Arachidonic Acid and the Brain (original) (raw)
Abstract
Kinetic methods in unanesthetized rodents have shown that turnover rates of arachidonic acid (AA) and docosahexaenoic acid (DHA) in brain membrane phospholipids are rapid and energy consuming and that phospholipase A2 (PLA2) and acyl-CoA synthetase enzymes that regulate turnover are specific for one or the other PUFA. Thus, AA turnover in brain phospholipids was reduced, and AA-selective cytosolic cPLA2 or acyl-CoA synthetase, as well as cyclooxygenase (COX)-2, were downregulated in brains of rats given drugs effective against bipolar disorder, whereas DHA turnover and expression of DHA-selective calcium-independent iPLA2 were unchanged. Additionally, the brain AA and DHA cascades can be altered reciprocally by dietary or genetic conditions. Thus, following 15 wk of dietary (n-3) PUFA deprivation, DHA loss from rat brain was slowed because of reduced iPLA2 and COX-1 expression, whereas AA-selective cPLA2, sPLA2, and COX-2 were upregulated, as were AA and docosapentaenoic acid concentrations. Measured rates of AA and DHA incorporation into brain represent their respective rates of metabolic consumption, because these PUFA are not synthesized de novo or converted significantly from their precursors in brain. In healthy human volunteers, positron emission tomography (PET) was used to show that the brain consumes AA and DHA at respective rates of 17.8 and 4.6 mg/d, whereas in patients with Alzheimer disease, AA consumption is elevated. In the future, PET could be used to relate human brain rates of AA and DHA consumption to liver PUFA metabolism and dietary PUFA intake.
Introduction
The conditionally essential PUFA arachidonic acid [AA,4 20:4(n-6)] and docosahexaenoic acid [DHA, 22:6(n-3)] make up ∼20% of fatty acids in the mammalian brain (1). They, as well as their respective shorter-chain precursors, linoleic acid [LA, 18:2(n-6)] and _α_-linolenic acid [_α_-LNA, 18:3(n-3)], cannot be synthesized de novo from 2-carbon fragments by vertebrate tissue, and biochemical pathways do not exist for their interconversion. Thus, they must be obtained from dietary sources. However, if sufficient LA or _α_-LNA is in a diet free of AA or DHA, respectively, normal brain DHA and AA concentrations can be maintained by elongation and desaturation of the precursors within the liver (2).
Multiple aspects of brain metabolism, function, and structure are thought to depend on having adequate brain concentrations of AA and DHA as well as on interactions among these PUFA and their metabolites (3). Furthermore, a number of human brain diseases, such as Alzheimer disease (4) and bipolar disorder (5), appear to involve disturbed PUFA metabolism. Thus, understanding the dynamics of brain PUFA metabolism could help to interpret brain function in health and disease.
Methods are available to quantitatively assess the kinetics and enzymatic regulation of brain PUFA metabolism in unanesthetized rodents and to image regional brain PUFA consumption in rodents and humans. This article briefly summarizes some of these methods and their applications, with a focus on AA metabolism.
Kinetic fatty acid methods
To measure brain PUFA kinetics and the enzymatic regulation of PUFA metabolism in rodents, a loosely restrained unanesthetized animal is injected intravenously with a radiolabeled PUFA bound to serum albumin. Labeled and unlabeled unesterified PUFA concentrations are measured in plasma at fixed times thereafter until the animal is killed. Its brain is removed after being subjected to high energy microwaving to instantaneously stop metabolism, or is removed and frozen. In the former case, lipids are extracted, and concentrations of labeled and unlabeled PUFA are measured by analytical procedures in brain unesterified fatty acid, acyl-CoA, and phospholipid pools. In the latter case, the frozen brain is cut into serial coronal slices on a freezing microtome, the slices are placed against X-ray film, and regional brain radioactivities are determined by quantitative autoradiography. The frozen brain also can be subjected to molecular or enzyme activity analysis. Another method involves injecting a radiolabeled PUFA intracerebrally, then measuring its specific activity in individual phospholipids of microwaved brain at different times thereafter, to determine loss half-lives and turnover rates of the unlabeled PUFA.
PUFA incorporation into stable brain lipids is unaffected by changes in cerebral blood flow, allowing us to image PUFA metabolism and signaling at rest and during functional activation in vivo, without correcting for concurrent variations in delivery by flow. Brain incorporation of AA and DHA also can be quantitatively imaged in human subjects using positron emission tomography (PET) and intravenously injected positron-emitting [1-11C]AA or [1-11C]DHA (6).
Compartmental representation of brain AA cascade
Pathways of plasma-brain exchange of AA and of its intravenously injected radiolabel (designated as AA*), as well as pathways and compartments of brain AA metabolism in what is termed the “arachidonic acid cascade” (7,8), are illustrated in Figure 1. DHA participates in a comparable cascade.
FIGURE 1 .

Model of brain AA cascade. AA, found at the _sn_-2 position of a phospholipid, is liberated by activation (star) of PLA2 at the synapse. A fraction of the liberated AA is converted to bioactive eicosanoids. The remainder diffuses to the endoplasmic reticulum while bound to a fatty acid binding protein (FABP), from where it is converted to arachidonoyl-CoA by acyl-CoA synthetase with the consumption of 2 ATP, then reesterified by an acyltransferase. Unesterified AA in the endoplasmic reticulum exchanges freely and rapidly with unesterified nonprotein-bound unesterified AA in plasma, into which labeled AA (AA*) has been injected. Equations for calculating incorporation coefficients k*, rates _J_in, and turnover of AA are shown in right lower corner. Adapted from Rapoport and Bosetti (26).
Circulating radiolabeled AA* will rapidly exchange with unesterified AA in the brain endoplasmic reticulum. Measuring the concentrations of AA and AA* in brain acyl-CoA and stable lipids in relation to integrated plasma specific activity can be used to calculate unesterified AA incorporation coefficients k*,
 |
(1) |
|---|
 is incorporated brain radioactivity at time of death T after tracer injection, and the denominator is the integral of plasma radioactivity
is incorporated brain radioactivity at time of death T after tracer injection, and the denominator is the integral of plasma radioactivity 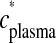 (plasma input function) between the start of injection at time t = 0 and t = T.
(plasma input function) between the start of injection at time t = 0 and t = T.
The rate of incorporation of the plasma unesterified unlabeled PUFA into brain, _J_in, equals the product of its unesterified, unlabeled plasma concentration _c_plasma and the incorporation coefficient k*,
 |
(2) |
|---|
The turnover rate of the fatty acid in phospholipids is _J_in divided by the fatty acid concentration _c_brain in stable brain lipid, multiplied by an arachidonyl-CoA dilution factor λ,
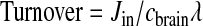 |
(3) |
|---|
Because AA cannot be synthesized de novo or be converted in the brain from its shorter-chain LA precursor (9,10), _J_in equals the quantity of AA that has been lost by metabolism, that is, the rate of AA consumption within brain (11). A similar consideration applies to DHA.
The brain AA cascade involves 2 major cycles (Fig. 1). One cycle is a continuous process of AA deacylation followed by reacylation in individual phospholipids, with short half-lives (hours) and high turnover rates (e.g., 15–30%/h) in rodent brain (12,13). The other is a cycle involving metabolic AA loss within brain, compensated for by AA replenishment from plasma, with half-lives of weeks (11,14).
In the first cycle, AA is released from the stereospecifically numbered (sn)-2 position of a synaptic membrane phospholipid by a phospholipase A2 (PLA2), whose activation is coupled to activation of any of a number of postsynaptic neuroreceptors by a G-protein or by calcium (15). A small fraction of the released AA (normally ∼4%) is lost by metabolism to eicosanoids or other metabolites or by _β_-oxidation after being transferred to mitochondria from the acyl-CoA pool. This transfer requires carnitine palmitoyltransferase, whose affinity for arachidonoyl-CoA and other long-chain acyl-CoAs, compared with the affinity of acyltransferase, determines whether the fatty acid will be largely _β_-oxidized in mitochondria or esterified into lysophospholipid (16). AA and DHA are largely reacylated in brain, whereas LA, _α_-LNA, and palmitic acid (16:0) are largely _β_-oxidized.
The larger fraction (∼96%) of released AA in brain will be reincorporated into phospholipid after diffusing to the unesterified AA pool in the endoplasmic reticulum with the help of a fatty acid binding protein, then being converted to arachidonoyl-CoA by an acyl-CoA synthetase with consumption of 2 ATP (8). For AA to be a substrate for cyclooxygenase (COX)-2, it first must be incorporated in phospholipid and then released by PLA2 (17,18).
Enzyme selectivity allows independent brain AA and DHA cascades
Three major PLA2 enzymes have been described in mammalian brain: 1) an AA-selective cytosolic cPLA2 (85 kDa, Type IV), which requires <1 _μ_mol/L Ca2+ for translocation to the membrane plus phosphorylation for activation; 2) an AA-selective secretory PLA2 (sPLA2 ) (14 kDa, Type IIA), which is also Ca2+ (20 mmol/L) dependent; and 3) a Ca2+-independent PLA2 (iPLA2) (101 kDa, Type VI) that is selective for DHA (19,20). cPLA2 colocalizes with and is coupled to COX-2 at postsynaptic sites, whereas iPLA2 is found at these sites and in astrocytes. Acyl-CoA synthetases also can be selective for AA or DHA (21).
As discussed in the following sections, the presence of AA- and DHA-selective enzymes within brain allows these PUFAs to recycle within phospholipids independently of each other and to be modified separately by drugs, genetics, disease, or diet (3). Thus, AA recycling and AA-selective cPLA2 were downregulated in rats given mood stabilizers and upregulated in rat models of neuroinflammation and excitotoxicity, while DHA recycling and DHA-selective iPLA2 were unchanged in these conditions (17,22–24). Additionally, reduced dietary (n-3) PUFA for 15 wk increased rat brain expression of cPLA2, sPLA2, and COX-2 while downregulating iPLA2 and COX-1 expression (25).
Selective targeting of the AA cascade by antimanic mood stabilizers
Bipolar disorder is a neuropsychiatric disease that consists of repeated cycles of manic and depressive episodes (bipolar I) or of hypomanic and depressive episodes (bipolar II). It affects 1–2% of the U.S. population, appears initially in young adults, and has a 10–20% lifetime incidence of suicide. Agents called “mood stabilizers” are used to treat the disease. Of these, lithium, carbamazepine (5-carbamoyl-5H-dibenz[b,f]azepine), and valproic acid (2-propylpentanoic acid) are FDA approved for treating bipolar mania, whereas lamotrigine [6-(2,3-dichlorophenyl)-1,2,4-triazine-3,5-diamine] is approved for bipolar depression and rapid cycling. The mechanisms of action of these agents are not agreed on, but experiments in unanesthetized rodents suggest that they commonly target the brain AA cascade (12,13,24,26).
Thus, lithium, valproic acid, or carbamazepine, when administered chronically to rats to produce plasma levels therapeutically relevant to bipolar disorder, reduced turnover of AA but not of DHA or palmitic acid in brain phospholipids (Tables 1 and 2). The reduced AA turnover produced by lithium and carbamazepine corresponded to reduced transcription of AA-selective cPLA2 and reduced expression of its transcription factor, activator protein-2. iPLA2 and sPLA2 were unaffected. On the other hand, the reduced AA turnover caused by valproic acid corresponded to its inhibiting AA conversion to arachidonoyl-CoA by an AA-selective microsomal acyl-CoA synthetase. Each of the 3 agents also reduced rat brain COX-2 activity and the concentration of prostaglandin E2 produced from AA via COX-2 (12,13,24,26). When reduced COX-2 transcription was the cause of reduced activity, it was related to reduced expression of the COX-2 transcription factor, NF-_κ_B (Table 2). Topiramate (2,3:4,5-di-O-isopropylidene-_β_-d-fructopyranose sulfamate), an anticonvulsant suggested by initial Phase II trials to work in bipolar disorder but later proven ineffective in phase III trials (27), had no affect on any of the above markers, whereas lamotrigine reduced transcription and expression of COX-2 without changing either AA or DHA turnover.
TABLE 1.
Chronic lithium administration reduces arachidonate turnover rate in rat brain phospholipids1
| Turnover,2%/h | ||||
|---|---|---|---|---|
| Fatty acid | _λ_acyl-CoA | PtdIns3 | ChoGpl3 | EtnGpl3 |
| Arachidonic acid | ||||
| Control | 0.04 ± 0.00 | 15.3 ± 0.4 | 18.3 ± 0.6 | 1.1 ± 0.1 |
| Lithium3 | 0.18 ± 0.02** | 2.6 ± 0.1** | 5.0 ± 0.3** | 0.2 ± 0.0** |
| DHA | ||||
| Control | 0.03 ± 0.00 | 17.7 ± 1.7 | 3.1 ± 0.4 | 1.2 ± 0.1 |
| Lithium3 | 0.03 ± 0.00 | 31.0 ± 9.0 | 4.5 ± 1.2 | 1.6 ± 0.4 |
| Palmitic acid | ||||
| Control | 0.02 ± 0.00 | 29.1 ± 2.6 | 7.0 ± 0.4 | 4.1 ± 0.4 |
| Lithium3 | 0.02 ± 0.00 | 26.0 ± 1.2 | 5.1 ± 0.3* | 3.5 ± 0.1 |
TABLE 2.
Effective antibipolar agents downregulate parts of rat brain AA cascade1
| Treatment | |||||
|---|---|---|---|---|---|
| Lithium2 | Carbamazepine2 | Valproate2 | Lamotrigine3 | Topiramate4 | |
| AA turnover | ↓ | ↓ | ↓ | NC | NC |
| DHA turnover | NC | NC | NC | — | NC |
| mRNA, protein, activity | |||||
| cPLA2 | ↓5 | ↓5 | NC | NC | NC |
| sPLA2 | NC | NC | NC | NC | NC |
| iPLA2 | NC | NC | NC | NC | NC |
| AA-selective acyl-CoA synthetase activity | NC | NC | ↓ | — | — |
| COX-2 protein and/or activity | ↓ | ↓ | ↓6 | ↓7 | NC |
| Prostaglandin E2 | ↓ | ↓ | ↓ | — | NC |
Downregulating the brain AA cascade may be one way in which the antimanic mood stabilizers work in bipolar disorder, in view of evidence that AA cascade enzymes—cPLA2, sPLA2, and COX-2—are overexpressed in the postmortem bipolar disorder brain (5).
Other examples of alterations of the AA but not DHA cascade are experimental neuroinflammation and excitotoxicity, where AA turnover and cascade enzymes cPLA2 and COX-2 are upregulated, whereas DHA turnover and iPLA2 expression are unchanged (17,22,23,25).
Upregulation of brain AA cascade by dietary (n-3) PUFA deprivation
The rat brain AA cascade is upregulated and the DHA cascade downregulated by reducing the dietary content of (n-3) PUFA, indicating reciprocal regulation of the AA and DHA cascades. Thus, feeding rats an (n-3) PUFA-deficient or (n-3) PUFA-adequate diet (containing no DHA and 0.2% or 4.6% _α_-LNA, respectively) for 15 wk postweaning reduced the brain concentration of esterified DHA by 30% while reciprocally increasing concentrations of esterified AA and the AA elongation product docosapentaenoic acid [DPA, 22:5(n-6)]. Phospholipid and neutral lipid concentrations were unchanged (2). The rats fed the deficient compared with the adequate diet demonstrated increased scores on tests of aggression and depression, which are like symptoms found in bipolar disorder (28).
The half-life of DHA loss from brain was prolonged in rats on the (n-3) PUFA-deficient diet (14) (Fig. 2). This prolongation reflected a transcriptional downregulation of the DHA-selective iPLA2 and of COX-1, whereas the AA-selective cPLA2, as well as sPLA2 and COX-2, were reciprocally upregulated (Fig. 3) (25). The latter changes were accompanied by elevated brain AA and DPA concentrations.
FIGURE 2 .

Prolongation of DHA half-life in brain by dietary (n-3) PUFA deprivation. [4,5-3H]DHA was injected into the brain after deprivation for 15 wk, and radioactivity from the tracer was followed in phospholipids for 60 d, from which half-lives t½ were calculated. _J_loss was calculated from half-lives as illustrated in the figure. The deprivation prolonged the DHA half-life from 33 to 90 d. Adapted from DeMar et al. (14).
FIGURE 3 .

Fifteen weeks of dietary (n-3) PUFA deprivation decreased rat frontal cortex iPLA2 and COX-1 protein but increased sPLA2, cPLA2, and COX-2 protein. Protein measured relative to actin. Adapted from Rao et al. (25).
Reciprocal changes in the brain AA and DHA cascade also occur in mice in which the gene for _α_-synuclein, which can be mutated in families with autosomal dominant Parkinson disease, is deleted. These mice have increased DHA turnover but reduced AA turnover in their brain phospholipids. They also have a marked reduction in brain arachidonoyl-CoA concentration and in microsomal acyl-CoA synthetase activity toward AA (29).
Quantifying regional PUFA consumption by the human brain
The regional rate of brain PUFA consumption equals _J_in, the product of the incorporation coefficient k* and the plasma concentration of unesterified PUFA (Eq. 2), because AA or DHA cannot be synthesized de novo to replace the PUFA that is lost by metabolism. Because of this, PET has been used to quantify _J_in in the human brain for both PUFA.
Horizontal brain sections showing AA and DHA incorporation coefficients k* (Eq. 1) in healthy human volunteers are presented in Figure 4. These scans were obtained by injecting [1-11C]AA or [1-11C]DHA intravenously and then measuring brain radioactivity with PET (30, J. C. Umhau, W. Zhou, R. E. Carson, S. I. Rapoport, A. Polozova, J. Demar, N. Hussein, A. K. Bhattacharjee, K. Ma, G. Esposito, S. Majchrzak, P. Herscovitch, W. C. Eckelman, K. A. Kurdziel, and N. Salem, Jr, unpublished material). When k* was summed over the entire brain and multiplied by the respective unesterified plasma concentration, _J_in for the whole brain equaled 17.8 mg/d for AA and 4.6 mg/d for DHA. The calculated rate of DHA consumption is 2.5–5% of the average daily dietary intake of EPA + DHA in the United States, 100–200 mg/d (31).
FIGURE 4 .

Horizontal brain images of incorporation coefficients k* for DHA and AA in healthy human volunteers, obtained with PET. Values of k* are corrected for partial volume errors. Plasma concentration of unesterified DHA was 2.63 ± 1.17 μmol/L for left scan; of unesterified AA, 3.8 ± 1.7 μmol/L for right scan. Rates of whole-brain daily consumption are given in the inset. Adapted from J. C. Umhau, W. Zhou, R. E. Carson, S. I. Rapoport, A. Polozova, J. Demar, N. Hussein, A. K. Bhattacharjee, K. Ma, G. Esposito, S. Majchrzak, P. Herscovitch, W. C. Eckelman, K. A. Kurdziel, and N. Salem, Jr, unpublished material and Giovacchini et al. (30).
Alzheimer disease is associated with neuroinflammation and excitotoxicity, and the postmortem Alzheimer brain demonstrates elevated expression of cPLA2, sPLA2, and COX-2 (32). Consistent with these findings and evidence that both excitotoxicity and neuroinflammation upregulated the rat brain AA cascade, PET demonstrated increased brain AA incorporation in patients with Alzheimer disease compared with healthy age-matched controls, particularly in neocortical regions reported to have activated microglia and high levels of inflammatory cytokines (4).
Discussion
During neurotransmission, the brain AA cascade is initiated when AA is released from synaptic membrane phospholipid by neuroreceptor-initiated activation of cPLA2 (15). In conditions such as neuroinflammation and excitotoxicity, additional AA is released at cytokine and glutamatergic N-methyl-d-aspartate receptors, and AA cascade enzymes are overexpressed.
Kinetic methods and models in unanesthetized rodents have been used to show that PUFA turnover in brain membrane phospholipids is rapid and energy consuming. AA and DHA recycling (deesterification-reesterification) in brain are independent processes that are catalyzed, respectively, by AA- and DHA-selective enzymes. The AA cascade can be targeted independently of the DHA cascade or altered in a reciprocal fashion to the DHA cascade by dietary (n-3) PUFA deprivation or genetic modulation.
For example, AA but not DHA turnover in brain membrane phospholipids is downregulated in unanesthetized rats that have been chronically administered lithium, carbamazepine, or valproic acid, mood stabilizers that are used to treat the mania of bipolar disorder. This downregulation correlates with reduced expression of AA-selective cPLA2 or acyl-CoA synthetase and of COX-2. Such effects are relevant to the therapeutic action of the agents, as the downregulated enzymes are overexpressed in postmortem brain from bipolar disease patients.
In the face of dietary (n-3) PUFA deprivation, homeostatic mechanisms slow brain DHA loss while increasing AA metabolism. Thus, in rats subjected to (n-3) PUFA deprivation for 15 wk, the half-life of DHA in brain phospholipids is prolonged, and the DHA-selective iPLA2 and COX-1 are transcriptionally downregulated. In contrast, expression of AA-selective enzymes cPLA2, sPLA2, and COX-2 is upregulated, as are concentrations of AA and its elongation product, DPA. These changes may exacerbate neuroinflammation and excitotoxicity.
PET has been used with positron-emitting tracers, [1-11C]AA or [1-11C]DHA, to quantitatively image regional rates of AA and DHA consumption by the normal human brain. AA consumption is increased in patients with Alzheimer disease compared with aged-matched healthy controls, likely because of neuroinflammation and excitotoxicity. In the future, PET could be used to determine brain rates of AA and DHA consumption in relation to human aging, brain or liver disease, or dietary PUFA intake.
Other articles in this symposium include references (33) and (34).
1
Published as a supplement to The Journal of Nutrition. Presented as part of the symposium “Dietary PUFA and the Aging Brain: Food for Thought” given at the 2008 Experimental Biology meeting on April 7, 2008, in San Diego, CA. The symposium was sponsored by the American Society for Nutrition and supported by an educational grant from Martek BioSciences Corporation. The symposium was chaired by Jay Whelan.
2
Supported entirely by the Intramural Program of the National Institute on Aging, NIH.
3
Author disclosures: S. I. Rapoport, no conflicts of interest.
4
Abbreviations used: AA, arachidonic acid; COX, cyclooxygenase; cPLA2, cytosolic phospholipase A2; DHA, docosahexaenoic acid; DPA, docosapentaenoic acid; iPLA2, calcium-independent PLA2; LA, linoleic acid; _α_-LNA, _α_-linolenic acid; NMDA, N-methyl-d-aspartate; PET, positron-emission tomography; PLA2, phospholipase A2; sn, stereospecifically numbered; sPLA2, secretory PLA2.
References
- 1.Contreras MA, Greiner RS, Chang MC, Myers CS, Salem N Jr, Rapoport SI. Nutritional deprivation of alpha-linolenic acid decreases but does not abolish turnover and availability of unacylated docosahexaenoic acid and docosahexaenoyl-CoA in rat brain. J Neurochem. 2000;75:2392–400. [DOI] [PubMed] [Google Scholar]
- 2.Igarashi M, DeMar JC Jr, Ma K, Chang L, Bell JM, Rapoport SI. Upregulated liver conversion of alpha-linolenic acid to docosahexaenoic acid in rats on a 15 week n-3 PUFA-deficient diet. J Lipid Res. 2007;48:152–64. [DOI] [PubMed] [Google Scholar]
- 3.Contreras MA, Rapoport SI. Recent studies on interactions between n-3 and n-6 polyunsaturated fatty acids in brain and other tissues. Curr Opin Lipidol. 2002;13:267–72. [DOI] [PubMed] [Google Scholar]
- 4.Esposito GI, Giovacchini G, Liow J-S, Bhattacharjee AK, Greenstein D, Schapiro M, Hallett M, Herscovitch P, Eckelman WC, et al. Imaging neuroinflammation in Alzheimer Disease with radiolabeled arachidonic acid and PET. J Nucl Med. 2008;49:1414–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rao JS, Kim EM, Lee HJ, Rapoport SI. Up-regulated arachidonic acid cascade enzymes and their transcription factors in post-mortem frontal cortex from bipolar disorder patients. Soc Neurosci Abstr. 2007;797.5/Z4.
- 6.Esposito G, Giovacchini G, Der M, Liow JS, Bhattacharjee AK, Ma K, Herscovitch P, Channing M, Eckelman WC, et al. Imaging signal transduction via arachidonic acid in the human brain during visual stimulation, by means of positron emission tomography. Neuroimage. 2007;34:1342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rapoport SI. In vivo fatty acid incorporation into brain phosholipids in relation to plasma availability, signal transduction and membrane remodeling. J Mol Neurosci. 2001;16:243–61. [DOI] [PubMed] [Google Scholar]
- 8.Robinson PJ, Noronha J, DeGeorge JJ, Freed LM, Nariai T, Rapoport SI. A quantitative method for measuring regional in vivo fatty-acid incorporation into and turnover within brain phospholipids: Review and critical analysis. Brain Res Brain Res Rev. 1992;17:187–214. [DOI] [PubMed] [Google Scholar]
- 9.DeMar JC Jr, Lee HJ, Ma K, Chang L, Bell JM, Rapoport SI, Bazinet RP. Brain elongation of linoleic acid is a negligible source of the arachidonate in brain phospholipids of adult rats. Biochim Biophys Acta. 2006;1761:1050–9. [DOI] [PubMed] [Google Scholar]
- 10.Holman RT. Control of polyunsaturated acids in tissue lipids. J Am Coll Nutr. 1986;5:183–211. [DOI] [PubMed] [Google Scholar]
- 11.Rapoport SI, Chang MC, Spector AA. Delivery and turnover of plasma-derived essential PUFAs in mammalian brain. J Lipid Res. 2001;42:678–85. [PubMed] [Google Scholar]
- 12.Chang MC, Bell JM, Purdon AD, Chikhale EG, Grange E. Dynamics of docosahexaenoic acid metabolism in the central nervous system: lack of effect of chronic lithium treatment. Neurochem Res. 1999;24:399–406. [DOI] [PubMed] [Google Scholar]
- 13.Chang MC, Grange E, Rabin O, Bell JM, Allen DD, Rapoport SI. Lithium decreases turnover of arachidonate in several brain phospholipids. Neurosci. Lett. 1996;220:171–4. Erratum in Neurosci Lett. 1997;222:141. [DOI] [PubMed] [Google Scholar]
- 14.DeMar JC Jr, Ma K, Bell JM, Rapoport SI. Half-lives of docosahexaenoic acid in rat brain phospholipids are prolonged by 15 weeks of nutritional deprivation of n-3 polyunsaturated fatty acids. J Neurochem. 2004;91:1125–37. [DOI] [PubMed] [Google Scholar]
- 15.Rapoport SI. In vivo approaches to quantifying and imaging brain arachidonic and docosahexaenoic acid metabolism. J Pediatr. 2003;143:S26–34. [DOI] [PubMed] [Google Scholar]
- 16.Gavino VC, Cordeau S, Gavino G. Kinetic analysis of the selectivity of acylcarnitine synthesis in rat mitochondria. Lipids. 2003;38:485–90. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberger TA, Villacreses NE, Hovda JT, Bosetti F, Weerasinghe G, Wine RN, Harry GJ, Rapoport SI. Rat brain arachidonic acid metabolism is increased by a 6-day intracerebral ventricular infusion of bacterial lipopolysaccharide. J Neurochem. 2004;88:1168–78. [DOI] [PubMed] [Google Scholar]
- 18.Laposata M, Majerus PW. Measurement of icosanoid precursor uptake and release by intact cells. Methods Enzymol. 1987;141:350–5. [DOI] [PubMed] [Google Scholar]
- 19.Strokin M, Sergeeva M, Reiser G. Docosahexaenoic acid and arachidonic acid release in rat brain astrocytes is mediated by two separate isoforms of phospholipase A2 and is differently regulated by cyclic AMP and Ca2+. Br J Pharmacol. 2003;139:1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dennis EA. Diversity of group types, regulation, and function of phospholipase A2. J Biol Chem. 1994;269:13057–60. [PubMed] [Google Scholar]
- 21.Van Horn CG, Caviglia JM, Li LO, Wang S, Granger DA, Coleman RA. Characterization of recombinant long-chain rat acyl-CoA synthetase isoforms 3 and 6: identification of a novel variant of isoform 6. Biochemistry. 2005;44:1635–42. [DOI] [PubMed] [Google Scholar]
- 22.Lee HJ, Rao JS, Chang L, Rapoport SI, Bazinet RP. Chronic N-methyl-d-aspartate administration increases the turnover of arachidonic acid within brain phospholipids of the unanesthetized rat. J Lipid Res. 2008;49:162–8. [DOI] [PubMed] [Google Scholar]
- 23.Rao JS, Ertley RN, Rapoport SI, Bazinet RP, Lee HJ. Chronic NMDA administration to rats up-regulates frontal cortex cytosolic phospholipase A2 and its transcription factor, activator protein-2. J Neurochem. 2007;102:1918–27. [DOI] [PubMed] [Google Scholar]
- 24.Rao JS, Lee HJ, Rapoport SI, Bazinet RP. Mode of action of mood stabilizers: is the arachidonic acid cascade a common target? Mol Psychiatry. 2008;13:585–96. [DOI] [PubMed] [Google Scholar]
- 25.Rao JS, Ertley RN, DeMar JC Jr, Rapoport SI, Bazinet RP, Lee H-J. Dietary n-3 PUFA deprivation alters expression of enzymes of the arachidonic and docosahexaenoic acid cascades in rat frontal cortex. Mol Psychiatry. 2007;12:151–7. [DOI] [PubMed] [Google Scholar]
- 26.Rapoport SI, Bosetti F. Do lithium and anticonvulsants target the brain arachidonic acid cascade in bipolar disorder? Arch Gen Psychiatry. 2002;59:592–6. [DOI] [PubMed] [Google Scholar]
- 27.Kushner SF, Khan A, Lane R, Olson WH. Topiramate monotherapy in the management of acute mania: results of four double-blind placebo-controlled trials. Bipolar Disord. 2006;8:15–27. [DOI] [PubMed] [Google Scholar]
- 28.Demar JC, Jr., Ma K, Bell JM, Igarashi M, Greenstein D, Rapoport SI. One generation of n-3 polyunsaturated fatty acid deprivation increases depression and aggression test scores in rats. J Lipid Res. 2006;47:172–80. [DOI] [PubMed] [Google Scholar]
- 29.Golovko MY, Rosenberger TA, Feddersen S, Faergeman NJ, Murphy EJ. Alpha-synuclein gene ablation increases docosahexaenoic acid incorporation and turnover in brain phospholipids. J Neurochem. 2007;101:201–11. [DOI] [PubMed] [Google Scholar]
- 30.Giovacchini G, Lerner A, Toczek MT, Fraser C, Ma K, DeMar JC, Herscovitch P, Eckelman WC, Rapoport SI, Carson RE. Brain incorporation of 11C-arachidonic acid, blood volume, and blood flow in healthy aging: a study with partial-volume correction. J Nucl Med. 2004;45:1471–9. [PubMed] [Google Scholar]
- 31.Kris-Etherton PM, Taylor DS, Yu-Poth S, Huth P, Moriarty K, Fishell V, Hargrove RL, Zhao G, Etherton TD. Polyunsaturated fatty acids in the food chain in the United States. Am J Clin Nutr. 2000;71:179S–88S. [DOI] [PubMed] [Google Scholar]
- 32.Sun GY, Horrocks LA, Farooqui AA. The roles of NADPH oxidase and phospholipases A2 in oxidative and inflammatory responses in neurodegenerative diseases. J Neurochem. 2007;103:1–16. [DOI] [PubMed] [Google Scholar]
- 33.Lukiw WJ, Bazan NG. Docosahexaenoic acid and the aging brain. J Nutr. 2008;138:2510–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whelan J. (n-6) and (n-3) polyunsaturated fatty acids and the aging brain: food for thought. J Nutr. 2008;138:2521–2. [DOI] [PubMed] [Google Scholar]