Draper-dependent glial phagocytic activity is mediated by Src and Syk family kinase signalling (original) (raw)
. Author manuscript; available in PMC: 2008 Aug 1.
Published in final edited form as: Nature. 2008 Apr 23;453(7197):935–939. doi: 10.1038/nature06901
Abstract
The cellular machinery promoting phagocytosis of corpses of apoptotic cells is well conserved from worms to mammals. An important component is the Caenorhabditis elegans engulfment receptor CED-1 (ref. 1) and its Drosophila orthologue, Draper2. The CED-1/Draper signalling pathway is also essential for the phagocytosis of other types of ‘modified self’ including necrotic cells3, developmentally pruned axons4,5 and dendrites6, and axons undergoing Wallerian degeneration7. Here we show that Drosophila Shark, a non-receptor tyrosine kinase similar to mammalian Syk and Zap-70, binds Draper through an immunoreceptor tyrosine-based activation motif (ITAM) in the Draper intracellular domain. We show that Shark activity is essential for Draper-mediated signalling events in vivo, including the recruitment of glial membranes to severed axons and the phagocytosis of axonal debris and neuronal cell corpses by glia. We also show that the Src family kinase (SFK) Src42A can markedly increase Draper phosphorylation and is essential for glial phagocytic activity. We propose that ligand-dependent Draper receptor activation initiates the Src42A-dependent tyrosine phosphorylation of Draper, the association of Shark and the activation of the Draper pathway. These Draper-Src42A-Shark interactions are strikingly similar to mammalian immunoreceptor-SFK-Syk signalling events in mammalian myeloid and lymphoid cells8,9. Thus, Draper seems to be an ancient immunoreceptor with an extracellular domain tuned to modified self, and an intracellular domain promoting phagocytosis through an ITAM-domain-SFK-Syk-mediated signalling cascade.
Developing tissues produce excessive numbers of cells and selectively destroy a subpopulation through programmed cell death to regulate growth. Rapid clearance of cell corpses is essential for maintaining tissue homeostasis and preventing the release of potentially cytotoxic or antigenic molecules from dying cells, and defects in cell corpse clearance are closely associated with autoimmune and inflammatory diseases10-13. In C. elegans the CED-1 receptor is expressed in engulfing cells, where it acts to recognize cell corpses and drive their phagocytosis1. CED-1 promotes engulfment through an intracellular NPXY motif, a binding site for proteins containing a phosphotyrosine-binding (PTB) domain, and a YXXL motif, a potential interaction site for proteins containing SH2 domains1. The PTB domain adaptor protein CED-6 can bind the NPXY motif of CED-1 (ref. 14), is required for cell corpse engulfment15 and acts in the same genetic pathway as CED-1 (ref. 16). CED-1 ultimately mediates actin-dependent cytoskeletal reorganization through the Rac1 GTPase17, and Dynamin modulates vesicle dynamics downstream of CED-1 during engulfment18, but the molecular signalling cascade that allows CED-1 to execute phagocytic events remains poorly defined.
Glia are the primary phagocytic cell type in the developing and mature brain. Glia rapidly engulf neuronal cell corpses produced during development, as well as neuronal debris generated during axon pruning19,20 or during Wallerian degeneration in the adult brain21. In Drosophila, glial phagocytosis of these engulfment targets requires Draper, the fly orthologue of CED-1 (refs 2, 4-6). Draper, like CED-1, contains 15 extracellular atypical epidermal growth factor (EGF) repeats, a single transmembrane domain, and NPXY and YXXL motifs in its intracellular domain (Fig. 1a)2. Drosophila Ced-6 is also required for the clearance of pruned axons4, indicating possible conservation of the interaction between CED-1 and CED-6 in flies, but additional signalling molecules acting downstream of Draper have not been identified.
Figure 1. Shark binds an ITAM in the Draper intracellular domain.

a, Draper contains an ITAM domain from Y934-L952 (YXXI-X11-YXXL). The requirements for the five tyrosine residues within and adjacent to this domain (shown) and Src were assayed in the yeast two-hybrid system23.
b, Lysates from yeast cultures in a were tested in quantitative β-galactosidase (β-Gal) assays. Error bars represent s.e.m.; n = 3; *, P < 0.05. c, S2 cells were transfected with pMT-Myc::Shark and pMT-Drpr-I constructs. Draper immunoprecipitates (IP) were analysed by western blotting (WB) with anti-phosphotyrosine (pTyr), anti-Myc and anti-HA antibodies. Vec., vector.
In a yeast two-hybrid screen for molecules interacting with the regulatory region of Shark22,23, we identified Draper. We found that when LexA-Shark, constitutively active Src kinase and AD-Draper are present, Shark and Draper interact physically (Fig. 1a). In the absence of Src kinase, Shark and Draper fail to interact, indicating that phosphorylation of Draper by Src may be essential for Shark-Draper interactions. We found that the Draper intracellular domain contains an ITAM (YXXI/L-X6-12-YXXL), a key domain found in many mammalian immunoreceptors including Fc, T-cell and B-cell receptors. SFKs phosphorylate the tyrosines in ITAM domains, thereby allowing ITAM association with SH2-domain-containing signal transduction proteins including Syk and Zap-70 (refs 9, 24). We therefore generated Y→F substitutions of the tyrosine residues within or near the Draper ITAM, and found that Tyr 949 and Tyr 934 were critical for robust Draper-Shark binding (Fig. 1a, b). These correspond to the consensus tyrosine residues in the predicted Draper ITAM (Fig. 1a). We next transfected plasmids with carboxy-terminally haemagglutinin-tagged Draper (Draper-HA) or with Draper-HA and Shark with an amino-terminal Myc tag (Myc-Shark) into Drosophila S2 cells, immunoprecipitated with anti-HA antibodies, and performed western blots with anti-phosphotyrosine, anti-Myc and anti-HA antibodies (Fig. 1c). We found that Myc-Shark co-immunoprecipitated with Draper-HA, and that anti-phosphotyrosine antibodies labelled a band corresponding to the position of Draper-HA that was absent in empty vector controls. Further, we found that a Y949F substitution markedly reduced Draper-Shark association (Fig. 1c). Taken together, these data indicate Draper and Shark can associate physically through the Draper ITAM domain.
We next sought to determine whether Shark is required for glial phagocytic activity in vivo. Severing adult Drosophila olfactory receptor neurons (ORNs) initiates Wallerian degeneration of ORN axons. Antennal lobe glia surrounding these severed axons respond to this injury by extending membranes towards severed axons and engulfing degenerating axonal debris7. These glia express high levels of Draper, and in draperΔ5 null mutants, glia fail to respond morphologically to axon injury, and severed axons are not cleared from the central nervous system (CNS)7. Thus, both the extension of glial membranes to severed axons and the phagocytosis of degenerating axonal debris require Draper signalling.
We explored whether Shark function in glia is essential for glial responses to axon injury by driving an upstream activating sequence (UAS)-regulated double-stranded RNA interference construct designed to target shark (sharkRNAi) with the glial-specific repo-Gal4 driver, severing ORN axons, and assaying the recruitment of Draper and green fluorescent protein (GFP)-labelled glial membranes to severed axons. Maxillary palp-derived ORN axons project to 6 of the roughly 50 glomeruli in the antennal lobe. Within hours after maxillary palps have been ablated in control animals, Draper immunoreactivity decorates severed axons projecting to (Fig. 2a, arrow) and within maxillary palp ORN-innervated glomeruli (Fig. 2a, b), and GFP-labelled glial membranes are recruited to these severed axons (Fig. 2c, d). Strikingly, knocking down Shark in glia completely suppressed these events (Fig. 2). We next severed antennal ORN axons; these axons project to about 44 of the 50 antennal lobe glomeruli. Antennal ablation therefore injures nearly all glomeruli in the antennal lobe and results in the majority of antennal lobe glia in control animals upregulating Draper (Fig. 2a, b, open arrowhead) and undergoing hypertrophy (Fig. 2c, d). We found that knocking down Shark in glia also blocked this glial response to axon injury (Fig. 2). Thus, Shark is essential for all axon-injury-induced changes in glial morphology and Draper expression.
Figure 2. Shark is required for recruitment of Draper and glial membranes to severed axons.

a, Control animals (yw;+/UAS-sharkRNAi) and those with glia-specific knockdown of shark (yw;repo-Gal4/UAS-sharkRNAi) were assayed for expression of Draper (red). Glial nuclei were stained with Repo (blue). Left, uninjured; centre, maxillary palp ablation (day 1); right, antennal ablation (day 1). Outlined, example of a maxillary palp-innervated glomerulus; arrow, nerve containing severed maxillary palp ORN axons; open arrowhead, antennal lobe glial cell; boxes, areas quantified in b. b, Quantification of data from a. Error bars represent s.e.m.; n ≥ 10. c, Glial membranes were labelled in control (yw;UAS-GFPS65T/+;repo-Gal4/+) or glial sharkRNAi animals (yw;UAS-GFPS65T/+;repo-Gal4/UAS-sharkRNAi) and assayed for morphology before or after injury (panel order as in a). Outlined, maxillary palp-innervated glomerulus; arrow, nerve containing severed maxillary palp ORN axons; boxes, areas used to quantify glial hypertrophy in d. d, Quantification data from c. Error bars represent s.e.m.; n ≥ 10.
To determine whether Shark is required for glial phagocytosis of severed axons we labelled a subset of maxillary palp ORN axons with mCD8::GFP, knocked down Shark function in glia, and assayed the clearance of severed axons. In control animals severed GFP-labelled ORN axonal debris was cleared from the CNS within 5 days (Fig. 3a, b). In contrast, glial-specific sharkRNAi potently suppressed the clearance of degenerating axons, with severed axons lingering in the CNS for at least 5 days (Fig. 3a, b). We then examined whether mutations in the shark gene affected the glial clearance of degenerating axons. The null allele of shark, shark1, is pupal lethal25. We therefore assayed glial responses to axon injury in shark1 heterozygous mutants, and tested for dominant genetic interactions between draperΔ5 and shark1. We found that both draperΔ5/+ and shark1/+ animals showed defects in glial phagocytic function: 5 days after injury, significant amounts of axonal debris remained within OR85e-innervated glomeruli (Fig. 3c, d) and in the maxillary nerve (Fig. 3c, e). Moreover, shark1/+; draperΔ5/+ animals showed a striking suppression of glial clearance of severed axons almost equivalent to that of draperΔ5 mutants (Fig. 3c-e). Thus, shark mutations dominantly suppress the glial clearance of degenerating ORN axons, and this phenotype is strongly enhanced by removing one copy of draper. These data, taken together with our sharkRNAi data, show that Shark is essential for the clearance of degenerating axons by glia.
Figure 3. Shark is required for glial clearance of severed axons from the CNS.

a, The axons of OR85e-expressing ORNs were labelled with mCD8::GFP in control (yw;OR85e-mCD8::GFP/+;repo-Gal4/+) and glial sharkRNAi (yw;OR85e-mCD8::GFP/+;repo-Gal4/UAS-sharkRNAi) animals, maxillary palps were ablated, and the clearance of severed ORN axons from the CNS was assayed with anti-GFP antibody stains (green). Maxillary nerves are indicated (arrowheads). b, Quantification of data from a. Error bars represent s.e.m.; n ≥ 10. c, OR85e-expressing ORN axons were labelled in control (yw;OR85e-mCD8::GFP/+) animals and in shark1 or draperΔ5 null mutant backgrounds, maxillary palps were ablated, and clearance was assayed at 5 days. d, Quantification of data from c. Error bars represent s.e.m.; n ≥ 10; *, P < 0.05; **, P < 0.001; ***, P < 0.0001. e, Quantification of brain hemispheres containing GFP-labelled ORN axonal debris along the maxillary nerve for genotypes described in c.
Is Shark required for the glial clearance of neuronal cell corpses? In embryonic stage 14-15 control animals we found 24.4 cell corpses per hemisegment (Table 1). In contrast, we found that shark1 null mutants showed a marked increase in CNS cell corpses, with null mutants containing almost twice as many corpses per hemisegment (43.3 cell corpses per hemisegment; Table 1). shark1/Df(2R)6063 mutants accumulated cell corpses at levels similar to those in shark1, indicating that this phenotype maps to shark. These cell corpse engulfment phenotypes are indistinguishable from that of draperΔ5 mutants (38.5 ± 1.68 cell corpses per hemisegment; Table 1). We conclude that Shark, like Draper, is also essential for the efficient clearance of embryonic neuronal cell corpses by glia.
Table 1.
Quantification of cell corpse engulfment defects in shark and draper mutants
| Genotype | Cell corpses per hemisegment | Hemisegments, n |
|---|---|---|
| yw;+/+ | 24.4±0.91) | 52 |
| yw;shark 1 | 43.3±1.88) | 50 |
| yw;shark 1/+ | 25.8±0.80) | 54 |
| yw;Df(2R)6063 | 39.0±2.07) | 51 |
| y w;Df(2R)6063/+ | 24.6±0.68) | 51 |
| y w;Df(2R)6063/shark 1 | 46.5±1.52) | 41 |
| y w;draperΔ5 | 38.5±1.68) | 52 |
Because we found that Shark binds Draper only in the presence of an active Src kinase in our two-hybrid assays, we screened Drosophila Src kinases for roles in glial phagocytic activity. Interestingly, we found that glia-specific knockdown of Src42A (src42ARNAi) potently suppressed glial phagocytic activity: in src42ARNAi animals, Draper was not recruited to severed maxillary palp axons (Fig. 4a, b); glial hypertrophy and upregulation of Draper after antennal ablation was blocked (Fig. 4a, c); and GFP-labelled severed axons lingered in the CNS for 5 days (Fig. 4d, e). Knockdown of two other Drosophila Src kinases, Btk29A and Src64B, had no effect on the glial phagocytosis of severed axons (Supplementary Fig. 1). Thus, Src42A seems to be essential for all morphological responses of glia to axon injury and for the efficient clearance of degenerating axonal debris from the CNS.
Figure 4. Src42Aa is required for glial responses to axon injury and modulates Draper phosphorylation status.
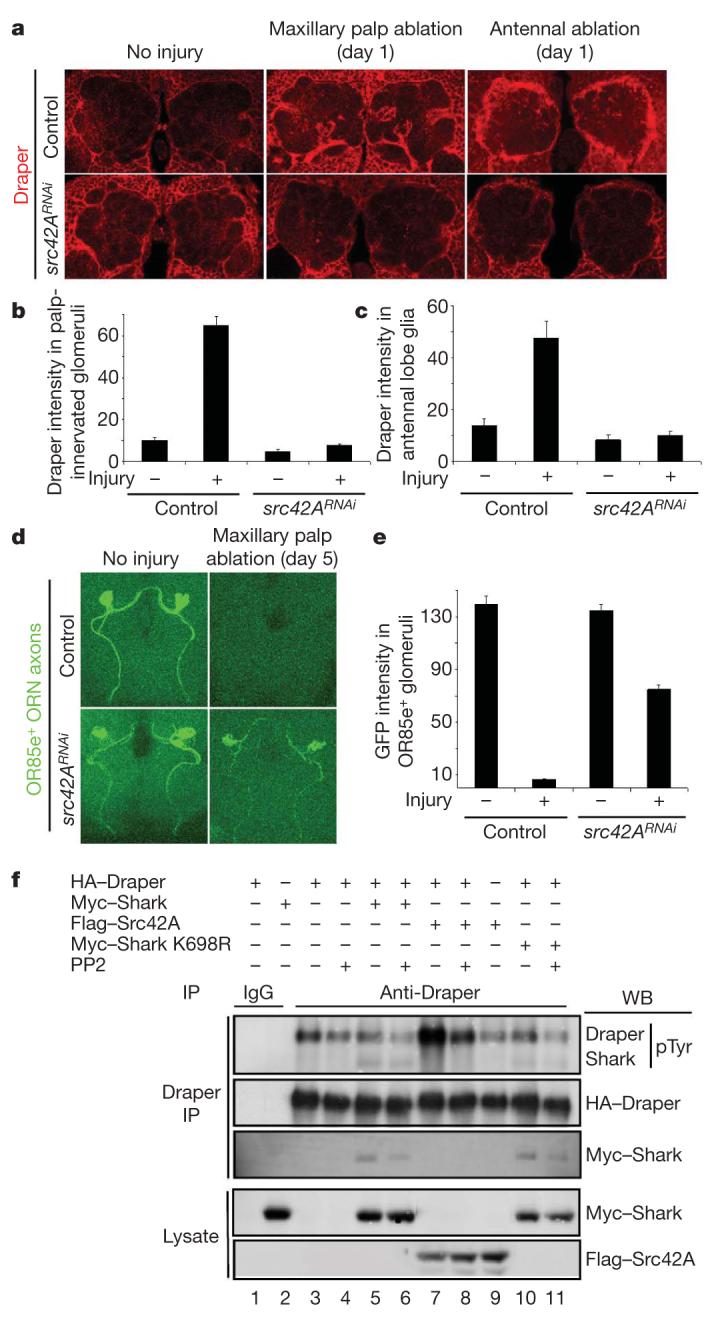
a, Control animals (yw;UAS-src42ARNAi/+, no driver) and those with glia-specific knockdown of src42A (yw;UAS-src42ARNAi/+;repo-Gal4/+) were assayed for injury-induced changes in glial Draper expression and for recruitment of Draper to severed axons (red). b, c, Quantification of data from a for palp-innervated glomeruli (b) and antennal lobe glia (c). Error bars represent s.e.m.; n ≥ 10. d, The axons of OR85e-expressing ORNs were labelled with mCD8::GFP in control (yw;OR85e-mCD8::GFP/+) and glial src42ARNAi (yw;OR85e-mCD8::GFP/UAS-src42ARNAi;repo-Gal4/+) animals, maxillary palps were ablated, and the clearance of severed ORN axons from the CNS was assayed with anti-GFP antibody stains (green) 5 days after injury. e, Quantification of data from d. Error bars represent s.e.m.; n ≥ 10. f, S2 cells were co-transfected with pMT-HA::Draper, pMT-Myc::Shark, pMT-Flag::Src42A, pMT-Myc::Shark K698R (kinase-dead) and pMT vector. After transfection and expression, some cells were incubated for 60 min with the SFK inhibitor PP2 (10 μM) before cell lysis. Anti-Draper and IgG control immunoprecipitates (IP) from cells were analysed by SDS-PAGE and western blotted (WB) with antibodies against pTyr, Draper, Myc and Flag.
We predicted that Draper phosphorylation status should be sensitive to the SFK inhibitor PP2. Indeed, addition of PP2 to S2 cultures led to a decrease in the phosphorylation of Draper (Fig. 4f, lanes 3 versus 4, 5 versus 6, 7 versus 8, and 10 versus 11) and Draper-Shark association (Fig. 4f, lanes 5 versus 6, and 10 versus 11). Strikingly, co-transfection of Draper and Src42A led to a marked increase in Draper phosphorylation (Fig. 4f, lane 7), which was PP2-sensitive (Fig. 4f, lane 8) and Draper-specific (Fig. 4f, lane 9). Draper-Shark interactions are not dependent on Shark kinase activity because kinase-dead Shark (Shark K698R) associates with Draper (Fig. 4f, lane 10). These data indicate that Src42A may phosphorylate the Draper intracellular domain, thereby increasing the association of Shark with Draper and the activation of downstream glial phagocytic signalling.
We have identified Shark and Src42A as novel components of the Draper pathway. One potential model for Draper-Shark-Src42A interactions is that Shark and Src42A drive the recruitment of Draper to engulfment targets. However, CED-1 has been shown to cluster around cell corpses even in the absence of its intracellular domain1. Moreover, Zap-70 and Syk bind phosphorylated ITAM domains in mammalian immunoreceptors when ITAM domains are phosphorylated by Src after ligand-dependent receptor clustering26-28. We therefore favour a model in which the engagement of Draper with its ligand (presumably presented by engulfment targets) promotes receptor clustering, tyrosine phosphorylation of Draper by Src42A, association of Shark, and activation of downstream phagocytic signalling events. Our work suggests that Draper is an ancient immunoreceptor in which the extracellular domain is tuned to recognize modified self and the intracellular domain signals through ITAM-Src-Syk-mediated mechanisms. This is the first identification of ITAM-Src-Syk signalling in invertebrates, and it suggests that a pathway similar to Draper-Ced-1 may ultimately have given rise to ITAM-based signalling cascades in mammalian myeloid and lymphoid cells, including those regulated by Fc, B-cell and T-cell receptors.
METHODS SUMMARY
Molecular biology and transgenics
To generate the sharkRNAi construct we amplified a 522-base-pair shark cDNA fragment (nucleotides 577-1098) from the BDGP complementary DNA LD41606, placing a 5′ _Nhe_I site and 3′ _Xba_I site, and cloned this into the _Nhe_I and _Xba_I sites in the pWiz UAS-RNAi vector to generate psharkRNAi-3. We next amplified the same 522-base-pair shark fragment but placed a 5′ _Xho_I site and 3′ _Bgl_II site, and cloned this fragment into psharkRNAi-3, to generate pUAS-sharkRNAi. The OR85e-mCD8::GFP line29 was a gift from B. Dickson. Transgenic Drosophila strains were generated by standard methods. The UAS-src42A, UAS-src64B and UAS-btk29A RNAi lines were obtained from the Vienna Drosophila Resource Center. The two-hybrid system used to identify Draper and the methods used to study its interaction in S2 cells have been described previously23. The Shark SH2-Ank-SH2-PRB region was fused to the DNA-binding domain (LexA) in the pGilda plasmid without Src (LexA-Shark) or with Src (LexA-Shark/Src). The activation domain (AD) in pB42AD was fused to the Draper intracellular domain (AD-Drpr) or to different Y→F Draper mutations (AD-DrprY_nnn_F). Transformed yeast were plated on Leu- plates for screening Leu-positive colonies. The anti-Draper antiserum used for immunoprecipitations was prepared by immunization of rabbits with GST fused to amino-acid residues 385-594 of the intracellular domain of Draper II.
Nerve injury assays and cell corpse quantification
Wallerian degeneration of ORN axons and quantification of clearance of axonal debris were performed as described previously7. Quantification of cell corpses was performed as described previously2.
Supplementary Material
Fig 1
Acknowledgements
This work was supported by National Institutes of Health grants (1R01NS053538 to M.R.F., and 1RO1GM55293 and 1RO1CA26504 to E.R.S), by an Albert Einstein Cancer Center Grant (PO3-13330 to E.R.S.), a Smith Family New Investigator Award (to M.R.F.) from the Smith Family Foundation, and a grant from the Christopher and Dana Reeves Foundation (to M.R.F). M.R.F. is an Alfred P. Sloan Research Fellow.
APPENDIX
METHODS
Yeast two-hybrid screen
The yeast two-hybrid screen was performed in accordance with the manufacturer’s instructions (Matchmaker Kit; Clontech). The cDNA encoding residues 10-468 of Shark was subcloned into the pGilda vector at the _Sma_l and _Bam_H1 sites to create the pLexA-Shark bait. To create the pLexA-Shark/Src bait encoding activated c-Src kinase driven by the ADH1 promoter, the cDNA encoding an activated c-Src-kinase domain was excised from the BTM116-Src vector30,31 as a _Bam_H1 fragment and subcloned into pLexA-Shark at the _Sac_1 site. A cDNA library from Drosophila embryos 0-21 h old cloned into pB42AD (Clontech) was used as the prey. The bait and prey plasmids were co-transformed into the Saccharomyces cerevisiae EGY48 (p8op-LacZ) host strain that carries LEU2 and lacZ under the control of LexA operators. Approximately 5 × 106 co-transformants were screened on _Ura_-, _His_-, _Trp_-, _Leu_-, 5-bromo-4-chloro-3-indolyl-β-d-galactoside plates. Seventeen independent transformants activated both the lacZ and LEU2 reporter genes. The pB42AD plasmids were individually isolated from each of the yeast transformants and sequenced. Three different overlapping sequences represented in 15 clones encompassed the Drpr I intracellular domain.
Schneider cell experiments
Drosophila Schneider (S2) cells were grown at room temperature (21 °C) in Schneider medium (Invitrogen) containing 10% fetal bovine serum (Invitrogen). cDNAs encoding Shark and Src42A, amino-terminally tagged with Myc and Flag, respectively, and Draper-1 and Draper-1 Y949F, carboxy-terminally tagged with HA, were cloned into the metallothionein promoter pMT/V5-His A vector (Invitrogen). For transfection, cells were plated in six-well plates (Falcon 3046; 2 × 106 cells per well; 2 ml) and cultured for 24 h. Cells were transfected with 2 μg of DNA per construct per well with the calcium phosphate procedure. The total DNA per transfection was kept constant by the addition of pMT vector DNA. After 15 h, induction of expression was initiated by the addition of 10 μl of 100 mM CuSO4 to each well and an additional 24-48 h was allowed for expression. Cells were harvested, lysed by vortex-mixing in 1% Nonidet P40, 10 mM Tris-HCl, 50 mM NaCl, 30 mM Na4P2O7, 50 mM NaF, 100 μM Na3VO4, 5 μM ZnCl2, 1 mM benzamidine, 10 μg ml-1 leupeptin and 10 μg ml-1 aprotinin pH 7.2 at 4 °C, and centrifuged at 13,000_g_ for 20 min; the supernatants were used for immunoprecipitation32. Immunoprecipitates were washed six times with lysis buffer without leupeptin and aprotinin and the immunoprecipitated proteins were eluted with SDS sample buffer and subjected to 7.5% SDS-PAGE, transfer and western blotting.
Induction of axon injury
ORN axon injury was induced by bilateral surgical ablation of third antennal segments and/or maxillary palps. After injury, animals were transferred to fresh food vials and aged at 25 °C.
Immunolabelling and confocal microscopy
Heads were severed from adult animals and fixed (1 × PBS/4% formaldehyde/0.1% Triton X-100) at room temperature for 20 min. Samples were then washed five times in PTx (1 × PBS/0.1% Triton X-100), brains were dissected at room temperature, and tissues were post-fixed (1 × PBS/4% formaldehyde/0.1% Triton X-100) for 15 min. Samples were then washed five times in PTx, blocked in PBTx (1 × PBS/0.1% Triton X-100/1% BSA) for 30 min and incubated overnight with primary antibodies in PBTx. The next day, samples were washed five times in PBTx, incubated with secondary antibodies (in PBTx) for 2 h at room temperature, washed five times with PBTx, and finally mounted in Bio-Rad antifade reagent.
Primary antibodies were used at the following dilutions: mouse anti-Repo, 1:10; rabbit anti-Draper, 1:500; mouse anti-GFP (Invitrogen), 1:500. Secondary antibodies (Jackson Immunoresearch) used for immunofluorescence were used at a dilution of 1:250; samples were mounted in Bio-Rad antifade reagent and viewed on a Zeiss LSM5 Pascal confocal microscope. For all experiments the entire antennal lobe was imaged in 1-2-μm steps and scored for relevant phenotypes. In experiments in which GFP or Draper intensity in different genetic backgrounds was quantified, samples were fixed and stained side by side, and we imaged them at the same time with standardized confocal settings for each series of experiments. Quantification of fluorescence intensity was performed on single Z sections in either the centre of the relevant glomerulus, at the same position of the maxillary nerve, or at the same dorsal position at the edge of the antennal lobe, with ImageJ software (http://rsb.info.nih.gov/ij/).
- 30.Keegan K, Cooper JA. Use of the two hybrid system to detect the association of the protein-tyrosine-phosphatase, SHPTP2, with another SH2-containing protein, Grb7. Oncogene. 1996;12:1537–1544. [PubMed] [Google Scholar]
- 31.Lioubin MN, et al. p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes Dev. 1996;10:1084–1095. doi: 10.1101/gad.10.9.1084. [DOI] [PubMed] [Google Scholar]
- 32.Li W, Yeung YG, Stanley ER. Tyrosine phosphorylation of a common 57-kDa protein in growth factor-stimulated and-transformed cells. J. Biol. Chem. 1991;266:6808–6814. [PubMed] [Google Scholar]
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Full Methodsand any associated references are available in the online version of the paper at www.nature.com/nature.
References
- 1.Zhou Z, Hartwieg E, Horvitz HR. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell. 2001;104:43–56. doi: 10.1016/s0092-8674(01)00190-8. [DOI] [PubMed] [Google Scholar]
- 2.Freeman MR, Delrow J, Kim J, Johnson E, Doe CQ. Unwrapping glial biology. Gcm target genes regulating glial development, diversification, and function. Neuron. 2003;38:567–580. doi: 10.1016/s0896-6273(03)00289-7. [DOI] [PubMed] [Google Scholar]
- 3.Chung S, Gumienny TL, Hengartner MO, Driscoll M. A common set of engulfment genes mediates removal of both apoptotic and necrotic cell corpses in C. elegans. Nature Cell Biol. 2000;2:931–937. doi: 10.1038/35046585. [DOI] [PubMed] [Google Scholar]
- 4.Awasaki T, et al. Essential role of the apoptotic cell engulfment genes draper and ced-6 in programmed axon pruning during Drosophila metamorphosis. Neuron. 2006;50:855–867. doi: 10.1016/j.neuron.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 5.Hoopfer ED, et al. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron. 2006;50:883–895. doi: 10.1016/j.neuron.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, Truman JW. Local caspase activity directs engulfment of dendrites during pruning. Nature Neurosci. 2006;9:1234–1236. doi: 10.1038/nn1774. [DOI] [PubMed] [Google Scholar]
- 7.MacDonald JM, et al. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 8.Fodor S, Jakus Z, Mocsai A. ITAM-based signaling beyond the adaptive immune response. Immunol. Lett. 2006;104:29–37. doi: 10.1016/j.imlet.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Underhill DM, Goodridge HS. The many faces of ITAMs. Trends Immunol. 2007;28:66–73. doi: 10.1016/j.it.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Hengartner MO. Programmed cell death in the nematode C. elegans. Recent Prog. Horm. Res. 1999;54:213–222. [PubMed] [Google Scholar]
- 11.Fadok VA, Bratton DL, Henson PM. Phagocyte receptors for apoptotic cells: recognition, uptake, and consequences. J. Clin. Invest. 2001;108:957–962. doi: 10.1172/JCI14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henson PM, Bratton DL, Fadok VA. Apoptotic cell removal. Curr. Biol. 2001;11:R795–R805. doi: 10.1016/s0960-9822(01)00474-2. [DOI] [PubMed] [Google Scholar]
- 13.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nature Rev. Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 14.Su HP, et al. Interaction of CED-6/GULP, an adapter protein involved in engulfment of apoptotic cells with CED-1 and CD91/low density lipoprotein receptor-related protein (LRP) J. Biol. Chem. 2002;277:11772–11779. doi: 10.1074/jbc.M109336200. [DOI] [PubMed] [Google Scholar]
- 15.Liu QA, Hengartner MO. Candidate adaptor protein CED-6 promotes the engulfment of apoptotic cells in C. elegans. Cell. 1998;93:961–972. doi: 10.1016/s0092-8674(00)81202-7. [DOI] [PubMed] [Google Scholar]
- 16.Ellis RE, Jacobson DM, Horvitz HR. Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics. 1991;129:79–94. doi: 10.1093/genetics/129.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kinchen JM, et al. Two pathways converge at CED-10 to mediate actin rearrangement and corpse removal in C. elegans. Nature. 2005;434:93–99. doi: 10.1038/nature03263. [DOI] [PubMed] [Google Scholar]
- 18.Yu X, Odera S, Chuang CH, Lu N, Zhou ZC. elegans Dynamin mediates the signaling of phagocytic receptor CED-1 for the engulfment and degradation of apoptotic cells. Dev. Cell. 2006;10:743–757. doi: 10.1016/j.devcel.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 19.Watts RJ, Schuldiner O, Perrino J, Larsen C, Luo L. Glia engulf degenerating axons during developmental axon pruning. Curr. Biol. 2004;14:678–684. doi: 10.1016/j.cub.2004.03.035. [DOI] [PubMed] [Google Scholar]
- 20.Awasaki T, Ito K. Engulfing action of glial cells is required for programmed axon pruning during Drosophila metamorphosis. Curr. Biol. 2004;14:668–677. doi: 10.1016/j.cub.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 21.Aldskogius H, Kozlova EN. Central neuron-glial and glial-glial interactions following axon injury. Prog. Neurobiol. 1998;55:1–26. doi: 10.1016/s0301-0082(97)00093-2. [DOI] [PubMed] [Google Scholar]
- 22.Ferrante AW, Jr, Reinke R, Stanley ER. Shark, a Src homology 2, ankyrin repeat, tyrosine kinase, is expressed on the apical surfaces of ectodermal epithelia. Proc. Natl Acad. Sci. USA. 1995;92:1911–1915. doi: 10.1073/pnas.92.6.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biswas R, Stein D, Stanley ER. Drosophila Dok is required for embryonic dorsal closure. Development. 2006;133:217–227. doi: 10.1242/dev.02198. [DOI] [PubMed] [Google Scholar]
- 24.Berton G, Mocsai A, Lowell CA. Src and Syk kinases: key regulators of phagocytic cell activation. Trends Immunol. 2005;26:208–214. doi: 10.1016/j.it.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Fernandez R, et al. The Drosophila shark tyrosine kinase is required for embryonic dorsal closure. Genes Dev. 2000;14:604–614. [PMC free article] [PubMed] [Google Scholar]
- 26.Monroe JG. ITAM-mediated tonic signalling through pre-BCR and BCR complexes. Nature Rev. Immunol. 2006;6:283–294. doi: 10.1038/nri1808. [DOI] [PubMed] [Google Scholar]
- 27.Pitcher LA, van Oers NS. T-cell receptor signal transmission: who gives an ITAM? Trends Immunol. 2003;24:554–560. doi: 10.1016/j.it.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Cox D, Greenberg S. Phagocytic signaling strategies: Fcγ receptor-mediated phagocytosis as a model system. Semin. Immunol. 2001;13:339–345. doi: 10.1006/smim.2001.0330. [DOI] [PubMed] [Google Scholar]
- 29.Couto A, Alenius M, Dickson BJ. Molecular, anatomical, and functional organization of the Drosophila olfactory system. Curr. Biol. 2005;15:1535–1547. doi: 10.1016/j.cub.2005.07.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig 1