CArG binding factor A (CBF-A) is involved in transcriptional regulation of the rat Ha-ras promoter (original) (raw)
Abstract
In the present study we identified a positive transcriptional element within the rat Ha-ras promoter previously known as Ha-ras response element (HRE) and identified a _trans_-acting factor that binds HRE sequences in rat mammary cells. To identify the binding protein we employed sequence specific DNA affinity chromatography. Amino acid sequence analysis of the affinity-purified proteins was performed by tandem mass spectroscopy. The results unexpectedly demonstrated that in rat mammary cells CArG box-binding factor A (CBF-A) is the major protein species that bind specifically to the rat and human HRE sequences with high affinity. The affinity of CBF-A binding to HRE was significantly higher than to the CArG box described as a recognition sequence for CBF-A protein. Transient transfection assays using reporter plasmids verified that mutations within the HRE that disrupt binding of CBF-A also reduced the activity of the rat Ha-ras promoter. Despite the fact that the HRE within the Ha-ras promoter resembles a binding site for Ets transcription factors, we did not detect the binding of Ets-related proteins to the rat HRE in BICR-M1Rk cells. We further demonstrated a correlation between the presence of HRE binding activity and induction of Ha-ras mRNA expression following serum stimulation in the mammary carcinoma cell line. Taken together, our results suggest that CBF-A may play an important role in transcriptional regulation of Ha-ras promoter activity during normal mammary cell growth and carcinogenesis.
INTRODUCTION
The Ras proteins are a closely related set of genes that encode membrane-associated proteins involved in cell proliferation and differentiation. The Ras proteins belong to the family of small GDP/GTP-binding proteins that transduce signals from activated cell surface tyrosine kinase receptors to the nucleus by activating a cascade of secondary messengers within the cytoplasm (1,2).
Activated Ras proteins are able to transform a number of immortalized cell lines in vitro, and decrease tumor latency and increase tumor frequencies in transgenic animals. However, transformation in these experimental models is usually associated with expression of activated Ha-ras alleles at levels that exceed those observed in most cancers (3).
When introduced into Rat 1 fibroblasts under the control of its own promoter, activated Ha-ras failed to transform immortalized Rat-1 fibroblasts (4). Furthermore, transformed clones arising during passage of these transfected cell populations invariably over-expressed the mutant allele as a result of either gene amplification or transcriptional deregulation. The latter studies are in accordance with the observations that mutant ras genes are frequently over-expressed in human tumors (5–7). A recent study of transgenic animals harboring an inducible Ha-ras transgene demonstrated that continued expression of the oncogene is necessary for the genesis and maintenance of solid tumors in vivo (8). A variety of in vitro transformation experiments have demonstrated that even wild-type Ras proteins have transforming potential when expressed above normal levels. An in vivo correlate of this observation is the finding that deregulation of Ha-ras pathways is frequently detected in human breast cancers, although ras gene mutations are rare (∼5%) in these tumors (9). Taken together, these studies support the hypothesis that deregulated expression of the mutant or wild-type Ras may be important for cancer development and maintenance in vivo. Understanding the mechanisms underlying ras deregulation therefore has implications for diagnosis and therapeutic intervention.
The Ha-ras proto-oncogene is constitutively expressed in all cell types and can be induced in response to a number of mitogenic stimuli (10). The rat and human Ha-ras promoters have been cloned and a number of regulatory elements identified (11,12). The Ha-ras promoter in both species is G+C rich and lacks a TATA box, features characteristic of constitutively expressed ‘housekeeping’ genes. Six GC boxes, two NF-1 binding sites and two potential AP-2 sites were identified within the upstream regulatory region of human Ha-ras. In addition, two copies of Ha-ras conserved sequence (HRC) and an Ha-ras element I (HRE-I) were identified in the human promoter (12). The individual GC boxes appear to have different effects on the promoter activity: only GC II, which binds SpI, shows a positive effect on Ha-ras promoter activity. The NF-I elements themselves have weak effects on the promoter activity. Deletion of the NF-I binding site along with the HRE and GC-II site decreases transcription by 2.5-fold in the context of the whole promoter (12). Overall, the rat and human Ha-ras promoters are highly conserved, sharing similar regulatory elements located in similar positions within the promoter relative to the start site. Only the Ha-ras element (HRE) site present in the human promoter, which is thought to be responsive to the Ets family of transcription factors (12), was not previously reported to have a counterpart in the rat promoter.
Our previous studies of carcinogen induced mammary tumors suggested carcinogen mediated effects on the Ha-ras promoter in vivo (13). Here we demonstrate that the region of the promoter sensitive to carcinogen treatment includes a positive transcriptional element identical to the HRE found in the human Ha-ras promoter, albeit in the inverted and complementary orientation. We demonstrate that the CArG Binding Factor-A (CBF-A) protein, originally defined by its ability to interact with CArG box, binds to both the human and rat HREs. CBF-A binds to the rat and human HRE with higher affinity than the CArG box, originally described as the recognition site for this protein. Furthermore, we failed to detect any Ets transcription factor binding to the rat HRE. These results indicated that in mammary cells, CBF-A is the major protein that binds to recognition sequences commonly accepted as Ets binding sites. CBF-A binding was correlated with increased Ha-ras promoter activity in mammary cells and there was a direct correlation between the presence of the HRE binding activity and induction of Ha-ras mRNA expression. Taken together, our results suggest that CBF-A mediated transactivation may play an important role in Ha-ras deregulation during carcinogenesis in rodents and humans.
MATERIALS AND METHODS
Cell culture, cell treatment and cell cycle analysis
The BICR-M1Rk rat mammary gland carcinoma cell line was grown in DMEM and 5% fetal calf serum (FCS; HyClone Laboratories, Inc., UT). Cell cultures were harvested during exponential growth or following appropriate treatment times. Following cell disruption, cytoplasmic fractions were used for RNA extraction (14). Released nuclei were used for protein extraction according to a previously described method (15). In some experiments, cells were serum starved for 48 h and stimulated with 5% FCS. At the indicated times after stimulation, cells were harvested for extraction of RNA and nuclear protein. Treatment with l-mimosine (Sigma, MO) was performed at a final concentration of 200 µg/ml for 10 h before as well as during serum stimulation. For cell cycle analysis, cell cultures were used at 70–80% confluence. After appropriate treatments, cultures were harvested by trypsinization, fixed in 35% ethanol, stained with propidium iodide and analyzed using a Becton Dickinson flow cytometer.
Electrophoretic Mobility Shift Assays (EMSA)
Oligonucleotides were purchased from Integrated DNA Technologies, Inc. (IDT, Inc., IA). For EMSA, double-stranded probe was labeled with polynucleotide kinase (NE Biolab, MA) and [α-32P]ATP (NEN Dupon, MA). Aliquots (∼10 µg) of protein from each nuclear extract were incubated for 30 min in binding buffer D (20 mM HEPES, pH 7.9, 20% glycerol, 100 mM KCl, 0.2 mM EDTA, 1.0 mM PMSF, 1 mM DTT) containing 0.5 nM of 5′-end-labeled probe, 1 µg of the non-specific binding inhibitor poly dI•dC (Sigma, MO) in a total volume 15 µl. In competition experiments, a 40- or 50-fold molar excess of unlabeled, double-stranded oligonucleotides were added to the reaction. The complexes formed were separated on 6–8% TBE polyacrylamide gels (Fisher Scientific, PA).
UV crosslinking
For UV crosslinking of oligonucleotide probes to specific binding proteins, EMSA reactions were subjected to UV irradiation for 30 min using a transilluminator (Fotodyne, Inc., WI). Protein–DNA complexes were boiled in sample buffer with 5% mercaptoethanol and separated by 10% SDS–PAGE. After electrophoresis, gels were dehydrated and subjected to autoradiography using Hyperfilm-MP (Amersham Life Science, Inc., IL).
Northern blot
Extracted RNA was dissolved in formamide. RNA (12 µg) was loaded on a 1.1% agarose gel containing formaldehyde. Following electrophoretic separation, RNA was transferred to the nylon membrane Hybond-N Plus (Amersham Life Science, Inc., IL) by electroblotting for 2 h at 1 mA in 25 mM sodium phosphate buffer, pH 6.5, using a transblot apparatus (BioRad, CA). The Ha-ras cDNA probe was labeled by random priming with [α-32P]dCTP (NEN Dupon, MA) and hybridized to the blotted membranes according to manufacturer’s recommendations (Amersham Life Science, Inc., IL).
Luciferase assay for Ha-ras promoter activity
Ha-ras promoter sequences were derived from the pNMU-1 plasmid (16) and inserted into the _Sma_I site of pGL2 plasmid. The wild-type promoter sequence at position –573 (CCGG) was replaced with GCGC using the Sculptor Kit (Amersham Life Science, Inc., IL) according to the manufacturer’s recommendation. The presence of the mutation within the promoter was verified by DNA sequencing. Transient transfection assays were performed using a modified method developed in our laboratory (17) to normalize for possible differences in transfection efficiencies of different DNAs. BICR-M1Rk cells were transfected in six-well plates with Lipofectamine Plus (Gibco BRL, MD) according to the manufacturer’s protocol. 8 and 24 h following addition of serum, cells were harvested, counted using a Coulter Counter (Coulter Electronics Ltd, UK) and lysed by three cycles of freezing and thawing in 25 mM Tris pH 8.0. Cytoplasmic fractions were used for the luciferase assay using standard procedure. Released nuclei were lysed in lysis buffer [1× AmplyTaq buffer II (Perkin Elmer) containing 1 mM MgCl2, 0.45% of Nonidet P-40 and 0.45% Tween 20] and digested with proteinase K (0.1 µg/µl) at 56°C for 1 h. Proteinase K was inactivated for 15 min at 94°C. To measure the transfected plasmid copy numbers in nuclei of transfected cells (17), a 2 µl aliquot of extracted DNA from each transfection was subjected to PCR amplification using 20 pM/µl of luciferase gene primers, pLZ1 (ATA CGC CCT GGT TC) and pLZ2 (CCC TGG TAA TCC GT). PCR reactions were carried out at 94°C for 35 s, 49°C for 35 s and 72°C for 40 s. Amplification was performed for eight cycles in a thermal cycler (Perkin Elmer Cetus) in the presence of 3 µCi of p32 dCTP (NEN Dupon, MA) per reaction in 25 µl total volume. Standards included DNA from untransfected cells and known copy number of the plasmid DNA. PCR products were separated on a 6% acrylamide gel (Fisher Scientific, PA) and quantitated by using a PhosphorImager (Molecular Dynamics, CA) analysis. Negative control reactions included water and cytoplasmic fraction from transfected cells. Activities of the promoter construct were plotted as luciferase values per plasmid copy number per cell number. Final results are presented as fold-activation of the wild-type or mutant promoter construct divided by the expression detected with the control pGL2 plasmid. Transfection experiments were performed at least four times using two independent plasmid preparations.
Protein purification
Protein purification was performed starting with ∼30 ml of a BICR-M1Rk wet cell pellet (800 g). Nuclear extracts were prepared as described above using 5–6 ml of cell pellet per preparation. The resulting nuclear extracts were clarified at 30 000 g and dialyzed against buffer D (see above). Each batch of nuclear extract was tested for binding activity using EMSA under the conditions described above. Biotinylated sense and unmodified antisense oligonucleotides (same as above) were obtained from Research Genetics (Huntsville, AL), annealed and attached to streptavidin-agarose (Pierce, IL) for use in affinity chromatography. Annealing of oligonucleotides was performed in excess of anti-sense strand to ensure complete annealing of the biotinylated strand. Affinity columns were similarly prepared using mutant oligonucleotides.
The individual batches of nuclear protein extracts were first incubated with poly dI•dC at 20 µg/ml to titrate non-specific DNA binding proteins, and centrifuged at 30 000 g to remove precipitates. To reduce the amount of non-specific DNA binding activity, the extracts were first passed over an affinity chromatography column generated with the mutant binding site. Column eluates were then passed over a column of the wild-type binding site to capture specific binding proteins. To ensure complete binding, the nuclear extract was passed over the column repeatedly overnight at 4°C using a peristaltic pump. Proteins bound to the wild-type column were then eluted with a 0.1 M step gradient of 0.2–1.0 M KCl. Collected fractions were dialyzed against buffer D and tested for binding activity by EMSA. All fractions showing specific binding were pooled and loaded on the specific column and eluted again as above. Positive fractions were concentrated (Millipore, MA), loaded on a 10% SDS gel and stained with Coomassie Brilliant Blue R-250 (BioRad laboratories, CA). Detected bands with estimated molecular weights of 42 and 44 kDa were excised. Protein from one-fifth of the most abundant band (42 kDa) was eluted and renatured (18). Resulting protein was used in UV crosslinking reactions to verify the presence of specific binding activity. The remainder of the 42 kDa band was used for peptide identification by capillary HPLC–mass spectrometry as described below. The 44 kDa band was forwarded to Harvard Microchemistry Laboratory directed by Dr W. Lane where protein identification was performed by microcapillary reverse-phase HPLC electrospray tandem mass spectrometry.
Protein identification by capillary HPLC–mass spectrometry
The protein band was excised from a one-dimensional preparative SDS–polyacrylamide gel and digested with 0.5 µg of Trypsin (Promega, Madison, WI). The digested peptide mixture was extracted and analyzed by a microcapillary LC system connected online to an electrospray ionization ion trap mass spectrometer (Finnigan-MAT, Model LCQ, San Jose, CA). Peptides were concentrated and separated on a micro C18 column with an inner diameter of 50 µm. Separation was accomplished by applying a gradient of 5–65% B over 20 min. The gradient was delivered by a Magic 2002 HPLC system (Michrom BioResource, Inc., Pleasanton, CA) and the flow delivered over the column was adapted with a pre-column flow splitter to 200 µl/min. Eluting peptides were introduced into the mass spectrometer by electrospray via a home built microESI ion source and analyzed by data dependent MS/MS (19,20). The collision induced dissociation spectra generated during the experiment were searched against protein as well as nucleotide databases using Sequest software to identify possible sequence matches.
RESULTS
Identification of the rat HRE
We first tested the hypothesis that the region of Ha-ras promoter around nucleotide –573, involved in the response to carcinogen treatment (13), is able to interact with proteins in vitro. This region of the Ha-ras promoter was found to include the nucleotide sequence GGAA. This sequence corresponds to the Ets transcription factor core binding site, albeit in the complimentary and inverse orientation. To determine whether any transcription factors can bind specifically to this region of the promoter, we performed EMSA using synthetic, double-stranded oligonucleotides (Fig. 1A) and nuclear extract from the BICR-M1Rk mammary carcinoma cell line. The results presented in Figure 1B provide evidence for specific binding of proteins from nuclear extracts to the HRE probe. The stable protein–DNA complexes formed were the result of sequence specific DNA binding, since a 40-fold molar excess of dsDNA probes (Mutants 1, 2 and 4) mutated within the putative Ets binding site (CCGGAA) failed to compete with wild-type probe (Fig. 1B). However, a 40-fold excess of dsDNA probes with mutation outside of the consensus CCGGAA Ets motif (Mutant 3) were as effective as the wild-type sequence in competition experiments. The human HRE possessing the same core sequence was also effective in competing for the binding activity. Unrelated promoter elements such as SP1, AP-1 and Stat5/6 binding sites failed to compete for binding to the HRE (not shown). Both the human and rat HRE probes formed similar protein(s) complexes with nuclear extracts from rat, mouse and human cells, as judged by mobility in EMSA gels (not shown).
Figure 1.

EMSA with rat HRE probe and competition experiment with mutant HRE, human HRE (hHRE) and EBS. Competitor DNAs were added in 40-fold molar excess. NE, nuclear extract from BICR-M1Rk. (A) Sequence of oligonucleotides used in the competition experiment. The conserved sequence CCGGAA found in the rat HRE and hHRE and the EBS is boxed. The summary of competition experiment results is on the right. Competition and absence of competition with the HRE probe are indicated by + and –, respectively. (B) EMSA and competition experiment with hHRE and the different mutant oligonucleotides (from 1 to 4) listed on (A). Specific DNA–protein complexes are shown with an arrow. (C) EMSA competition experiment with EBS or rat HRE in the presence of excess rat HRE or EBS respectively. C1 and C2 DNA–protein complexes are indicated. The rat HRE probe without nuclear extract added is not shown.
To determine if the proteins bound to the rat HRE were members of the Ets transcription factor family, we performed competitive binding experiments with an oligonucleotide probe comprising the binding site for the Drosophila melanogaster E74 Ets transcription factor. The E74 probe (EBS) forms two distinct EMSA complexes (C1 and C2) with mammary cell nuclear extracts (Fig. 1C). The rat HRE probe was able to compete effectively with the E74 probe, although the rat HRE affinity for protein complex C1 was higher than for the complex C2 (Fig. 1C). In the inverse experiments (Fig. 1C), labeled rat HRE probe formed predominantly complex C1, while complex C2 was very weak or undetectable. An excess of unlabeled oligonucleotide corresponding to the E74 binding site efficiently abrogated rat HRE binding. The latter result is consistent with the notion that the proteins bound to the rat HRE could be members of the Ets protein family, or at least compete for binding to the same DNA sequences.
To investigate the role of rat HRE in the context of the promoter in vivo, we performed a transient transfection assay using wild-type and mutant rat Ha-ras promoter linked to the luciferase reporter gene. A double mutation that disrupts the Msp1 site at position –573 was introduced into the HRE using the mutant oligonucleotide (Mutant 2; see Fig. 1A). In transient transfection experiments in BICR-M1Rk cells the wild-type promoter showed strong (33-fold) activation 8 h following serum stimulation relative empty vector, and a 3.9-fold increase in activity relative to the mutant promoter (Fig. 2). At 24 h after serum stimulation the activity of the wild-type promoter was lower compared to an 8 h time point, but it remained 3-fold more active compared to mutant promoter. The activity of the mutant promoter at 24 h was only slightly higher than the activity of the empty vector. Thus, in the context of the whole promoter, the HRE has a strong positive effect on the rat Ha-ras promoter activity in mammary cells and correlates with increased HRE binding activity following serum stimulation (see Fig. 6).
Figure 2.

Wild-type Ha-ras promoter is 3–3.9-fold more active compared to the mutant promoter. BICR-M1Rk cells were transfected with 1 µg of plasmid DNA in serum free conditions and luciferase activity was measured after 8 and 24 h following serum stimulation. Normalization for transfection efficiency was performed as described in Materials and Methods. At the top of the figure is a schematic presentation of the constructs showing wild-type and mutant sequences in the Ha-ras promoter linked to the luciferase gene. Error bars represent standard deviations.
Figure 6.
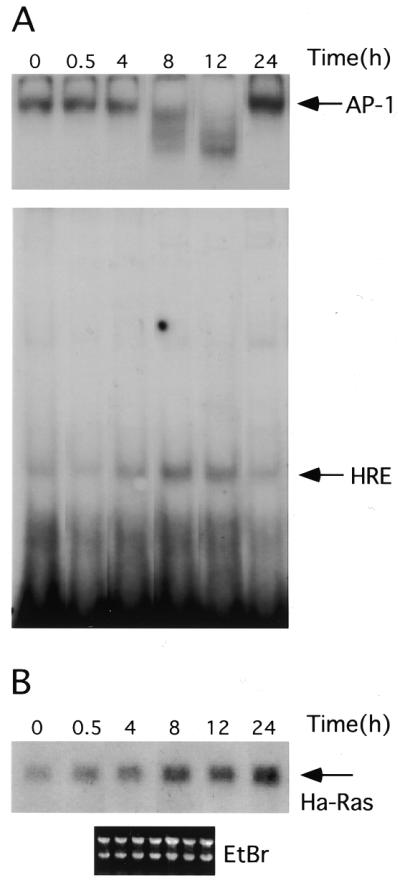
HRE–CBF-A binding activity correlates with Ha-ras mRNA expression. Serum deprived cells were stimulated with 5% calf serum. RNA and nuclear proteins were extracted at indicated time points from the same culture (see Materials and Methods for details). (A) Nuclear proteins were used in EMSA with AP-1 (top, only DNA–protein complexes are shown with arrow) and rat HRE probes (bottom). (B) RNAs extracted from cells at the same time points following serum stimulation were separated on the 1.1% agarose gel, blotted and probed with rat Ha-ras cDNA. Bottom, ethidium bromide staining of the membrane following RNA transfer demonstrates equal RNA loading. For every time point shown on the figure we also performed controls using nuclear extract (A) and RNA (B) from serum deprived cells harvested at a given time. The level of binding activity to the AP-1 and HRE probes and the level of Ha-ras expression did not differ from the zero time points in these samples. For simplicity these controls were removed from the final figure using image analysis software.
We next used UV irradiation to cross-link the specific binding proteins to the DNA probe in order to estimate the approximate molecular weight of the specific binding protein. Analyses of the cross-linked products by SDS–polyacrylamide gel electrophoresis demonstrated that the protein bound to the DNA probe has an estimated molecular weight of ∼51–52 kDa. Assuming the probe bound to the protein was single-stranded, the latter result suggested that the protein alone is ∼43–44 kDa in size (data not shown and see Fig. 3B).
Figure 3.

(A) Coomassie Brilliant Blue stained gel following purification of the DNA binding protein by affinity chromatography. Migration of the molecular weight standards is shown on the left. The most abundant species are labeled p42 and p44. Bands of lower intensity which were also subjected to protein identification by micro-HPLC–mass spectrometry are indicated by short arrows. (B) Comparison of radiolabeled, UV crosslinked HRE–protein complexes from whole nuclear extract (NE) with the HRE–p42 complex. The p42 protein was recovered from gel shown on (A). BSA, bovine serum albumin.
Taken together, these experiments suggested that the HRE from rat and human Ha-ras promoter is a specific binding site for an Ets related transcription factor present in mammary cell lines. However, Ets-1 and -2 antibodies (21) or Ets 1/Ets 2 antibodies (Santa Cruz, CA) designed to recognize a broad spectrum of Ets related proteins failed to super-shift the complexes formed between the HRE oligonucleotide probe and the mammary cell nuclear extracts (not shown). These results suggested that the HRE binding proteins in mammary cells were either novel members of the Ets transcription factor family or unrelated proteins that recognize the same DNA sequences as Ets proteins. We therefore performed experiments to identify and clone the HRE binding proteins.
Purification of the protein and protein identification
The HRE binding protein was extensively purified using sequence specific DNA affinity chromatography as described in Materials and Methods. Bound proteins were eluted from the column with a KCl step gradient and fractions assayed for HRE binding by EMSA. Most of the binding activity eluted in 0.7–1.0 M KCl (not shown). Fractions with binding activity were pooled, concentrated and analyzed on a 12% SDS gel. Two main bands with estimated molecular weights of 42 and 44 kDa were detected in the fractions with binding activity (Fig. 3A). To confirm that the most abundant protein band contained the HRE binding protein, the 42 kDa band was excised from a Coomassie Brilliant Blue stained gel and protein was eluted from one-fifth of the gel slice. Following renaturation, the eluted proteins were incubated with radiolabeled HRE probe and any resulting protein–DNA complexes were crosslinked with UV light. The protein eluted from 42 kDa bands formed a stable complex with the HRE probe that was indistinguishable from the crosslinked complexes formed with the protein in total nuclear extract as shown by SDS–PAGE (Fig. 3B). Since the amount of the protein in the 44 kDa band was significantly lower than in the former band, we did not perform crosslinking experiments to conserve protein for further analysis. In summary, the crosslinking experiment above showed that the protein in the 42 kDa protein preparation contained a HRE binding protein whose molecular weight is consistent with that estimated by UV crosslinking.
Independent protein analyses identified similar polypeptides present in both the 42 and 44 kDa proteins (summarized in Table 1). The polypeptides identified corresponded to sequences detected in the previously identified mouse CBF-A (22). Together the polypeptides identified in our analysis encompassed almost 36% of the CBF-A amino acid sequence. Moreover, MS/MS analysis of these two and three additional bands with low intensity staining from the preparative protein gel (Fig. 3A, short arrows) failed to detect any Ets related proteins (data not shown). Together these results suggested that the CBF-A or a closely related protein was responsible for most of the HRE binding activity detected in mammary cells.
Table 1. List of the overlapping polypeptides identified by micro HPLC–mass spectrometry of the 42 and 44 kDa proteins isolated by affinity chromatography.
| MFVGGLSWDTSK |
|---|
| MFVGGLSWDTSKK |
| MFVGGLSWDTSKKDLKDYFTK |
| DLKDYFTK |
| SRGFGFILFK |
| GFGFILFK |
| IFVGGLNPEATEEK |
| IFVGGLNPEATEEKIR |
| GGLNPEATEEK |
| IREYFGQFGEIEAIELPIDPK |
| EYFGQFGEIEAIELPIDPK |
| GFVFITFKEEDPVKK |
| GFVFITFKEEDPVK |
| FHTVSGSK |
| EVYQQQQYGSGGR |
CBF-A interacts with rat HRE with greater affinity as compared to CArG-box
To verify that CArG binding protein interacts with rat and human HRE, we employed an anti-CBF-A antibody kindly provided by T. Leandersson (Lund University, Sweden: 23). First, we verified that the polyclonal antibody against mouse CBF-A would cross-react with the rat protein. Rat CBF-A was cloned by PCR from BICR-M1Rk cells, in vitro translated and detected by western blot (not shown). EMSA demonstrated that the antibody completely and specifically abrogated the interaction of nuclear protein with the rat (Fig. 4A) or human (not shown) HRE probes. These results provided direct evidence that CBF-A was responsible for the HRE binding activity observed in mammary cells.
Figure 4.

(A) Anti CBF-A antibody completely abrogated protein binding to the rat HRE and human HRE (not shown) probes. EMSA was performed as described in Materials and Methods. Anti CBF-A antibody or normal serum were added as indicated, + and –, respectively. NE, nuclear extract from BICR-M1Rk cells. (B) Comparison of the binding affinity of CBF-A to the rat HRE and CArG box. Competitor oligonucleotides, rat HRE, CArG and EBS were added to the binding reaction in 50-fold molar excess. Specific HRE–protein complex are indicated with arrow. (C) Comparison of rat HRE probe, EBS and CArG box oligonucleotide sequences. Conserved sequence is boxed.
We next compared the relative affinities of CBF-A protein for rat HRE and the CArG-box originally described as its recognition sequence. A 50-fold molar excess of unlabeled, double-stranded oligonucleotides corresponding to CArG box (22), rat HRE or an Ets binding site (EBS) were used as competitors in binding experiments using labeled rat HRE probe and analyzed by EMSA. While the addition of 50-fold molar excess of cold rat HRE and EBS efficiently diminished DNA–protein binding, addition of same molar excess of the CArG box oligomer demonstrated only partial competition with the rat HRE (Fig. 4B). The latter results suggested that CBF-A binds to the HRE with higher affinity than the CArG box originally used to isolate the CBF-A. Similarly, CBF-A is able to bind the EBS with higher affinity than the CArG box. From these competition experiments (also see Fig. 1), we suggest that sequence CCGGAA is important for high affinity CBF-A binding to DNA.
In a previous study it was shown that antibody against CBF-A recognizes two protein species in cell extracts by western blot (23). However, the authors demonstrated that only the lower molecular weight protein was found to interact with the A-T rich region of the pd element within the SP6 k promoter. To determine if rat HRE interacts with one or both protein species, we probed a western blot containing protein fractions eluted from affinity column (see Materials and Methods for details) with the CBF-A antibody on the western blot. Figure 5 demonstrates that the same two protein species found in the nuclear extract of mammary cells (p44 and p42) are detected in protein fractions eluted from the affinity column. However, protein ratio (p44/p42) is reduced compared to nuclear extract before loading on the column. In our hands, under normal growth conditions this ratio is 0.5–1.2. This experiment together with microcapillary reverse-phase HPLC electrospray tandem mass spectrometry provided the evidence that slower migrating protein (44 kDa) is related to CBF-A.
Figure 5.
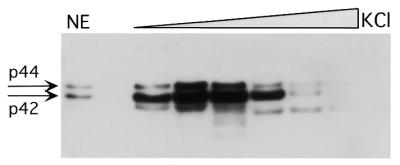
Two protein species (p42 and p44) interact with rat HRE. The western blot was performed with the CBF-A antibody against HRE binding proteins eluted from two sequential affinity columns with 0.1 M KCl step gradient. Aliquots (15 µl) from each dialyzed fraction were used. NE, nuclear extract before loading on the affinity column. The two protein bands, p42 and p44, are indicated by arrows.
Establishing a correlation between CBF-A binding to HRE and Ha-ras mRNA expression
Previous studies have demonstrated that serum stimulation of cells arrested in G0 leads to induction of Ha-ras expression (24). To establish a correlation between HRE binding activity and Ha-ras mRNA expression, BICR-M1Rk cells were serum deprived for 48 h to induce growth arrest, and then stimulated with serum. To control for activities among different preparations of extracted nuclear proteins, we assayed each extract for serum responsive binding activity to the AP-1 recognition sequence. At the specified time points following serum induction, cells were harvested for extraction of RNA and nuclear proteins. The AP-1 binding was observed in serum starved cells (Fig. 6) and was increased by 24 h post induction. Serum transiently stimulated binding to the HRE between 4 and 12 h, and decreased by 24 h. Interestingly, binding to the rat HRE was maximal at 8 and 12 h after treatment, at which time AP-1 binding activities were transiently reduced to somewhat lower levels. Reasons for modification of the AP-1 binding remain unknown and beyond the scope of this paper. As expected, serum stimulated Ha-ras mRNA expression (Fig. 6B), and the increase in RNA levels detected at 8 h, corresponded with the time of maximal CBF-A binding to the HRE. Reduced CBF-A–HRE binding at the 24 h time point suggested that Ha-ras expression at later time points following serum stimulation depend on other transcription factors and/or RNA stability.
To expand this observation we stimulated cells with serum in the presence or absence of l-mimosine, a p53 independent inducer of the cyclin dependent kinase inhibitor, p21 waf-1 (25). Induction of p21 waf-1 by l-mimosine was confirmed by western blot (not shown). As expected from the experiment above, treatment of cells with serum in the absence of l-mimosine, induced binding of CBF-A to the HRE (Fig. 7A), and resulted in elevated Ha-ras mRNA expression (Fig. 7B) by 12 h after serum stimulation. At the same time, we observed accumulation of cells in the S phase of the cell cycle, with a concomitant decrease in the G1/S ratio (Fig. 7C). l-mimosine treatment abrogated stimulation of CBF-A binding to the HRE sequence, and the levels of Ha-ras mRNA expression in cells stimulated with serum in the presence of l-mimosine were significantly lower compared to levels in cells stimulated with serum only (Fig. 7B). As predicted, in the presence of l-mimosine, cells were arrested in G1 phase resulting in an increased G1/S ratio. Together, these results demonstrated a correlation between increased CBF-A binding to the rat HRE and stimulation of Ha-ras expression.
Figure 7.
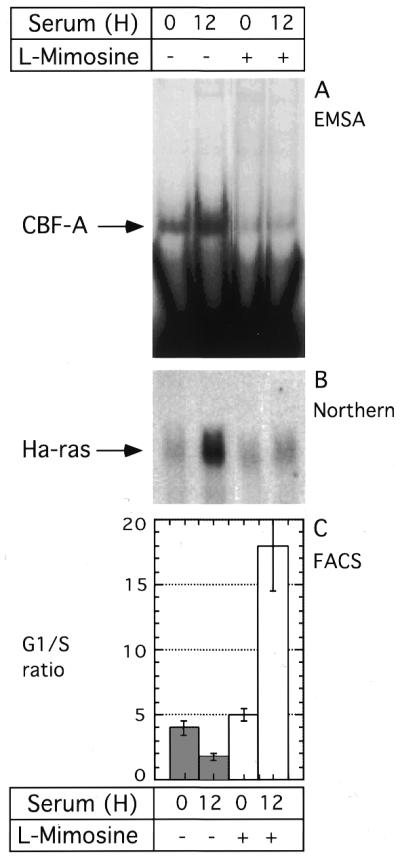
l-mimosine inhibits CBF-A binding to the rat HRE and expression of the Ha-ras mRNA. Serum deprived cells were stimulated with serum or serum plus l-mimosine (200 µg/ml). 12 h later, nuclear proteins and RNA were extracted from the same culture. At each time point, a duplicate cell culture was taken for cell cycle analysis by fluorescence activated cell sorting. (A) EMSA of the CBF-A binding to rat HRE probe. (B) Northern blot analysis of the RNA probed with Ha-ras cDNA. Equal loading was verified by ethidium bromide staining of RNA following transfer to the membrane (not shown). (C) Cell cycle analysis shown the average G1/S ratio from three independent measurements. Error bars represent standard deviations. Open and closed bars, cells untreated and treated with l-mimosine, respectively.
DISCUSSION
In our previous study we found that the rat Ha-ras promoter between positions –582 and –563 is involved in response to specific carcinogen treatment (13). In the present study we asked if this region of the Ha-ras promoter can bind specifically with any _trans_-acting transcription factor and if the binding of factors can regulate expression of the oncogene in vitro. We determined that double-stranded oligodeoxynucleotides corresponding to the rat HRE were able to bind specifically with transcription factors present in nuclear extracts from a rat mammary tumor cell line. Our result suggested that rat and human HRE interact with members of the Ets transcription factor family. HRE–protein binding appears to be highly conserved among different species since similar binding activities were detected in nuclear extracts from a variety of rat, mice and human cell lines using either the human or rat HRE elements as probe (not shown).
Ets phosphoproteins play an important role in the control of cell growth and development (26–30). Ets binding sites have been identified in several oncogene responsive promoters (28,29,31). A number of studies have shown that Ets related transcription factors may play an important role in ras mediated signal transduction and involved in regulation of a number of genes downstream of Ras (28). It is thus reasonable to posit that Ets related proteins, or proteins that compete with Ets proteins for specific binding sites, could play an important role in Ha-ras mediated cell transformation.
In transient transfection assay we found that at 8 h after serum stimulation wild-type promoter construct showed strong 33-fold higher luciferase activity compared to construct lacking the HRE. The activity of the wild-type promoter in transient transfection assay correlated well with binding activity of the CBF-A to the target sequence (Figs 2 and 6). We concluded that despite the different relative positions of HRE within rat and human Ha-ras promoters, they are functionally equivalent. Our results are consistent with others showing that deletion of HRE from human Ha-ras promoter results in 2-fold drop in the promoter activity following transfection in HeLa cells (12). To further characterize the HRE binding proteins from mammary cells, we employed affinity purification of the protein followed by protein identification by micro HPLC–mass spectroscopy. Sequence analysis of the two most abundant proteins, with approximate molecular weights of 42 and 44 kDa, unexpectedly matched sequences corresponding to mouse CBF-A. We confirmed interaction of CBF-A with rat and human HRE in EMSA using CBF-A specific antibody. Our results also provided evidence that the slower migrating protein species (p44) detected on the western blot is a CBF-A related protein. The p44 is able to interact with the DNA target, although with lower affinity compared to the p42. We support the suggestion that the slower migrating protein is a post-translationally modified form of the CBF-A protein or an RNA splice form of slightly higher molecular weight (23). Identified peptides encompassed almost 36% of the CBF-A amino acid sequence suggesting a high level of homology between mouse and rat proteins. Indeed, rat and mouse cDNA for CBF-A are very conserved (S.Kamada and T.Miwa, EMBL accession no. D90151; A.Mikheev, L.Jing and H.Zarbl, EMBL accession no. AF216753). In an attempt to detect any putative Ets related protein, we performed sequence analysis of additional bands of very low intensity detected on preparative Coomassie Blue stained gel. We failed to detect polypeptides corresponding to Ets related proteins. Together these results suggest that Ets related proteins are probably not involved in interaction with HRE site of the rat Ha-ras promoter in BICR-M1Rk mammary carcinoma cell line and that CBF-A is indeed the major binding HRE factor in these cells.
The CBF-A was discovered by screening an expression library with the CArG box DNA fragment as a probe (22). The CArG box sequence was initially described in a number of genes showing muscle tissue specific expression (32–40). For example, it was shown that the serum response factor (SRF) can interact with the CArG box and activate transcription of muscle-specific genes and immediate-early genes, such as fos (33,38,41). CBF-A is a protein with a calculated molecular weight of 31 kDa and migrates with an apparent molecular weight of 42 kDa in SDS–PAGE. The protein has an RPN domain that is thought to be involved in the binding to nucleic acid (42). The RPN domain is common to heterogeneous nuclear ribonucleoproteins (hnRPN) A/C types involved in splicing, transport and protection of RNA (43). CBF-A was initially found to be a transcriptional repressor (22). However, our study shows that CBF-A is a transcriptional activator of Ha-ras in transient transfection assays. The discrepancy with published results is not surprising since the CArG regulatory element can interact with a number of other transcription factors, including Ets related factors Elk-1 and SAP-1 (44,45), E12, NF-IL-6 (46) and HMG-I family proteins (47). It is therefore plausible that CBF-A complexes with, or replaces, other transcriptional factors depending on the context of the CArG box. For example, functional antagonism between SRF and YY1 proteins at CArG elements has been described (48). Likewise, studies have demonstrated that protein–protein interactions affect transcription from CArG box (47). For example, in the Arabidopsis APETALA3 gene, individual CArG boxes within a tandem array of three, have opposite regulatory effects on the promoter activity (40). While the first two CArG boxes are positive regulatory elements, the third has a negative effect on the promoter activity.
It was also noted previously that CBF-A is able to interact with single-stranded DNA (22). Binding of CBF-A to the single-stranded form of the A-T rich pd element is stronger compared to the double-stranded form (23). In our experiment we failed to detect any single-stranded DNA binding activity (not shown). We speculate that CBF-A may demonstrate different functional specificity, depending on the affinity of the interaction with target DNA and/or interactions with other factors. CBF-A modulation of transcription from the CarG, as well as other elements, may therefore be gene and cell type specific.
In our competition experiments, the affinities of CBF-A for the rat HRE and Ets binding sites (E74) were clearly higher than its affinity for the interaction with the CArG box, CC(AT)6GG. Comparison of human and rat HRE, E74 and CArG box sequences suggests that the sequence CCGGAA is important for high affinity binding of CBF-A to DNA. Since this sequence is frequently present in a number of binding sites for Ets proteins, we suggest a potential role of CBF-A in the regulation of Ets responsive promoters. Our suggestion is supported by the fact that CBF-A is able to bind Ets related proteins in vitro (23). The functional significance of CBF-A and Ets protein interaction is not clear. Since a number of Ets proteins are involved in the regulation of different genes, the role of CBF-A may be widespread. CArG binding factor A was found to be overexpressed in NIH 3T3 cells transformed with _ets_-1 and _ets_-2 genes (49). This result suggests possible co-operation of Ets proteins and CBF-A in cell transformation. Our preliminary results suggested that the CBF-A protein undergoes posttranslational modification which is required for binding activity. Ectopic overexpression of the CBF-A is not sufficient to induce efficient interaction to the target DNA and fails to induce Ha-ras expression.
In summary our results demonstrated the high affinity interaction of the CArG binding factor (CBF-A) with the HRE present in the Ha-ras promoters of both rodents and humans. Furthermore, the HRE is a strong positive regulatory element in the Ha-ras gene. Contrary to expectations, we did not find any Ets related proteins capable of high affinity binding to the HRE in mammary cells. The correlation of CBF-A binding to the HRE and Ha-ras mRNA expression suggests that CBF-A may be involved in control of cell cycle and carcinogenesis in mammary cells.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to T. Leandersson for anti-CBF-A antibody, Arun Seth for generous gifts of Ets-1 and Ets-2 antibody, P. Lampe, A. McShea and Sherry McLaughlin for help and encouragement. The US Army Medical Research and Materiel Command under DAMD17-98-1-8086, and the National Science Foundation, Science and Technology Center for Molecular Biotechnology and NIH Research Resource for Comprehensive Biology supported this work.
DDBJ/EMBL/GenBank accession no. AF216753
REFERENCES
- 1.Downward J. (1997) Curr. Biol., 7, R258–R260. [DOI] [PubMed] [Google Scholar]
- 2.Lloyd A.C. (1998) Curr. Opin. Genet. Dev., 8, 43–48. [DOI] [PubMed] [Google Scholar]
- 3.Hua V.Y., Wang,W.K. and Duesberg,P.H. (1997) Proc. Natl Acad. Sci. USA, 94, 9614–9619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finney R.E. and Bishop,J.M. (1993) Science, 260, 1524–1527. [DOI] [PubMed] [Google Scholar]
- 5.Yaginuma Y., Yamashita,K., Kuzumaki,N., Fujita,M. and Shimizu,T. (1992) Gynecol. Oncol., 46, 45–50. [DOI] [PubMed] [Google Scholar]
- 6.Bredel M. and Pollack,I.F. (1999) Brain Res. Brain Res. Rev., 299, 232–249. [DOI] [PubMed] [Google Scholar]
- 7.Gohring U.J., Schöndorf,T., Kiecker,V.R., Becker,M., Kurbacher,C. and Scharl,A. (1999) Tumour Biol., 20, 173–183. [DOI] [PubMed] [Google Scholar]
- 8.Chin L., Tam,A., Pomerantz,J., Wong,M., Holash,J., Bardeesy,N., Shen,Q., O’Hagan,R., Pantginis,J., Zhou,H., Horner,J.W.,II., Cordon-Cardo,C., Yancopoulos,G.D. and DePinho,R.A. (1999) Nature, 400, 468–472. [DOI] [PubMed] [Google Scholar]
- 9.Clark G.J. and Der,C.J. (1995) Breast Cancer Res. Treat., 35, 133–144. [DOI] [PubMed] [Google Scholar]
- 10.McCormick F. (1995) Mol. Reprod. Dev., 42, 500–506. [DOI] [PubMed] [Google Scholar]
- 11.Damante G., Filetti,S. and Rapoport,B. (1987) Proc. Natl Acad. Sci. USA, 84, 774–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee W. and Keller,E.B. (1991) J. Mol. Biol., 220, 599–611. [DOI] [PubMed] [Google Scholar]
- 13.Jin Z., Houle,B., Mikheev,A.M., Cha,R. and Zarbl,H. (1996) Cancer Res., 56, 4927–4935. [PubMed] [Google Scholar]
- 14.Chomczynski P. and Sacchi,N. (1987) Anal. Biochem., 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 15.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sukumar S., Notario,V., Martin-Zanca,D. and Barbacid,M. (1983) Nature, 306, 658–661. [DOI] [PubMed] [Google Scholar]
- 17.Bahramian M.B. and Zarbl,H. (1994) PCR Methods Appl., 4, 145–153. [DOI] [PubMed] [Google Scholar]
- 18.Sealy L., Malone,D. and Pawlak,M. (1997) Mol. Cell. Biol., 17, 1744–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ducret A., Van Oostveen,I., Eng,J.K., Yates,J.R.,III and Aebersold,R. (1998) Protein Sci., 7, 706–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gygi S., Han,D., Gingras,A., Sonenberg,N. and Aebersold,R. (1999) Electrophoresis, 20, 310–319. [DOI] [PubMed] [Google Scholar]
- 21.Venanzoni M.C., Robinson,L.R., Hodge,D.R., Kola,I. and Seth,A. (1996) Oncogene, 12, 1199–1204. [PubMed] [Google Scholar]
- 22.Kamada S. and Miwa,T. (1992) Gene, 119, 229–236. [DOI] [PubMed] [Google Scholar]
- 23.Bemark M., Olsson,H., Heinegård,D. and Leanderson,T. (1998) J. Biol. Chem., 273, 18881–18890. [DOI] [PubMed] [Google Scholar]
- 24.Kelleher M.D., Naureckas,E.T., Solway,J. and Hershenson,M.B. (1995) Am. J. Respir. Cell Mol. Biol., 12, 19–26. [DOI] [PubMed] [Google Scholar]
- 25.Alpan R.S. and Pardee,A.B. (1996) Cell Growth Differ., 7, 893–901. [PubMed] [Google Scholar]
- 26.Bradford A.P., Conrad,K.E., Wasylyk,C., Wasylyk,B. and Gutierrez-Hartmann,A. (1995) Mol. Cell. Biol., 15, 2849–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wasylyk B., Hahn,S.L. and Giovane,A. (1993) Eur. J. Biochem., 211, 7–18. [DOI] [PubMed] [Google Scholar]
- 28.Wasylyk B., Hagman,J. and Gutierrez-Hartmann,A. (1998) Trends Biochem. Sci., 23, 213–216. [DOI] [PubMed] [Google Scholar]
- 29.Sharrocks A.D., Brown,A.L., Ling,Y. and Yates,P.R. (1997) Inter. J. Biochem. Cell Biol., 29, 1371–1387. [DOI] [PubMed] [Google Scholar]
- 30.Oikawa T., Yamada,T., Kihara-Negishi,F., Yamamoto,H., Kondoh,N., Hitomi,Y. and Hashimoto,Y. (1999) Cell Death Differ. 6, 599–608. [DOI] [PubMed] [Google Scholar]
- 31.Galang C.K., García-Ramírez,J., Solski,P.A., Westwick,J.K., Der,C.J., Neznanov,N.N., Oshima,R.G. and Hauser,C.A. (1996) J. Biol. Chem., 271, 7992–7998. [DOI] [PubMed] [Google Scholar]
- 32.Catala F., Wanner,R., Barton,P., Cohen,A., Wright,W. and Buckingham,M. (1995) Mol. Cell. Biol., 15, 4585–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galvagni F., Cartocci,E. and Oliviero,S. (1998) J. Biol. Chem., 273, 33708–33713. [DOI] [PubMed] [Google Scholar]
- 34.Mack C.P. and Owens,G.K. (1999) Circ. Res., 84, 852–861. [DOI] [PubMed] [Google Scholar]
- 35.Madsen C.S., Regan,C.P. and Owens,G.K. (1997) J. Biol. Chem., 272, 29842–29851. [DOI] [PubMed] [Google Scholar]
- 36.Molkentin J.D., Jobe,S.M. and Markham,B.E. (1996) J. Mol. Cell. Cardiol., 28, 1211–1225. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu R.T., Blank,R.S., Jervis,R., Lawrenz-Smith,S.C. and Owens,G.K. (1995) J. Biol. Chem., 270, 7631–7643. [DOI] [PubMed] [Google Scholar]
- 38.Soulez M., Tuil,D., Kahn,A. and Gilgenkrantz,H. (1996) Biochem. Biophy. Res. Commun., 219, 418–422. [DOI] [PubMed] [Google Scholar]
- 39.Zilberman A., Dave,V., Miano,J., Olson,E.N. and Periasamy,M. (1998) Circ. Res., 82, 566–575. [DOI] [PubMed] [Google Scholar]
- 40.Tilly J.J., Allen,D.W. and Jack,T. (1998) Development, 125, 1647–1657. [DOI] [PubMed] [Google Scholar]
- 41.Martin K.A., Gualberto,A., Kolman,M.F., Lowry,J. and Walsh,K. (1997) DNA Cell Biol., 16, 653–661. [DOI] [PubMed] [Google Scholar]
- 42.Gorlach M., Wittekind,M., Beckman,R.A., Mueller,L. and Dreyfuss,G. (1992) EMBO J., 11, 3289–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dreyfuss G., Matunis,M.J., Piñol-Roma,S. and Burd,C.G. (1993) Annu. Rev. Biochem., 62, 289–321. [DOI] [PubMed] [Google Scholar]
- 44.Hipskind R.A., Rao,V.N., Mueller,C.G., Reddy,E.S. and Nordheim,A. (1991) Nature, 354, 531–534. [DOI] [PubMed] [Google Scholar]
- 45.Dalton S. and Treisman,R. (1992) Cell, 68, 597–612. [DOI] [PubMed] [Google Scholar]
- 46.Metz R. and Ziff,E. (1991) Oncogene, 6, 2165–2178. [PubMed] [Google Scholar]
- 47.Chin M.T., Pellacani,A., Wang,H., Lin,S.S., Jain,M.K., Perrella,M.A. and Lee,M.E. (1998) J. Biol. Chem., 273, 9755–9760. [DOI] [PubMed] [Google Scholar]
- 48.Gualberto A., LePage,D., Pons,G., Mader,S.L., Park,K., Atchison,M.L. and Walsh,K. (1992) Mol. Cell. Biol., 12, 4209–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robinson L., Panayiotakis,A., Papas,T.S., Kola,I. and Seth,A. (1997) Proc. Natl Acad. Sci. USA, 94, 7170–7175. [DOI] [PMC free article] [PubMed] [Google Scholar]