MBP GacB WT (0.5 μM) was incubated in a 50 μL solution of 50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 100 mM NaCl, and 5 mM dithiothreitol, with both substrates (ATP and GTP) at 1 mM each and 1 μM of candidate activators (cAMP, cGMP, adenosine, AMP, ADP, or none). Reaction mixtures were incubated at 37 °C for 6 h, heated to 95 °C for 30 s to denature the protein, then centrifuged for 3 min at 13,000 rpm to remove precipitate. For LC-MS analysis, 20 μL of the sample was injected, and starting materials and products were confirmed by mass and quantified by peak area at 254 nm absorbance.
Inhibition assays were performed following the same protocol as above, with the following changes. 6xHis-MBP GacB WT (0.5 μM) was first incubated in reaction buffer with 10 μM of candidate inhibitors (cGAMP, cdiG, cdiA, or none) for 10 min at room temperature. Then substrate (1 mM ATP) was added to start the reaction. Signal was measured by ion extraction of respective CDN m/z values: 659 m/z cdiA, 675 m/z cGAMP, 691 m/z cdiG. The IC50 value for GacB was obtained using this protocol, but with varied amounts of cdiG between 0–100 μM.
LC-MS Analysis of In Vitro Enzyme Activity Assays
LC-MS analysis of in vitro enzyme activity assays was performed on an Agilent 6120 Quadrupole MS with an Agilent 1260 Infinity HPLC equipped with a multi-wavelength detector (MWD). Samples were separated on a Poroshell 120 EC C18 column (50 mm length × 4.6 mm internal diameter, 2.7 μm particle size, Agilent) at a flow rate of 0.4 mL/min. The solvent system (aqueous 10 mM NH4OAc, 0.1% AcOH as Solvent A and MeOH as Solvent B) and gradient was based on the procedure developed by Burhenne et al. (Burhenne and Kaever, 2013). Under these conditions, cyclic dinucleotides eluted between 10–12 min with an elution peak order of cdiG, cGAMP, and cdiA observed at 254 nm. For phosphodiesterase assays, linear products eluted in a similar range of retention times. Molecular assignment was made through analysis of the mass spectra. MS was conducted in positive ion mode with an electrospray ion source (3000 V, 35 psig, 350 °C) and mass range of 150–1000 m/z. MH+ peaks were detected with the following m/z values: cGAMP 675, cdiG 691, cdiA 659, pGpA/pApG 693, pGpG 709, GMP 364, AMP 348.
Activity Assay for GacB using Radiolabeled NTPs
EC50 measurements were performed using 6xHis-MBP GacB R292A, a mutant that was predicted to be an I-site mutant. This mutant was chosen to remove possible feedback inhibition during the reaction, but since then has been shown to retain cyclic di-GMP binding. GacB R292A (50 nM) was incubated in a solution of 50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 100 mM NaCl, and 5 mM dithiothreitol with 50 μM GTP, ~10 μCi of [α−32P]-GTP, and 0–5 μM of cAMP. Reaction mixtures were incubated at room temperature for 90 min., then treated with ~10 U of Calf Intestinal Alkaline Phosphatase (NEB) for 20 min at RT to degrade unreacted NTPs. An aliquot of the reaction mixture (0.5 μL) was spotted on a PEI-cellulose F thin-layer chromatography plate (Millipore), allowed to dry, then run on the plate using 1.5 M KH2PO4, pH 3.8 following the protocol developed by Kranzusch et al. (Kranzusch et al., 2014). Plates were dried, exposed to a Phosphor-image screen (GE Healthcare), and analyzed using a Typhoon scanner (GE Healthcare). Signal intensity was measured by ImageQuant software.
Thermal Shift Assay
Thermal shift assays (Huynh and Partch, 2015) were used to assess binding between 6xHis-MBP GacB1−180 WT GAF domain and candidate activator ligands, as well as between 6xHis-MBP GacB WT (full length) and cyclic dinucleotides. Samples were prepared at 4 °C prior to the thermal denaturation procedure. Assays were performed in a 96-well PCR plate using the CFX96 Real-Time Thermal Cycler (Bio-Rad). A reaction master mixture was prepared by mixing protein (1 μM final) and Sypro Orange (1:2500 dilution of 5000x stock, Invitrogen) in buffer containing 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 10 mM MgCl2, and 5 mM DTT. 49 μL aliquots were distributed to individual wells where 1 μL of ligand was added and plates were sealed. Ligands for activator screening (cAMP, adenosine, AMP, ADP, ATP, GTP, or none) were used at a final concentration of 1 μM whereas ligands for inhibition assays (cdiG, cGAMP, cdiA, or none) were used at 1 or 10 μM. Final concentrations of components in 50 μL reaction mixtures were 1 μM protein, 2x Sypro orange, and buffer containing 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 10 mM MgCl2, and 5 mM DTT. Reaction mixtures were simultaneously denatured with a gradient of 1 °C/min from 4–95 °C. Fluorescence was recorded using the CFX96 FRET channel, which measures broad emission signal in the absence of filters. ΔTm values were calculated as a difference in Tm between a +ligand and -ligand control.
Activity Assay for Phosphodiesterases using LC-MS
The protocol was adapted from previously published methods for PmxA (Skotnicka, Smaldone, et al., 2016). Briefly, enzyme (10 μM final concentration) was incubated at 30 °C for 5 min in buffer containing 50 mM Tris-HCl pH 8.0, 300 mM NaCl, and 10 mM MgCl2. Then, 50 μM substrate (cGAMP, cdiG, or cdiA) was added and the mixture was incubated at 30 °C for 4 h. Reactions were stopped by addition of an equal volume of 0.5 M EDTA, pH 8.0. Samples were incubated at 95 °C for 30 s to denature the enzyme, centrifuged for 3 min at 13,000 rpm to remove precipitate, and 20 μL of the sample was analyzed by LC-MS. Peak assignments were made based on m/z values. For experiments testing enzyme activity in presence of other divalent metals, 10 mM of each metal (MnCl2, CaCl2, FeSO4, NiSO4) was substituted for MgCl2 in the reaction buffer.
Myxococcus xanthus Phenotypic Assays
To construct the in-frame deletion strains, in-frame deletion mutation cassettes were amplified with polymerase chain reaction (PCR) using chromosomal DNA as template, digested and inserted into plasmid pBJ113. All constructs were confirmed by DNA sequencing. To generate mutant strains, transformants were obtained by homologous recombination as previously described (Bustamante et al., 2004), using electroporation of M. xanthus DZ2 cells with 4 μg of plasmid DNA. Mutants were confirmed by PCR and sequencing of the gene of interest. T4P-dependent motility and fruiting body formation were assayed on CYE plates containing 0.5% (w/v) agar and CF plates containing 1.5% (w/v) agar as described, respectively (Bustamante et al., 2004; Nan et al., 2010). For T4P-dependent motility, colony expansion was determined as the increase in colony diameter after 24 h incubation at 32 °C. For fruiting body formation, cells were incubated at 32 °C for 72 h and 120 h under low (0 M NaCl) and high (0.15 M NaCl) osmolarity, respectively. A Nikon SMZ1000 microscope and an OMAX A3590U digital camera were used to capture the images of fruiting bodies.
Fluorescent Biosensor Screening Assay for Phosphodiesterases
Chemically competent E. coli BL21 (DE3) Star cells (Life Technologies) were co-transformed with different combinations of biosensor-synthase plasmid (pET-Duet with synthase in multicloning site 1 and biosensor in multicloning site 2) and candidate PDE plasmid (pCOLA-Duet with PDE gene cloned into multicloning site 2). The screen for cGAMP PDE activity used biosensor Gm790P1–4ΔA-Spinach2 (Kellenberger et al., 2015) and synthase GacA from G. sulfurreducens (Hallberg et al., 2016). The screen for cyclic di-GMP PDE activity used biosensor Dp17-Spinach2 (Wang et al., 2016) and synthase WspR from P. fluorescens (De et al., 2008). Single colonies from LB/Carb/Kan plates (50 μg/mL each antibiotic) were used to inoculate 500 μL starter cultures in non-inducing media (Studier, 2005) with antibiotics in a 96-deep well culture plate (VWR). The 96-well starter cultures were grown 16–20 h at 37 °C with vigorous shaking at 325 rpm. After non-inducing growth, a 1:100 dilution of starter culture was added to ZYP autoinduction media modified to contain 1% α-lactose, which was found to improve expression and fluorescence signal. Culture volumes at a ratio of ~1:7 media:headspace (300 μL media in 2200 μL wells) were found to be critical for efficient induction of expression. Cells were grown under inducing conditions for 16–20 h at 37 °C with vigorous shaking at 325 rpm, then diluted 1:70 in 1x PBS, pH 7.4 with 50 μM DFHBI-1T for analysis by flow cytometry. Cellular fluorescence was measured for 50,000 cells at a flow rate of 25 μL/min using the Attune NxT Flow Cytometer (CIRM/QB3 Facility, UC Berkeley) on the BL1 channel (Ex. 530 +/− 30 nm, PMT V=540). Data was analyzed with FlowJo software (version 10.5.3). Successive polygon gates were applied to isolate single E. coli cells. A final bisecting gate was applied to separate cells into bright versus dark populations. This bisecting gate isolated the individual bright (Mean Fluorescence Intensity ~55,000 AU) or dark (Mean Fluorescence Intensity ~1,500 AU) peak in the fluorescence histogram and was uniformly applied across all replicates for a given CDN tested. Results are reported as both representative fluorescence histograms and averages of % cells in the bright population.
Bioinformatic Search for Other cGAMP Phosphodiesterases
A tBLASTn search of the NCBI database nucleotide collection was performed for homologs of the PmxA HD-GYP domain (amino acids 367–562). Search parameters excluded Myxococcales and environmental samples. 14,326 of a maximum threshold of 20,000 unique sequences were returned. DNA sequences were translated using EMBOSS Transeq and multiple sequence alignments were created with MUSCLE. A Python-based program using Biopython (Cock et al., 2009) was developed to analyze alignment data of HD-GYP domains and identify sequences with specific key residues. 11,504 sequences were found to have an aligned RxxX motif. These sequences were sorted based on the identity of the fourth residue in the motif and ranked by total abundance as shown in Fig. 8. Sequences with Gln(Q) or Asn(N) at the fourth residue were selected and hand-curated based on conservation of active site residues, species, and lack of transmembrane regions for ease of expression and screening in vitro.
To obtain full-length protein sequences, a BLASTp search requiring 100% match was performed using the tBLASTn hits to obtain the corresponding NCBI protein accession IDs, which were used to compile a FASTA file for all GAP candidates (RxxQ and RxxN in separate files, see Supplementary Files) containing protein ID, description, organism, and protein coding sequence. For protein domain analysis, the Batch Web CD-Search Tool using the two FASTA files as inputs was used to search the NCBI conserved domain database (Marchler-Bauer et al., 2017). The results were tabulated as shown in Table S1.
Supplementary Material
Supp info
ACKNOWLEDGEMENTS
The authors would like to acknowledge Dr. Mary West of the CIRM/QB3 Shared Stem Cell Facility at UC Berkeley for use of the Attune flow cytometer. We thank Dr. Wenjun Zhang and her lab for allowing TAW to share lab space at UC Berkeley, as well as Andrew Dippel for helpful discussion of cyclic dinucleotide signaling proteins.
FUNDING
This work was supported by the National Science Foundation [grant number 1716256, 1915466 to M.C.H.]; National Institutes of Health [grant number GM124589 to M.C.H., GM129000 to B.N.]; and UC Berkeley College of Chemistry undergraduate summer research awards to J.J.P., L.J., and W.A.A.
Footnotes
SUPPLEMENTARY DATA
Supplementary Data are available online. The data that support the findings of this study are available from the corresponding author upon reasonable request.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Aravind L, Ponting CP (1997). The GAF domain: an evolutionary link between diverse phototransducing proteins. Trends in Biochemical Sciences, 22(12), 458–459. [DOI] [PubMed] [Google Scholar]
- 2.Bellini D, Caly DL, Mccarthy Y, Bumann M, An SQ, Dow JM, et al. (2014). Crystal structure of an HD-GYP domain cyclic-di-GMP phosphodiesterase reveals an enzyme with a novel trinuclear catalytic iron centre. Molecular Microbiology, 91(1), 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burhenne H, Kaever V (2013). Quantification of Cyclic Dinucleotides by Reversed-Phase LC-MS/MS. Methods in Molecular Biology, 1016, 27–37. [DOI] [PubMed] [Google Scholar]
- 4.Bustamante VH, Martínez-Flores I, Vlamakis HC, Zusman DR (2004). Analysis of the Frz signal transduction system of Myxococcus xanthus shows the importance of the conserved C-terminal region of the cytoplasmic chemoreceptor FrzCD in sensing signals. Molecular Microbiology, 53(5), 1501–1513. [DOI] [PubMed] [Google Scholar]
- 5.Christen B, Christen M, Paul R, Schmid F, Folcher M, Jenoe P, et al. (2006). Allosteric control of cyclic di-GMP signaling. Journal of Biological Chemistry, 281(42), 32015–32024. [DOI] [PubMed] [Google Scholar]
- 6.Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, et al. (2009). Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25(11), 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen D, Melamed S, Millman A, Shulman G, Oppenheimer-Shaanan Y, Kacen A, et al. (2019). Cyclic GMP–AMP signalling protects bacteria against viral infection. Nature. [DOI] [PubMed] [Google Scholar]
- 8.Corrigan RM, Gründling A (2013). Cyclic di-AMP: Another second messenger enters the fray. Nature Reviews Microbiology, 11(8), 513–524. [DOI] [PubMed] [Google Scholar]
- 9.da Costa Vasconcelos FN, Maciel NK, Favaro DC, de Oliveira LC, Barbosa AS, Salinas RK, et al. (2017). Structural and Enzymatic Characterization of a cAMP-Dependent Diguanylate Cyclase from Pathogenic Leptospira Species. Journal of Molecular Biology, 429(15), 2337–2352. [DOI] [PubMed] [Google Scholar]
- 10.Davies BW, Bogard RW, Young TS, Mekalanos JJ (2012). Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell, 149(2), 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De N, Pirruccello M, Krasteva PV, Bae N, Raghavan RV, Sondermann H (2008). Phosphorylation-independent regulation of the diguanylate cyclase WspR. PLoS Biology, 6(3), 601–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng M, Tao J, Chao E, Ye Z, Jiang Z, Yu J, et al. (2018). Novel Mechanism for Cyclic Dinucleotide Degradation Revealed by Structural Studies of Vibrio Phosphodiesterase V-cGAP3. Journal of Molecular Biology, 430(24), 5080–5093. [DOI] [PubMed] [Google Scholar]
- 13.Enomoto G, Nomura R, Shimada T, Ni-Ni-Win, Narikawa R, Ikeuchi M (2014). Cyanobacteriochrome SesA Is a Diguanylate Cyclase That Induces Cell Aggregation in Thermosynechococcus. Journal of Biological Chemistry, 289(36), 24801–24809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao J, Tao J, Liang W, Zhao M, Du X, Cui S, et al. (2015). Identification and characterization of phosphodiesterases that specifically degrade 3’3’-cyclic GMP-AMP. Cell Research, 25(5), 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hallberg ZF, Chan CH, Wright TA, Kranzusch PJ, Doxzen KW, Park JJ, et al. (2019). Structure and mechanism of a Hypr GGDEF enzyme that activates cGAMP signaling to control extracellular metal respiration. eLife, 8, 1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hallberg ZF, Su Y, Kitto RZ, Hammond MC (2017). Engineering and In Vivo Applications of Riboswitches. Annual Review of Biochemistry, 86(1), 515–539. [DOI] [PubMed] [Google Scholar]
- 17.Hallberg ZF, Wang XC, Wright TA, Nan B, Ad O, Yeo J, et al. (2016). Hybrid promiscuous (Hypr) GGDEF enzymes produce cyclic AMP-GMP (3′, 3′-cGAMP). Proceedings of the National Academy of Sciences, 113(7), 1790–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hammer BK, Bassler BL (2009). Distinct sensory pathways in Vibrio cholerae El Tor and classical biotypes modulate cyclic dimeric GMP levels to control biofilm formation. Journal of Bacteriology, 91(1), 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heikaus CC, Pandit J, Klevit RE (2009). Cyclic Nucleotide Binding GAF Domains from Phosphodiesterases: Structural and Mechanistic Insights. Structure, 17(12), 1551–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huynh K, Partch CL (2015). Analysis of protein stability and ligand interactions by thermal shift assay. Current Protocols in Protein Science, 79, 28.9.1–28.9.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kellenberger CA, Wilson SC, Hickey SF, Gonzalez TL, Su Y, Hallberg ZF, et al. (2015). GEMM-I riboswitches from Geobacter sense the bacterial second messenger cyclic AMP-GMP. Proceedings of the National Academy of Sciences, 112(17), 5383–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimura Y, Mishima Y, Nakano H, Takegawa K (2002). An adenylyl cyclase, CyaA, of Myxococcus xanthus functions in signal transduction during osmotic stress. Journal of Bacteriology, 184(13), 3578–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimura Y, Ohtani M, Takegawa K (2005). An adenylyl cyclase, CyaB, acts as an osmosensor in Myxococcus xanthus. Journal of Bacteriology, 187(10), 3593–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kranzusch PJ, Lee ASY, Wilson SC, Solovykh MS, Vance RE, Berger JM, et al. (2014). Structure-Guided Reprogramming of Human cGAS Dinucleotide Linkage Specificity. Cell, 158(5), 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Little R, Dixon R (2003). The amino-terminal GAF domain of Azotobacter vinelandii NifA binds 2-oxoglutarate to resist inhibition by NifL under nitrogen-limiting conditions. Journal of Biological Chemistry, 278(31), 28711–28718. [DOI] [PubMed] [Google Scholar]
- 26.Lovering AL, Capeness MJ, Lambert C, Hobley L, Sockett RE (2011). The structure of an unconventional HD-GYP protein from bdellovibrio reveals the roles of conserved residues in this class of Cyclic-di-GMP phosphodiesterases. mBio, 2(5), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S et al. (2017). CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res, 45(D1), D200–D203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minasov G, Padavattan S, Shuvalova L, Brunzelle JS, Miller DJ, Baslé A, et al. (2009). Crystal structures of Ykul and its complex with second messenger cyclic Di-GMP suggest catalytic mechanism of phosphodiester bond cleavage by EAL domains. Journal of Biological Chemistry, 284(19), 13174–13184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miner KD, Kurtz DM (2016). Active Site Metal Occupancy and Cyclic Di-GMP Phosphodiesterase Activity of Thermotoga maritima HD-GYP. Biochemistry, 55(6), 970–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nan B, Mauriello EMF, Sun IH, Wong A, Zusman DR (2010). A multi-protein complex from Myxococcus xanthus required for bacterial gliding motility. Molecular Microbiology, 76(6), 1539–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nelson JW, Sudarsan N, Phillips GE, Stav S, Lünse CE, McCown PJ, et al. (2015). Control of bacterial exoelectrogenesis by c-AMP-GMP. Proceedings of the National Academy of Sciences, 112(17), 5389–5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paul R, Abel S, Wassmann P, Beck A, Heerklotz H, Jenal U (2007). Activation of the diguanylate cyclase PleD by phosphorylation-mediated dimerization. Journal of Biological Chemistry, 282(40), 29170–29177. [DOI] [PubMed] [Google Scholar]
- 33.Reinders A, Hee CS, Ozaki S, Mazur A, Boehm A, Schirmer T, et al. (2015). Expression and genetic activation of cyclic di-GMP-specific phosphodiesterases in Escherichia coli. Journal of Bacteriology, 198(3), 448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rinaldo S, Paiardini A, Stelitano V, Brunotti P, Cervoni L, Fernicola S, et al. (2015). Structural basis of functional diversification of the HD-GYP domain revealed by the Pseudomonas aeruginosa PA4781 protein, which displays an unselective bimetallic binding site. Journal of Bacteriology, 197(8), 1525–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Römling U, Galperin MY, Gomelsky M (2013). Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiology and Molecular Biology Reviews, 77(1), 1–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Römling U, Liang Z, Dow J (2017). Progress in Understanding the Molecular Basis Underlying Functional Diversification of Cyclic Dinucleotide Turnover Proteins. Journal of Bacteriology, 199(5), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sardiwal S, Kendall SL, Movahedzadeh F, Rison SCG, Stoker NG, Djordjevic S (2005). A GAF domain in the hypoxia/NO-inducible Mycobacterium tuberculosis DosS protein binds haem. Journal of Molecular Biology, 353(5), 929–936. [DOI] [PubMed] [Google Scholar]
- 38.Sarenko O, Klauck G, Wilke FM, Pfiff V, Eal G, Richter AM, et al. (2017). More than Enzymes That Make or Break Cyclic Di-GMP — Local Signaling in the. mBio, 8(5), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skotnicka D, Petters T, Heering J, Hoppert M, Kaever V, Søgaard-Andersen L (2016). Cyclic dI-GMP regulates type IV pilus-dependent motility in Myxococcus xanthus. Journal of Bacteriology, 198(1), 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skotnicka D, Smaldone GT, Petters T, Trampari E, Liang J, Kaever V, et al. (2016). A Minimal Threshold of c-di-GMP Is Essential for Fruiting Body Formation and Sporulation in Myxococcus xanthus. PLoS Genetics, 12(5), 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song W, Strack RL, Svensen N, Jaffrey SR (2014). Plug-and-play fluorophores extend the spectral properties of spinach. Journal of the American Chemical Society, 136(4), 1198–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Studier FW (2005). Protein production by auto-induction in high density shaking cultures. Protein Expression and Purification, 41(1), 207–234. [DOI] [PubMed] [Google Scholar]
- 43.Tang K, Knipp M, Liu BB, Cox N, Stabel R, He Q, et al. (2015). Redox-dependent ligand switching in a sensory heme-binding GAF domain of the cyanobacterium nostoc sp. PCC7120. Journal of Biological Chemistry, 290(31), 19067–19080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tudor JJ, Davis JJ, Panichella M, Zwolak A (2008). Isolation of predation-deficient mutants of Bdellovibrio bacteriovorus by using transposon mutagenesis. Applied and Environmental Microbiology, 74(17), 5436–5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Villapakkam AC, Handke LD, Belitsky BR, Levdikov VM, Wilkinson AJ, Sonenshein AL (2009). Genetic and biochemical analysis of the interaction of Bacillus subtilis CodY with branched-chain amino acids. Journal of Bacteriology, 191(22), 6865–6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang XC, Wilson SC, Hammond MC (2016). Next-generation fluorescent RNA biosensors enable anaerobic detection of cyclic di-GMP. Nucleic Acids Research, 44(17), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whiteley AT, Eaglesham JB, de Oliveira Mann CC, Morehouse BR, Lowey B, Nieminen EA, et al. (2019). Bacterial cGAS-like enzymes synthesize diverse nucleotide signals. Nature, 567(7747), 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yeo J, Dippel AB, Wang XC, Hammond MC (2018). In Vivo Biochemistry: Single-Cell Dynamics of Cyclic Di-GMP in Escherichia coli in Response to Zinc Overload. Biochemistry, 57(1), 108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yeo J, Wang XC, Hammond MC (2017). c-di-GMP Signaling Methods and Protocols. 1657th ed. Sauer K, ed. New York, NY: Humana Press. [Google Scholar]
- 50.Zhu D, Wang L, Shang G, Liu X, Zhu J, Lu D, et al. (2014). Structural Biochemistry of a Vibrio cholerae Dinucleotide Cyclase Reveals Cyclase Activity Regulation by Folates. Molecular Cell, 55(6), 931–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp info