The Extracellular Signal-Regulated Kinase Cascade Is Required for NMDA Receptor-Independent LTP in Area CA1 But Not Area CA3 of the Hippocampus (original) (raw)
Abstract
Activation of extracellular signal-regulated kinase (ERK) has been shown to be necessary for NMDA receptor-dependent long-term potentiation (LTP). We studied the role of ERK in three forms of NMDA receptor-independent LTP: LTP induced by very high-frequency stimulation (200 Hz-LTP), LTP induced by the K+channel blocker tetraethylammonium (TEA) (TEA-LTP), and mossy fiber (MF) LTP (MF-LTP). We found that ERK was activated in area CA1 after the induction of both 200 Hz-LTP and TEA-LTP and that this activation required the influx of Ca2+ through voltage-gated Ca2+ channels. Inhibition of the ERK signaling cascade with either PD 098059 or U0126 prevented the induction of both 200 Hz-LTP and TEA-LTP in area CA1. In contrast, neither PD 098059 nor U0126 prevented MF-LTP in area CA3 induced by either brief or long trains of high-frequency stimulation. U0126 also did not prevent forskolin-induced potentiation in area CA3. However, incubation of slices with forskolin, an activator of the cAMP-dependent protein kinase (PKA) cascade, did result in increases in active ERK and cAMP response element-binding protein (CREB) phosphorylation in area CA3. The forskolin-induced increase in active ERK was inhibited by U0126, whereas the increase in CREB phosphorylation was not, which suggests that in area CA3 the PKA cascade is not coupled to CREB phosphorylation via ERK. Overall, our observations indicate that activation of the ERK signaling cascade is necessary for NMDA receptor-independent LTP in area CA1 but not in area CA3 and suggest a divergence in the signaling cascades underlying NMDA receptor-independent LTP in these hippocampal subregions.
Keywords: NMDA receptor-independent LTP, 200 Hz-LTP, TEA-LTP, mossy fiber LTP, extracellular signal-regulated kinase, hippocampus, learning, memory
Long-term potentiation (LTP) is an activity-dependent, persistent enhancement of synaptic strength widely studied as a cellular model for learning and memory. In area CA1 of the hippocampus, the most commonly studied forms of LTP are induced by either one or multiple trains of high-frequency (100 Hz) stimulation. These forms of LTP require postsynaptic Ca2+ influx (Lynch et al., 1983) and are dependent on NMDA receptor activation (Collingridge et al., 1983). Other forms of LTP that require postsynaptic Ca2+ influx but not the activation of NMDA receptors also have been described in area CA1. For example, NMDA receptor-independent LTP can be induced with either bath application of the K+ channel blocker tetraethylammonium (TEA) (TEA-LTP) (Aniksztejn and Ben-Ari, 1991) or very high-frequency (200 Hz) stimulation (200 Hz-LTP) (Grover and Teyler, 1990). TEA-LTP was shown to be blocked by flunarizine (Aniksztejn and Ben-Ari, 1991) and nifedipine (Huang and Malenka, 1993; Powell et al., 1994; but seeHuber et al., 1995a). Similarly, 200 Hz-LTP was shown to be blocked by nifedipine (Grover and Teyler, 1990). These findings indicate that both TEA-LTP and 200 Hz-LTP are dependent on the activation of voltage-gated Ca2+ channels.
Similar to NMDA receptor-independent LTP in area CA1, LTP at the mossy fiber input to area CA3 (MF-LTP) is NMDA receptor independent (Harris and Cotman, 1986). Although there is disagreement whether induction of MF-LTP is localized either presynaptically (Zalutsky and Nicoll, 1990) or postsynaptically (Jaffe and Johnston, 1990; Yeckel et al., 1999), it is likely that the activation of voltage-gated Ca2+ channels is necessary for induction of MF-LTP (Jaffe and Johnston, 1990; Castillo et al., 1994; Kapur et al., 1998). Thus, 200 Hz-LTP, TEA-LTP, and MF-LTP are dependent on Ca2+ influx through voltage-gated Ca2+ channels.
Relatively little is known about the signaling cascades required for the induction and expression of NMDA receptor-independent forms of LTP. In contrast, there has been much progress in determining the signaling cascades involved in NMDA receptor-dependent LTP in area CA1 (Roberson et al., 1996). Many of the cascades involved in the induction of NMDA receptor-dependent LTP may converge on and activate extracellular signal-regulated kinase 2 [ERK2; also referred to as p42 mitogen-activated protein kinase (p42 MAPK)] (Roberson et al., 1999). In support of this idea, it was shown that NMDA receptor-dependent LTP in area CA1 is associated with an increase in active ERK2 (English and Sweatt, 1996). In addition, NMDA receptor-dependent LTP was blocked by inhibition of MAP kinase kinase (MEK), the dual-specific kinase that activates ERK2 (English and Sweatt, 1997; Impey et al., 1998), which indicates that the activation of ERK2 is required for the induction of this form of LTP.
In the present study, we tested the hypothesis that the activation of ERK is associated with 200 Hz-LTP and TEA-LTP in area CA1. We also tested the hypothesis that the activation of ERK is necessary for the induction of 200 Hz-LTP and TEA-LTP in area CA1 and of MF-LTP in area CA3. The results of these studies should provide insight into the signaling cascades involved in the various forms of NMDA receptor-independent LTP in the hippocampus.
MATERIALS AND METHODS
Preparation of hippocampal slices and induction of LTP. For 200 Hz-LTP and TEA-LTP, hippocampal slices were prepared as described previously (Klann, 1998). The slices were perfused for 1 hr with a standard saline solution for area CA1 recordings containing (in mm): 124 NaCl, 4.4 KCl, 26 NaHCO3, 10 d-glucose, 2 CaCl2, and 2 MgCl2, gassed with 95% O2/5% CO2, pH 7.4, in an interface tissue slice chamber at 30–32°C. Responses to Schaffer collateral stimulation in area CA1 were monitored for at least 20 min before the delivery of LTP-inducing high-frequency stimulation (HFS). Test stimuli (50 μsec) were given at a current (30–50 μA) that produced 50% of the maximum initial slope of the extracellular field EPSP (fEPSP). Responses to test stimuli were measured every 2.5 min as an average of four individual traces (0.1 Hz). Unless otherwise noted, 50 μmd,l-2-amino-5-phosphonovaleric acid (APV) was present in the recording solution for the duration of all of the experiments.
200 Hz-LTP was induced with three 1 sec trains of stimuli (200 Hz), given 2.5 min apart with the use of a current (60–100 μA) that elicited the maximum fEPSP. TEA-LTP was induced with a 10 min application of 25 mm TEA. In both types of experiments, responses to test stimuli were measured every 2.5 min as an average of four individual traces (0.1 Hz) for 45 min after either the final train of HFS or the washout of TEA. Post-HFS and post-TEA responses were elicited by the same test stimulation intensity applied before either HFS or TEA.
For studies of MF-LTP and associational/commissural LTP in area CA3, hippocampal slices were prepared as described previously (Urban and Barrionuevo, 1996). Slices were transferred to an incubation chamber containing a standard saline solution for area CA3 recordings containing (in mm): 125 NaCl, 2 KCl, 26 NaHCO3, 10 dextrose, 1 CaCl2, and 6 MgCl2, gassed with 95% O2/5% CO2, pH 7.4, at room temperature. After incubation, slices were transferred to a recording chamber and submerged in the standard saline solution for area CA3 recordings, with the exception that the divalent ion concentrations were 2.5 mm CaCl2 and 1 mm MgCl2. The temperature of the recording chamber was 30–34°C. Associational/commissural LTP was induced as described in Results (see Fig. 7 legend).
Fig. 7.

U0126 blocks associational/commissural LTP but not MF-LTP in area CA3. A, MF input. Hippocampal slices were incubated with 20 μm U0126 for 30 min before recording baseline mossy fiber fEPSP amplitudes. U0126 (indicated by the_horizontal_ bar) remained in the perfusing solution for an additional 10 min before and for 30 min after the delivery of L-HFS (indicated by the arrow). The NMDA receptor antagonist APV (50 μm) was present in the perfusing solution (also indicated by the horizontal bar) for the duration of the experiment. MF-LTP was not blocked by U0126. B, Associational/commissural (A/C) input. Open circles_are ensemble averages indicating the potentiation of the slope of the fEPSP observed after the delivery of HFS (indicated by the_arrow) in the presence of 0.33% DMSO (indicated by the_horizontal_ bar; n = 4). Open squares are ensemble averages indicating that potentiation is blocked after HFS in the presence of 20 μm U0126 (indicated by the_horizontal_ bar; n = 4). U0126 significantly blocked LTP 45 min after the final train of HFS (p < 0.001 by paired Student's_t_ test). Error bars in all _panels_indicate SEM.
MF-LTP was induced either with brief HFS (B-HFS) consisting of 8 pulses at 100 Hz repeated eight times at 5 sec intervals or with long HFS (L-HFS) consisting of 100 pulses at 100 Hz repeated three times at 10 sec intervals (Urban and Barrionuevo, 1996). The magnitude of LTP was calculated by dividing the average fEPSP amplitude of 10 responses evoked 15–20 min after the HFS by the average amplitude of responses evoked in the 5 min before the delivery of the HFS.
For all of the LTP experiments described above, PD 098059 and U0126 were dissolved in DMSO and diluted in the standard saline solutions to give the desired final concentration (usually 50 μm PD 098059 and 20 μm U0126). In addition, DMSO (either 0.33 or 0.50%) was present during either HFS or TEA application in all of the control LTP experiments. In control experiments, we observed that 20 min incubations of hippocampal slices with either 50 μm PD 098059 or 20 μm U0126 resulted in significant decreases in active ERK1 and ERK2 in area CA1 when compared with control slices incubated for 20 min with DMSO (PD 098059 = 36 ± 8% of control; n = 3; U0126 = 33 ± 4% of control; n = 3).
Analysis of active ERK. In the 200 Hz-LTP and TEA-LTP experiments, slices were removed from the recording chamber either 2.5, 10, or 25 min after either the last HFS or the washout of TEA. The slices were frozen, and the CA1 region between the stimulating and recording electrodes was microdissected and homogenized in ice-cold buffer (50 mm Tris-HCl, pH 7.5, 1 mm EDTA, 1 mm EGTA, 2 mm sodium pyrophosphate, 4 mm_p_-nitrophenyl phosphate, 1 mm sodium orthovanadate, 100 ng/ml leupeptin, 100 ng/ml aprotinin, and 10 μm benzamidine). After homogenization, the protein concentration was measured by the method ofBradford (1976) using bovine serum albumin as the standard. Equivalent amounts of protein for each sample were resolved in 10% SDS-polyacrylamide gels (SDS-PAGE), blotted electrophoretically to Immobilon membranes, and incubated in Tris-buffered saline with Tween 20 (50 mm Tris-HCl, pH 7.5–8.0, 150 mm NaCl, and 0.1% Tween 20) containing 3% bovine serum albumin for 30 min.
Blots were incubated with an active ERK polyclonal antibody (1:5000 dilution; Promega, Madison, WI), followed by incubation with horseradish peroxidase-linked goat anti-rabbit IgG (1:4000 dilution; Amersham, Arlington Heights, IL), and developed using enhanced chemiluminescence (ECL; Amersham). The blots then were incubated in stripping buffer (62 mm Tris-HCl, pH 6.8, 2% SDS, and 100 mm β-mercaptoethanol) for 30 min at 50°C, followed by incubation in Tris-buffered saline with Tween 20 containing 3% bovine serum albumin for 30 min. The blots then were incubated with an ERK polyclonal antibody (1:1000 dilution; Upstate Biotechnology, Lake Placid, NY) that binds to ERK regardless of phosphorylation at the dual phosphorylation site. After incubation with the ERK polyclonal antibody, the blots were incubated with the horseradish peroxidase-linked goat anti-rabbit IgG (1:4000 dilution; Amersham) and developed using ECL. Densitometric analysis of active ERK immunoreactivity and ERK immunoreactivity was conducted using NIH Image software. Active ERK immunoreactivity was normalized to total ERK protein levels using ERK immunoreactivity.
Analysis of phospho-cAMP response element-binding protein. Control slices and slices incubated with 50 μm forskolin for 20 min were incubated in either the presence or absence of 20 μm U0126. Slices were homogenized and split into two aliquots. One aliquot was analyzed for ERK and active ERK as described above. For phospho-cAMP response element-binding protein (phospho-CREB), the homogenates were sonicated, and proteins were resolved via SDS-PAGE and blotted electrophoretically to Immobilon membranes as described above.
Blots were incubated with a phospho-CREB polyclonal antibody (1:500 dilution; Upstate Biotechnology), followed by incubation with horseradish peroxidase-linked goat anti-rabbit IgG, and developed with ECL. The blots were stripped as described above and incubated with a CREB polyclonal antibody (0.5 mg/ml; Upstate Biotechnology), followed by incubation with horseradish peroxidase-linked goat anti-rabbit IgG, and developed using ECL. Densitometric analysis of phospho-CREB and CREB immunoreactivity was conducted as described above. Phospho-CREB immunoreactivity was normalized to protein levels using CREB immunoreactivity.
RESULTS
ERK in NMDA receptor-independent LTP in area CA1
We hypothesized that 200 Hz-LTP, an NMDA receptor-independent form of LTP (Grover and Teyler, 1990), is associated with an increase in active ERK. The CA1 subregion between the stimulating and recording electrodes was analyzed for active ERK 2.5, 10, and 25 min after the final train of 200 Hz HFS. Healthy slices from the same hippocampus, maintained in the same recording chamber for the length of each experiment, were used for control slices. We observed a statistically significant increase in active ERK1 and active ERK2 (ERK1/ERK2) 10 min after the final train of HFS (Fig. 1; ERK1 = 151 ± 9% of control; ERK2 = 155 ± 7% of control; n = 4). We observed no significant alterations in active ERK1/ERK2 either 2.5 min (Fig. 1; ERK1 = 122 ± 22% of control; ERK2 = 130 ± 32% of control;n = 6) or 25 min (ERK1 = 102% of control;n = 2; ERK2 = 97% of control; n = 2) after the final train of HFS. These data suggest that 200 Hz-LTP is associated with a transient increase in active ERK1/ERK2.
Fig. 1.

200 Hz-LTP is associated with an increase in active ERK1/ERK2. A, Representative ERK Western blots of area CA1 subregions from control slices and slices given three 1 sec trains of HFS (200 Hz). The slices given 200 Hz HFS were analyzed either 2.5 or 10 min after the final train of HFS and compared with control slices from the same recording chamber. The α-ERK antibody detects ERK1 and ERK2 independent of phosphorylation state. The α-dual-P-ERK antibody detects the dually phosphorylated, active forms of ERK1 and ERK2. B, Normalized active ERK1 immunoreactivity 2.5 min (n = 6) and 10 min (n = 4) after the final train of HFS.C, Normalized active ERK2 immunoreactivity 2.5 min (n = 6) and 10 min (n = 4) after the final train of HFS. Error bars in B and_C_ indicate SEM; * denotes statistical significance compared with control (p < 0.05 by paired Student's t test).
To strengthen the association between 200 Hz-LTP and the increase in active ERK1/ERK2, we delivered the LTP-inducing stimulation in the presence of the voltage-gated Ca2+ channel antagonist nifedipine (10 μm). Nifedipine, which has been shown to block 200 Hz-LTP (Cavus and Teyler, 1996), prevented the increase in active ERK1/ERK2 measured 10 min after the final train of HFS (Fig. 2; ERK1 = 108 ± 12% of control; ERK2 = 104 ± 5% of control; n = 4). These data demonstrate that Ca2+influx through L-type voltage-gated Ca2+channels is necessary for ERK1/ERK2 activation in response to 200 Hz stimulation.
Fig. 2.

Nifedipine blocks the increase in active ERK1/ERK2 associated with 200 Hz-LTP. A, Representative Western blot of the area CA1 subregion from a control slice and a slice given the 200 Hz-LTP-inducing HFS in the presence of 10 μmnifedipine. The slice was analyzed 10 min after the final train of HFS.B, Normalized active ERK1/ERK2 immunoreactivity 10 min after the delivery of 200 Hz-LTP-inducing HFS (n = 4). Error bars indicate SEM.
To determine whether the activation of ERK1/ERK2 was necessary for the induction of 200 Hz-LTP in area CA1, we tested whether inhibitors of MEK, the dual-specific protein kinase that activates ERK1/ERK2, could block the expression of 200 Hz-LTP. HFS consisting of three 1 sec trains (200 Hz), delivered in the presence of the drug vehicle (0.33% DMSO), resulted in a slowly rising potentiation of the fEPSP slope that was stable 45 min after the last train of stimulation (fEPSP slope = 151 ± 11% of control; n = 6). Delivery of LTP-inducing stimulation in the presence of the MEK inhibitor PD 098059 (50 μm) (Alessi et al., 1995) resulted in a slowly rising potentiation of the slope of the fEPSP for the first 20 min after the final train of HFS, similar to that observed in slices incubated with drug vehicle (Fig.3A). However, 20 min after the final train of HFS, potentiation began to decay in slices incubated with PD 098059, reaching baseline levels by 45 min after HFS (Fig.3A; fEPSP slope = 104 ± 7% of control;n = 6). Similarly, delivery of LTP-inducing stimulation in the presence of U0126 (20 μm), a recently described MEK inhibitor (Favata et al., 1998; Roberson et al., 1999), resulted in a normal initial LTP, with a decaying potentiation observed beginning 20 min after HFS that reached near baseline levels 45 min after HFS (Fig. 3A; fEPSP slope = 109 ± 7% of control; n = 6). In control experiments, we found that a 20 min application of either PD 098059 or U0126 to hippocampal slices had no effect on baseline, NMDA receptor-independent synaptic responses (Fig. 3B). These data demonstrate that the activation of ERK1/ERK2 is critical for the induction of 200 Hz-LTP in area CA1.
Fig. 3.
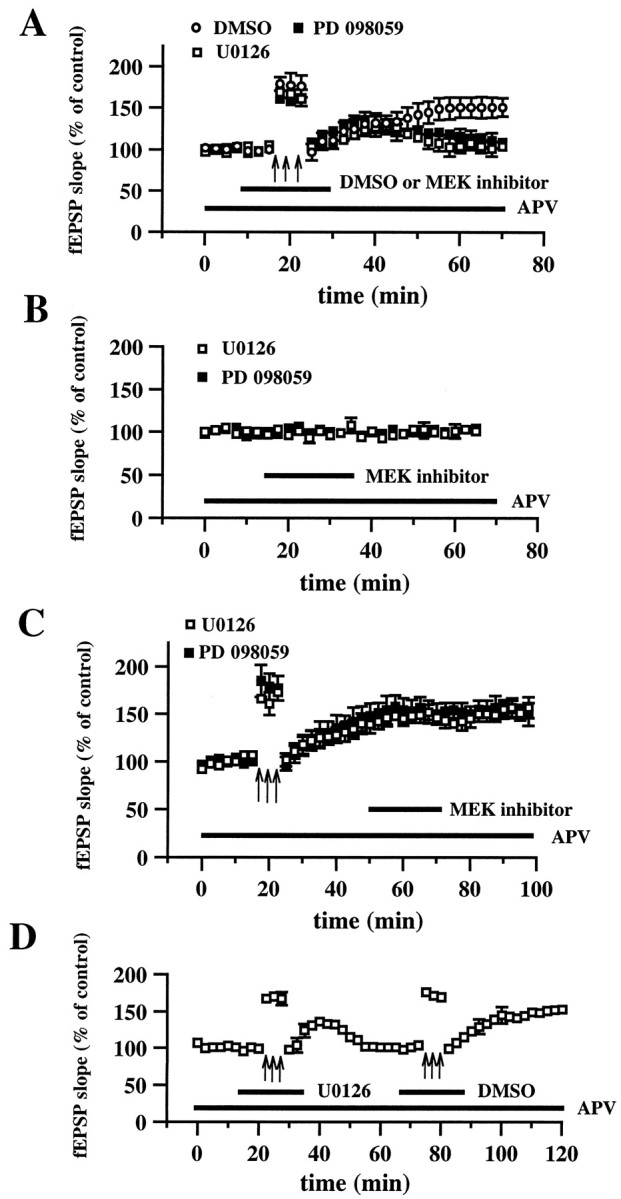
Effect of U0126 and PD 098059 on 200 Hz-LTP and NMDA receptor-independent synaptic transmission. A, Blockade of 200 Hz-LTP by either U0126 or PD 098059.Open circles are ensemble averages of the fEPSP slope from slices given 200 Hz-LTP-inducing HFS (indicated by the_arrows_) in the presence of 0.33% DMSO (n = 6). Open squares_are ensemble averages of the fEPSP slope from slices given 200 Hz-LTP-inducing HFS in the presence of 20 μm U0126 (n = 6). Closed squares are ensemble averages from slices given 200 Hz-LTP-inducing HFS in the presence of 50 μm PD 098059 (n = 6). DMSO, U0126, or PD 098059 was present in the perfusing solution 7.5 min before, during, and 7.5 min after the delivery of the HFS (total time in the solution was 20 min, indicated by the horizontal bar). Both MEK inhibitors significantly blocked LTP 45 min after the final train of HFS (p < 0.001 by paired Student's_t test). B, Effect of MEK inhibitors on NMDA receptor-independent synaptic transmission. Baseline responses were recorded for 15 min before the slices were perfused for 20 min (indicated by the horizontal bar) with either 20 μm U0126 (open squares; n = 4) or 50 μm PD 098059 (closed squares; n = 4). Responses were recorded for an additional 30 min after the washout of each compound.C, Effect of MEK inhibitors on established 200 Hz-LTP. Baseline responses were recorded for 17.5 min before the delivery of HFS (indicated by the_arrows_). Twenty-five minutes after the delivery of HFS, slices were perfused with either 20 μm U0126 (open squares; n = 4) or 50 μm PD 098059 (closed squares; n = 4) for 20 min. Responses were recorded for 30 min after the washout of each compound.D, Induction of 200 Hz-LTP after the washout of U0126. Delivery of 200 Hz-LTP-inducing HFS (indicated by the_arrows_ on the left) in the presence of 20 μm U0126 (indicated by the horizontal bar) resulted in the blockade of LTP. Forty minutes after the washout of U0126, 200 Hz-LTP-inducing HFS (indicated by the_arrows_ on the right) was delivered in the presence of 0.33% DMS0 (indicated by the horizontal bar), resulting in LTP. The NMDA receptor antagonist APV (50 μm) was present in the perfusing solution (indicated by the horizontal bar) in all experiments in A–D. Error bars in all _panels_indicate SEM.
Persistent protein kinase activity is thought to underlie the expression of NMDA receptor-dependent LTP (Malinow et al., 1988; Klann et al., 1991; Wang and Feng, 1992). Our biochemical analysis suggested that ERK1/ERK2 was transiently, but not persistently, activated during 200 Hz-LTP. To test this idea further, we examined the effect of either PD 098059 or U0126 on established 200 Hz-LTP. In these experiments, 200 Hz-LTP was elicited, and either PD 098059 or U0126 was applied to the hippocampal slices for 20 min beginning 25 min after the last train of HFS. Neither PD 098059 (fEPSP slope = 157 ± 11% of control;n = 4) nor U0126 (150 ± 12% of control;n = 4) had a significant effect on previously established 200 Hz-LTP measured 30 min after the washout of the inhibitors (Fig. 3C). These results suggest that persistent activation of ERK1/ERK2 is not necessary for the expression of 200 Hz-LTP.
To ensure that the blockade of 200 Hz-LTP by U0126 was not caused by deterioration of the slices because of the inhibitor, we determined whether 200 Hz-LTP could be induced in slices after washout of U0126. Stimulation of 200 Hz was delivered in the presence of U0126, and 20 min after the HFS, the potentiation began to decay, reaching baseline levels by 45 min after HFS (Fig. 3D). LTP-inducing stimulation then was delivered in the presence of the drug vehicle, resulting in a slowly rising potentiation that was stable 40 min after the last train of stimulation (fEPSP slope = 153 ± 4% of control; n = 4). These data demonstrate that the blockade of 200 Hz-LTP by the MEK inhibitors is not attributable to nonspecific effects on slice viability and are consistent with the idea that activation of ERK1/ERK2 is necessary for the induction of 200 Hz-LTP in area CA1.
Bath application of hippocampal slices with the K+ channel blocker TEA results in an NMDA receptor-independent, voltage-gated Ca2+channel-dependent form of LTP in area CA1 (Aniksztejn and Ben-Ari, 1991). Because of the similarities between 200 Hz-LTP and TEA-LTP, we hypothesized that TEA-LTP also was associated with an increase in active ERK1/ERK2. We found that TEA-LTP was associated with a significant increase in active ERK1/ERK2 10 min after the washout of TEA (Fig. 4; ERK1 = 138 ± 5% of control; ERK2 = 148 ± 11% of control; _n_= 4). We observed no significant alterations in active ERK1/ERK2 either 2.5 min (Fig. 4; ERK1 = 114 ± 5% of control; ERK2 = 122 ± 11% of control; n = 4) or 25 min (ERK1 = 105% of control; ERK2 = 98% of control;n = 2) after the washout of TEA. These data demonstrate that TEA-LTP is associated with a transient increase in active ERK1/ERK2 and suggest that 200 Hz-LTP and TEA-LTP share similar biochemical signaling mechanisms.
Fig. 4.

TEA-LTP is associated with an increase in active ERK1/ERK2. A, Representative ERK Western blots of area CA1 subregions from control slices and slices exposed to 25 mm TEA for 10 min. The slices exposed to TEA were analyzed either 2.5 or 10 min after the final train of HFS and compared with control slices from the same animal in an adjacent recording chamber.B, Normalized active ERK1 immunoreactivity 2.5 min (n = 6) and 10 min (n = 4) after the washout of TEA. C, Normalized active ERK2 immunoreactivity 2.5 min (n = 6) and 10 min (n = 4) after the washout of TEA. Error bars in_B_ and C indicate SEM; * denotes statistical significance compared with control (p < 0.05 by paired Student's_t_ test).
To strengthen the association between TEA-LTP and the increase in active ERK1/ERK2, slices were incubated with TEA in the presence of nifedipine, which has been shown to block the TEA-LTP (Huang and Malenka, 1993; Powell et al., 1994). Similar to 200 Hz-LTP, nifedipine prevented the increase in active ERK1/ERK2 measured 10 min after the washout of TEA (ERK1 = 109 ± 9% of control; ERK2 = 103 ± 10% of control; n = 3). These data demonstrate that stimulation of voltage-gated Ca2+ channels is necessary for the increase in active ERK1/ERK2 associated with TEA-LTP.
To determine whether the increase in active ERK1/ERK2 associated with TEA-LTP was necessary for the induction of TEA-LTP, hippocampal slices were incubated with 25 mm TEA in the presence of either 50 μm PD 098059 or 20 μm U0126. In each group of experiments, we observed a transient depression of the slope of the fEPSP during the TEA application (Fig.5A). After washout of TEA, slices incubated with the drug vehicle (0.33% DMSO) exhibited a rising potentiation of the fEPSP slope that was faster than that observed in the 200 Hz-LTP experiments. The potentiation peaked at ∼20 min and was stable 45 min after the washout of TEA (Fig. 5A; fEPSP slope = 196 ± 13% of control; n = 6). Slices incubated with TEA in the presence of either PD 098059 or U0126 exhibited a fast-rising potentiation for ∼20 min, similar to that observed in slices incubated with drug vehicle (Fig. 5A). However, at this time the potentiation of the fEPSP slope began to decay in the slices incubated with the MEK inhibitors, reaching near baseline levels 45 min after the washout of TEA (Fig. 5A; for PD 098059, fEPSP slope = 114 ± 15% of control;n = 6; for U0126, fEPSP slope = 121 ± 6% of control; n = 6). In contrast to the effects on 200 Hz-LTP, neither PD 098059 nor U0126 completely blocked the induction of TEA-LTP because some potentiation could be observed at the 45 min time point. These data indicate that the increase in active ERK1/ERK2 associated with TEA-LTP is not absolutely required for TEA-LTP but is necessary for the full expression of TEA-LTP.
Fig. 5.
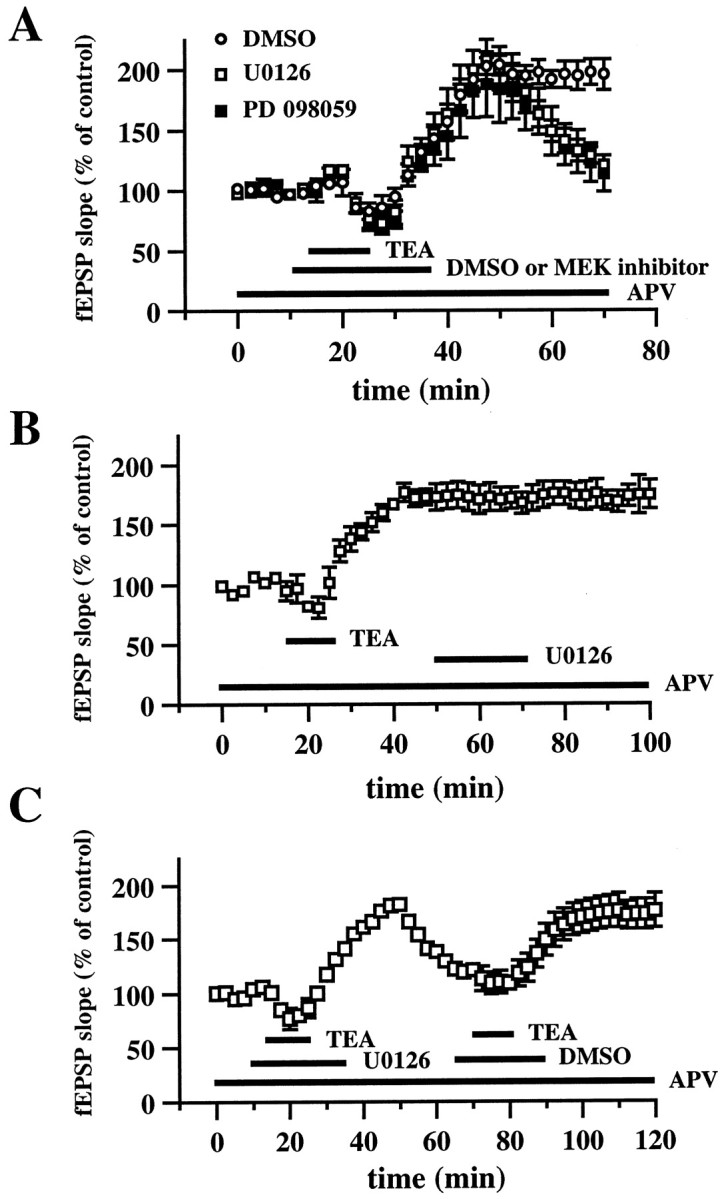
Effect of U0126 and PD 098059 on TEA-LTP.A, Ensemble averages of fEPSP slopes from slices exposed to 25 mm TEA for 10 min (indicated by the_horizontal_ bar) in the presence of 0.33% DMSO (open circles; n_= 6), 20 μm U0126 (open squares; n = 6), or 50 μm PD 098059 (closed squares; n = 6). DMSO, U0126, or PD 098059 was present in the perfusing solution 5 min before, during, and 10 min after the washout of TEA (total time in the solution was 20 min, indicated by the horizontal bar). Both MEK inhibitors significantly blocked LTP 45 min after the washout of TEA (p < 0.001 by paired Student's_t test). B, Effect of U0126 on established 200 Hz-LTP. Baseline responses were recorded for 15 min before application of TEA (indicated by the horizontal bar). Twenty-five minutes after TEA application, slices were perfused with 20 μm U0126 (n = 4) for 20 min. Responses were recorded for 30 min after the washout of U0126. C, Induction of TEA-LTP after the washout of U0126. Application of TEA (indicated by the horizontal bar) in the presence of 20 μm U0126 (indicated by the horizontal bar) resulted in the blockade of LTP. Thirty-five minutes after the washout of U0126, TEA was applied (indicated by the horizontal bar) in the presence of 0.33% DMS0 (indicated by the_horizontal_ bar), resulting in LTP. The NMDA receptor antagonist APV (50 μm) was present in the perfusing solution (indicated by the horizontal bar) in all experiments in A–C. Error bars in all panels indicate SEM.
To determine whether the transient activation of ERK1/ERK2 was necessary for the expression of TEA-LTP, we examined the effect of U0126 on established TEA-LTP. TEA-LTP was elicited, and U0126 was applied to the hippocampal slices for 20 min beginning 25 min after the washout of TEA. U0126 had no significant effect on previously established TEA-LTP measured 30 min after washout of the inhibitors (Fig. 5B; fEPSP slope = 175 ± 12% of control;n = 4). These results suggest that persistent activation of ERK1/ERK2 is not necessary for the expression of TEA-LTP.
To ensure that the blockade of TEA-LTP by U0126 was not caused by deterioration of the slices because of the inhibitor, we determined whether TEA-LTP could be induced in slices after the washout of U0126. TEA was applied to slices in the presence of U0126, and 20 min after the washout of TEA, the potentiation began to decay (Fig.5C). TEA then was reapplied to the slices in the presence of the drug vehicle, which resulted in a slowly rising potentiation that was stable 40 min after the washout of TEA (fEPSP slope = 176 ± 16% of control; n = 4). These data illustrate that the blockade of TEA-LTP by U0126 is not attributable to nonspecific effects on the health of the slice and are consistent with the idea that activation of ERK1/ERK2 is necessary for the induction of TEA-LTP in area CA1.
ERK in mossy fiber LTP
An additional form of NMDA receptor-independent LTP in the hippocampus is MF-LTP in area CA3. Similar to 200 Hz-LTP and TEA-LTP in area CA1, MF-LTP is dependent on activation of voltage-gated Ca2+ channels (Jaffe and Johnston, 1990;Castillo et al., 1994). Therefore, we hypothesized that MF-LTP also was associated with an increase in active ERK1/ERK2. We found that MF-LTP induced with L-HFS (Zalutsky and Nicoll, 1990; Urban and Barrionuevo, 1996) was not associated with significant increases in active ERK2 measured 10 min after the final train of stimulation (126 ± 32% of control; n = 4). It should be noted that we were unable to detect ERK1 consistently in the small amount of CA3 tissue dissected from the region between the stimulating and recording electrodes (data not shown). However, because we detected LTP-associated increases in active ERK2 in two of the four experiments, we proceeded to test the hypothesis that activation of ERK1/ERK2 is necessary for MF-LTP.
As a first test of the hypothesis that activation of ERK1/ERK2 is necessary for MF-LTP, we delivered L-HFS (Zalutsky and Nicoll, 1990;Urban and Barrionuevo, 1996) in either the presence or absence of the MEK inhibitors. Control experiments in which L-HFS was delivered in the presence of the drug vehicle (0.50% DMSO) resulted in LTP at mossy fiber synapses 40 min after the final train of HFS (fEPSP amplitude = 156 ± 19% of control; n = 5). Slices given L-HFS in the presence of either PD 098059 or U0126 also exhibited MF-LTP 40 min after HFS (Fig.6A; for PD 098059, fEPSP amplitude = 206 ± 18% of control; n = 3; for U0126, fEPSP amplitude = 170 ± 30% of control;n = 4). We proceeded to determine whether activation of ERK1/ERK2 was necessary for MF-LTP induced with a B-HFS (Jaffe and Johnston, 1990; Urban and Barrionuevo, 1996). Control experiments in which B-HFS was delivered in the presence of the drug vehicle (0.50% DMSO) resulted in LTP at mossy fiber synapses 40 min after the final train of HFS (fEPSP amplitude = 128 ± 11% of control;n = 4). Again, neither PD 098059 nor U0126 blocked MF-LTP measured 40 min after the final train of HFS (Fig.6B; for PD 098059, fEPSP amplitude = 124 ± 12% of control; n = 4; for U0126, fEPSP amplitude = 154 ± 9% of control; n = 5). These data suggest that activation of ERK1/ERK2 is not necessary for the induction of MF-LTP induced by either L-HFS or B-HFS.
Fig. 6.
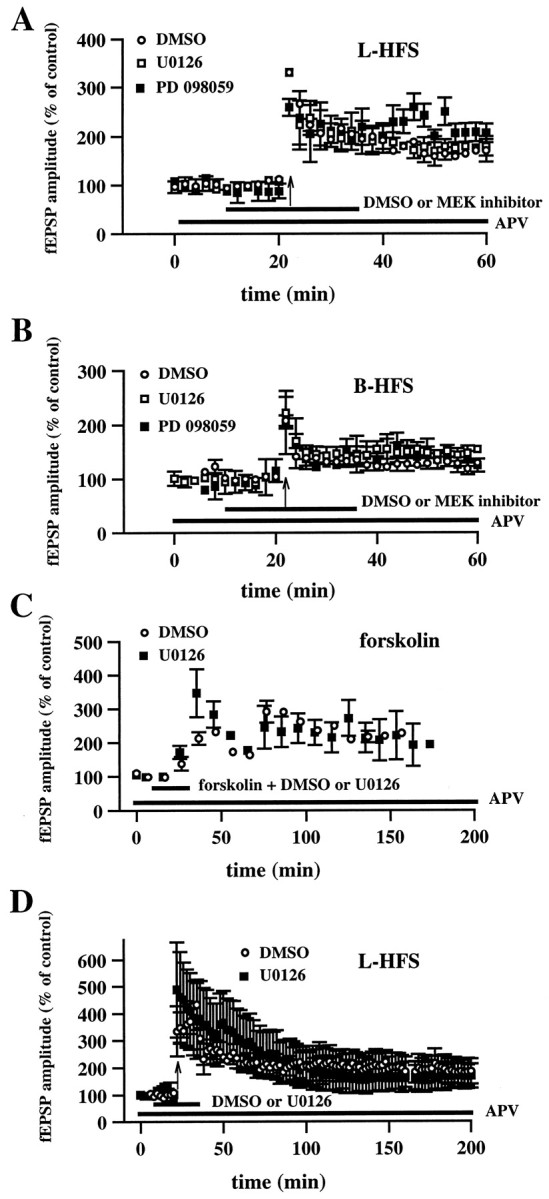
Effect of U0126 and PD 098059 on MF-LTP.A, MEK inhibitors do not block LTP induced by L-HFS (100 pulses at 100 Hz). Open circles are ensemble averages indicating the potentiation of mossy fiber fEPSP amplitudes observed after L-HFS in the presence of 0.5% DMSO (n = 5). Open squares_are ensemble averages indicating that potentiation is observed after L-HFS in the presence of 20–50 μm U0126 (n = 4). Closed squares are ensemble averages indicating that potentiation is observed after L-HFS in the presence of 20–50 μm PD 098059 (n = 3). Either DMSO or the MEK inhibitor was present for 15 min before and 15 min after the HFS, as indicated by the horizontal bar_on the graph. The arrow indicates the time at which HFS was delivered. LTP was not blocked by either of the inhibitors (p > 0.3 by paired Student's_t test). B, Experiments are similar to those in A except that mossy fiber LTP was induced by B-HFS (8 pulses at 100 Hz; repeated 10 times at 5 sec intervals). Again, the MEK inhibitors (DMSO,n = 4; U0126, n = 5; PD 098059,n = 4) had no significant effect on LTP (p > 0.3 by paired Student's_t test). C, Slices were exposed to 50 μm forskolin for 20 min (indicated by the_horizontal_ bar) in the presence of either 0.5% DMSO (open circles;n = 4) or 20 μm U0126 (closed squares; n = 4). U0126 had no significant effect on forskolin-induced potentiation (p > 0.3 by paired Student's_t_ test). D, Experiments are similar to those in A except that LTP was followed for 3 hr after the L-HFS. Again, LTP was not affected by the addition of U0126 (p > 0.3 by Student's _t_test; DMSO, n = 4; U0126, n = 4). The NMDA receptor antagonist APV (50 μm) was present in the perfusing solution (indicated by the horizontal bar) in all experiments in A–D. Error bars in all panels indicate SEM.
Forskolin is a diterpene that directly activates adenylyl cyclase (Seamon and Daly, 1986), producing cAMP and the subsequent activation of cAMP-dependent protein kinase (PKA). In addition, forskolin-induced activation of the PKA-signaling cascade has been shown to be coupled to the activation of ERK1/ERK2 in the CA1 area of hippocampal slices (Roberson et al., 1999). Finally, forskolin has been shown to induce potentiation that occludes MF-LTP induced by L-HFS (Weisskopf et al., 1994). Taken together, these studies prompted us to examine whether inhibition of ERK activation with either PD 098059 or U0126 prevents forskolin-induced potentiation at MF synapses. In the presence of DMSO, incubation of hippocampal slices with 50 μm forskolin for 20 min produced robust potentiation measured 40 min after the washout of forskolin (fEPSP amplitude = 228 ± 8% of control;n = 4). Similar to MF-LTP induced with either B-HFS or L-HFS, U0126 did not block forskolin-induced potentiation (Fig.6C; fEPSP amplitude = 208 ± 55% of control;n = 4). These data indicate that the ERK-signaling cascade is not necessary for forskolin-induced potentiation at mossy fiber synapses.
We also determined whether U0126 could reduce potentiation at MF synapses measured 3 hr after the induction of LTP with L-HFS. In control experiments, MF-LTP induced by L-HFS in the presence of DMSO exhibited LTP 3 hr after the final train of HFS (fEPSP amplitude = 182 ± 55% of control; n = 4). Slices given L-HFS in the presence of U0126 also exhibited LTP at the 3 hr time point (fEPSP amplitude = 169 ± 45% of control; _n_= 4). Similarly, U0126 had no effect on forskolin-induced potentiation measured 2 hr after the washout of forskolin (Fig. 6C). These data suggest that activation of the ERK-signaling cascade is not necessary for the late expression of either MF-LTP induced by L-HFS or forskolin-induced potentiation at mossy fiber synapses.
In the experiments shown in Figure 6, the MEK inhibitors were incubated with hippocampal slices for 10–15 min before delivery of the HFS. Although the duration of this incubation was sufficient for the inhibitors to block NMDA receptor-independent LTP in area CA1 (Figs. 3,5), it is possible that the 10–15 min incubation was insufficient for the inhibitors to block MEK in area CA3. To address this possibility, we incubated hippocampal slices with U0126 for 40 min (30 min before and 10 min after recording baseline synaptic responses) before and 40 min after delivery of L-HFS to the mossy fiber input to area CA3. Again, we observed that U0126 did not block MF-LTP measured 30 min after the final train of HFS (Fig.7A; fEPSP amplitude = 178 ± 25% of control; n = 4). These data are consistent with the idea that activation of ERK1/ERK2 is not necessary for the induction of MF-LTP.
To ensure that U0126 was effective in inhibiting MEK in area CA3, we determined whether activation of ERK1/ERK2 was necessary for associational/commissural LTP, an NMDA receptor-dependent form of LTP observed in area CA3 (Zalutsky and Nicoll, 1990). LTP-inducing stimulation delivered to the associational/commissural input in the presence of DMSO resulted in LTP measured 45 min after the final train of HFS (fEPSP slope = 155 ± 7% of control;n = 4). In contrast, delivery of LTP-inducing stimulation to associational/commissural input in the presence of U0126 resulted in the blockade of LTP in area CA3 measured 45 min after the final train of HFS (fEPSP slope = 105 ± 4% of control;n = 4). These data indicate that U0126 is effective in inhibiting MEK in area CA3 (also see Fig.8A,B) and also demonstrate that the ERK-signaling cascade, although not required for MF-LTP, is required for associational/commissural LTP in area CA3.
Fig. 8.
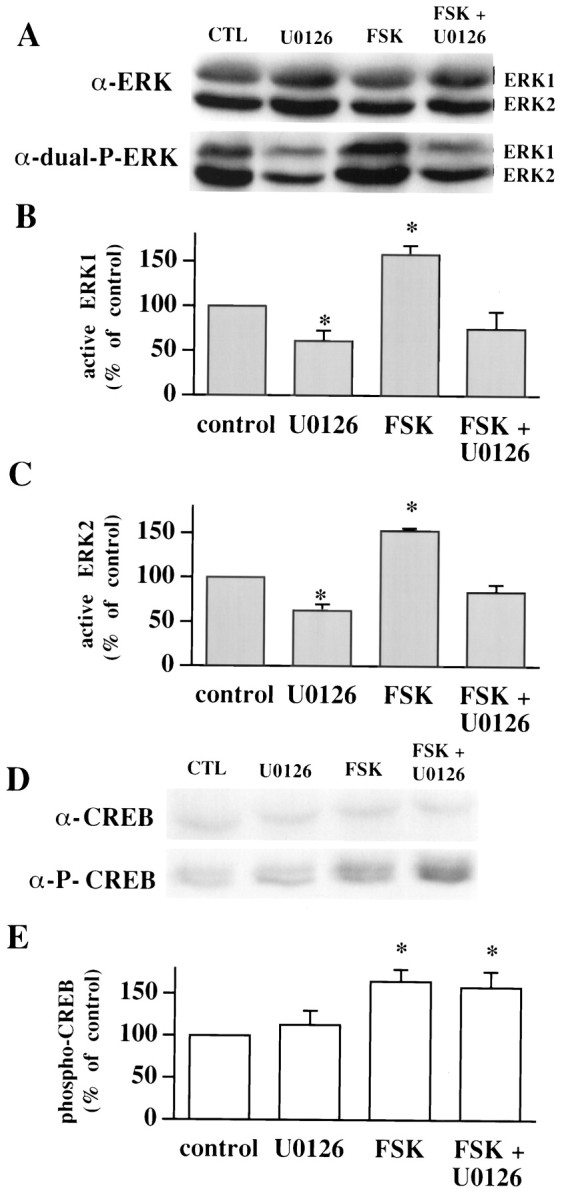
U0126 blocks forskolin-induced increases in active ERK1/ERK2 but not phospho-CREB in hippocampal area CA3.A, Representative ERK Western blots of area CA3 subregions from control slices (CTL), slices exposed to 20 μm U0126, slices exposed to 50 μmforskolin (FSK), and slices exposed to forskolin and U0126. B, Normalized active ERK1 immunoreactivity for each experimental condition (n = 4).C, Normalized active ERK2 immunoreactivity for each experimental condition (n = 4). D, Representative CREB Western blots of area CA3 subregions from control slices, slices exposed to 20 μm U0126, slices exposed to 50 μm forskolin, and slices exposed to forskolin and U0126. E, Normalized phospho-CREB immunoreactivity for each experimental condition (n = 4). Error bars in_B_, C, and E indicate SEM; * denotes statistical significance compared with control (p < 0.05 by paired Student's_t_ test with the Bonferroni correction factor).
In area CA1, the ERK cascade couples PKA to the phosphorylation of CREB (Roberson et al., 1999). We hypothesized that the reason we observed no effect of the MEK inhibitors on potentiation at MF synapses was that the activation of the ERK cascade is not coupled to the phosphorylation of CREB in area CA3. To test this hypothesis we incubated hippocampal slices with forskolin and examined the activation of ERK1/ERK2 as well as the phosphorylation of CREB in area CA3. Hippocampal slices incubated with 50 μm forskolin for 20 min exhibited an increase in active ERK1/ERK2 in area CA3 (Fig. 8A–C; ERK1 = 158 ± 10% of control; ERK2 = 153 ± 3% of control; n = 4). The forskolin-induced increase in active ERK1/ERK2 was blocked when slices were co-incubated with 20 μm U0126 (Fig. 8A–C; ERK1 = 75 ± 19% of control; ERK2 = 84 ± 8% of control; n = 4). In the same slices, forskolin also elicited an increase in CREB phosphorylation (Fig.8D,E; 165 ± 14% of control; _n_= 4). However, U0126 did not inhibit the forskolin-induced increase in CREB phosphorylation (Fig. 8D,E; 158 ± 17% of control; n = 4). These data suggest that the ERK cascade does not couple PKA to CREB phosphorylation in hippocampal area CA3.
DISCUSSION
The ERK-signaling cascade has been shown to be involved in synaptic plasticity and in memory formation in both invertebrates and mammals. In invertebrates, the ERK cascade has been shown to be necessary for long-term facilitation in Aplysia (Martin et al., 1997) and Pavlovian conditioning in Hermissenda(Crow et al., 1998). In mammals, the ERK cascade has been shown to be necessary for NMDA receptor-dependent LTP in area CA1 (English and Sweatt, 1997; Atkins et al., 1998) and the dentate gyrus of the hippocampus (Coogan et al., 1999). In addition, two NMDA receptor-independent forms of LTP in the dentate gyrus of the hippocampus, TEA-LTP and LTP induced by the metabotropic glutamate receptor agonist _S_-dihydrophenylglycine, were shown to be dependent on ERK (Coogan et al., 1999). Finally, the ERK cascade has been shown to be required for contextual and auditory fear conditioning (Atkins et al., 1998; Schafe et al., 1999), conditioned taste aversion (Berman et al., 1998), and long-term spatial learning (Blum et al., 1999).
The goal of this study was to examine the role of ERKs in NMDA receptor-independent forms of synaptic plasticity in the hippocampus. Toward this end, we used inhibitors of MEK, the dual specificity kinase that phosphorylates and activates ERK1/ERK2, to determine whether activation of ERK was necessary for LTP in area CA1 induced by either 200 Hz HFS or bath application of TEA. In addition, we investigated the role of ERKs in MF-LTP induced in area CA3 by either B-HFS, L-HFS, or bath application of forskolin.
Activation of ERK is necessary for NMDA receptor-independent LTP in area CA1
We observed that both 200 Hz-LTP and TEA-LTP in area CA1 were associated with an increase in active ERK1 and ERK2 (Figs. 1, 4). Incubation of slices with either PD 098059 or U1026 during the HFS prevented 200 Hz-LTP observed 45 min after the 200 Hz stimulation, but not the initial, slowly rising potentiation (Fig. 3). Similarly, incubation of slices with either MEK inhibitor during TEA application inhibited TEA-LTP observed 45 min after washout of TEA, but not the initial slowly rising potentiation (Fig. 5). These findings are similar to studies of NMDA receptor-dependent LTP (English and Sweatt, 1997), suggesting that regardless of the initial biochemical mechanisms triggered immediately after the LTP-inducing stimulation, activation of the ERK-signaling cascade during the induction of LTP is required for the expression of LTP in area CA1. In contrast to 200 Hz-LTP and TEA-LTP, NMDA receptor-dependent LTP appears to be associated with the activation of only ERK2 and not of ERK1 (English and Sweatt, 1996). Because both 200 Hz-LTP (Grover and Teyler, 1990) and TEA-LTP (Aniksztejn and Ben-Ari, 1991; Huang and Malenka, 1993;Powell et al., 1994; but see Huber et al., 1995a) are dependent on activation of voltage-gated Ca2+ channels, differences in either the subcellular localization of Ca2+ influx or intracellular Ca2+ concentrations between NMDA receptor-dependent and -independent LTP might account for the differential activation of the ERK isoforms.
Previous studies have shown that membrane depolarization stimulates the ERK-signaling cascade in pheochromocytoma 12 cells (Rosen et al., 1994). In hippocampal slices, depolarization induced with a brief pulse of KCl, shown previously to result in LTP (Fleck et al., 1992), results in activation of ERK2 (Baron et al., 1996). Blockade of voltage-gated Ca2+ channels by nifedipine also blocked depolarization-induced increases in active ERK (Rosen et al., 1994;Baron et al., 1996). In agreement with these observations, we observed that nifedipine blocked the increase in active ERK1/ERK2 associated with both 200 Hz-LTP and TEA-LTP. Thus, influx of Ca2+ through voltage-gated Ca2+ channels is the initial trigger for the activation of ERK signaling during 200 Hz-LTP and TEA-LTP in area CA1.
Compared with NMDA receptor-dependent LTP in area CA1, little is known about the signaling cascades that link the activation of voltage-gated Ca2+ channels to the activation of ERK after induction of NMDA receptor-independent LTP in area CA1. 200 Hz-LTP has been reported to be reduced by tyrosine kinase inhibitors (Cavus and Teyler, 1996) but not by H-7, a broad spectrum serine/threonine kinase inhibitor (Grover and Teyler, 1995; Cavus and Teyler, 1996). In contrast, TEA-LTP has been reported to be blocked by K-252a, a broad spectrum serine/threonine kinase inhibitor, as well as by KN-62, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Huber et al., 1995b). In addition, TEA-LTP is associated with an increase in autonomous PKC activity (Powell et al., 1994). Thus, it is possible that 200 Hz-LTP and TEA-LTP have different upstream signaling cascades capable of activating the ERK cascade. For example, the activation of ERK that accompanies 200 Hz-LTP might be dependent on a neurotrophic receptor/Raf-1/MEK-signaling cascade. In agreement with this idea, neurotrophins, which bind to receptor tyrosine kinases, can either potentiate synaptic transmission in area CA1 (Kang and Schuman, 1995) or enhance the response of hippocampal synapses to tetanic stimulation (Figurov et al., 1996). Voltage-gated Ca2+ channels appear to play a role in neurotrophin-induced potentiation because nifedipine can attenuate this potentiation (Kang et al., 1995). Taken together, these data are consistent with the possibility that 200 Hz-LTP is associated with activation of a neurotrophic receptor/Raf-1/MEK/ERK-signaling cascade. It will be of interest to determine whether such a cascade is activated by 200 Hz-LTP and whether a CaMKII/PKC/Raf-1/MEK/ERK-signaling cascade is activated by TEA-LTP.
The downstream effectors of ERK in either 200 Hz-LTP or TEA-LTP are unknown. However, recent studies suggest that one effector may be CREB. A variety of neurotransmitter receptors have been shown to be coupled to ERK activation in area CA1 via either PKC or PKA (Roberson et al., 1999). Activation of either PKC or PKA in area CA1 increases the phosphorylation of CREB with activation of ERK acting as a critical intermediary (Roberson et al., 1999). Because TEA-LTP in area CA1 has been shown to be accompanied by increased autonomous PKC activity (Powell et al., 1994), it is possible that PKC could be an upstream transducer for ERK activation and the subsequent phosphorylation of CREB. This possibility and downstream effectors of ERK in 200 Hz-LTP remain to be studied.
The ERK-signaling cascade is not required for MF-LTP in area CA3
In contrast to other forms of synaptic plasticity in invertebrates and mammals, we found that MF-LTP in area CA3, induced by either B-HFS, L-HFS, or forskolin, did not require ERK signaling (Fig. 6). In area CA1, activation of PKA by forskolin elicits an ERK-dependent increase in CREB phosphorylation (Roberson et al., 1999), which has been hypothesized to be necessary for PKA regulation of gene expression in LTP (Impey et al., 1998; Roberson et al., 1999). Forskolin has been shown to elicit an increase in CREB phosphorylation in area CA3 (Fig.8D,E) (Roberson et al., 1999). However, in contrast to the findings in area CA1, the MEK inhibitor U0126 did not prevent forskolin-induced phosphorylation of CREB (Fig.8D,E). Thus, ERK activation is not likely to be necessary for forskolin-induced CREB phosphorylation in area CA3.
Application of forskolin to hippocampal slices completely occludes MF-LTP induced by L-HFS (Weisskopf et al., 1994), which suggests that these two forms of MF-LTP share similar biochemical signaling cascades. The experimental conditions that result in the forskolin-induced enhancement in CREB phosphorylation (Fig. 8D,E) also result in the potentiation of synaptic transmission in area CA3 (Fig.6C). Therefore, it seems likely that MF-LTP induced by L-HFS is accompanied by increased CREB phosphorylation. CREB can be phosphorylated directly by both PKA and Ca2+/calmodulin-dependent kinases (Dash et al., 1991). Furthermore, MF-LTP induced by L-HFS is dependent on PKA (Huang et al., 1994; Weisskopf et al., 1994; Yeckel et al., 1999), and MF-LTP is blocked in mice with genetic ablations of either the Cβ1 or RIβ subunits of PKA (Huang et al., 1995). Taken together, these data suggest that MF-LTP induced by either forskolin or L-HFS may be dependent on the phosphorylation of CREB by PKA, bypassing the need for the ERK/RSK2/CREB-signaling cascade that is likely to be necessary for NMDA receptor-dependent LTP in area CA1 (Roberson et al., 1999).
Recently it was reported that increased phosphorylation of ERK was restricted to CA1/CA2 neurons after behavioral training in a spatial memory task (Blum et al., 1999). Interestingly, in studies with genetically altered mice, it was shown that mice with deficient LTP in area CA1 show spatial memory deficits, whereas mice with deficient LTP in either area CA3 or the dentate gyrus do not (Huang et al., 1995;Nosten-Bertrand et al., 1996). Herein, we have shown that NMDA receptor-independent LTP in area CA1 is dependent on the ERK-signaling cascade, whereas NMDA receptor-independent LTP in area CA3 is not. Thus, our data are consistent with the idea that ERK-dependent LTP in area CA1 is more critical for spatial memory than is MF-LTP in area CA3.
In conclusion, we have shown that two forms of NMDA receptor-independent LTP in hippocampal area CA1 are associated with increases in active ERK that are necessary for the induction of these forms of synaptic plasticity. In contrast, we found that the ERK-signaling cascade is not required for MF-LTP in hippocampal area CA3, regardless of the induction paradigm. Future studies will be necessary to delineate the signal transduction pathway and downstream effectors of ERK in area CA1 and the signaling cascade(s) responsible for CREB phosphorylation in area CA3.
Footnotes
This work was supported by National Institutes of Health Grants NS34007 (E.K.) and NS24288 (G.B.). N.N.U. was supported by a Howard Hughes Medical Institute Predoctoral Fellowship. We thank Dr. Edda Thiels for helpful comments on this manuscript. We also thank Drs. A. Christine Tabaka, Jia-Sheng Yan, and Christopher Teleha for synthesizing U0126.
B.I.K. and N.N.U. contributed equally to this work.
Correspondence should be addressed to Dr. Eric Klann, Department of Neuroscience, University of Pittsburgh, 446 Crawford Hall, Pittsburgh, PA 15260. E-mail: klann@brain.bns.pitt.edu.
Dr. Urban's present address: Max-Planck Institut für medizinische Forschung, Abteilung Zellphysiologie, 29 Jahnstrasse, D69120 Heidelberg, Germany.
REFERENCES
- 1.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 2.Aniksztejn L, Ben-Ari Y. Novel form of long-term potentiation produced by a K+ channel blocker in the hippocampus. Nature. 1991;349:67–69. doi: 10.1038/349067a0. [DOI] [PubMed] [Google Scholar]
- 3.Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- 4.Baron C, Benes C, Van Tan H, Fagard R, Roisin M-P. Potassium chloride pulse enhances mitogen-activated protein kinase activity in rat hippocampal slices. J Neurochem. 1996;66:1005–1010. doi: 10.1046/j.1471-4159.1996.66031005.x. [DOI] [PubMed] [Google Scholar]
- 5.Berman DE, Hazvi S, Rosenblum K, Seger R, Dudai Y. Specific and differential activation of mitogen-activated protein kinases by unfamiliar taste in the insular cortex of the behaving rat. J Neurosci. 1998;18:10037–10044. doi: 10.1523/JNEUROSCI.18-23-10037.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blum S, Moore A, Adams F, Dash P. A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J Neurosci. 1999;19:3535–3544. doi: 10.1523/JNEUROSCI.19-09-03535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradford MM. Rapid and sensitive method for quantification of microgram quantities of protein using the principle of protein-dye binding. Anal Biochem. 1976;72:248–252. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 8.Castillo PE, Weisskopf MG, Nicoll RA. The role of calcium channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron. 1994;12:261–269. doi: 10.1016/0896-6273(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 9.Cavus I, Teyler T. Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. J Neurophysiol. 1996;76:3038–3047. doi: 10.1152/jn.1996.76.5.3038. [DOI] [PubMed] [Google Scholar]
- 10.Collingridge GL, Kehl SJ, McClennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol (Lond) 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coogan AN, O'Leary DM, O'Connor JJ. P42/44 MAP kinase inhibitor PD98059 attenuates multiple forms of synaptic plasticity in rat dentate gyrus in vitro. J Neurophysiol. 1999;81:103–110. doi: 10.1152/jn.1999.81.1.103. [DOI] [PubMed] [Google Scholar]
- 12.Crow T, Xue-Bian JJ, Siddiqi V, Kang Y, Neary JT. Phosphorylation of mitogen- activated protein kinase by one-trial and multi-trial classical conditioning. J Neurosci. 1998;18:3480–3487. doi: 10.1523/JNEUROSCI.18-09-03480.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dash PK, Karl KA, Colicos MA, Prywes R, Kandel ER. cAMP response element-binding protein is activated by Ca2+/calmodulin as well as cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1991;88:5061–5065. doi: 10.1073/pnas.88.11.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.English JE, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long-term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- 15.English JE, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long-term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 16.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 17.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 18.Fleck MW, Palmer AM, Barrionuevo G. Potassium-induced long-term potentiation in rat hippocampal slices. Brain Res. 1992;580:100–105. doi: 10.1016/0006-8993(92)90931-x. [DOI] [PubMed] [Google Scholar]
- 19.Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- 20.Grover LM, Teyler TJ. Different mechanisms may be required for maintenance of NMDA receptor-dependent and independent forms of long-term potentiation. Synapse. 1995;19:121–133. doi: 10.1002/syn.890190208. [DOI] [PubMed] [Google Scholar]
- 21.Harris EW, Cotman CW. Long-term potentiation of guinea pig mossy fiber responses is not blocked by N-methyl-d-aspartate antagonists. Neurosci Lett. 1986;70:132–137. doi: 10.1016/0304-3940(86)90451-9. [DOI] [PubMed] [Google Scholar]
- 22.Huang Y-Y, Malenka RC. Examination of TEA-induced synaptic enhancement in area CA1 of the hippocampus: the role of voltage-dependent Ca2+ channels in the induction of LTP. J Neurosci. 1993;13:568–576. doi: 10.1523/JNEUROSCI.13-02-00568.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang YY, Li XC, Kandel ER. cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell. 1994;79:69–79. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- 24.Huang YY, Kandel ER, Varshavsky L, Brandon EP, Qi M, Idzera RL, McKnight GS, Bourtchuladze R. A genetic test of the effects of mutations in PKA on mossy fiber LTP and its relation to spatial and contextual learning. Cell. 1995;83:1211–1222. doi: 10.1016/0092-8674(95)90146-9. [DOI] [PubMed] [Google Scholar]
- 25.Huber KH, Mauk MD, Kelly PT. Distinct LTP induction mechanisms: contribution of NMDA receptors and voltage-dependent calcium channels. J Neurophysiol. 1995a;73:270–279. doi: 10.1152/jn.1995.73.1.270. [DOI] [PubMed] [Google Scholar]
- 26.Huber KH, Mauk MD, Kelly PT. LTP induced by activation of voltage-dependent Ca2+ channels requires protein kinase activity. NeuroReport. 1995b;6:1281–1284. doi: 10.1097/00001756-199506090-00013. [DOI] [PubMed] [Google Scholar]
- 27.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DS. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 28.Jaffe D, Johnston D. Induction of long-term potentiation at hippocampal mossy fiber synapses follows a Hebbian rule. J Neurophysiol. 1990;64:948–960. doi: 10.1152/jn.1990.64.3.948. [DOI] [PubMed] [Google Scholar]
- 29.Kang H, Schuman E. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 30.Kang H, Welcher AA, Shelton D, Schuman EM. Characterization of neurotrophin-induced synaptic potentiation in area CA1 of the rat hippocampus. Soc Neurosci Abstr. 1995;21:599. [Google Scholar]
- 31.Kapur A, Yeckel MF, Gray R, Johnston D. L-type calcium channels are required for one form of hippocampal mossy fiber LTP. J Neurophysiol. 1998;79:2181–2190. doi: 10.1152/jn.1998.79.4.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klann E. Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. J Neurophysiol. 1998;80:452–457. doi: 10.1152/jn.1998.80.1.452. [DOI] [PubMed] [Google Scholar]
- 33.Klann E, Chen S-J, Sweatt JD. Persistent protein kinase activation in the maintenance phase of long-term potentiation. J Biol Chem. 1991;266:24253–24256. [PubMed] [Google Scholar]
- 34.Lynch G, Larson J, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305:719–721. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- 35.Malinow R, Madison DV, Tsien RW. Persistent protein kinase activity underlying long-term potentiation. Nature. 1988;335:820–824. doi: 10.1038/335820a0. [DOI] [PubMed] [Google Scholar]
- 36.Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- 37.Nosten-Bertrand M, Errington ML, Murphy KP, Tokugawa Y, Barboni E, Kozlova E, Michalovich D, Morris RG, Silver J, Stewart CL, Bliss TV, Morris RJ. Normal spatial learning despite regional inhibition of LTP in mice lacking Thy-1. Nature. 1996;379:826–829. doi: 10.1038/379826a0. [DOI] [PubMed] [Google Scholar]
- 38.Powell CM, Johnston D, Sweatt JD. Autonomously active protein kinase C in the maintenance of N-methyl-d-aspartate receptor-independent long-term potentiation. J Biol Chem. 1994;269:27958–27963. [PMC free article] [PubMed] [Google Scholar]
- 39.Roberson ED, English JE, Sweatt JD. A biochemist's view of long-term potentiation. Learn Mem. 1996;3:1–24. doi: 10.1101/lm.3.1.1. [DOI] [PubMed] [Google Scholar]
- 40.Roberson ED, English JE, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosen LB, Ginty DD, Weber MJ, Greenberg ME. Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron. 1994;12:1207–1221. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 42.Schafe GE, Nadel NV, Sullivan GM, Harris A, LeDoux JE. Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn Mem. 1999;6:97–110. [PMC free article] [PubMed] [Google Scholar]
- 43.Seamon KB, Daly JW. Forskolin: its biological and chemical properties. Adv Cyclic Nucleotide Protein Phosphorylation Res. 1986;20:1–150. [PubMed] [Google Scholar]
- 44.Urban NN, Barrionuevo G. Induction of Hebbian and non-Hebbian mossy fiber long-term potentiation by distinct patterns of high-frequency stimulation. J Neurosci. 1996;16:4293–4299. doi: 10.1523/JNEUROSCI.16-13-04293.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J-H, Feng D-P. Postsynaptic protein kinase C essential to induction and maintenance of long-term potentiation in the hippocampal CA1 region. Proc Natl Acad Sci USA. 1992;89:2576–2580. doi: 10.1073/pnas.89.7.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- 47.Yeckel MF, Kapur A, Johnston D. Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat Neurosci. 1999;2:625–633. doi: 10.1038/10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zalutsky RA, Nicoll RA. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]