The lymphangiogenic growth factors VEGF-C and D are ligands for the integrin α9β1 (original) (raw)
. Author manuscript; available in PMC: 2006 Feb 15.
Published in final edited form as: J Biol Chem. 2004 Dec 6;280(6):4544–4552. doi: 10.1074/jbc.M412816200
Summary
Mice homozygous for a null mutation of the integrin α9 subunit die 6–12 days after birth from bilateral chylothoraces suggesting an underlying defect in lymphatic development. However, until now the mechanisms by which the integrin α9β1 modulates lymphangiogenesis have not been described. In this study we show that adhesion to and migration on the lymphangiogenic vascular endothelial growth factors (VEGF)-C and D are α9β1 dependent. Mouse embryonic fibroblasts and human colon carcinoma cells (SW-480) transfected to express α9β1 adhered and/or migrated on both growth factors in a concentration-dependent fashion, and both adhesion and migration were abrogated by anti-α9β1 function blocking antibody. In SW-480 cells, which lack cognate receptors for VEGFC and D, both growth factors induced α9β1-depedent Erk and paxillin phosphorylation. Human microvascular endothelial cells, which express both α9β1 and VEGF-R3, also adhered to and migrated on both growth factors, and both responses, were blocked by anti-α9β1 antibody. Furthermore, in a solid phase binding assay recombinant VEGF-C and D bound to purified α9β1 integrin in a dose- and cation-dependent fashion showing that VEGF-C and VEGF-D are ligands for the integrin α9β1. The interaction between α9β1 and VEGF-C and/or D may begin to explain the abnormal lymphatic phenotype of the α9 knockout mice.
Introduction
Integrins are heterodimeric transmembrane proteins, which serve as receptors for a variety of spatially fixed extracellular ligands (1). By virtue of their dual roles in adhesion and signaling, and because of their close association with the actin cytoskeleton, integrins play important roles in regulating cell shape and cell migration (2,3). In vertebrates there are 8 identified integrin β subunits and 18 α subunits that form at least 25 different heterodimers (4). The integrin α9 subunit forms a single heterodimer with β1 and is expressed in epithelial cells, smooth and skeletal muscle, neutrophils and a subset of endothelial cells (5,6). In vitro, the principal demonstrated function of α9β1 is acceleration of cell migration, an effect that depends on unique sequences within the α9 cytoplasmic domain (7,8).
In a previous study we inactivated the α9 subunit in mice, in order to better understand the function of α9β1. In these mice lymph leaked into the pleural space (chylothorax) and the mice died 6–12 days after birth. This phenotype was an unexpected finding which indicated that lymph vessel development and/or function was abnormal (9). On gross inspection, the thoracic duct and peripheral lymphatic vessels were present, but their integrity was compromised, as evidenced by chylothoraces and edema of the thoracic dermis, skeletal muscle and pleural surface. To date the molecular mechanisms underlying α9β1’s role in lymphatic development and/or function remain unexplained.
The vascular endothelial growth factors (VEGF)1-C and D are important mediators of lymphatic development (10,11). VEGF-C and D constitute a subfamily of VEGF proteins characterized by 48% overall homology, receptor specificity (VEGF-R3) and highly homologous cysteine-rich C-terminal regions (11,12). These growth factors are secreted as pro-proteins and, after enzymatic cleavage to their mature form, (13–15) signal through VEGF receptors 2 (VEGF-R2) and 3 (VEGF-R3), inducing angiogenesis and lymphangiogenesis, respectively (16–22). The importance of these VEGF proteins in lymphangiogenesis was demonstrated by their transgenic over expression in skin, resulting in dermal lymphatic hyperplasia which could be blocked by soluble VEGF-R3-Ig (23).
Therefore, in this study, we hypothesized that the lymphatic abnormality in α9 knockout mice could be explained by an interaction between α9β1 and VEGF-C and/or D. In order to address this question we used α9-transfected cell lines and primary microvascular endothelial cells to assess in vitro cell adhesion, migration and receptor signaling and purified α9β1 and VEGF-C or D protein for solid phase binding assays.
We found that α9-transfected cells and primary microvascular endothelial cells, which endogenously express α9β1, utilize α9β1 to adhere to and migrate on VEGF-C and D. This effect was inhibited by the specific α9β1-blocking antibody, Y9A2 and siRNA silencing of α9β1 protein expression. Furthermore, VEGF-C and D directly bound to α9β1 in a solid phase protein binding assay and activated α9β1 signaling, as evidenced by Erk 1/2 and paxillin phosphorylation that was inhibited by anti-α9β1 antibody. These novel findings therefore identify the growth factors VEGF-C and D as ligands for α9β1, and provide a potential explanation for the abnormal lymphatic phenotype of the α9 knockout mouse.
Experimental Procedures
Materials
Human VEGF-C and D were purchased from R&D Systems. Rabbit anti-human antibody to VEGF-C was purchased from IBL (Gunma, Japan). Rabbit polyclonal antibody to VEGF-R3 (M-20) and rabbit anti-human antibody to VEGF-D (sc-13085) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-VEGF-R3 antibody, 9D9f9 was a kind gift of Dr K. Alitalo (University of Helsinki, Helsinki, Finland). The VEGF-R3 blocking chemical, MAZ-51 was purchased from Alexis Biochemicals (San Diego, CA). Rabbit polyclonal anti-paxillin, anti-paxillin pY31 and anti-Erk1/2 pTpY185/187 were purchased from Biosource (Camarillo, CA). Mouse anti-Erk2 was purchased from BD Biosciences (San Jose, CA) and anti-phosphotyrosine antibody (4G10) was purchased from Upstate (Lake Placid, NY). Peroxidase-conjugated goat anti-rabbit, goat anti-mouse IgG, phycoerythrin conjugated goat anti-mouse and biotinylated rat anti-mouse antibodies and streptavidin-HRP were purchased from Jackson Immunoresearch Laboratories (West Grove, PA).
Cells and Cell Culture
α9 and mock-transfected mouse embryonic fibroblasts (MEF) and SW-480 cells were made as previously described (8,24). Cells were grown in Dulbecco’s minimum essential medium (DMEM) supplemented with 10% fetal calf serum (FCS, HyClone, UT), 1% penicillin/streptomicin (Sigma, St Louis, MO) and stable clones maintained with 2.5 μg/mL puromycin (Invitrogen, Carlsbad, CA; MEF,) or 1 mg/mL G418 (Sigma, SW-480). Primary adult human dermal microvascular endothelial cells (HMVEC, Cambrex, East Rutherford, NJ) were grown in cell specific growth factor-supplemented nutrient media (Cambrex, EBM-2). Schneider Drosophila S2 cells (Invitrogen), used for production of recombinant VEGF-C and D proteins were grown in Schneider S2 media (Invitrogen) supplemented with 10% fetal calf serum, 1% penicillin/streptomicin at 27°C and no CO2. Stable clones were maintained in 25 μg/mL blasticidin (Invitrogen).
Synthesis and Purification of VEGF-C and D
The cDNA’s encoding the fully processed and active forms of VEGF-C (Accession No. NM-005429) and D (Accession No. NM-004469) were expressed in Schneider Drosophila S2 cells (Invitrogen). The following primers were used to introduce 5’ NcoI and 3’ XbaI restriction sites for cloning into the multiple cloning site of the insect expression vector pCoBlast/MT/BiP/V5-His (Invitrogen): VEGF-D: 5’: 5’-gcagccatggtttgcggcaactttc-3’, 3’: 5’-cgatctagatcttctgataattgagtatg-3’ and VEGF-C: 5’: 5’-gcgaccatgggcacattataatacag-3’, 3’: 5’-cggtctagaacgtctaataatggaatg-3’. S2 cells were transfected by calcium phosphate precipitation. Individual clones were isolated by limiting dilution in 25 μg/mL blasticidin and screened by western blotting cell supernatants with anti-V5 antibody conjugated to horseradish peroxidase (V5-HRP, Invitrogen).
Transfected S2 cells were initially grown in 15 cm culture plates (Corning, Corning, NY) and then transferred to 1 L spinner flasks (Corning) for 5 days in reduced serum conditions. Protein secretion was induced with 500 μM CuSO4 (Sigma) for 5 days. The supernatant was collected following centrifugation (3000 x g for 10 min at 4 0C), filtered, dialyzed and concentrated (Vivaflow 200, VivaScience Inc., Carlsbad, CA) in binding buffer (0.5 M NaCl, 50 mM Na2PO4, pH 8.0). Concentrated supernatant containing VEGF-C or D was then applied to a Ni++ column (ProBond, Invitrogen), washed with binding buffer containing 50 μM imidazole to remove contaminating proteins, and bound VEGF was eluted with buffer containing 300 μM imidazole. The eluents were then dialyzed against 150 mM NaCl/20 mM Tris-HCl/pH 7.5. Purity was assessed by silver staining (GelCode SilverSNAP, Pierce, Rockford, IL).
Immunoprecipitation, SDS-PAGE and Western Blot Analysis
For immunoprecipitation of VEGF-R3, HMVEC’s were grown in 6-well plates with full growth media until 70% confluent and subsequently in basal media with 0.1% FBS for 4 hr. VEGF-C or D was then added to the cells for 5 min, the cells washed with PBS/Na orthovanadate (NaV 10 mM) and then lysed with buffer containing 20 mM Tris pH-7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton-X, 0.5% sodium deoxycholate, 10% glycerol, 25 mM NaF, 1 mM PMSF, 1 mM NaV, and protease inhibitors (Complete Mini EDTA-free, Roche, Indianapolis, IN). Lysates were centrifuged at 14,000 g for 10 min at 40C and the pre-cleared lysates immunoprecipitated with 2 μg of rabbit anti-VEGF-R3 antibody bound to 30 μL of protein-A Sepharose beads (Amersham, Piscataway, NJ). The beads were washed five times with lysis buffer, resuspended in Laemmli sample buffer, boiled at 95 0C for 6 min, resolved on 8% SDS-PAGE under reducing conditions and transferred to PVDF membranes (Immobilon-P, Millipore, Billerica, MA). The membrane was blocked in 5% milk/Tris buffered saline with 0.1% Tween (TBST) for 1 h at room temperature and then probed with anti-phosphotyrosine Ab
For immunblotting of VEGF-C, VEGF-D, α9β1, Erk 1/2 and paxillin, proteins were resuspended in Laemmli sample buffer and resolved on 15%, 10% or 8% SDS-PAGE, before transfer to PVDF membrane. The membrane was probed with the appropriate specific primary antibodies, washed 3 times with TBST, and subsequently probed with HRP conjugated secondary antibodies and developed using chemiluminescence (ECL, Amersham).
For analysis of Erk 1/2 and paxillin phosphorylation, SW-480 cells were grown to 70–80% confluency in full growth media and then treated with 20 μg/mL of cyclohexamide. Cells were trypsinised, kept in suspension for 2 h in 6-well plates coated with 1% BSA (Sigma). Subsequently, 1 x 106 cells/mL, unntreated or pretreated with relevant blocking antibodies, were seeded into separate 6-well plates coated with either 1% BSA, VEGF-C or D (3 μg/mL) or Tnfn3RAA (2.5 μg/mL). Tnfn3RAA is an α9β1 specific ligand, that is a recombinant form of the third fibronectin type 3 repeat of tenascin C in which the arginine-glycine-aspartic acid sequence (RGD) is mutated to RAA, as previously described (25,26). After 60 min, cells were washed with PBS/NaV 1mM and then lysed with buffer containing 20 mM Tris pH-7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton-X, 25 mM NaF, 10% glycerol, 1 mM PMSF, 1 mM NaV, and protease inhibitors (Complete Mini EDTA-free, Roche, Indianapolis, IN). Lysates were centrifuged at 14,000 g at 40C for 10 min, and Western blotting performed as outlined above.
Flow Cytometry
Cultured cells were trypsinized, washed with phosphate buffered saline (PBS), blocked with normal goat serum at 4 0C for 10 min, incubated with primary antibody for 20 min at 4 0C, and then with phycoerythrin conjugated goat anti-mouse antibody. Labeled cells were suspended in PBS and fluorescence determined for 5,000 cells with a flow cytometer (Becton Dickinson FACSort, San Jose, CA). Primary mouse monoclonal antibody Y9A2 [Wang, 1996 #34 (10 ug/mL), was used to detect α9β1 and 9D9f9 (gift from Dr K. Alitalo, 5 ug/mL) to detect VEGF-R3.
Adhesion Assay
96-well non-tissue culture flat bottomed microtiter plates (ICN, Linbro/Titertek, Aurora, Ohio) were coated with VEGF-C or D at 4 0C overnight and then blocked with 1% bovine serum albumin (BSA, Sigma) in DMEM for 30 min at 370C. After trypsinisation cells were incubated with or without the α9β1 blocking antibody, Y9A2, (20 μg/mL) for 30 min on ice and 5 x 104 cells/well were seeded (8). Control wells were treated with 1% BSA in DMEM. The plates were centrifuged (topside up) at 10 x g for 5 min and incubated for 1 h (MEF) or 2 h (HMVEC) at 37 0C. Non-adherent cells were removed by centrifugation (top-side down) for 5 min at 48 x g and adherent cells fixed and stained with 1% formaldehyde, 0.5% crystal violet, 20% methanol for 30 min after which wells were washed 3 times with PBS. Following solubilization in 2% Triton X-100, the number of adherent cells was evaluated by measuring absorbance at 595 nm in a microplate reader (SpectraMax 190, Molecular Devices, Sunnyvale, CA).
Migration Assay
The under surface of 12-well, 8 micron Transwell plates (Corning Costar, Cambridge, MA) were coated with relevant ligand (VEGF-C or D) or 1% BSA as a binding control at 4 0C overnight. Wells were then washed with PBS and blocked with 1% BSA in DMEM or PBS for 60 min at 37 0C. After trypsinization, 5 x 104 cells in DMEM were incubated with or without Y9A2 and/or MAZ51 for 30 min on ice and then seeded into the top chamber of the Transwell (8). 1% fetal calf serum was added to the bottom well, to serve as a chemoattractant, and the plates were incubated at 37 0C for 3 h. Cells that migrated and adhered to the bottom surface of the Transwell membrane were fixed with Diff-Quik fixative (Pierce) and the non-adherent cells gently removed from the top with a Q-tip. After air drying, the membranes were stained with DiffQuik (Pierce), washed in water, air-dried and the membranes cut from the Transwell and mounted onto glass slides. Cells were counted in 10 high power (25X) fields for each condition.
RNA silencing of α9β1
HMVEC were grown in 10 cm dishes with full growth media (Cambrex) until 70% confluent. Cells were then washed twice with serum free media and transfected with siRNA (20μM, Ambion, #86946) targeted to exon 4 of the α9 integrin using siPort Amine (Ambion) in OptiMem media. Cell media was replaced with full growth media 6 hr after transfection and daily after this. Transfection efficiency was assessed by flow cytometry analysis of α9β1 expression 48–72 hr after transfection and compared to mock and untransfected cells of the same passage. Cell adhesion assays using VEGF-C or VEGF-D as substrate were performed using transfected cells in which α9β1 was successfully silenced and compared to untransfected and transfected cells in which α9β1 was not silenced.
Production of Non-blocking Monoclonal Antibody to α9β1
A non-blocking antibody to α9β1 was produced for detection of integrin bound to VEGF protein in binding assays. Murine L2 cells (ATCC), transfected with human α9, were injected into mice every 14 days for a total of 3 injections. 3 days after the final injection, lymphocytes were harvested and spin-combined with SP2/0 myeloma cells as previously described (27). After addition of selection media (RPMI-1640, 10% FCS, 1% penicillin/streptomycin, 1% sodium pyruvate, L-glutamine), single clones were selected and supernatants screened by differential flow cytometry of mock- and α9-transfected cells. Antibody was purified by gradient anion exchange.
Metabolic Labeling of Cells and Immunoprecipitation
To determine if A9A1 was able to immunoprecipitate α9β1 integrin efficiently, α9 transfected SW-480 cells were grown for 4 h in methionine-free DMEM supplemented with 1% penicillin/streptomycin and then incubated with 0.5 mCi 35S for 24 h. Lysates of cells labeled with 35S methionine were then pre-cleared with protein-A-Sepharose for 1 h at 4 0C. The pre-cleared supernatant was incubated with 20 μg of primary antibody, A9A1, at 4 0C overnight. The beads and bound protein were washed 4 times with buffer containing 100 mM Tris, 300 mM NaCl, 1% Triton X-100, 0.1% SDS and then boiled in reducing Laemmli’s buffer at 95 0C for 6 min. Protein samples were separated by 8% SDS-PAGE and autoradiography performed after 1 week at –80 0C.
Immuno-affinity Purification of Human Integrin α9β1
α9-transfected SW-480 cells (24) were lysed with buffer containing 100 mM N-octylglucosylpyranoside (ICN), 150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 1 mM Mg and CaCl2, 1 mM NaV, 1 mM polymethyl sodium fluoride (PMSF) overnight at 4 0C. After centrifugation (12,000 x g, 15 min) and filtering, lysate containing α9β1 was applied to an α9 antibody (A9A1) column. The column was washed with wash buffer (20 mM N-octylglucosylpyranoside, 150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 2 mM Mg and 0.1 mM CaCl2) and a second time with wash buffer containing 500 mM NaCl. Bound integrin was eluted in 150 mM NaCl at pH 3.0 into microcentrifuge tubes containing Tris pH 9.0, to bring the final pH to 7.4. Eluents were analyzed for α9β1 by Western blotting with the rabbit polyclonal antibody, 1057 (5).
Immuno-affinity Purification of Human Integrin αvβ6
Secreted αvβ6 integrin was collected from conditioned media of CHO cells stably transfected with truncated forms of each subunit of the integrin, as previously described (28). The integrin was purified using immunoaffinity chromatography on columns with αvβ6 antibody, R6G9, covalently cross-linked to protein A-Sepharose beads. Bound proteins were eluted with 100 mM glycine, pH 3 into 1/20 volume of 1M Na2PO4, pH 8 (29).
Binding Assay
Direct binding of VEGF-C and D to α9β1 was assessed by a solid-phase binding assay in non-tissue coated 96-well microtiter plates (Nunc ImmunoPlate, Naperville. IL). Recombinant VEGF-C or D (5 μg/mL) was attached to the plates and purified α9β1 added for 2 h at room temperature in the presence or absence of 10 mM EDTA. Following 5 washes with PBS/1% BSA/0.05% Tween, the extent of α9β1 binding was detected using A9A1 antibody (20 μg/mL, 1 h at 37 0C). After incubation with biotinylated rat anti-mouse antibody (1:250, 1 h, room temperature), streptavidin-HRP was added for 20 min at room temperature followed by 3,3’,5,5’ tetramethylbenzidine substrate solution (BD Biosciences Pharmingen, San Jose , CA). Absorbance was then measured at 450 nm with a microplate reader (Molecular Devices). The functional integrity of purified α9β1 integrin was assessed in a similar assay using the α9β1 specific ligand, Tnfn3RAA (5 μg/mL), as previously described (25,26).
Statistical Methods
For statistical analysis of adhesion and migration assays comparisons were made by analysis of variance with a post-hoc Tukey’s test. Statistical significance was assumed at p ≤ 0.05, with respect to a two-tailed probability distribution. Data are presented as mean values ± SEM unless otherwise stated, from at least 3 separate experiments.
Results
Production and Purification of VEGF-C and D
Fully processed VEGF-C and D proteins were purified from transfected Drosophila S2 cells. Western blots (Fig. 1A) of secreted and purified VEGF-C (top panel) and D (bottom panel) with anti-V5 antibody (left panels) demonstrate proteins with the expected molecular mass for VEGF-C (or D) tagged with V5 and His (~ 23–24 kDa). In addition, immunoblotting with VEGF-C and D specific antibodies (right panels) showed the V5-tagged proteins were indeed VEGF-C and D. The purity of each recombinant protein was determined by silver staining of 15% SDS-PAGE gels as shown in Fig. 1B. VEGF-C and D-V5 induced phosphorylation of their cognate receptor, VEGF-R3. Fig. 1C shows the expected VEGF-R3 isoforms and confirms the activity of the purified VEGF proteins.
FIG. 1. Production and purification of VEGF-C and D proteins.
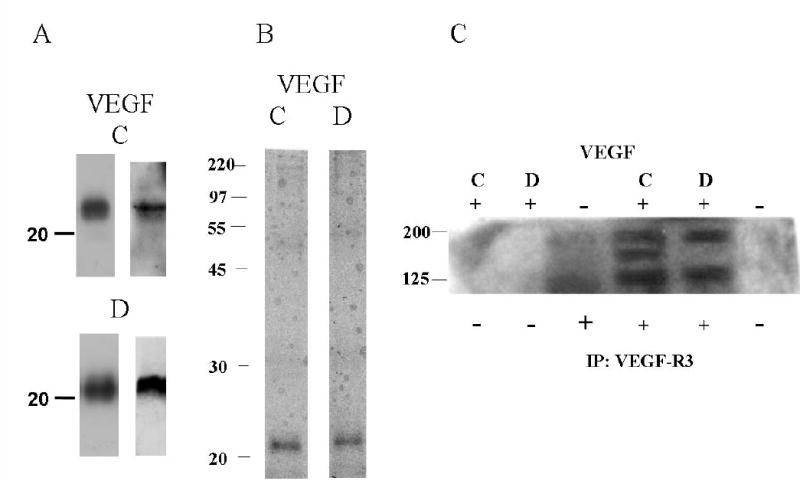
VEGF-C and D were purified from supernatant of transfected Drosphila S2 cells by Ni++ affinity chromatography. Proteins were separated by 15% SDS-PAGE under reducing conditions, and analyzed by (A) Western Blot analysis with V5 antibody (left panels) and VEGF-C and D specific antibodies (right panels), and (B) silver staining. (C) Phosphotyrosine blot from control HMVEC and HMVEC incubated with V5-tagged VEGFC or D. Lysates were immunoprecipitated with either no antibody or antibody against VEGFR-3, separated by 8% SDS-PAGE under reducing conditions and Western blotted with an anti-phosphotyrosine antibody. Molecular mass markers (kDa) are indicated to the left of all panels.
Cell Adhesion to VEGF-C and D is α9β1-Dependent
Adhesion assays were performed using α9- and mock-transfected MEF and SW-480 cells that do not express VEGF-R2 or 3, (11,30) (data not shown). Flow cytometry with the anti-α9β1 antibody, Y9A2, showed robust expression of α9β1 in both α9-transfected cell types and no expression in mock-transfectants (Fig. 2A). α9-transfected MEF demonstrated a concentration dependent adhesion to both VEGF-C and D (Figs. 2B and 2C). This effect was abolished by the α9β1 blocking antibody, Y9A2 (p<0.05). In contrast, mock-transfected MEF did not adhere to either VEGF protein above background levels of attachment to BSA coated wells. Similarly, α9β1-dependent adhesion on both VEGF-D (Fig. 2D) and VEGF-C (data not shown) was demonstrated in SW-480 cells.
FIG. 2. α9-transfected MEF and SW-480 cells adhere to VEGF-C and D.
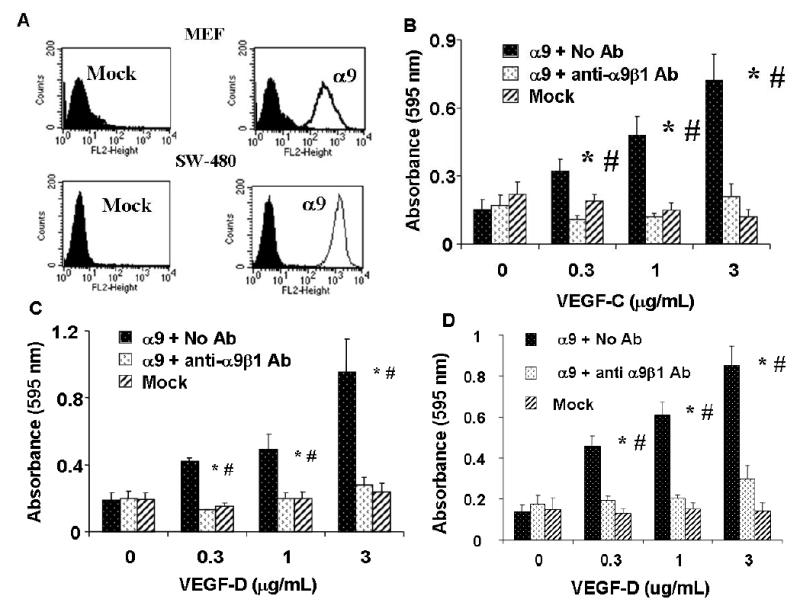
(A) Flow cytometry analysis of mock (left) and α9 (right) transfected MEF (upper panel) and SW-480 cells (lower panel) used in cell adhesion and migration assays and for α9β1 purification (SW-480’s). For detection of α9β1 integrin, cells were labeled with the α9β1-specific antibody, Y9A2. Purified VEGF-C or D was used as substrate for adhesion assays with mock (diagonal bars) or α9 (black bars) transfected MEF (B & C) or SW-480’s (D) in the absence or presence (white bars) of the α9β1-blocking antibody, Y9A2. Cells were allowed to adhere to wells coated with a range of VEGF concentrations and then washed, fixed and stained with crystal violet. Adhesion is expressed as absorbance at 595 nm. * = p<0.05 compared to cells treated with Y9A2, # = p<0.05 compared to 0 ug/mL VEGF
Binding of VEGF-C or D activates α9β1 integrin
We have previously shown that in SW-480 cells, activation of α9β1 integrin by established ligands results in phosphorylation of Erk 1/2 and paxillin. To assess whether binding of VEGF-C or D similarly results in α9β1 induced signaling, phosphorylation of these proteins was compared in mock- and α9-transfected SW-480 cells in the presence or absence of α9β1 blocking antibody. Fig. 3A shows that after 15 minutes of α9-transfectants binding to adherent Tnfn3RAA (lanes 3 & 4), VEGF-C (Lanes5 & 6) or D (Lanes 7 & 8), Erk 1/2 phosphorylation was increased in an α9-dependent fashion. Similar to 1% BSA (Lane 1), this response to VEGF was not seen in mock transfected cells (Lane 2). Paxillin phosphorylation was also induced after 15 min in α9-transfectants and was inhibited by α 9β1-blocking antibody (Fig. 3B).
FIG. 3. Binding of VEGF-C and D to α9β1 activates the integrin.
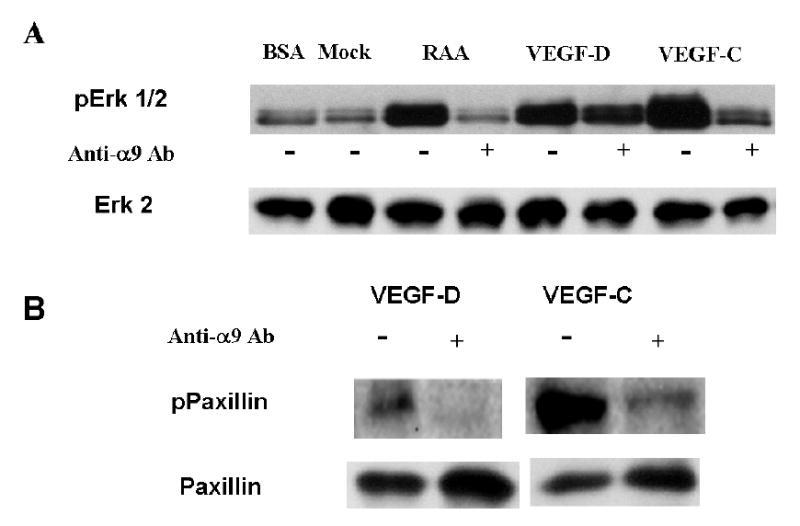
Mock- or α9-transfected SW-480 cells were plated on various proteins for 15 min and cell lysates obtained. Following BCA protein quantification equal amounts of protein were separated under reducing conditions on 10% SDS-PAGE. (A) SW-480 cells (upper panel) plated for 15 min on 1% BSA, Tnfn3RAA (α9 specific ligand), VEGF-D or VEGF-C in the presence or absence of α9-blocking antibody (20 μg/mL) and immunoblotted with phospho-Erk 1/2 (upper panel). The PVDF membrane was re-probed with total Erk 2 to ensure equal protein loading (lower panel). (B) SW-480 cells plated for 15 min on VEGF-C or VEGF-D in the presence or absence of α9-blocking antibody (20 μg/mL) and immunoblotted with phosphopaxillin (upper panel). The PVDF membrane was re-probed with total paxillin to ensure equal protein loading (lower panel).
α9β1-Dependent Cell Adhesion is Demonstrable in Primary Endothelial Cells
To verify the biological significance of the results obtained with transfected cell lines, and to understand the role of α9β1 in adhesion to VEGF-C and D in the presence of their cognate receptor, VEGF-R3, HMVEC were also studied. HMVEC were found to express both VEGF-R3 and α9β1 as measured by flow cytometry (Figs. 4A and B). Essentially, all of the cells expressed VEGF-R3 (Fig. 4A), belying their lymphatic origin, whilst the expression of α9β1 varied from 40–55% (Fig. 4B) depending on the lot purchased, and decreased with increasing passage number. As a result, all experiments were performed between passages 3–7. Like α9- transfected cell lines, HMVEC demonstrated concentration dependent adhesion, to both VEGF-C (Fig. 4C) and D (Fig. 4D). Adhesion to both substrates was inhibited by α9β1 blocking antibody, Y9A2**,** (p<0.05) to the levels measured in wells coated with 1% BSA, demonstrating that the presence of VEGF-R3 is not sufficient to mediate cell adhesion to VEGF-C or D.
FIG. 4. Primary endothelial cell adhesion to VEGF-C and D is α9β1 dependent.
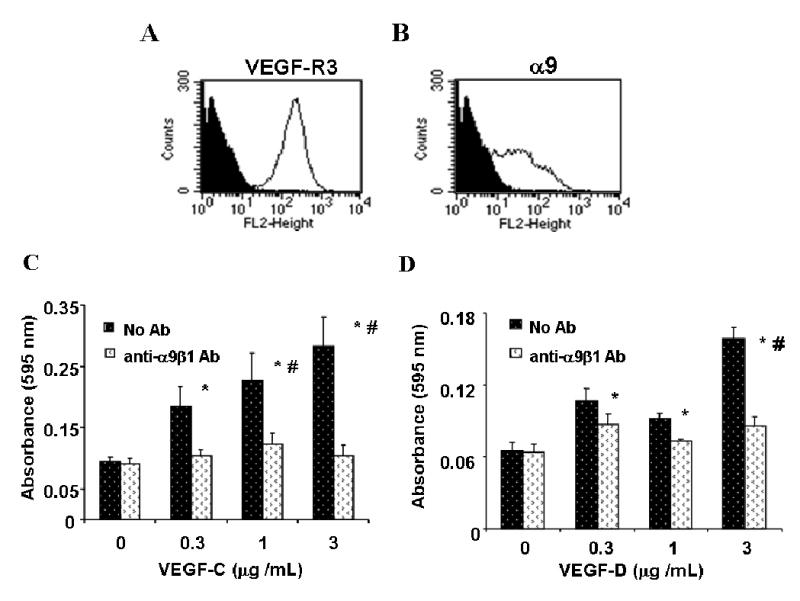
Flow cytometry analysis of HMVEC labeled with 9D9f9 (A) and Y9A2 (B) to measure expression of VEGF-R3 and α9β1, respectively. Shaded areas represent cells labeled with no primary antibody and the unshaded area those labeled with the primary antibody, as indicated. Purified VEGF-C (C) and D (D) were used as substrate for adhesion assays with HMVEC in the absence (dark bars) or presence (white bars) of the α9β1-blocking antibody, Y9A2. Cells were processed as described in Fig. 2 above. * = p<0.05 compared to cells treated with Y9A2, # = p<0.05 compared to 0 ug/mL VEGF
Similar results were obtained when α9β1 was silenced using siRNA transfection (Fig. 5). The effectiveness of siRNA directed against exon 4 of α9 (alpha9 siRNA) was assessed by measuring α9 expression on the cell surface by flow cytometry (Fig 5A). The expression of α9 in these cells was compared against cells transfected with mock siRNA and cells not treated with siRNA (No siRNA). Adhesion assays were then performed using these cell populations. Fig 5B shows that knockdown of α9 in HMVEC cells significantly inhibits cell adhesion on both VEGF-C and VEGF-D. Mock siRNA cells adhered to VEGF-C or VEGF-D substrate to the same degree as non-transfected HMVEC.
FIG. 5. siRNA silencing of α9 inhibits cell adhesion to VEGF-C and D.
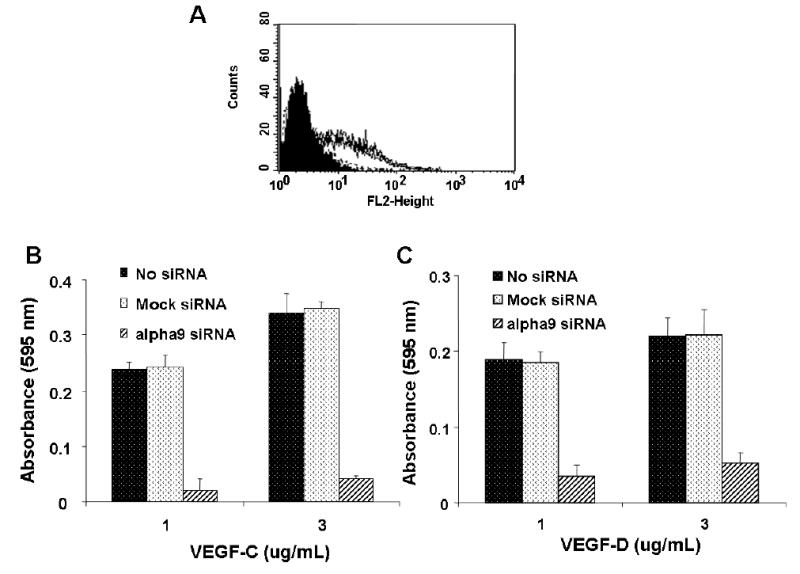
(A) Flow cytometry analysis of α9 expression in HMVEC, using the α9 specific antibody Y9A2, after no transfection, mock siRNA transfection (overlapping lines) or effective α9 siRNA transfection (shaded area). Unstained cells are also shown as a shaded area. VEGF-C (B) and D (C) were used as substrate for adhesion assays with α9 siRNA transfected (diagonal bars), mock transfected (white bars) or untransfected HMVEC (black bars).
Cell Migration on VEGF-C and D is also α9β1-Dependent
We have previously shown that one of the principal functions of α9β1 is enhancement of cell migration. Since migration of lymphatic endothelial cells into mesenchyme enriched for VEGF-C (48) and/or D is potentially an important step in lymphangiogenesis, we next sought to determine the role of α9β1 in migration on VEGF-C and D. α9-transfected MEF migrated on VEGF-C (Fig. 6A and D) (Fig. 6B) in a concentration-dependent manner, whereas mock-transfected cells did not migrate on any concentration above values seen for migration on BSA. This effect depended on ligation of α9β1, since migration on VEGF-C or D was inhibited by α9β1 blocking antibody. Similar results were obtained for HMVEC, which also demonstrated concentration-dependent migration on VEGF-C (Fig. 6C) and D (Fig. 6D) that was inhibited by α9β1 blocking antibody.
FIG. 6. Cell migration on VEGF-C and D is α9β1-dependent.
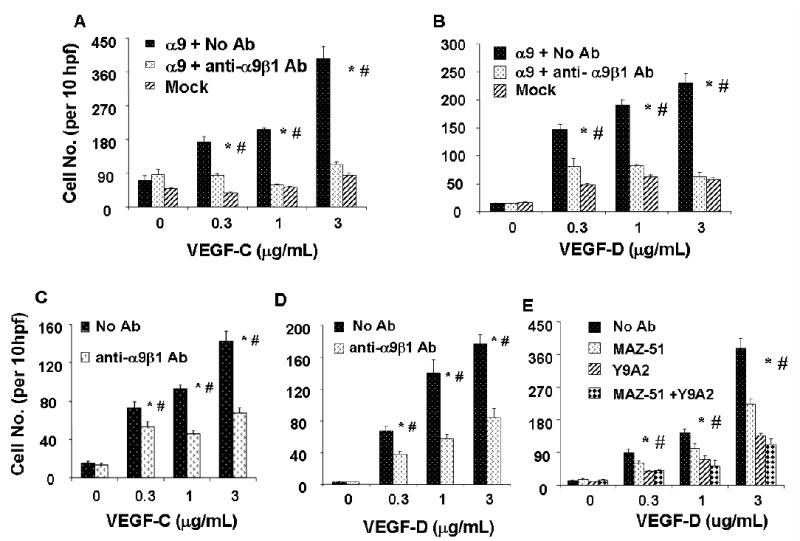
Migration assays on VEGF-C or D, coated on the lower surface of a Transwell at various concentrations, were performed with 1% fetal calf serum as a chemotactic factor. VEGF-C (A) or VEGF-D (B) was used as substrate for migration assays with mock (diagonal bars) or α9 (dark bars) transfected MEF in the absence or presence (white bars) of Y9A2. Similar assays were performed in HMVEC on VEGF-C (C) or VEGF-D (D) substrate in the absence (dark bars) or presence (white bars) of the α9β1-blocking antibody. (E) VEGF-D was used as substrate for migration assays with HMVEC in the absence (dark bars) or presence (white bars) of the α9β1-blocking antibody, Y9A2 and/or the VEGF-3 blocking drug, MAZ-51. * = p<0.05 compared to cells treated with Y9A2, # = p<0.05 compared to cells treated with MAZ-51 alone.
To asses the relative contribution of VEGF-R3 and α9β1 to HMVEC cell migration, cells were treated with α9β1 blocking antibody and/or the VEGF-R3 blocking chemical, MAZ51 (49). Fig. 6E shows that blocking either α9β1 or VEGF-R3 significantly inhibits cell migration. These results suggest that both α9β1 and VEGF-R3 are required for maximal cell migration on immobilized VEGF.
Production of Non-blocking α9β1 Antibody
A mouse anti-human monoclonal antibody (A9A1) was produced to serve as a detection antibody for α9β1 in VEGF-C or D solid phase binding assays. Flow cytometry using the antibody A9A1 shows detection of α9β1 integrin on α9-transfected MEF to a similar level as that of Y9A2 an established α9β1 integrin antibody (Fig. 7A). To further characterize its specificity, A9A1 was used to immunoprecipitate lysates of 35S-methionine labeled α9-transfected SW-480 cells. Fig. 7B shows an autoradiograph of immunoprecipitated protein samples separated by 8% SDS-PAGE where the α9 and β1 bands of the heterodimer are clearly seen. Fig. 7C shows that in contrast to Y9A2, a blocking antibody to α9β1, A9A1 did not inhibit adhesion of α9-transfected MEF to the α9β1–specific ligand Tnfn3RAA. This antibody was thus suitable for use in α9β1-VEGF binding assays.
FIG. 7. A9A1, a specific non-blocking antibody to α9β1.
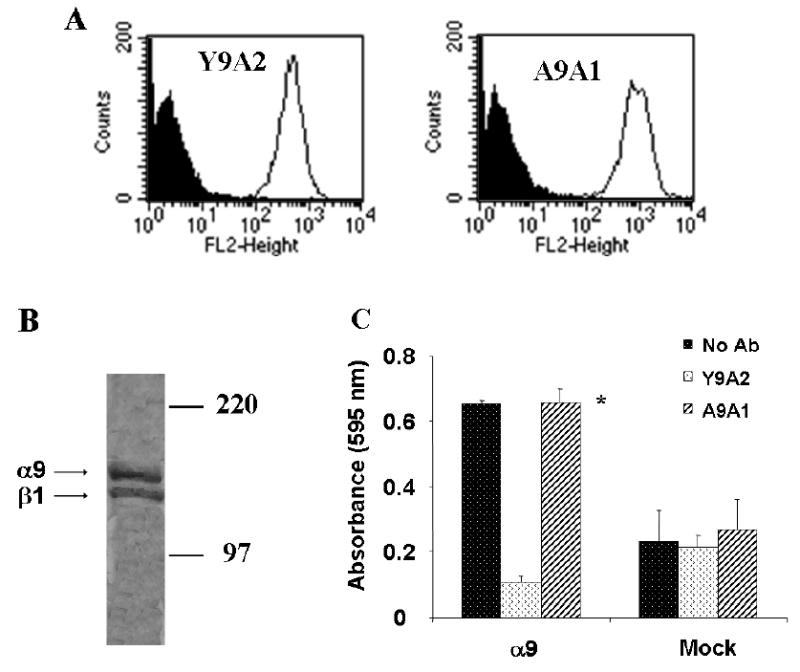
(A) Flow cytometry analysis of α9 (line) and mock transfected (shaded area) MEF using the α9 antibody, A9A1 (right) compared to the α9β1 specific antibody, Y9A2 (left). (B) Autoradiograph of 35S labelled α9 transfected SW-480 cell lysates immunoprecipitated with 20μg of A9A1. The observed bands represent the α9 and β1 subunits of the integrin. (C) Tnfn3RAA, an α9-specific ligand, was used as substrate for adhesion assays in α9-transfected MEF in the absence (dark bars) or presence (diagonal bars) of A9A1 antibody and compared to the blocking antibody, Y9A2 (white bars). * = p<0.05 compared to cells treated with Y9A2
Purification of Active Human α9β1 Integrin
To determine whether α9β1 directly binds to VEGF-C and/or D, solid phase binding assays were performed using purified VEGF-C or D and α9β1 purified from α9-transfected SW-480 cells (24) using an A9A1 affinity column. Figs. 8A and B show a Coomassie stain (reduced, 8% SDS-PAGE) and immunoblot of eluted α9β1 (α9 ~160 kDa, β1 ~ 130 kDa). This purified integrin was still capable of binding to the α9β1-specific ligand, Tnfn3RAA, an effect that was inhibited by 10 mM EDTA (Fig. 8C).
FIG. 8. Purification of active α9β1.
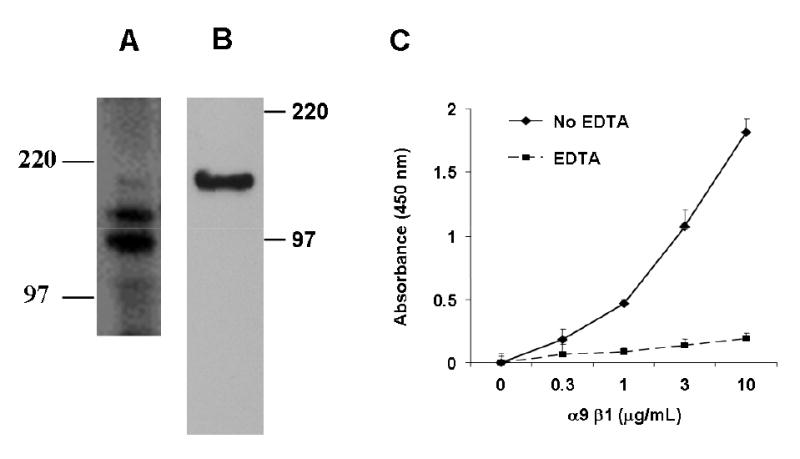
α9β1 integrin was purified from cell lysates of α9-transfected SW-480 cells by affinity chromatography using an α9β1 antibody (A9A1) column. Proteins under reducing conditions were separated by 8% SDS-PAGE and analyzed by (A) silver staining and (B) Western Blot analysis with 1057, rabbit polyclonal antibody to α9β1. Molecular mass markers (kDa) are indicated. (C) Tnfn3RAA, an α9-specific ligand, was used as substrate for binding assays with purified α9β1 in the absence (diamonds) or presence (squares) of 10 mM EDTA.
VEGF-C and D Bind the Integrin α9β1
Solid phase binding assays demonstrated robust concentration-dependent adhesion of α9β1 to 5μg/mL of both VEGF-C or D, (Figs. 9A and 9B). In both cases, binding was completely inhibited by chelating divalent cations (EDTA, 10 mM), as expected for authentic integrin-ligand interactions. The irrelevant integrin, αvβ6, (28) showed no binding to either VEGF-C or D.
FIG. 9. VEGF-C and D bind directly to the integrin α9β1.
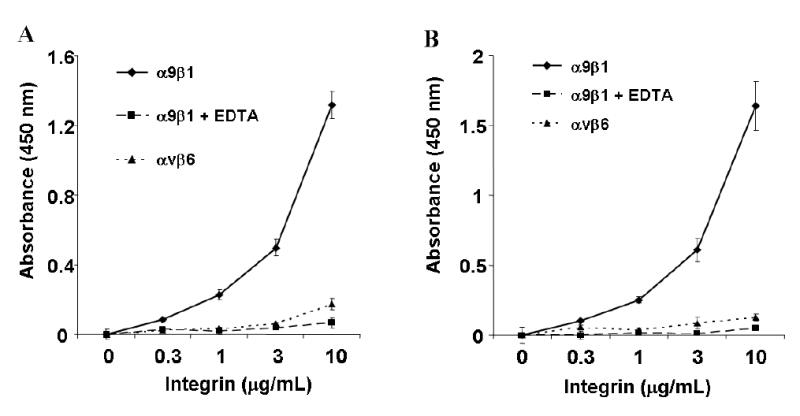
Purified VEGF-C (A) or D (B) was used for solid phase binding assays with purified α9β1 at various concentrations in the absence (diamonds) or presence (squares) of 10 mM EDTA. Similar assays were performed using purified αvβ6, an irrelevant integrin, (triangles).
Discussion
In this study we found that α9-transfected cells (lacking VEGF receptors) adhere and migrate on fully processed VEGF-C and D. These cellular responses require functional α9β1 since they are inhibited by a blocking antibody. These results alone suggest that an interaction between α9β1 and the VEGF-C and D proteins can affect cell behavior in the absence of the growth factors’ cognate receptor, VEGF-R3. Importantly, α9β1 was also shown to mediate adhesion and migration on VEGFC and D in primary human endothelial cells that co-express α9β1 and VEGF-R3. Inhibition of migration by α9β1 antibody and/or VEGF-R3 inhibition suggests that VEGF-R3 is not sufficient to mediate maximal migration on VEGF-C or D. Solid phase binding assays of purified VEGF-C or D and α9β1 verified that these cell functions are a result of VEGF-C and D binding α9β1 in an integrin-specific and cation-dependent manner. Moreover, binding of VEGF-C or D to α9β1 leads to induction of downstream signals, as demonstrated by α9β1 dependent Erk 1/2 and paxillin phosphorylation. The finding that growth factors (VEGF-C and D) bind to an integrin (α9β1) highlights a novel and important mechanism of integrin-growth factor interaction.
A number of previous studies have described cooperative interactions between integrins and growth factors, (reviewed in, (31,32)). Growth factor ligation of its cognate receptor has been shown to lead to close physical association with integrins. For example, ligation of the PDGF receptor not only allowed its co-immunoprecipitation with the αvβ3 integrin, but also potentiated αvβ3-dependent cell migration (33). In addition, input from VEGF-R2, stimulated by VEGF-A, has been shown to activate the αv integrins, αvβ3 and αvβ5, and the β1 integrins, α5β1 and α2β1 (34).
Activated integrins have also been shown to modulate growth factor protein and receptor expression and signaling. For example, integrin α6β4 increases VEGF-A translation in breast carcinoma cells through inactivation of the translational repressor, 4E-BP1 (35). In addition, integrin-matrix interactions modulate expression of VEGF and FGF receptor 1 and 2 expression on HMVEC (36). The functional importance of growth factor/integrin interactions has also been demonstrated in vivo where blocking antibodies to αvβ3 and αvβ5 integrins inhibit basic fibroblast growth factor (bFGF) and VEGF induced angiogenesis, respectively (37). Finally, mice lacking the integrin αvβ5 are specifically protected from the increases in vascular permeability induced by VEGF-A, again presumably through cross-talk with VEGF-R2 (38).
In the current study, we describe a novel mechanism of growth factor-integrin interaction; direct ligation of an integrin by a growth factor. This interaction thus provides a mechanism for VEGF-C and D to directly affect cell behavior (i.e. cell adhesion and migration) even in the absence of their cognate receptor, VEGF-R3. Furthermore, even in cells expressing the cognate receptor, ligation of the integrin α9β1 is required for stable cell adhesion to these growth factors and substantially enhances cell migration across them. Our results do not exclude the possibility that α9β1 may modulate cell function by activating another receptor capable of binding VEGF or inhibiting a VEGF-R3 suppressor. However, taken together these results strongly suggest that VEGF-C and D directly bind to α9β1. We speculate that simultaneous binding of both α9β1 and VEGF-R3 by VEGF-C and/or D, which is present at high concentration in the extracellular matrix, results in co-clustering of both receptors facilitating cooperative interactions between the two receptors, as demonstrated by our cell migration data. The specific binding sites on VEGF-C and D and their relationship to the binding sites on other α9β1 ligands, such as tenascin-C and osteopontin, are as yet undetermined. Due to the lack of a conserved binding sequence for known α9β1 ligands, determination of this site for VEGF-C or D would be difficult. Since the VEGF family members share a VEGF homology domain our results suggests that other VEGF family member proteins may also interact with α9β1. It is also clear that in addition to integrins, other cell surface proteins can modulate VEGF responses, such as the other VEGF-receptors (39) and the neuropilins (40). The potential interactions between these modulating proteins and α9β1 remain to be determined.
Normal vessel development requires the coordinated function of both cellular and extracellular regulatory and effector proteins to ensure an optimal milieu for correct vessel morphogenesis and function (41). The molecular regulators of angiogenesis include the VEGF family of proteins and receptors (VEGF-R1, 2, 3), acidic and basic fibroblast growth factor, angiopoietins and Tie receptors, ephrins, integrins and matrix metalloproteases (10,42). The molecules that regulate lymphangiogenesis have not been as well characterized. However, it is clear that VEGF-C and D are key modulators of lymphatic development and function (21,22), although their relative roles remain to be determined (19). These growth factors induce lymphatic endothelial cell growth, survival and migration in vitro (16) and lymphangiogenesis in vivo (43), mediated at least in part through their activation of VEGF-R3. In addition, loss of function mutations in VEGF-R3 have been described in a significant subgroup of patients with congenital human lymphedema (44).
The α9β1 integrin has now been described to interact with a relatively large number of ligands including tenascin C , osteopontin, VCAM-1, coagulation factor XIII and several members of the ADAMs family of transmembrane metalloproteinases (7,24,45–47). The biological significance of α9β1 interactions with most of these ligands remains to be determined. In contrast, we now show that the ligands VEGF-C and D play a role in endothelial cell adhesion and migration, key cellular functions required for lymphangiogenesis. The most dramatic phenotypic feature of α9 null mice is the presence of bilateral chylothoraces, suggesting a functional defect in the collecting lymphatics of the thorax. Subtle edema and the accumulation of lymphocytes around some peripheral lymphatics provides further support for a role of this integrin in lymphangiogenesis (9). The phenotype of VEGF-D null mice is yet to be published, however it is clear from E10.5 VEGF-C heterozygote mice that VEGF-C is expressed in mesenchymal cells surrounding the jugular vein toward which endothelial cells, already with a lymphatic differentiation (Prox-1 positive) must migrate (48). Given the specialized role that α9β1 plays in accelerated cell migration (8), we speculate that abnormal migration on VEGF-C and/or D might be responsible for the functional defects that occur in α9 null mice. However, the fact that lymphatic vessels are formed in these mice suggests that either the presence of VEGF-R3, or some other unidentified receptor(s), can partially compensate for the loss of α9β1.
In summary, the findings of this paper describe a novel integrin-growth factor interaction whereby the lymphangiogenic proteins VEGF-C and D bind the integrin α9β1. Although the precise mechanisms by which binding of VEGF-C and/or D to α9β1 contributes to lymphatic development remain to be fully elucidated, these data strongly support a role for this interaction in explaining the lymphatic defects in α9 knockout mice.
Acknowledgments
We thank Dr Kari Alitalo (University of Helsinki, Helsinki, Finland) for the generous gift of the VEGF-R3 antibody, 9D9f9.
Footnotes
*
This work was supported by a Mayo Foundation Scholarship (NEV), and by R01 HL64353 from the National Heart Lung and Blood Institute (DS)
1
The abbreviations used are: VEGF, vascular endothelial growth factor; VEGF-R, vascular endothelial growth factor receptor; MEF, mouse embryonic fibroblasts; HMVEC, human microvascular endothelial cells; BSA, bovine serum albumin; RGD, arginine-glycine-aspartic acid; FGF, fibroblast growth factor; Tnfn3RAA, α9 specific ligand, recombinant third fibronectin repeat of tenascin C in which RGD is mutated to RAA
References
- 1.Hynes RO. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 2.Friedl P. Curr Opin Cell Biol. 2004;16:14–23. doi: 10.1016/j.ceb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 4.Hynes RO. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 5.Palmer EL, Ruegg C, Ferrando R, Pytela R, Sheppard D. J Cell Biol. 1993;123:1289–1297. doi: 10.1083/jcb.123.5.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tarui T, Miles LA, Takada Y. J Biol Chem. 2001;276:39562–39568. doi: 10.1074/jbc.M101815200. [DOI] [PubMed] [Google Scholar]
- 7.Taooka Y, Chen J, Yednock T, Sheppard D. J Cell Biol. 1999;145:413–420. doi: 10.1083/jcb.145.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Young BA, Taooka Y, Liu S, Askins KJ, Yokosaki Y, Thomas SM, Sheppard D. Mol Biol Cell. 2001;12:3214–3225. doi: 10.1091/mbc.12.10.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang XZ, Wu JF, Ferrando R, Lee JH, Wang YL, Farese RV, Jr, Sheppard D. Mol Cell Biol. 2000;20:5208–5215. doi: 10.1128/mcb.20.14.5208-5215.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alitalo K, Carmeliet P. Cancer Cell. 2002;1:219–227. doi: 10.1016/s1535-6108(02)00051-x. [DOI] [PubMed] [Google Scholar]
- 11.Jussila L, Alitalo K. Physiol Rev. 2002;82:673–700. doi: 10.1152/physrev.00005.2002. [DOI] [PubMed] [Google Scholar]
- 12.Ferrara N, Gerber HP, LeCouter J. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 13.Achen MG, Jeltsch M, Kukk E, Makinen T, Vitali A, Wilks AF, Alitalo K, Stacker SA. Proc Natl Acad Sci U S A. 1998;95:548–553. doi: 10.1073/pnas.95.2.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joukov V, Sorsa T, Kumar V, Jeltsch M, Claesson-Welsh L, Cao Y, Saksela O, Kalkkinen N, Alitalo K. Embo J. 1997;16:3898–3911. doi: 10.1093/emboj/16.13.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McColl BK, Baldwin ME, Roufail S, Freeman C, Moritz RL, Simpson RJ, Alitalo K, Stacker SA, Achen MG. J Exp Med. 2003;198:863–868. doi: 10.1084/jem.20030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makinen T, Veikkola T, Mustjoki S, Karpanen T, Catimel B, Nice EC, Wise L, Mercer A, Kowalski H, Kerjaschki D, Stacker SA, Achen MG, Alitalo K. Embo J. 2001;20:4762–4773. doi: 10.1093/emboj/20.17.4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mandriota SJ, Jussila L, Jeltsch M, Compagni A, Baetens D, Prevo R, Banerji S, Huarte J, Montesano R, Jackson DG, Orci L, Alitalo K, Christofori G, Pepper MS. Embo J. 2001;20:672–682. doi: 10.1093/emboj/20.4.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marconcini L, Marchio S, Morbidelli L, Cartocci E, Albini A, Ziche M, Bussolino F, Oliviero S. Proc Natl Acad Sci U S A. 1999;96:9671–9676. doi: 10.1073/pnas.96.17.9671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rissanen TT, Markkanen JE, Gruchala M, Heikura T, Puranen A, Kettunen MI, Kholova I, Kauppinen RA, Achen MG, Stacker SA, Alitalo K, Yla-Herttuala S. Circ Res. 2003;92:1098–1106. doi: 10.1161/01.RES.0000073584.46059.E3. [DOI] [PubMed] [Google Scholar]
- 20.Skobe M, Hamberg LM, Hawighorst T, Schirner M, Wolf GL, Alitalo K, Detmar M. Am J Pathol. 2001;159:893–903. doi: 10.1016/S0002-9440(10)61765-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, Jackson DG, Nishikawa S, Kubo H, Achen MG. Nat Med. 2001;7:186–191. doi: 10.1038/84635. [DOI] [PubMed] [Google Scholar]
- 22.Szuba A, Skobe M, Karkkainen MJ, Shin WS, Beynet DP, Rockson NB, Dakhil N, Spilman S, Goris ML, Strauss HW, Quertermous T, Alitalo K, Rockson SG. Faseb J. 2002;16:1985–1987. doi: 10.1096/fj.02-0401fje. [DOI] [PubMed] [Google Scholar]
- 23.Veikkola T, Jussila L, Makinen T, Karpanen T, Jeltsch M, Petrova TV, Kubo H, Thurston G, McDonald DM, Achen MG, Stacker SA, Alitalo K. Embo J. 2001;20:1223–1231. doi: 10.1093/emboj/20.6.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yokosaki Y, Palmer EL, Prieto AL, Crossin KL, Bourdon MA, Pytela R, Sheppard D. J Biol Chem. 1994;269:26691–26696. [PubMed] [Google Scholar]
- 25.Prieto AL, Edelman GM, Crossin KL. Proc Natl Acad Sci U S A. 1993;90:10154–10158. doi: 10.1073/pnas.90.21.10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yokosaki Y, Matsuura N, Higashiyama S, Murakami I, Obara M, Yamakido M, Shigeto N, Chen J, Sheppard D. J Biol Chem. 1998;273:11423–11428. doi: 10.1074/jbc.273.19.11423. [DOI] [PubMed] [Google Scholar]
- 27.Wang A, Yokosaki Y, Ferrando R, Balmes J, Sheppard D. Am J Respir Cell Mol Biol. 1996;15:664–672. doi: 10.1165/ajrcmb.15.5.8918373. [DOI] [PubMed] [Google Scholar]
- 28.Weinacker A, Chen A, Agrez M, Cone RI, Nishimura S, Wayner E, Pytela R, Sheppard D. J Biol Chem. 1994;269:6940–6948. [PubMed] [Google Scholar]
- 29.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 30.Witmer AN, Dai J, Weich HA, Vrensen GF, Schlingemann RO. J Histochem Cytochem. 2002;50:767–777. doi: 10.1177/002215540205000603. [DOI] [PubMed] [Google Scholar]
- 31.Smyth SS, Patterson C. J Cell Biol. 2002;158:17–21. doi: 10.1083/jcb.200202100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamada KM, Even-Ram S. Nat Cell Biol. 2002;4:E75–76. doi: 10.1038/ncb0402-e75. [DOI] [PubMed] [Google Scholar]
- 33.Borges E, Jan Y, Ruoslahti E. J Biol Chem. 2000;275:39867–39873. doi: 10.1074/jbc.M007040200. [DOI] [PubMed] [Google Scholar]
- 34.Byzova TV, Goldman CK, Pampori N, Thomas KA, Bett A, Shattil SJ, Plow EF. Mol Cell. 2000;6:851–860. [PubMed] [Google Scholar]
- 35.Chung J, Bachelder RE, Lipscomb EA, Shaw LM, Mercurio AM. J Cell Biol. 2002;158:165–174. doi: 10.1083/jcb.200112015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsou R, Isik FF. Mol Cell Biochem. 2001;224:81–89. doi: 10.1023/a:1011947301849. [DOI] [PubMed] [Google Scholar]
- 37.Brooks PC, Montgomery AM, Rosenfeld M, Reisfeld RA, Hu T, Klier G, Cheresh DA. Cell. 1994;79:1157–1164. doi: 10.1016/0092-8674(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 38.Eliceiri BP, Puente XS, Hood JD, Stupack DG, Schlaepfer DD, Huang XZ, Sheppard D, Cheresh DA. J Cell Biol. 2002;157:149–160. doi: 10.1083/jcb.200109079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeng H, Dvorak HF, Mukhopadhyay D. J Biol Chem. 2001;276:26969–26979. doi: 10.1074/jbc.M103213200. [DOI] [PubMed] [Google Scholar]
- 40.Whitaker GB, Limberg BJ, Rosenbaum JS. J Biol Chem. 2001;276:25520–25531. doi: 10.1074/jbc.M102315200. [DOI] [PubMed] [Google Scholar]
- 41.Pepper MS. Arterioscler Thromb Vasc Biol. 2001;21:1104–1117. doi: 10.1161/hq0701.093685. [DOI] [PubMed] [Google Scholar]
- 42.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Nature. 2000;407:242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 43.Oh SJ, Jeltsch MM, Birkenhager R, McCarthy JE, Weich HA, Christ B, Alitalo K, Wilting J. Dev Biol. 1997;188:96–109. doi: 10.1006/dbio.1997.8639. [DOI] [PubMed] [Google Scholar]
- 44.Karkkainen MJ, Ferrell RE, Lawrence EC, Kimak MA, Levinson KL, McTigue MA, Alitalo K, Finegold DN. Nat Genet. 2000;25:153–159. doi: 10.1038/75997. [DOI] [PubMed] [Google Scholar]
- 45.Eto K, Puzon-McLaughlin W, Sheppard D, Sehara-Fujisawa A, Zhang XP, Takada Y. J Biol Chem. 2000;275:34922–34930. doi: 10.1074/jbc.M001953200. [DOI] [PubMed] [Google Scholar]
- 46.Majumdar M, Tarui T, Shi B, Akakura N, Ruf W, Takada Y. J Biol Chem. 2004;279:37528–37534. doi: 10.1074/jbc.M401372200. [DOI] [PubMed] [Google Scholar]
- 47.Yokosaki Y, Matsuura N, Sasaki T, Murakami I, Schneider H, Higashiyama S, Saitoh Y, Yamakido M, Taooka Y, Sheppard D. J Biol Chem. 1999;274:36328–36334. doi: 10.1074/jbc.274.51.36328. [DOI] [PubMed] [Google Scholar]
- 48.Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, Jeltsch M, Jackson DG, Talikka M, Rauvala H, Betsholtz C, Alitalo K. Nat Immunol. 2004;5:74–80. doi: 10.1038/ni1013. [DOI] [PubMed] [Google Scholar]
- 49.Kirkin V, Mazitschek R, Krishnan J, Steffen A, Waltenberger J, Pepper MS, Giannis A, Sleeman JP. J Biol Chem. 2001;268:5530–5540. doi: 10.1046/j.1432-1033.2001.02476.x. [DOI] [PubMed] [Google Scholar]