Cerebral Ischemia and Seizures Induce Tyrosine Phosphorylation of PYK2 in Neurons and Microglial Cells (original) (raw)
Abstract
The nonreceptor tyrosine kinase PYK2 represents a stress-sensitive mediator of c-Jun N-terminal kinase and p38 mitogen-activated protein kinase (MAPK) signaling pathways in many cell types. In the present study, we assessed the tyrosine phosphorylation of PYK2 under normal and pathological conditions in the CNS. We generated a polyclonal antibody that selectively recognizes tyrosine-phosphorylated PYK2 at its major autophosphorylation site. By using this antibody, we demonstrate that the phosphorylation profile of PYK2 after focal cerebral ischemia is biphasic. The first phase occurs within 1 hr, when most of the phospho-PYK2 immunoreactivity was observed in cortical neurons, whereas 24–72 hr after ischemia, a striking induction of phospho-PYK2 immunoreactivity was evident in microglia around the necrotic infarcted area. Double-immunostaining analysis using both anti-phospho-PYK2 antibody and antibody against the double-phosphorylated active form of p38MAPK revealed that the two phosphorylated protein kinases exhibit strikingly similar distribution patterns after ischemia. A short time after ischemia, phosphorylation of p38MAPK was evident in the cortical neurons as demonstrated by both immunohistochemistry and immunoblotting analysis, whereas 24–72 hr after ischemia, phospho-p38MAPK was found in activated microglia and colocalized with phospho-PYK2. In contrast to cortical neurons, basal phospho-PYK2 immunoreactivity was observed in hippocampal pyramidal neurons, which was markedly decreased after kainate acid-induced status epilepticus. However, 24 hr after the epileptic onset, a pronounced upregulation of PYK2 and phospho-PYK2 immunoreactivities was evident in microglial cells, as demonstrated by double-immunostaining with the microglial marker OX42. These results provide, for the first time, in situ localization of tyrosine-phosphorylated PYK2 in neuronal stress pathways in the adult rat brain and are consistent with the role of PYK2 as an upstream regulator of p38MAPK signaling cascades in response to stress signals.
Keywords: PYK2, tyrosine phosphorylation, p38MAPK, cerebral ischemia, epilepsy, stress signals, microglia
Many tyrosine kinases stimulate signaling cascades that lead to activation of mitogen-activated protein kinase (MAPK) pathways. In vertebrates, there are at least three different MAPKs that convey distinct biological responses: extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38MAPK (Lewis et al., 1998). Whereas ERK signaling is generally involved in the control of cell proliferation and differentiation, JNK and p38MAPK signaling pathways mediate responses to various forms of cellular stress (Pettmann and Henderson, 1998). In the nervous system, the MAPK pathway, mediated by ERK activation, may function to regulate synaptic transmission through long-term changes in protein synthesis and gene expression in the nucleus. On the other hand, activation of the JNK and p38MAPK pathways is closely related to apoptosis and is associated with a variety of pathological events in the CNS. In the case of Alzheimer's disease for example, the immunoreactivity for c-Jun is elevated in association with the pathological state of the neurons (Cotman, 1995). Therefore, signaling pathways that induce activation of MAPKs may play an important role in the physiological and pathological responses of the nervous system.
The nonreceptor tyrosine kinase PYK2 acts as an upstream regulator of MAPK signaling pathways in response to diverse cellular stimuli. In the nervous system, it is highly sensitive to depolarization and can link calcium influx events to ERK activation (Lev et al., 1995). G-protein-coupled receptors, such as those of bradykinin or lysophosphatidic acid, stimulate ERK activation by a mechanism that involves PYK2 autophosphorylation, association with the tyrosine kinase Src, recruitment of the Grb2/Sos complex, and subsequent activation of the Ras/ERK signaling pathway (Dikic et al., 1996). On the other hand, stress signals, such as UV irradiation or hyperosmotic shock, inflammatory cytokines, or activation of the angiotensin II receptor, induce tyrosine phosphorylation of PYK2, activation of the small GTP-binding proteins Cdc42 and Rac, and subsequently, activation of JNK (Tokiwa et al., 1996; Yu et al., 1996). PYK2 was also implicated as an upstream regulator of p38MAPK in response to certain cytotoxic agents, such as methylmethane sulfonate and hyperosmolarity (Pandey et al., 1999).
In addition to the nervous system, PYK2 is highly expressed in hematopoietic and epithelial cells. In many hematopoietic cells, PYK2 is tyrosine-phosphorylated in response to integrin engagement, is localized in focal adhesions, and is associated with cytoskeletal proteins such as paxillin (Gismondi et al., 1997). The role of PYK2 in the integrin signaling pathway is not understood at present, although many studies describe the role of its most related kinase, focal adhesion kinase (FAK), in the integrin-mediated signaling cascade. FAK was shown to be required for integrin-mediated cell survival and proliferation and was implicated in cell migration (Hanks and Polte, 1997; Schlaepfer and Hunter, 1998). However PYK2, but not FAK, is activated in response to stress signals and appears to be capable of triggering JNK phosphorylation (Girault et al., 1999).
In the present study, we assessed the tyrosine phosphorylation of PYK2 in stress-mediated pathways in the CNS. By using antibody that specifically recognizes tyrosine-phosphorylated PYK2, we provide evidence that PYK2 is rapidly phosphorylated in cortical neurons after cerebral ischemia. Tyrosine-phosphorylated PYK2 was upregulated in activated microglia after focal cerebral ischemia and epilepsy. Furthermore, we demonstrate that tyrosine-phosphorylated PYK2 is colocalized with activated p38MAPK, in both neuron and microglia, after ischemia. We propose that PYK2 is a stress-sensitive mediator of the p38MAPK pathway in vivo after cerebral ischemia.
MATERIALS AND METHODS
Animals. All animal experiments were performed in conformity with the institutional and National Institutes of Health guidelines. Male Wistar rats (weight of 250–300 gm) were obtained from the Animal Breeding Unit of the Weizmann Institute of Science and maintained under controlled environmental conditions (air-conditioned quarters, ambient temperature of 22°C, illumination between 5:00 A.M. and 7:00 P.M., and relative humidity 45–55%). Food and water were available ad libitum.
Middle cerebral artery occlusion. A focal cerebral ischemia model was applied under chloral hydrate (350 mg/kg body weight, i.p.) anesthesia by intraluminal occlusion of the right middle cerebral artery (MCA) as described previously (Longa et al., 1989; Nakano et al., 1990). The right common, internal, and external carotid arteries were exposed surgically, extracranial branches of the internal and external carotid arteries were occluded, and the right common carotid artery was clamped. A silicon-coated 4/0 nylon thread was introduced into the lumen of the internal carotid artery at the level at which the MCA branches out. The clip on the common carotid artery was released. Rats were divided into two general groups: short-time ischemia group and long-time ischemia group. Rats of the short-time ischemia group were killed at 15 min (n = 5), 30 min (n = 6), and 1 hr (n = 5) after MCA occlusion (MCAO). Rats of the long-time ischemia group were reperfused at 1 hr after MCAO by removing the filament from the artery and were then killed at 6 (n = 5), 24 (n = 5), 48 (n = 5) or 72 (n = 6) hr. Sham rats, which received the same surgical procedure except the introduction of the nylon thread into the artery, were killed at 30 min (n = 2) or 48 hr (n = 2) after surgery.
Kainate acid treatment. Rats were injected intraperitoneally with kainate acid (KA) (12 mg/kg body weight; Sigma). Rats showing typical KA-induced limbic seizures for 1–1.5 hr upon recovery were killed at 6 (n = 3), 24 (n = 3) or 72 (n = 4) hr after KA administration. Control rats (n = 2) were injected with saline and killed 24 hr later.
Antibodies, immunoprecipitation, and immunoblotting analysis. Anti-phospho-PYK2 antibody was raised in rabbits immunized with a keyhole limpet hemocyanin-conjugated phosphopeptide (CSIESDIYpAEIPDET) corresponding to amino acids 395–409 of PYK2. Three sequential steps of affinity purification were performed using a nonphosphorylated peptide coupled to Sepharose 4B, followed by a phosphotyrosine (PTYR) column, and subsequently by an affinity column containing the phosphorylated peptide. The specificity of the anti-phospho-PYK2 antibody was analyzed by Western blotting, immunoprecipitation, or immunofluorescence analysis. Anti-PYK2 antibody was raised in rabbits immunized with a maltose-binding protein fusion protein containing amino acids 285–455 of PYK2 and was affinity-purified by using the cognate antigen immobilized on Sepharose beads. Anti-phospho-p38MAPK and anti-phospho-ERK monoclonal antibodies were obtained from Sigma. Anti-phospho-p38MAPK antibody was directed against the highly conserved Thr-Gly-Tyr motif corresponding to the phosphorylated p38MAPK kinase activation loop. Anti-OX42 monoclonal antibody was purchased from Serotec (Oxford, UK). This antibody recognizes the complement receptor type 3 (CR3) and specifically labels the plasma membrane of the microglial cells (Ling et al., 1990). Anti-PTYR antibody was purchased from Upstate Biotechnology (Lake Placid, NY). Anti-phospho-JNK monoclonal antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-p38MAPK polyclonal antibody was kindly provided by Dr. R. Zeger (Department of Bioregulation, Weizmann Institute of Science, Rehovot, Israel). Immunoprecipitation and immunoblotting were performed as described previously (Lev et al., 1995).
Immunohistochemistry. Rats were anesthetized as described above and perfused transcardially with heparinized saline, followed by 4% paraformaldehyde in 0.1 m phosphate buffer for 45 min. Brains were removed and post-fixed for 48–72 hr at 4°C with a fixative solution containing 1% paraformaldehyde and 20% sucrose. Sections (50 μm) were cut on a microtome and stored in PBS at 4°C as free-floating sections. Sections were incubated with 3% H2O2 in methanol for 10 min to quench endogenous peroxidase activity and then incubated in blocking buffer solution (1% glycine, 10% goat serum, 2% bovine serum albumin, and 0.1% Triton X-100 in PBS) for 2 hr at 22°C. Sections were incubated overnight at 4°C with anti-PYK2 antibody (3 μg/ml), anti-phospho-PYK2 antibody (1.25 μg/ml), or anti-OX42 antibody (2.5 μg/ml) in blocking buffer. After washing with PBS–0.1% Triton X-100, immunoperoxidase histochemistry was performed using the ABC method (Vector Laboratories, Burlingame, CA). The sections were washed three times with PBS–0.1% Triton X-100 and mounted onto gelatin-coated slides, dried overnight at room temperature, dehydrated, and coverslipped.
Confocal immunofluorescence microscopy. Sections were permeabilized and blocked in blocking buffer as described above. Anti-PYK2 (3 μg/ml) or anti-phospho-PYK2 (1.25 μg/ml) polyclonal antibodies were incubated overnight at 4°C together with either anti-PTYR (0.12 μg/ml), anti-OX42 (2.5 μg/ml), anti-p38MAPK (1 μg/ml), anti-phospho-ERK (2 μg/ml), or anti-phospho-JNK (2.5 μg/ml) monoclonal antibodies. After washing with PBS–Triton X-100, the sections were incubated with a mixture of Alexa donkey anti-mouse IgG (1 μg/ml; Molecular Probes, Eugene, OR) and Cy3-conjugated goat anti-rabbit IgG (2.0 μg/ml; The Jackson Laboratory, Bar Harbor, ME). The sections were washed with PBS and coverslipped with fluorescence mounting medium. Sections were observed under a confocal laser microscope (Zeiss 510; Zeiss, Oberkochen, Germany) equipped with filters for fluorescein and Cy3 epifluorescence. Both 488 and 543 nm laser lines were used for excitation, and 505–530 nm bandpass and 560 nm long-pass filters were used for emission. Image analysis was performed using the standard system operating software provided with the Zeiss 510 microscope (version 2.01).
RESULTS
Specificity of anti-phospho-PYK2 antibody
PYK2 is phosphorylated at multiple sites, including tyrosine 402, 579, 580, and 881 (Schlaepfer and Hunter, 1998). Tyrosine 402 appears to be the major autophosphorylation site and was shown to mediate the interaction with Src homology 2-containing proteins, including the tyrosine kinases Src and Lck (Dikic et al., 1996; Berg and Ostergaard, 1997). To probe tyrosine-phosphorylated PYK2 in situ, we generated anti-phosphopeptide antibody that selectively recognizes tyrosine-phosphorylated PYK2 at its major autophosphorylation site, tyrosine 402. The specificity of this antibody was assessed by Western blotting, immunoprecipitation, and immunofluorescence analysis. The results shown in Figure1 demonstrate the specificity of this antibody by Western blot analysis. We have shown previously that PYK2 is tyrosine-phosphorylated in response to membrane depolarization (Lev et al., 1995). Therefore, PYK2 was immunoprecipitated from either quiescent PC12 cells or cells that were treated with a high concentration of KCl (75 mm) to induce membrane depolarization. PYK2 immunoprecipitates were immunoblotted with anti-PYK2, anti-PTYR, or anti-phospho-PYK2 antibodies. As shown in Figure 1, anti-PYK2 antibody recognizes PYK2 immunoprecipitated from both quiescent and KCl-stimulated PC12 cells, whereas anti-PTYR antibody recognizes only PYK2 immunoprecipitated from KCl-stimulated PC12 cells. Several tyrosine-phosphorylated proteins that coimmunoprecipitate with PYK2 in response to membrane depolarization are shown in the anti-PTYR immunoblot. In contrast, anti-phospho-PYK2 antibody exclusively recognizes the tyrosine-phosphorylated PYK2, thus demonstrating that this antibody is specific for tyrosine-phosphorylated PYK2. It does not react with any other tyrosine-phosphorylated protein, including the most related kinase FAK, and it recognizes a single protein on a Western blot of brain lysate. Preincubation of anti-phospho-PYK2 antibody with the phosphopeptide immunogen (300 nm) virtually abolished its immunoreactivity in immunoprecipitation, immunoblotting, and immunofluorescence analysis (data not shown).
Fig. 1.

Specificity of anti-phospho PYK2 antibody by Western blot analysis. Shown are PYK2 immunoprecipitates from either quiescent PC12 cells or cells that were treated with 75 mmKCl for 10 min at 37°C. PYK2 immunoprecipitates were resolved on SDS-PAGE and analyzed by immunoblotting using anti-PYK2, anti-PTYR, or anti-phospho-PYK2 antibodies. Anti-PYK2 antibody recognizes PYK2 immunoprecipitated from both quiescent and KCl-stimulated PC12 cells (left), whereas anti-PTYR antibody recognizes PYK2, as well as several coimmunoprecipitated proteins only in response to KCl treatment. On the other hand, anti-phospho-PYK2 antibody recognizes only PYK2 immunoprecipitated from cells that were treated with KCl. No other tyrosine-phosphorylated proteins that coimmunoprecipitate with PYK2 are recognized by anti-phospho-PYK2 antibody (middle vs right).
Immunolocalization of phosphorylated PYK2 within 1 hr after focal cerebral ischemia
It was shown previously that PYK2 is tyrosine-phosphorylated in response to diverse stress signals, such as UV irradiation, hyperosmotic shock, tumor necrosis factor α (TNFα), and β-amyloid peptide (Tokiwa et al., 1996; Combs et al., 1999). Although these studies demonstrate phosphorylation of PYK2 in cultured cells, tyrosine phosphorylation of PYK2 in response to pathological events in the brain has not yet been described. Focal cerebral ischemia provides a suitable model for the pathophysiological investigation of ischemic stroke (Kawamura et al., 1991). We have adapted this model to determine the time course and localization of phosphorylated PYK2 by immunohistochemical analysis. Focal cerebral ischemia was induced by MCAO using an intraluminal thread technique (Longa et al., 1989), as described in Material and Methods. Brain sections of animals killed at 15 min, 30 min, or 1 hr after MCAO were immunostained with anti-PYK2, anti-phospho-PYK2, or anti-PTYR antibodies. The results shown in Figure2 demonstrate that phospho-PYK2 and PTYR immunoreactivities were barely detectable in nonischemic brain sections, whereas strong PYK2 immunoreactivity was evident in the parietal cortex, especially in cortical lamina V. It is noteworthy that some cells localized near the outer margin of the cortex were faintly immunostained with anti-PTYR but not with anti-phospho-PYK2 antibodies (data not shown). These cells, which exhibit typical morphological characteristics of resting microglia, were immunostained in the contralateral side as well, suggesting that tyrosine-phosphorylated proteins are localized in resting microglia. These results are consistent with previous studies demonstrating phosphotyrosine immunoreactivity in the ramified microglia in the rat brain (Tillotson and Wood, 1989; Karp et al., 1994). However, 15 min after ischemia, the immunoreactivity of phospho-PYK2, as well as of phosphotyrosine, was already increased. Intense phospho-PYK2 and PTYR immunoreactivities were detected and colocalized in cortical neurons in lamina V. Maximum immunoreactivity of phospho-PYK2 was observed at 30 min and was slightly decreased 30 min later. Intense immunoreactivity of PTYR was observed 1 hr after ischemia and was evident in both neurons and microglial cells (Fig. 2). Interestingly, at this time (1 hr after ischemia), no detectable immunoreactivity of either phospho-PYK2 or PYK2 was observed in microglial cells, suggesting that the level of PYK2 expression and its tyrosine phosphorylation are quite low in resting microglia. These results also demonstrate the specificity of the anti-phospho-PYK2 antibody toward tyrosine-phosphorylated PYK2. This specificity was further confirmed by preincubation of anti-phospho-PYK2 or anti-PTYR with the phosphopeptide immunogen, which virtually abolished the immunoreactivity of phospho-PYK2 but partially inhibited the immunoreactivity of anti-PTYR (data not shown).
Fig. 2.
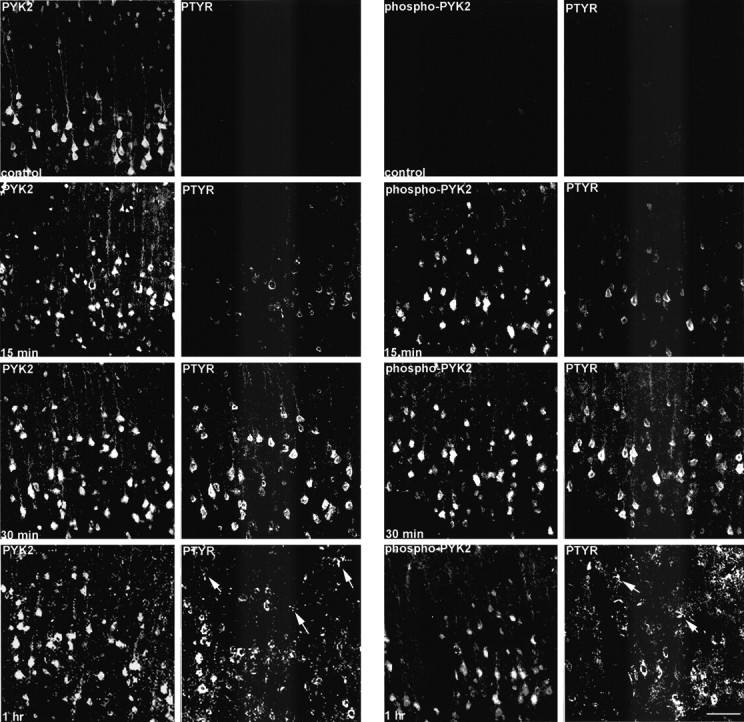
Enhanced phospho-PYK2 immunoreactivity in cortical neurons after focal cerebral ischemia. Coronal brain sections (50 μm) of animals killed at the indicated time points after MCAO were double-immunostained with anti-PTYR monoclonal antibody and with either anti-PYK2 (left) or anti-phospho-PYK2 (right) polyclonal antibodies. Shown are confocal images of lamina V neurons within the infarct cortex. In the control sections, a strong PYK2 immunoreactivity was visualized, whereas no detectable immunoreactivity of either phospho-PYK2 or PTYR was detected. At 15 min after ischemia, intense phospho-PYK2 and PTYR immunoreactivities were observed in the neurons, which were further increased at 30 min and slightly decreased at 1 hr. PTYR-positive microglial cells (arrows) were observed 1 hr after MCAO. Scale bar, 50 μm.
Increase in phospho-PYK2 immunoreactivity in microglia after cerebral ischemia
To further characterize the distribution of phospho-PYK2 after MCAO, we immunostained brain sections of animals killed at 6, 24, 48, or 72 hr after cerebral ischemia with anti-phospho-PYK2 antibody. One day after ischemia, a global decrease of phospho-PYK2 immunoreactivity in cortical neurons was observed, whereas a striking increase of phospho-PYK2 immunoreactivity was seen in the striatum. The immunoreactivity of phospho-PYK2 was further enhanced over 48–72 hr after ischemia and localized in cells that exhibit morphological characteristics of microglia. Microglia can be distinguished from other glia of the CNS by immunostaining with the monoclonal antibody OX42 against CR3 (Flaris et al., 1993). We used this antibody to assess the distribution and the morphological changes of microglia before and after cerebral ischemia. In nonischemic sections, OX42 immunoreactivity was detected in cells that exhibit highly arborized cell processes typical for resting microglia (McRae et al., 1998), whereas no detectable immunoreactivity of either phospho-PYK2 (Fig.3) or PYK2 (data not shown) was observed in the control striatum. The phospho-PYK2-positive cells shown at 6 hr after ischemia exhibit very similar morphology to OX42-positive cells. At 24–72 hr after ischemia, dramatic morphological changes were observed in the microglia, as demonstrated by OX42 immunostaining. Shortening of cell processes and loss of secondary arborizations were detected 24–48 hr after ischemia, whereas a complete loss of cellular processes and rounding of the cell body typical for phagocytes were visualized 72 hr after ischemia. These morphological changes are very typical for activated microglia (Gehrmann et al., 1995). Thus, activated microglia, but not resting or ramified ones, can be labeled by anti-phospho-PYK2 antibody. The increase in PYK2 immunoreactivity, as well as in phospho-PYK2 immunoreactivity, suggests that there is an upregulation of PYK2 protein, as well as of its phosphorylation, during microglial activation. To confirm the localization of phospho-PYK2 in activated microglia, an indirect immunofluorescence analysis was performed using anti-phospho-PYK2 and anti-OX42 antibodies. Confocal images demonstrate that phospho-PYK2 is colocalized with OX42 in activated microglia (Fig. 4). However, the subcellular distribution of these two proteins was different; OX42 immunostaining was localized in the cell periphery as expected for a membrane bound protein, whereas phospho-PYK2 was found in the cytosol.
Fig. 3.

Immunoreactivity of phospho-PYK2 and OX42 within the core infract (striatum) after focal cerebral ischemia. Coronal brain sections (50 μm) of animals killed at the indicated time points after MCAO were immunostained with either anti-OX42 (top row) or anti-phospho-PYK2 (bottom row) antibody. No detectable immunoreactivity of phospho-PYK2 was observed in the control sections, whereas OX42-positive resting microglial cells were randomly distributed in the striatum. At 6 hr after MCAO, phospho-PYK2 immunoreactivity was visualized in microglia-like cells in the affected striatum. Note that the morphology of phospho-PYK2-positive cells resembles the microglia, as demonstrated by OX42 immunostaining. A further increase in phospho-PYK2 immunoreactivity was observed over 24–72 hr after MCAO. The number of microglial cells and their morphology were dramatically changed during this period, as demonstrated by OX42 immunostaining. By 72 hr, the core infarction area was filled with amoeboid-like microglial cells. Scale bar, 50 μm.
Fig. 4.

Colocalization of phospho-PYK2 and OX42 in microglial cells after focal cerebral ischemia. Coronal brain sections (50 μm) of animals killed at 72 hr after MCAO were double-immunostained with anti-phospho-PYK2 and anti-OX42 antibodies. Shown are confocal images of phospho-PYK2 (red) and OX42 (green) immunostaining and the merged image. Colocalization is shown in yellow. As demonstrated, phospho-PYK2 appears mainly in the cytosol, whereas OX42 appears in the cell periphery. Scale bar, 25 μm.
Colocalization of phospho-PYK2 and activated p38MAPK after cerebral ischemia
It was shown previously that p38MAPK is activated in microglia after global forebrain ischemia (Walton et al., 1998) and that PYK2 represents a stress-sensitive mediator of the p38MAPK signaling pathway in response to certain cytotoxic agents (Pandey et al., 1999). Therefore, we examined the time course of p38MAPK and PYK2 activation in the rat brain after ischemia. Brain sections of animals killed at different time points after MCAO as indicated in Figure5 were double-immunostained with anti-phospho-p38MAPK and anti-phospho-PYK2 antibodies. The anti-phospho-p38MAPK antibody specifically targets the double-phosphorylated active form of p38MAPK (Derijard et al., 1995;Ichijo, 1999). As shown in Figure 5, no detectable immunostaining of phospho-p38MAPK was observed in nonischemic cortical neurons (lamina V), similar to the results obtained with anti-phospho-PYK2 antibody (Figs. 2, 5). However, phospho-p38MAPK immunoreactivity was already detected in cortical neurons 15 min after focal cerebral ischemia, which reached its maximal levels at 30 min after ischemia, and then it slightly subsided. The striking similarity between p38MAPK and phospho-PYK2 immunoreactivity is demonstrated in Figure 5. The increase in PYK2 and p38MAPK phosphorylation could also be demonstrated by biochemical studies (Fig. 6). PYK2 and p38MAPK were immunoprecipitated from either control or ischemic cortex extracts. The immunoprecipitates were then subjected to Western blot analysis, using anti-phospho-PYK2, anti-PYK2, anti-p38MAPK, or anti-phospho-p38MAPK antibody. An increase in PYK2 and p38MAPK phosphorylation was already observed 15 min after MCAO (Fig. 6). The phosphorylation of both PYK2 and p38MAPK was sustained for 1 hr and slightly decreased 6 hr after MCAO. No detectable changes in either PYK2 or p38MAPK expression level was observed in the cortex (Fig.6).
Fig. 5.

Colocalization of phospho-PYK2 and phospho-p38MAPK in cortical neurons after focal cerebral ischemia. Coronal brain sections (50 μm) of animals killed at the indicated time points after MCAO were double-immunostained with anti-phospho-p38MAPK monoclonal antibody and anti-phospho-PYK2 polyclonal antibody. Shown are confocal images of phospho-PYK2 (red) and phospho-p38MAPK (green) immunoreactivity in lamina V neurons of the cortex. Phospho-PYK2 and p38MAPK were barely detectable in the control cortex, whereas intense phospho-PYK2 and p38MAPK immunoreactivities were visualized in the cortex at 15 min after ischemia. These immunoreactivities were further increased at 30 min. Scale bar, 50 μm.
Fig. 6.

Increased phosphorylation of PYK2 and p38MAPK in the cortex after ischemia. Shown are PYK2 and p38MAPK immunoprecipitates prepared from homogenates of either control animals or animals that were exposed to MCAO for the indicated time. PYK2 and p38MAPK immunoprecipitates were resolved on SDS-PAGE, transferred to nitrocellulose, and immunoblotted with anti-PYK2, anti-phospho-PYK2, anti-p38MAPK, or anti-phospho-p38MAPK antibodies. An increase in PYK2 and p38MAPK phosphorylation was observed in the cortex at 15 min after MCAO (top row). The phosphorylation of PYK2 was sustained for 1 hr after MCAO and dramatically decreased at 6 hr, whereas slight decrease of p38MAPK phosphorylation was evident 6 hr after MCAO. No detectable changes of either PYK2 or p38MAPK expression levels was observed during the first 6 hr after MCAO (bottom row).
To further characterize the localization of activated p38MAPK after MCAO, double-immunofluorescence analysis was performed at 6–72 hr after MCAO using both anti-phospho-PYK2 and anti-phospho-p38MAPK antibodies. In the striatum, no detectable phospho-p38MAPK immunoreactivity was observed in microglia cells in nonischemic sections, similar to the results obtained with anti-phospho-PYK2 antibody (Fig. 2). However, enhanced phospho-PYK2 and phospho-p38MAPK immunoreactivities were visualized in the activated microglia 24–72 hr after ischemia. A representative result from this set of experiments is shown in Figure 7, demonstrating the colocalization of phospho-PYK2 and phospho-p38MAPK in activated microglia 72 hr after MCAO. Interestingly, no detectable phospho-ERK or phospho-JNK immunoreactivity was observed after MCAO using the same experimental conditions (data not shown). These results are consistent with previous studies obtained after global forebrain ischemia in which phospho-p38MAPK immunoreactivity was observed in microglia in a region adjacent to CA1 neurons, but no detectable immunoreactivity for either phospho-ERK1/2 or phospho-JNK was observed (Walton et al., 1998).
Fig. 7.

Colocalization of phospho-PYK2 and phospho-p38MAPK in microglial cells after focal cerebral ischemia. Coronal brain sections (50 μm) of animals killed at 72 hr after MCAO were double-immunostained with anti-phospho-p38MAPK monoclonal antibody and anti-phospho-PYK2 polyclonal antibody. Shown are confocal images of phospho-PYK2 and phospho-p38MAPK immunoreactivities within the infarct striatum in microglial cells 72 hr after ischemia. Colocalization appears in yellow. As shown, the amoeboid-like microglial cells are double-immunostained with anti-phospho-PYK2 and anti-phospho-p38MAPK antibodies. Scale bar, 50 μm.
Increase in phospho-PYK2 immunoreactivity after KA-induced status epilepticus
To further characterize the distribution of phospho-PYK2 in the brain under pathological conditions, we immunohistochemically investigated the changes in PYK2 phosphorylation in the hippocampus after systemic injections of KA to rats. As shown previously, KA treatment induces status epilepticus 1–1.5 hr after injection and therefore can provide a chronic model of epilepsy (Kim et al., 1996). Brain samples were obtained at 6, 24, and 72 hr after the first onset of seizure and immunostained with either anti-PYK2 or anti-phospho-PYK2 antibodies. In control hippocampus, PYK2 immunoreactivity was abundantly visualized in the hippocampal pyramidal neurons and granule cells of the dentate gyrus. PYK2 immunoreactivity was homogeneously distributed throughout the neuronal somata and processes (Figs.8, 9). In contrast, phospho-PYK2 was mainly localized in the neuronal somata. After KA treatment, dramatic changes in PYK2 and phospho-PYK2 immunoreactivity were observed in the CA1, CA2, and CA3 pyramidal neurons. Figure 8 shows the immunoreactivities of PYK2 and phospho-PYK2 in the CA1 field. Similar results were obtained in other fields (CA2, CA3, and dentate gyrus) (data not shown). The level of PYK2 and phospho-PYK2 immunoreactivity in the hippocampal pyramidal neurons decreased markedly 6 hr after epileptic onset and further subsided at 24–72 hr. Some cells that exhibited microglia-like morphology were faintly immunostained with either anti-PYK2 or anti-phospho-PYK2 antibodies at 24 hr (Fig. 8). PYK2- and phospho-PYK2-positive microglia-like cells exhibit a “rod-like” shape and accumulated at the pyramidal layers 72 hr after epileptic onset, whereas very faint PYK2 or phospho-PYK2 immunoreactivity was visualized in pyramidal neurons at this time point (Fig. 8). To confirm the localization of PYK2 and phospho-PYK2 in microglial cells 72 hr after KA injection, double-immunofluorescence analysis was performed using anti-OX42 monoclonal antibody and either anti-PYK2 or phospho-PYK2 polyclonal antibody. Figure 9 shows the colocalization of PYK2 or phospho-PYK2 with OX42 immunoreactivity in the microglial cells 72 hr after seizure. In the control hippocampus, OX42-positive microglial cells were uniformly distributed throughout the hippocampus, exhibiting typical resting microglial morphology. However, 72 hr after seizure, microglial cells immunostained with anti-OX42 antibody accumulated in the pyramidal layer, striatum radiatum, and dentate hilus. Most of the OX42-positive cells, but not all, demonstrated PYK2 or phospho-PYK2 immunoreactivity (Fig. 9). This result indicates that KA-induced status epilepticus leads to upregulation of PYK2 and phospho-PYK2 in microglial cells in a time-dependent manner, similar to the results obtained after ischemia.
Fig. 8.

Tyrosine phosphorylation of PYK2 within the hippocampus after KA-induced epilepsy. Coronal brain sections (50 μm) of animals killed at the indicated time points were immunostained with either anti-PYK2 (top row) or with anti-phospho-PYK2 (bottom row) antibody. Shown are photomicrographs of the hippocampal CA1 field. Intense PYK2 and moderate phospho-PYK2 immunoreactivity were detected in the CA1 pyramidal neurons of the control hippocampus. PYK2 and phospho-PYK2 immunoreactivities were dramatically decreased in the CA1 pyramidal neurons 6 hr after epileptic onset and were barely detected at 72 hr. However, PYK2 and phospho-PYK2 immunoreactivities were evident in microglia-like cells at 24–72 hr after KA injection (arrows). At 72 hr after KA injection, pronounced immunoreactivity of PYK2 and phospho-PYK2 was observed in rod-like microglial cells. Scale bar, 50 μm.
Fig. 9.
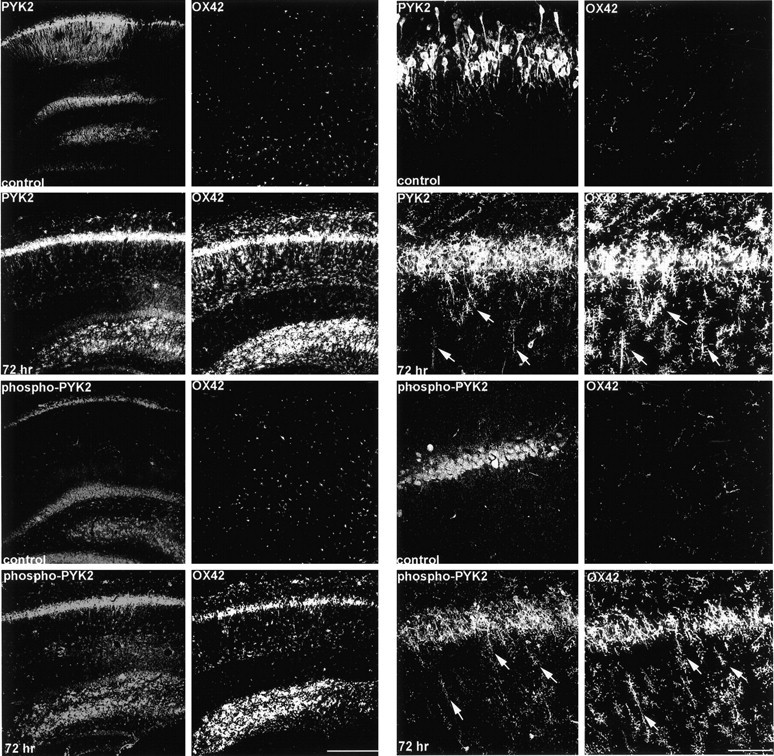
Colocalization of phospho-PYK2 and OX42 in the microglial cells after KA-induced epilepsy. Coronal brain sections (50 μm) of either control animals or animals killed at 72 hr after epileptic onset were double-immunostained with anti-OX42 monoclonal antibody and with either anti-PYK2 or anti-phospho-PYK2 polyclonal antibody. As shown in low-power confocal images (left panel), intense PYK2 and moderate phospho-PYK2 immunoreactivities were visualized in CA1 pyramidal neurons and granule cells of the dentate gyrus in the control hippocampus. Microglial cells immunostained with OX42 are uniformly distributed throughout the control hippocampus, and no colocalization with either PYK2 or phospho-PYK2 was observed. At 72 hr after seizure, OX42-positive microglial cells were accumulated in the CA1 pyramidal layer, striatum radiatum, and dentate hilus. Enhanced PYK2 and phospho-PYK2 immunoreactivities were observed in microglial cells 72 hr after seizure. High-power confocal images (right panel) demonstrate the dramatic changes in PYK2, phospho-PYK2, and OX42 immunoreactivities in the CA1 field. At 72 hr, most of the OX42-positive rod-like microglial cells (arrows) exhibit intense PYK2 and phospho-PYK2 immunoreactivity. Scale bars:left, 200 μm; right, 50 μm.
DISCUSSION
The nonreceptor tyrosine kinase PYK2 is highly expressed in the CNS (Menegon et al., 1999), although its cellular function under physiological and pathological conditions has not been elucidated yet. In the present study, we determined the cellular distribution of tyrosine-phosphorylated PYK2 in response to cerebral ischemia and epilepsy, using antibody that selectively recognizes tyrosine-phosphorylated PYK2 at its major autophosphorylation site, tyrosine 402. We provide evidence that PYK2 undergoes rapid tyrosine phosphorylation in cortical neurons after focal cerebral ischemia (Fig.2). Tyrosine phosphorylation of PYK2 reached maximal levels at 30 min and began to decline 1 hr after ischemia (Fig. 2). It was shown previously that persistent neuronal depolarization and elevation of intracellular calcium concentration are induced by cerebral ischemia (Arias et al., 1999; Koponen et al., 1999; Zhang and Lipton, 1999). It was also shown that PYK2 is tightly regulated by intracellular calcium concentration and rapidly undergoes tyrosine phosphorylation in response to membrane depolarization (Siciliano et al., 1994; Lev et al., 1995). Thus, elevation of the intracellular calcium concentration induced by cerebral ischemia might be crucial for tyrosine phosphorylation of PYK2 in neurons a short time after ischemia. Interestingly, tyrosine phosphorylation of PYK2 slightly subsided 1 hr after ischemia, demonstrating a transient phosphorylation of PYK2 in cortical neurons after focal cerebral ischemia. Because calcium plays a critical role in neuronal cell death, the transient phosphorylation of PYK2 as well as of p38MAPK (Figs. 5, 6) might be related to neuronal apoptosis and death. Indeed, the p38MAPK signaling pathway is implicated in neuronal cell death and apoptosis in response to a variety of pathological conditions in the nervous system (Behrens et al., 1999; Nakahara et al., 1999). Therefore, it is reasonable to assume that PYK2 acts as an upstream mediator of p38MAPK cascades, linking calcium influx to neuronal cell apoptosis and death.
In contrast to the immunoreactivity of phospho-PYK2 obtained in cortical neurons, a pronounced increase of phospho-PYK2 immunoreactivity was evident in the striatum 6 hr after ischemia (Fig.3). Double-immunofluorescence labeling using anti-phospho-PYK2 and anti-OX42 antibodies demonstrated that phospho-PYK2 is localized in microglia (Fig. 4). The elevated phospho-PYK2 immunoreactivity was accompanied by morphological changes in microglia. It was shown previously that microglia, the resident brain macrophages, strongly respond to cerebral ischemia (Schroeter et al., 1999). Activation of microglia in response to injury, inflammation, or neuronal degeneration coincides with microglial migration to the site of injury, proliferation in the area of neurodegeneration, and releasing of cytokines (Kreutzberg, 1996). Therefore, migration, proliferation, and cytokine secretions are crucial for the microglial activation process and physiological function.
Migration of microglia was shown to be regulated by β chemokines, such as macrophage inflammatory proteins, monocyte chemoattractant proteins, and RANTES (regulated on activation normal T cell expressed). These chemokines also induced a reorganization of the actin cytoskeleton, which is required for cell locomotion (Cross and Woodroofe, 1999). Whether PYK2 is involved in microglial migration is not clear at present. The enhanced tyrosine phosphorylation of PYK2 in microglia after cerebral ischemia obtained in this study (Fig. 3), together with previous reports demonstrating the activation of PYK2 by chemokines (Dikic et al., 1998; Ganju et al., 1998a,b), and its association with cytoskeletal proteins such as paxillin (Ganju et al., 1998), may support this possibility.
In contrast to migration, microglial proliferation was shown to be regulated by colony stimulating factors (CSFs), such as granulocyte-macrophage-CSF or macrophage-CSF (M-CSF) (Giulian and Ingeman, 1988; Lee et al., 1994). Recent studies have demonstrated that PYK2 is also activated by M-CSF in macrophages and that a dominant interfering mutant of PYK2 partially inhibits MAPK activation in response to M-CSF (Hatch et al., 1998). These results suggest that PYK2 might be involved in microglial proliferation mediated by activation of the ERK/Ras signaling pathway. However, after focal cerebral ischemia, we could not detect the immunoreactivity of phosphorylated ERK but rather detected the immunoreactivity of phosphorylated p38MAPK (Fig. 5). Four p38MAPK homologs, p38α, p38β, p38γ, and p38δ, were cloned in mammals. Although they exhibit different tissue distribution, all of them contain the Thr-Gly-Tyr motif. p38αMAPK is activated by physical and chemical stresses, as well as by lipopolysaccharide and various cytokines. It appears to play an essential role in regulating inflammatory responses, cytokine secretion, and cell apoptosis (Ono and Han, 2000). The striking similarity between phospho-PYK2 and phospho-p38MAPK immunoreactivities after cerebral ischemia (Figs. 5, 7) suggests that PYK2 is an upstream regulator of p38MAPK. Indeed, it was shown recently that overexpression of PYK2 leads to activation of p38MAPK, whereas a dominant negative mutant of PYK2 inhibits p38MAPK activation in response to cytotoxic agents (Pandey et al., 1999). However, we cannot exclude the possibility that activation of p38MAPK is independent of PYK2 tyrosine phosphorylation and is actually mediated by a different upstream regulator.
Microglial cells are able to produce and respond to a wide range of inflammatory stimuli, including TNFα, interferon-γ (INF-γ), interleukin-1 (IL-1), and IL-6 (Gehrmann and Kreutzberg, 1995;Benveniste, 1997). Because p38MAPK is activated by IL-1 and TNFα in many cell types, it was proposed that p38MAPK is involved in microglial activation (Walton et al., 1998). In light of the presented data, we suggest that PYK2 is also involved in microglial activation, probably upstream to p38MAPK. TNFα (Tokiwa et al., 1996) and INF-γ (Takaoka et al., 1999) were shown previously to induce tyrosine phosphorylation of PYK2. Tyrosine-phosphorylated PYK2 was also observed in microglia_in vitro_ in response to fibrilla amyloid peptides. It was suggested that activation of PYK2 by amyloid fibril is required for neurotoxin production, and therefore it might be involved in the pathophysiology of Alzheimer's disease (Combs et al., 1999). This hypothesis can be experimentally addressed in vivo using the anti-phospho-PYK2 antibody that we describe in this study.
An increase in phospho-PYK2 immunoreactivity was also observed in microglia after KA-induced status epilepticus (Figs. 8, 9), suggesting that tyrosine phosphorylation of PYK2 in microglia is a general phenomenon that can be attributed to microglial activation. Although PYK2 is highly expressed in several macrophage, monocyte, and microglial cell lines (Hatch et al., 1998; Combs et al., 1999; our unpublished results), neither PYK2 nor phospho-PYK2 immunoreactivity was detected in resting microglia in vivo (Figs. 2, 3, 8,9). These results suggest, that during the activation of microglia, both PYK2 expression and its tyrosine phosphorylation are upregulated_in vivo_. The different expression of PYK2 in resting microglia in vivo compared with the microglial cell line may be related to the basal activation state of the microglial cell lines because of the immortalization process. PYK2 is not the only protein that is upregulated in activated microglia, several additional proteins, such as the major histocompatibility complex type I and II molecules, the phago lysosomal marker ED1, and several adhesion proteins, including the integrin adhesion molecules leukocyte function antigen-1 (LFA-1), very late antigen-4, and the ligand for LFA-1, intercellular adhesion molecule-1, have been shown previously to be upregulated at different stages of microglial activation (Hailer et al., 1997; Schroeter et al., 1999). The upregulation of these adhesion molecules is involved in the phagocytosis activity of activated microglia. Because PYK2 is activated in different cell types by cell adhesion to the extracellular matrix, it may participate in signaling pathways downstream to integrin during microglial activation. This hypothesis should be experimentally addressed.
The main role of microglia in the adult CNS is to respond to the disruption of brain homeostasis attributable to direct damage of neurons, neuronal degeneration, or through disease (Gehrmann and Kreutzberg, 1995). Therefore, elucidating the signaling pathways involved in microglial activation will contribute to a better understanding of neuronal cell survival and death under physiological and pathological conditions.
In the present study, PYK2 was shown to be tyrosine-phosphorylated in activated microglia, elicited by two pathological events: ischemia and epilepsy. We demonstrated tyrosine phosphorylation of PYK2 in situ using anti-phospho-PYK2 antibody. This antibody can be used to determine the localization of tyrosine-phosphorylated PYK2 in the CNS under various physiological or pathological conditions and to dissect the signaling pathways in situ applying both antibodies against activated downstream targets, such as phospho-MAPKs, specific inhibitors, or animal models.
Footnotes
This work was supported by the Carl and Micaela Einhorn-Dominic Institute funds for brain research. Sima Lev is incumbent of the Helena Rubinstein Career Development Chair. We thank Shari Carmon for technical assistance and Dr. R. Zeger for the anti-MAPKs antibodies. We also thank V. Teichberg, B. Attali, and I. Nevo for critical reading of this manuscript.
Correspondence should be addressed to Dr. Sima Lev, Department of Neurobiology, Weizmann Institute of Science, 76100 Rehovot, Israel. E-mail: sima.lev@weizmann.ac.il.
REFERENCES
- 1.Arias RL, Tasse JR, Bowlby MR. Neuroprotective interaction effects of NMDA and AMPA receptor antagonists in an in vitro model of cerebral ischemia. Brain Res. 1999;816:299–308. doi: 10.1016/s0006-8993(98)01051-8. [DOI] [PubMed] [Google Scholar]
- 2.Behrens MM, Strasser U, Koh JY, Gwag BJ, Choi DW. Prevention of neuronal apoptosis by phorbol ester-induced activation of protein kinase C: blockade of p38 mitogen-activated protein kinase. Neuroscience. 1999;94:917–927. doi: 10.1016/s0306-4522(99)00212-2. [DOI] [PubMed] [Google Scholar]
- 3.Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med. 1997;75:165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- 4.Berg NN, Ostergaard HL. T cell receptor engagement induces tyrosine phosphorylation of FAK and Pyk2 and their association with Lck. J Immunol. 1997;159:1753–1757. [PubMed] [Google Scholar]
- 5.Combs CK, Johnson DE, Cannady SB, Lehman TM, Landreth GE. Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of β-amyloid and prion proteins. J Neurosci. 1999;19:928–939. doi: 10.1523/JNEUROSCI.19-03-00928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cotman CWAA. A potential role for apoptosis in neurodegeneration and Alzheimer's disease. Mol Neurobiol. 1995;10:19–45. doi: 10.1007/BF02740836. [DOI] [PubMed] [Google Scholar]
- 7.Cross AK, Woodroofe MN. Chemokines induce migration and changes in actin polymerization in adult rat brain microglia and a human fetal microglial cell line in vitro. J Neurosci Res. 1999;55:17–23. doi: 10.1002/(SICI)1097-4547(19990101)55:1<17::AID-JNR3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 8.Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, Davis RJ. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 9.Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- 10.Dikic I, Dikic I, Schlessinger J. Identification of a new Pyk2 isoform implicated in chemokine and antigen receptor signaling. J Biol Chem. 1998;273:14301–14308. doi: 10.1074/jbc.273.23.14301. [DOI] [PubMed] [Google Scholar]
- 11.Flaris NA, Densmore TL, Molleston MC, Hickey WF. Characterization of microglia and macrophages in the central nervous system of rats: definition of the differential expression of molecules using standard and novel monoclonal antibodies in normal CNS and in four models of parenchymal reaction. Glia. 1993;7:34–40. doi: 10.1002/glia.440070108. [DOI] [PubMed] [Google Scholar]
- 12.Ganju RK, Brubaker SA, Meyer J, Dutt P, Yang Y, Qin S, Newman W, Groopman JE. The alpha-chemokine, stromal cell-derived factor-1 alpha, binds to the transmembrane G-protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. J Biol Chem. 1998a;273:23169–23175. doi: 10.1074/jbc.273.36.23169. [DOI] [PubMed] [Google Scholar]
- 13.Ganju RK, Dutt P, Wu L, Newman W, Avraham H, Avraham S, Groopman JE. Beta-chemokine receptor CCR5 signals via the novel tyrosine kinase RAFTK. Blood. 1998b;91:791–797. [PubMed] [Google Scholar]
- 14.Gehrmann J, Kreutzberg GW. Microglia in experimental neuropathology. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; Oxford: 1995. pp. 883–904. [Google Scholar]
- 15.Gehrmann J, Banati RB, Wiessner C, Hossmann KA, Kreutzberg GW. Reactive microglia in cerebral ischaemia: an early mediator of tissue damage? Neuropathol Appl Neurobiol. 1995;21:277–289. doi: 10.1111/j.1365-2990.1995.tb01062.x. [DOI] [PubMed] [Google Scholar]
- 16.Girault JA, Costa A, Derkinderen P, Studler JM, Toutant M. FAK and PYK2/CAKbeta in the nervous system: a link between neuronal activity, plasticity and survival? Trends Neurosci. 1999;22:257–263. doi: 10.1016/s0166-2236(98)01358-7. [DOI] [PubMed] [Google Scholar]
- 17.Gismondi A, Bisogno L, Mainiero F, Palmieri G, Piccoli M, Frati L, Santoni A. Proline-rich tyrosine kinase-2 activation by beta 1 integrin fibronectin receptor cross-linking and association with paxillin in human natural killer cells. J Immunol. 1997;159:4729–4736. [PubMed] [Google Scholar]
- 18.Giulian D, Ingeman JE. Colony-stimulating factors as promoters of ameboid microglia. J Neurosci. 1988;8:4707–4717. doi: 10.1523/JNEUROSCI.08-12-04707.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hailer NP, Bechmann I, Heizmann S, Nitsch R. Adhesion molecule expression on phagocytic microglial cells following anterograde degeneration of perforant path axons. Hippocampus. 1997;7:341–349. doi: 10.1002/(SICI)1098-1063(1997)7:3<341::AID-HIPO8>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 20.Hanks SK, Polte TR. Signaling through focal adhesion kinase. BioEssays. 1997;19:137–145. doi: 10.1002/bies.950190208. [DOI] [PubMed] [Google Scholar]
- 21.Hatch WC, Ganju RK, Hiregowdara D, Avraham S, Groopman JE. The related adhesion focal tyrosine kinase (RAFTK) is tyrosine phosphorylated and participates in colony-stimulating factor-1/macrophage colony-stimulating factor signaling in monocyte-macrophages. Blood. 1998;91:3967–3973. [PubMed] [Google Scholar]
- 22.Ichijo H. From receptors to stress-activated MAP kinases. Oncogene. 1999;18:6087–6093. doi: 10.1038/sj.onc.1203129. [DOI] [PubMed] [Google Scholar]
- 23.Karp HL, Tillotson ML, Soria J, Reich C, Wood JG. Microglial tyrosine phosphorylation systems in normal and degenerating brain. Glia. 1994;11:284–290. doi: 10.1002/glia.440110310. [DOI] [PubMed] [Google Scholar]
- 24.Kawamura S, Yasui N, Shirasawa M, Fukasawa H. Rat middle cerebral artery occlusion using an intraluminal thread technique. Acta Neurochir (Wien) 1991;109:126–132. doi: 10.1007/BF01403007. [DOI] [PubMed] [Google Scholar]
- 25.Kim HC, Pennypacker KR, Bing G, Bronstein D, McMillian MK, Hong JS. The effects of dextromethorphan on kainic acid-induced seizures in the rat. Neurotoxicology. 1996;17:375–385. [PubMed] [Google Scholar]
- 26.Koponen S, Keinanen R, Roivainen R, Hirvonen T, Narhi M, Chan PH, Koistinaho J. Spreading depression induces expression of calcium-independent protein kinase C subspecies in ischaemia-sensitive cortical layers: regulation by N-methyl-d-aspartate receptors and glucocorticoids. Neuroscience. 1999;93:985–993. doi: 10.1016/s0306-4522(99)00166-9. [DOI] [PubMed] [Google Scholar]
- 27.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 28.Lee SC, Liu W, Brosnan CF, Dickson DW. GM-CSF promotes proliferation of human fetal and adult microglia in primary cultures. Glia. 1994;12:309–318. doi: 10.1002/glia.440120407. [DOI] [PubMed] [Google Scholar]
- 29.Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J. Protein tyrosine kinase PYK2 involved in Ca(2+)-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- 30.Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- 31.Ling EA, Kaur LC, Yick TY, Wong WC. Immunocytochemical localization of CR3 complement receptors with OX-42 in amoeboid microglia in postnatal rats. Anat Embryol (Berl) 1990;182:481–486. doi: 10.1007/BF00178913. [DOI] [PubMed] [Google Scholar]
- 32.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniotomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 33.McRae A, Ling EA, Schubert P, Rudolphi K. Properties of activated microglia and pharmacologic interference by propentofylline. Alzheimer Dis Assoc Disord. 1998;12:S15–S20. [PubMed] [Google Scholar]
- 34.Menegon A, Burgaya F, Baudot P, Dunlap DD, Girault JA, Valtorta F. FAK+ and PYK2/CAKbeta, two related tyrosine kinases highly expressed in the central nervous system: similarities and differences in the expression pattern. Eur J Neurosci. 1999;11:3777–3788. doi: 10.1046/j.1460-9568.1999.00798.x. [DOI] [PubMed] [Google Scholar]
- 35.Nakahara S, Yone K, Sakou T, Wada S, Nagamine T, Niiyama T, Ichijo H. Induction of apoptosis signal regulating kinase 1 (ASK1) after spinal cord injury in rats: possible involvement of ASK1-JNK and -p38 pathways in neuronal apoptosis. J Neuropathol Exp Neurol. 1999;58:442–450. doi: 10.1097/00005072-199905000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Nakano S, Kogure K, Fujikura H. Ischemia-induced slowly progressive neuronal damage in the rat brain. Neuroscience. 1990;38:115–124. doi: 10.1016/0306-4522(90)90378-h. [DOI] [PubMed] [Google Scholar]
- 37.Ono K, Han J. The p38 signal transduction pathway activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 38.Pandey P, Avraham S, Kumar S, Nakazawa A, Place A, Ghanem L, Rana A, Kumar V, Majumder PK, Avraham H, Davis RJ, Kharbanda S. Activation of p38 mitogen-activated protein kinase by PYK2/related adhesion focal tyrosine kinase-dependent mechanism. J Biol Chem. 1999;274:10140–10144. doi: 10.1074/jbc.274.15.10140. [DOI] [PubMed] [Google Scholar]
- 39.Pettmann B, Henderson CE. Neuronal cell death. Neuron. 1998;20:633–647. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- 40.Schlaepfer DD, Hunter T. Integrin signalling and tyrosine phosphorylation: just the FAKs? Trends Cell Biol. 1998;8:151–157. doi: 10.1016/s0962-8924(97)01172-0. [DOI] [PubMed] [Google Scholar]
- 41.Schroeter M, Jander S, Witte OW, Stoll G. Heterogeneity of the microglial response in photochemically induced focal ischemia of the rat cerebral cortex. Neuroscience. 1999;89:1367–1377. doi: 10.1016/s0306-4522(98)00398-4. [DOI] [PubMed] [Google Scholar]
- 42.Siciliano JC, Gelman M, Girault JA. Depolarization and neurotransmitters increase neuronal protein tyrosine phosphorylation. J Neurochem. 1994;62:950–959. doi: 10.1046/j.1471-4159.1994.62030950.x. [DOI] [PubMed] [Google Scholar]
- 43.Takaoka A, Tanaka N, Mitani Y, Miyazaki T, Fujii H, Sato M, Kovarik P, Decker T, Schlessinger J, Taniguchi T. Protein tyrosine kinase Pyk2 mediates the Jak-dependent activation of MAPK and Stat1 in IFN-γ, but not IFN-α, signaling. EMBO J. 1999;18:2480–2488. doi: 10.1093/emboj/18.9.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tillotson ML, Wood JG. Phosphotyrosine antibodies specifically label ameboid microglia in vitro and ramified microglia in vivo. Glia. 1989;2:412–419. doi: 10.1002/glia.440020604. [DOI] [PubMed] [Google Scholar]
- 45.Tokiwa G, Dikic I, Lev S, Schlessinger J. Activation of Pyk2 by stress signals and coupling with JNK signaling pathway. Science. 1996;273:792–794. doi: 10.1126/science.273.5276.792. [DOI] [PubMed] [Google Scholar]
- 46.Walton KM, DiRocco R, Bartlett BA, Koury E, Marcy VR, Jarvis B, Schaefer EM, Bhat RV. Activation of p38MAPK in microglia after ischemia. J Neurochem. 1998;70:1764–1767. doi: 10.1046/j.1471-4159.1998.70041764.x. [DOI] [PubMed] [Google Scholar]
- 47.Yu H, Li X, Marchetto GS, DyR, Hunter D, Calvo B, Dawson TL, Wilm M, Anderegg RJ, Graves LM, Earp HS. Activation of a novel calcium-dependent protein-tyrosine kinase. Correlation with c-Jun N-terminal kinase but not mitogen-activated protein kinase activation. J Biol Chem. 1996;271:29993–29998. doi: 10.1074/jbc.271.47.29993. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y, Lipton P. Cytosolic Ca2+ changes during in vitro ischemia in rat hippocampal slices: major roles for glutamate and Na+-dependent Ca2+ release from mitochondria. J Neurosci. 1999;19:3307–3315. doi: 10.1523/JNEUROSCI.19-09-03307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]