KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma (original) (raw)
. Author manuscript; available in PMC: 2014 Jul 8.
Published in final edited form as: Nat Rev Cancer. 2010 Sep 3;10(10):683–695. doi: 10.1038/nrc2899
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is characterized by near-universal mutations in KRAS and frequent deregulation of crucial embryonic signalling pathways, including the Hedgehog (Hh) and Wnt–β-catenin cascades. The creation of mouse models that closely resemble the human disease has provided a platform to better understand when and in which cell types these pathways are misregulated during PDAC development. Here we examine the central part that KRAS plays in the biology of PDAC, and how the timing and location of Hh and Wnt–β-catenin signalling dictate the specification and oncogenic properties of PDAC.
Tumours frequently display inappropriate activation of signalling pathways that are essential for embryonic development and tissue homeostasis. Not only do such pathways contribute to the ability of tumour cells to proliferate and evade cell death, but they also alter cell plasticity. Pancreatic ductal adenocarcinoma (PDAC) fits this pattern and commonly displays reactivation of embryonic signalling pathways, such as transforming growth factor-β (TGFβ), Notch, Hedgehog (Hh), and Wnt–β-catenin signalling (see REF. 1 for a review of the molecular genetics of PDAC).
Unlike human tumours, such as medulloblastoma in which aberrant Hh signalling is sufficient for disease development2,3, or colon cancer4 in which deregulated Wnt–β-catenin can represent an initiating event, genetic experiments have revealed that the misregulation of Hh and Wnt–β-catenin signalling alone is not sufficient to drive PDAC development. Instead, analysis of PDAC mouse models driven by targeted pancreatic expression of oncogenic KRAS suggest that both temporal and spatial control of Hh and Wnt–β-catenin activity are involved in specifying a cell lineage that can progress to PDAC. We review the ability of KRAS to alter cell fate in the pancreas and how the timing and location of Hh and Wnt–β-catenin signalling contribute to PDAC development.
Mutant KRAS drives PDAC development
Substantial efforts have been applied to determining the molecular underpinnings of PDAC. Although some (~2–10%) PDACs seem to be associated with hereditary factors5,6, most are associated with high-frequency somatic mutations in a subset of genes, including those that encode the small GTPase protein KRAS7, and the tumour suppressors INK4A8, p53 (REFS 9,10) and SMAD4 (REF. 11). Of these frequently observed alterations, it is key to note that KRAS mutation is nearly universal (>95%) in human PDAC. The KRAS mutations found in PDAC result in a protein locked in a constitutively active state, unable to hydrolyse GTP, thus promoting persistent signalling to downstream effectors (reviewed in REF. 12). Although large-scale genomic studies are expanding knowledge of the wider landscape of mutations found in PDAC13, investigating the function of such ‘classical’ genes listed above in cell culture and animal models has considerably advanced insights into PDAC maintenance and progression. PDAC is associated with non-invasive, preneoplastic lesions that are thought to be precursors to the disease14 (BOX 1). Pancreatic intraepithelial neoplasias (PanINs) are the most common and most widely studied putative precursors. They are histologically classified into three stages of increasing cellular and nuclear atypia15 (FIG. 1). Molecular studies have shown that the PanIN stage correlates with increasing mutation frequency and variety16. For example, PanIN1 lesions frequently possess mutated KRAS (estimates suggest 15–40%17) but less often harbour mutations in p53 or SMAD4. PanIN3 lesions are more likely to express mutated KRAS, p53 and SMAD4 (REFS 18,19).
Box 1. KRAS necessity and sufficiency in the stepwise progression to pancreatic ductal adenocarcinoma.
Mucinous cystic neoplasm (MCN), intraductal papillary mucinous neoplasm (IPMN) and pancreatic intraepithelial neoplasia (PanIN)1 are thought to represent precursor stages for pancreatic ductal adenocarcinoma (PDAC). This view is supported by KRAS-driven mouse models in which such preneoplastic lesions precede the development of PDAC (TABLE 1). Indeed, in models that develop PanINs (the most commonly observed putative precursor lesion in humans) and PDAC, PanIN lesions increase in severity and PDAC becomes more prevalent as mice age. Therefore, in this Review, we refer to this cell fate as the PanIN–PDAC lineage (indicated as precursor–PDAC lineage in the figure below). However, other mouse models suggest that IPMNs and MCNs could be parallel routes to PDAC development. All three lesions share expression of ductal markers and therefore would require reprogramming into a ductal lineage if they emerge from non-ductal compartments, such as acini, centroacinar cells or endocrine cells.
Even though mutant KRAS is sufficient to initiate the PanIN–PDAC lineage in mice, there is some evidence that it may not be necessary. Although frequently observed, KRAS mutation is not universal in early human PanINs. Furthermore, in mice, lesions resembling early-stage PanINs can develop owing to Hedgehog ligand overexpression during pancreatic development57 and chronic inflammation120. KRAS mutation becomes increasingly frequent in advanced PanINs and PDAC, leading to an important but unresolved question as to when deregulated KRAS activity becomes necessary for disease progression.
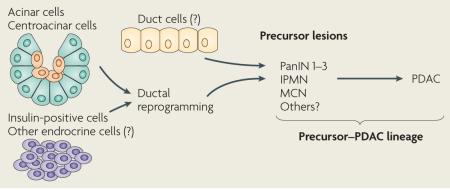
Figure 1. KRAS is a master regulator of pancreatic ductal adenocarcinoma initiation and progression.

Constitutively active KRAS (caused by KrasG12D or KrasG12V mutations) is sufficient to initiate the development of pancreatic intraepithelial neoplasia (PanIN) and pancreatic ductal adenocarcinoma (PDAC). PanINs are classified into three stages of increasing cellular atypia and, in humans, have been found to possess increasing numbers of mutations (common mutations are indicated in boxes). Changes in the epithelium are matched by desmoplastic changes in the stroma. In mouse models, the human PanIN spectrum followed by progression to PDAC has been recapitulated by activating mutant KRAS in embryonic pancreatic progenitors. Eliminating tumour suppressors commonly inactivated in the human disease dramatically decreases PDAC latency (a limited set of examples is indicated). Mouse models in which KRAS is activated specifically in some adult cell types have shown that both acini and insulin-positive cells can give rise to PanINs and, in some cases, PDAC depending on tissue damage and tumour suppressor inactivation. For these cell types, reprogramming into a ‘ductal’ cell type is required to assume the PanIN–PDAC lineage. Question marks are shown for centroacinar and duct cells as they have not been specifically assessed for their ability to be reprogrammed into a lineage capable of becoming PDAC under the control of KRAS. However, until specific targeting has been achieved, they cannot be ruled out as sources of the precursor–PDAC lineage. Figure is modified, with permission, from REF. 128 © (2000) American Association of Cancer Research.
Owing to its near universal frequency in PDAC, mutation of KRAS was proposed as an initiating genetic lesion in this disease. However, initial efforts to audit the sufficiency of mutant KRAS to initiate PDAC progression were stymied by the limitations of transgenic approaches. expression of mutant KRAS under acinar and ductal promoters resulted in ductal lesions reminiscent of PanINs and mixed acinar and ductal carcinomas20, or periductal inflammation21, respectively. However, neither model resulted in PDAC or a faithful recapitulation of the stepwise progression of precursor lesions. Although it is unclear why these models failed to recapitulate human disease progression, they may have been hampered by hyperphysiological KRAS output, or activation of KRAS in an inappropriate cell type or developmental stage. The ability of mutant KRAS to drive PDAC was not successfully investigated until the development of a Cre-inducible conditional allele (lox-stop-lox KrasG12D (LSL-KrasG12D)) targeted to the endogenous Kras locus22, thus allowing expression of constitutively active KRAS under temporal and spatial control. This tool eliminated possible issues of confounding cellular responses to overexpression, as transcription of the mutant Kras allele depends on the activity of the endogenous Kras promoter. Initially, mice expressing the LSL-KrasG12D allele were crossed to mice that expressed Cre recombinase under the control of promoters of the key pancreatic progenitor genes: pancreatic and duodenal homeobox 1 (Pdx1) and p48 (also known as Ptf1a), thus targeting mutant KRAS to most cells in the developing pancreas. A small number of Pdx1-Cre;LSL-KrasG12D and p48-Cre;LSL-KrasG12D mice (TABLE 1) developed PDAC over the course of 1 year23. Furthermore, early-stage PanINs were universally penetrant, and lesions resembling the entire human PanIN spectrum were observed with increasing age.
Table 1.
Mouse models of _Kras_-driven pancreatic ductal adenocarcinoma
| Genetic model | Phenotype | Refs |
|---|---|---|
| Pdx1-Cre;LSL-KrasG12D p48Cre;LSL-KrasG12D | Universally penetrant PanIN development. Age-dependent increase inlesion severity and occasional PDAC with long latency | 23 |
| Pdx1-Cre;LSL-KrasG12D;Cdkn2aflox/flox | Accelerated PanIN and PDAC development | 31 |
| Pdx1-Cre;LSL-KrasG12D;Trp53R172H/+ | Accelerated development of PanIN and metastatic PDAC. PDAC wasroutinely focal, well differentiated, with extensive genomic instability | 33 |
| Pdx1-Cre;LSL-KrasG12D;Smad4flox/flox p48-Cre;LSL-KrasG12D;Smad4flox/flox | Development of IPMN and PDAC | 34 |
| p48-Cre;LSL-KrasG12D;Tgfbr2flox/flox | Accelerated PanIN and PDAC development. Latency further decreasedwith nullizygosity compared with heterozygosity | 35 |
| Pdx1-Cre;LSL-KrasG12D;Trp53flox/flox | Accelerated PanIN and well-differentiated PDAC development | 32 |
| Pdx1-Cre;LSL-KrasG12D;Ink4aflox/flox | Accelerated development of PanIN and poorly differentiated PDAC | 32 |
| p48-Cre;LSL-KrasG12D;Smad4flox/flox | Development of MCN and PDAC. Latency shortened with nullizygosity | 36 |
| Pdx1-Cre;LSL-KrasG12D;Smad4flox/flox | Accelerated PanIN, IPMN and PDAC development | 37 |
| Nestin-Cre;LSL-KrasG12D | Development of exocrine-derived PanINs | 129 |
| p48-Cre;LSL-KrasG12D;Ela-Tgfa Ela-CreERT;LSL-KrasG12D;Ela-Tgfa | Accelerated PanIN, IPMN and PDAC development | 116 |
| Ela-Tta;tetO-Cre;LSL-KrasG12V | Development of acinar- and centroacinar-derived PanIN and PDACwhen KrasG12V activated during development or postnatally. Chronicpancreatitis required for PanIN and PDAC development when _KrasG12V_activated in adult cells | 24 |
| Ela-CreERT2;LSL-KrasG12D Mist1-CreERT2;LSL-KrasG12D | Development of acinar-derived PanIN | 25 |
| Pdx1-CreERT;LSL-KrasG12D;R26-NotchNIC Ela-CreERTLSL-KrasG12D;R26-NotchNIC | Accelerated development of acinar-derived PanIN | 26 |
| Mist1-CreERT2;LSL-KrasG12D;Mist1−/− | Accelerated development of acinar-derived PanIN | 27 |
| Pdx1-Cre;LSL-KrasG12D;Tif1γflox/flox | Rapid development of IPMN | 38 |
| Ela-CreERT;CAG-lox-GFP-lox-KrasG12V | Rapid development of chronic pancreatitis, PanIN, cystic papillarycarcinoma and PDAC | 28 |
Recently, these original models have been modified to begin to determine which pancreatic cell types can develop into PDAC when mutant KRAS is expressed. by using strategies that make use of inducible Cre, which allows the activation of KrasG12D in specific populations of adult cells, it has become evident that although PDAC displays ductal characteristics, it may not necessarily emanate from the duct compartment24–30 (FIG. 1; TABLE 1). Therefore, mutant KRAS is a crucial determinant of the PanIN–PDAC ‘lineage’ and is capable of driving pancreatic cells from terminal differentiation into a duct-like fate that can ultimately give rise to PDAC.
Additionally, KRAS-driven models have been combined with loss-of-function alleles of the most commonly inactivated tumour suppressors, including Ink4a, Trp53 and various components of the TGFβ signalling cascade31–38 (the models are summarized in TABLE 1), revealing that these pathways constrain KRAS-directed PDAC development. Interestingly, eliminating different tumour suppressors can dramatically alter the type of precursor lesion that develops and the ultimate differentiation state of the malignant disease. It is important to note that cells in these models are subjected to simultaneous activation of KRAS and loss of tumour suppressor function. This is likely to contrast to human tumour progression in which KRAS mutations seem to occur early in disease development, and cells subsequently undergo selection pressure and accumulate progressive tumour suppressor loss. The observation that enforcing tumour suppressor loss, out of the order that may occur in the spontaneous development of human disease, changes the course of PDAC development suggests that the disease depends on specific and sequential tuning of signalling pathways, in a similar fashion to normal development. Characterization of human tumours and mouse models indicate that this progression depends not just on compromising tumour suppressors, but also on the development of aberrant activity of other signalling pathways, including those that dictate developmental processes. The remainder of this Review focuses on two of these pathways: Hh and Wnt–β-catenin signalling.
Hh signalling in PDAC
Hh signalling plays a crucial part in embryonic development and has been implicated in homeostasis (impacting regeneration39,40 and stem cell maintenance41–43) and disease development in adult organs (for reviews on the biochemistry and developmental biology dependent on the pathway, see REFS 44,45). Hh signalling is mediated by a family of three, often tissue-specific, secreted ligands (Sonic hedgehog (SHH), Indian hedgehog (IHH) and Desert hedgehog (DHH)). Hh ligands activate signalling in target cells by binding to the 12-pass transmembrane receptor patched (PTC). In the absence of ligand, PTC represses the activity of a 7-pass transmembrane receptor, smoothened (SMO). ligand binding inactivates PTC, resulting in the activation of SMO. SMO activation, in turn, stimulates downstream intracellular components of Hh signalling that lead to the cytoplasmic accumulation and nuclear localization of active forms of the Gli transcription factors. Three Gli family members are found in mammals, GlI1, GlI2 and GlI3, and each possess unique properties. GlI1 is only present in an active form and GlI2 also functions predominately as an activator, as its repressor form is less stable than its activating form46, whereas GlI3 functions mainly as a repressor, as its repressor form is more stable than its transient full-length form47. Changing this balance of activating and repressing Gli proteins can lead to the induction of transcriptional targets that drive Hh-dependent phenotypes, such as proliferation through expression of cyclin D1 (REF. 48) and MYC49, evasion of apoptosis by promoting expression of BCL2 (REF. 50) and cell differentiation through the expression of Forkhead family transcription factors51,52. Moreover, the magnitude and duration of Hh signalling can alter owing to changes to positive (such as GlI1) and negative regulators (such as PTC) of the pathway.
The role of Hh signalling in tumorigenesis has emerged in parallel with an understanding of Hh biochemistry and its contribution to normal development. Deregulated Hh signalling in tumours can be approximately classified into two types depending on the level in the signalling cascade at which the pathway is activated (reviewed by REF. 53). The first class is defined by cell-autonomous mutations in key regulatory proteins; for example, inactivating mutations in PTC or activating mutations in SMO, such as those observed in basal cell carcinoma54,55 and medulloblastoma56. The second class of Hh-driven tumours, including cancers of the breast, colon, prostate and PDAC, is characterized by inappropriate ligand expression. Aberrant Hh ligand expression is observed at a high frequency in human PDAC (~75%) and is detectable throughout disease progression, beginning in early PanINs57, an expression pattern that is recapitulated in mutant KRAS-driven mouse models33. Hh signalling in PDAC cells was initially thought to be activated in an autocrine fashion. However, although chemical inhibitors targeting the pathway at the level of SMO (such as cyclopamine) or Hh ligand decrease the tumour burden and metastasis in xenotransplanted primary human tumours and cell lines, and SMO inhibition affects survival and tumour development in mutant KRAS-mutant mouse models58–61, recent evidence indicates that epithelial PDAC cells do not respond to Hh ligand and are refractory to ligand inhibition62,63. Instead, Hh signalling in PDAC seems to involve a ligand-dependent component in the tumour microenvironment in which the classical cascade is activated and a ligand-independent module in the tumour epithelium in which Gli activity is deregulated (FIG. 2).
Figure 2. Hedgehog signalling in pancreatic ductal adenocarcinoma.

Although the pancreatic ductal adenocarcinoma (PDAC) epithelium overexpresses Hedgehog (Hh), ligand-dependent canonical signalling is activated in stromal cells, including cancer-associated fibroblasts, infiltrating bone marrow-derived cells and subsets of endothelial cells, through the patched (PTC)–smoothened (SMO) axis. In turn, these cells directly proliferate or produce factors that might enhance tumour cell growth (potentially through secreted growth factors or by changing extracellular matrix (ECM) composition) and angiogenesis in a paracrine fashion (the factors that have been identified are indicated). Furthermore, cancer-associated fibroblasts and other Hh-responsive cells might produce cytokines and other molecules that communicate with infiltrating immune cells. Conversely, autocrine activation through this canonical pathway does not seem to occur in the tumour epithelium. Gli activity is maintained in part by activation of GLI1 through alternative signalling pathways, such as mutant KRAS expression and transforming growth factor-β (TGFβ) signalling. ANGPT1, angiopoietin 1; IGF, insulin-like growth factor; MMP9, matrix metalloproteinase 9; VEGFA, vascular endothelial growth factor A.
A robust desmoplastic response is one hallmark of PDAC. The desmoplastic stroma of PDAC consists of a complex array of cell types, including cancer-associated fibroblasts (CAFs), inflammatory cells and tumour-associated vasculature. evidence is mounting that paracrine Hh signalling plays a crucial part in supporting pro-tumorigenic communication between tumour epithelium and stroma, especially with respect to CAFs. CAFs are widely recognized as promoters of tumorigenesis64. Reconstitution experiments, in which tumour cells from various cancer types are admixed with CAFs and grown as xenografts, have shown that CAFs can promote transformation of immortalized epithelium (for example, SV40-immortalized prostate cells65) and enhance tumour cell growth66. PDAC-derived CAFs have been shown to have similar enhancing effects on the growth of xenotransplanted PDAC cells67–69. In support of a paracrine role for Hh ligand in promoting tumour growth, Yauch and colleagues62 showed that stromal cells recruited to xenografts of human PDAC cell lines had active Gli signalling, and xenografts treated with a SMO inhibitor displayed a decrease in the expression of mouse Hh target genes, but not in the expression of transplant-derived, human genes. Using mouse embryonic fibroblasts (MEFs) as a proxy for CAFs in reconstitution experiments, Yauch and colleagues62 demonstrated that SMO-deficient MEFs were significantly less efficient at promoting tumour growth than SMO-expressing cells. Furthermore, bailey and colleagues70 showed that co-transplantation of human pancreatic fibroblasts with transformed, SHH-overexpressing human duct cells led to increased growth of xenografts. Although paracrine Hh signalling clearly contributes to PDAC maintenance, the exact nature of the paracrine functioning, CAF-derived factors remains unclear. However, profiling experiments suggest that they may include some known pro-tumorigenic factors such as the insulin-like growth factors62. Hh ligand signalling may not only drive CAFs to produce pro-tumorigenic factors, but may also support an activated CAF phenotype, characterized by the expression of smooth muscle actin — a marker of the myofibroblast state — and the production of extracellular matrix proteins such as fibronectin and collagen I71, the latter of which has been shown to enhance PDAC cell proliferation and invasiveness72–75. Treating human pancreatic fibroblasts with SHH increases smooth muscle actin expression, and overexpression of SHH ligand in xenotransplanted cells enhances collagen I and fibronectin expression in recruited host fibroblasts71. Furthermore, Hh ligand promotes CAF proliferation and stimulates CAF migration71. Therefore, epithelial-derived Hh ligands may initiate a feed-forward loop that supports CAFs and stimulates the production of factors that act on tumour cells (FIG. 2). As Hh ligand expression increases as the severity of PanINs progresses57, this may be a mechanism by which CAFs are expanded and maintained during PDAC progression.
Recent reports indicate that the role of paracrine Hh signalling may not be limited to the tumour cell–CAF axis, and that paracrine Hh ligand signalling may also affect the tumour-associated vasculature. bailey and colleagues70 showed that expression of SHH increased angiogenesis in xenografts of transformed human pancreatic ductal cells. This effect depended in part on ligand signalling, as functional inhibition of Hh ligands with a blocking antibody blocked lymphangiogenesis in xenotransplanted tumour cells expressing Hh ligand70. Indeed, this effect may be direct owing to the ability of Hh ligands to activate migration and expansion of lymphatic endothelial cells70. It may also occur through indirect mechanisms, such as the induction of pro-angiogenic factors; for example, insulin-like growth factor 1 (IGF1) and angiopoietin 1 (REF. 76) produced by bone marrow-derived cells recruited to the tumour microenvironment, and vascular endothelial growth factor A (VEGFA) and matrix metalloproteinase 9 (MMP9)70 produced by CAFs.
The effect of SMO inhibition on disease progression in KRAS-driven mouse models further reflects the importance of paracrine Hh ligand signalling in PDAC. Olive and colleagues61 recently demonstrated that treating Pdx1-Cre;LSL-KrasG12D;Trp53R172H/+ or Pdx1-Cre;LSL-KrasG12D;Trp53R270H/+ mice with a SMO antagonist and gemcitabine, a first-line PDAC chemotherapeutic agent77,78, not only increased survival and decreased metastasis, but also significantly decreased the fibroblastic component of the tumours. Interestingly, SMO inhibitor treatment transiently increased the density of the tumour vasculature, and increased the concentration of gemcitabine reaching the tumour cells. Although this is a surprising finding given the data that indicate a proangiogenic role for stromal Hh signalling, it might reflect the observation that the apparent pro-tumorigenic hypovascularity observed in PDAC depends on a balance between the stromal and epithelial compartments. Nonetheless, these studies suggest that stromal Hh signalling may be a useful therapeutic target that could be explored in parallel with other drugs.
If ligand-mediated, canonical signalling is absent in the PDAC epithelium, does the Hh pathway have a direct role in the evolution of the tumour cells? It probably does, as several studies have shown that active Gli proteins support the proliferation and viability of epithelial PDAC cells79–81. Currently two signalling cascades have been implicated in supporting this non-canonical activation of Gli signalling. Targeting mutant KRAS directly by small interfering RNA (siRNA) or by targeting its downstream RAF–MAPK effector pathway with chemical inhibitors decreases the transcription of Gli-target genes and tumour cell growth81,82. Also, blocking TGFβ signalling in PDAC cells inhibits Gli activity and cell growth83. Regulation of Gli signalling by TGFβ raises questions regarding how the relative concentrations of the three different forms of Gli proteins contribute to Gli output. Treating cell lines derived from KRAS-driven mouse PDAC with TGFβ increases expression of GlI1 and GlI3, and GlI3 is induced to greater levels than GlI1 (REF. 81). This is surprising, as GlI3 predominantly acts as a repressor of Gli targets. However, sequencing analysis has shown that GLI3 and the related gene GLI4 are mutated at a high frequency in PDAC13. Therefore, regulation of epithelial Gli signalling might not only occur through non-canonical upstream signals, but also may involve novel interactions and functions of the Gli proteins.
Hh signalling and PDAC development
As Hh signalling seems to be important in PDAC progression and maintenance, a key question is how it contributes to disease initiation. Several genetic approaches have been used to determine how modulating Hh signalling alone, as well as in the context of mutant KRAS, changes the course of PDAC inception (these models are summarized in TABLE 2). A relationship between Hh signalling and PDAC initiation was first implicated in experiments that set out to determine how ligand-mediated Hh signalling directs gastrointestinal development. Several developmental studies illustrated that normal gut development is dependent on the tightly controlled expression of Hh ligands. For example, transgenic mice expressing SHH during early pancreas development under the control of the Pdx1 promoter (Pdx1-Shh)84 display near-complete pancreatic agenesis and, in place of a normal pancreas, possess duct structures embedded in an expanded mesenchymal compartment that expresses molecular markers characteristic of tumour-associated stroma84. Interestingly, the ductal remnants morphologically resemble early human PanIN1–2 lesions, and several of these transgenic mice also developed spontaneous, pancreas-specific KRAS mutations, suggesting that inappropriate ligand expression may promote changes in tissue architecture and signalling involved in PDAC initiation57. However, these animals died shortly after birth (3 weeks), which prevented evaluation of the ability of Hh ligand signalling alone to drive PDAC. To avoid the developmental consequences of aberrant ligand signalling, models aimed at testing if the intracellular modules of the Hh signalling cascade could drive PDAC were developed. First, mice carrying the CLEG2 transgene, which allows Cre-inducible, conditional expression of a dominantly active GlI2 protein lacking the amino-terminus repressor domain, were crossed with mice expressing a Pdx1-Cre driver85. Approximately one-third of Pdx1-Cre;CLEG2 mice develop pancreatic tumours85 which consist of undifferentiated, spindle-shaped cells that do not molecularly or histologically resemble PDAC. Therefore, although Gli signalling is potently oncogenic in the pancreas, alone it is inefficient in driving PDAC development.
Table 2.
Mouse models of Hedgehog and β-catenin deregulation in the pancreas
| Genetic model | Phenotype | Refs |
|---|---|---|
| Hedgehog | ||
| Pdx1-Shh | Atrophic pancreas displaying lesions reminiscent of PanIN1 and 2 embeddedin intestinal-like stroma | 57 |
| Pdx1-Cre;CLEG2 | Development of large, undifferentiated pancreatic tumours | 85 |
| Pdx1-Cre;CLEG2;LSL-KrasG12D | Accelerated PanIN development and undifferentiated ‘CLEG2-like’ tumours | 85 |
| CAGGS-CreER;R26-SmoM2 | Development of MCN-like lesions | 130 |
| p48-Cre;LSL-KrasG12D; Trp53flox/+;Smoflox/flox | No difference in PanIN and PDAC development compared to p48-Cre; LSL-KrasG12D;Trp53flox/+ mice | 81 |
| p48-Cre;LSL-KrasG12D; SmoM2 | No acceleration of PanIN development compared to p48-Cre;LSL-KrasG12D mice | 63 |
| β-catenin | ||
| Pdx1-CreLate; Ctnnb1exon3/+ | Acinar proliferation and postnatal pancreatomegaly. No tumour development | 102 |
| Pdx1-Cre;Apcflox/flox | Acinar proliferation and postnatal pancreatomegaly. No tumour development | 103 |
| p48-Cre;Ctnnb1exon3/+ | Development of tumours resembling human solid pseudopapillary tumour | 104 |
| p48-Cre;Ctnnb1exon3/+; LSL-KrasG12D | Development of tumours resembling human intraductal tubular tumour | 104 |
Given the high frequency of activating KRAS mutations in PDAC and that mutations in KRAS and persistent Gli activity are found in fully transformed PDAC lines, the ability of KRAS and Gli signalling to synergize to drive PDAC initiation and development was determined by intercrossing Pdx1-Cre;CLEG2 mice with LSL-KrasG12D animals. Within 3 to 6 weeks of birth, these animals rapidly developed PanIN lesions, including lesions that possessed characteristics of advanced grade 2 and 3 lesions85. PanIN lesions in this model also displayed aberrant Hh ligand expression and were accompanied by an expanded, proliferative stromal compartment, further linking Hh ligand signalling with stromal activation. However, the Pdx1-Cre;CLEG2; LSL-KrasG12D mice did not develop PDAC and instead developed an undifferentiated tumour of unclear origin, similar to that observed in Pdx1-Cre;CLEG2 mice.
The fact that PanINs in the Pdx1-Cre;CLEG2; LSL-KrasG12D model invariably displayed aberrant Hh ligand expression further suggested a role for ligand-dependent signalling in PDAC development. To determine if this role depended on ligand-dependent signalling in epithelial cells, Steveaux and colleagues generated p48-Cre; LSL-KrasG12D;Smoflox/flox mice81, therefore rendering pancreatic progenitors insensitive to Hh ligand. Surprisingly, PanIN and PDAC development progressed at an equivalent rate in these mice as in wild-type SMO controls. Similarly, both genotypes developed persistent epithelial ligand overexpression and equivalent levels of Hh-target gene activation. In addition, inhibition of GlI1 in cell lines derived from _Smo_-deficient and _Smo_-wild-type tumours led to increased apoptosis and decreased cell growth. Therefore, this model suggests that aberrant ligand expression and epithelial Gli signalling contribute to PDAC initiation and progression, but are uncoupled and evolve independently.
Taken together, these mouse models suggest that non-canonical epithelial Gli signalling and stromal ligand-dependent signals synergize with KRAS to drive PanIN initiation and progression to PDAC. However, important questions still remain. Hh ligand is clearly involved in instructing the tumour microenvironment, although which stromal Hh-target genes are key for the maintenance of CAFs and other components of the tumour stroma and which factors exert effects on the epithelium, including impacting differentiation, are mainly unknown. Furthermore, epithelial Gli activity seems to be involved in PDAC progression and develops owing to the activity of pathways other than the canonical Hh ligand–SMO-dependent pathway. A better understanding of how these non-canonical pathways (including KRAS and TGFβ signalling) activate Gli signalling and which epithelial Gli targets are crucial for maintenance of tumour biology might provide additional therapeutic targets, in parallel with factors activated by Hh ligand stimulation in stromal cells. Another therapeutic possibility is to determine other developmental signalling pathways that may interact in parallel with Hh signalling. One such candidate pathway that has been shown to have substantial effects on PDAC initiation and progression is Wnt–β-catenin signalling.
Wnt–β-catenin signalling in PDAC
Like Hh signalling, Wnt–β-catenin signalling is an important embryonic signalling pathway that is required for the proliferation, morphogenesis and differentiation of several organs (for a review, see REF. 86). β-catenin signal transduction has been excellently reviewed elsewhere87 (summarized in FIG. 3a). briefly, Wnt ligands bind to receptors of the Frizzled family of proteins and co-receptors. ligand binding results in the inactivation of a complex of cytoplasmic proteins (including adenomatous polyposis coli (APC) and axin) that promote the proteasomal degradation of β-catenin, resulting in its cytoplasmic accumulation and nuclear localization. In the nucleus, β-catenin binds to TCF/LEF factors to activate target genes. Nineteen Wnt ligands have been identified in mammals that are known to activate canonical (β-catenin-dependent) and non-canonical (β-catenin-independent) signal transduction cascades on binding to the Frizzled–LRP co-receptor complex. In this section, we focus on the canonical pathway, as this pathway has been implicated in PDAC development. However, non-canonical Wnts such as WNT5a are deregulated in PDAC and might have a role in PDAC biology88,89.
Figure 3. Canonical Wnt–β-catenin signalling in pancreatic ductal adencocarcinoma.
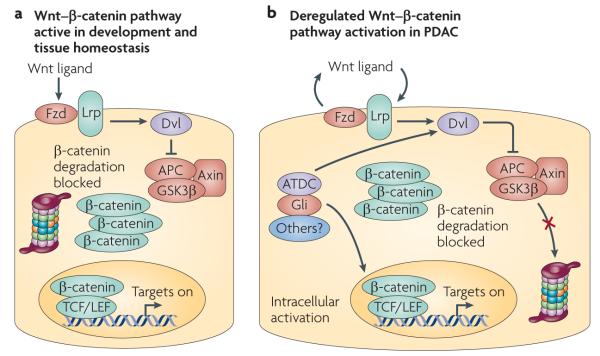
a | In normal development, canonical Wnt–β-catenin signalling depends on secreted ligands (Wnt ligands) that activate receptors (Frizzled (Fzd)–Lrp complex) that block the proteosomal degradation of β-catenin promoted by the destruction complex (comprising adeomatous polyposis coli (APC), axin, glycogen synthase kinase 3β (GSK3β) and other proteins) through activation of Dishevelled (Dvl). b | β-catenin accumulation is frequently observed in pancreatic ductal adenocarcinoma (PDAC). Accumulated β-catenin can translocate into the nucleus and activate target genes in concert with TCF/LEF co-factors. Presently, the dominant mechanism of persistent β-catenin accumulation and activity in PDAC is unclear. There is evidence for both autocrine (owing to epithelial-derived Wnt ligands) and cell-autonomous activation (through Gli signalling and genes such as ataxia telangiectasia group D-associated (ATDC), which activates Dvl). There may also be contributions from the stromal cells and extracellular matrix that may promote β-catenin accumulation.
Mutations in genes encoding regulatory proteins of the canonical signalling cascade are associated with many tumour types and in some cases, such as colon cancer, can act as initiating genetic lesions. loss-of-function mutations in negative regulators, such as APC4, and gain-of-function mutations in β-catenin itself90 activate constitutive, ligand-independent β-catenin signalling in a similar fashion to PTC and SMO mutations. like these Hh pathway mutations, genetic lesions in classical β-catenin regulatory modules are rare in PDAC91–93. Therefore, the role of β-catenin in PDAC has been controversial.
However, recent studies have begun to cement an important role for Wnt–β-catenin signalling in PDAC. Although β-catenin accumulation is not a universal characteristic of this disease, both nuclear (10–60%) and cytoplasmic accumulation (25–65%) of β-catenin are observed in PanIN lesions and PDAC94–96. Functional evidence is also accumulating that implicates a supporting role for β-catenin in PDAC maintenance and progression. Inhibiting β-catenin with siRNA substantially compromises PDAC proliferation and increases apoptosis96. Furthermore, increased levels of cytoplasmic and nuclear β-catenin correlate with PanIN grade and the development of invasive PDAC94,97. Recently, ataxia telangiectasia group D-associated protein (ATDC, also known as TRIM29) has been shown to contribute to the progressive accumulation of β-catenin and persistent activation of β-catenin-target genes in PDAC97. Inhibiting ATDC function in PDAC cell lines decreases β-catenin activity, tumour growth and metastasis. Therefore, genes such as ATDC might comprise a set of non-classical players that influence oncogenic β-catenin activity in PDAC in the absence of mutations normally observed in other β-catenin-driven tumours. Interestingly, accumulation of β-catenin mediated by ATDC97 leads to increased expression of MYC, a key node in β-catenin-driven transformation in other tissues. There is also emerging evidence that β-catenin accumulation and signalling could be increased through paracrine signalling which occurs in the PDAC micro-environment. Recent studies have shown that PDAC cells admixed with CAFs grown in three-dimensional extracellular matrix plugs display increased cytoplasmic β-catenin accumulation98, and growth on collagen I promotes nuclear β-catenin accumulation and activation of known β-catenin-target genes75.
With the exception of the specific examples discussed above, the general mechanism by which β-catenin accumulation occurs in PDAC is poorly understood (summarized in FIG. 3b). For example, although β-catenin accumulation correlates with disease severity, it is unclear whether accumulation depends on an autocrine response to Wnt ligands overexpressed by tumour cells. In support of this, canonical Wnt ligands (including WNT3 and WNT8b) are strongly expressed in PDAC samples96, and enforced expression of secreted frizzled receptor protein (SFRP, an inhibitor of Wnt ligands) decreases β-catenin transcriptional activity in a subset of human PDAC lines99. However, overexpression of DKK1, another inhibitor of secreted Wnt ligands, is associated with more advanced PanINs and PDAC, and increases PDAC cell growth and motility100. Furthermore, which of the vast number of β-catenin target genes are crucial for PDAC maintenance and progression are currently unknown. Therefore, although classical β-catenin oncogenic targets might be important in PDAC, more work is required to determine whether and at what level β-catenin signalling should be targeted for potential therapeutic effects.
β-catenin is insufficient to initiate PDAC
Increasing evidence indicates that β-catenin signalling contributes to PDAC maintenance. by contrast, the role of β-catenin function during PDAC initiation has only recently been analysed. Three studies have investigated the ability of uncontrolled β-catenin signalling to transform cells in the pancreas. Our studies have used a β-catenin allele in which the third exon (encoding phopshorylation sites important for protein degradation) is floxed and can be eliminated through Cre-mediated recombination101, resulting in enforced, conditional β-catenin stabilization and signalling. When crossed to a Cre driver line that expresses Cre in all pancreatic progenitor cells — Pdx1-CreEarly — stabilized β-catenin impaired pancreatic development, resulting in severe exocrine agenesis and the formation of large cysts102. Interestingly, this effect on pancreatic development was stage- and cell type-dependent. Crossing the stabilized β-catenin allele with another Cre driver line, Pdx1-CreLate, in which Cre expression is delayed and restricted to acini and endocrine cells, resulted in normal pancreatic development. Surprisingly, postnatal expansion of the exocrine pancreas was observed in ageing mice that correlated with increased acinar proliferation and accumulation of β-catenin. Interestingly, although β-catenin accumulation persisted in older mice, no tumours were observed. Strom and colleagues103 generated Pdx1-Cre mice expressing floxed alleles of the crucial destruction complex component APC (Pdx1-Cre;Apcflox/flox), thereby enforcing canonical Wnt signalling in cells of the developing pancreas. These mice also had increased postnatal pancreatic mass that correlated with enhanced exocrine proliferation, age-dependent accumulation of nuclear β-catenin, and increased expression of β-catenin-target genes; yet, these mice also failed to develop tumours by 1 year of age. Therefore, this work supported the ability of β-catenin to potently activate proliferation in the exocrine pancreas and again demonstrated that the transforming capacity of β-catenin is tightly regulated in pancreatic cells.
Additional studies have shown that enforced β-catenin signalling can induce pancreatic transformation when activated at an appropriate developmental stage. Activation of stabilized β-catenin using a p48-Cre driver (p48-Cre;Ctnnb1exon3/+) resulted not only in acinar proliferation and activation of some β-catenin-target genes, but also the development of large benign tumours that resemble human solid pseudopapillary tumours (SPTs)104. These SPT-like tumours, which in humans have been shown to carry activating mutations in β-catenin105, are morphologically and molecularly distinct from PDAC. Taken together, these models suggest that β-catenin signalling is a crucial mediator of exocrine proliferation and, although it can induce pancreatic tumorigenesis in a temporal and cell-type-dependent manner, it does not seem to be sufficient to initiate PDAC.
Given its predominant role in initiating PDAC, an important question is whether KRAS can synergize with β-catenin signalling to drive PDAC development, as observed in other tumour types such as colon cancer106–108. Surprisingly, p48-Cre;Ctnnb1exon3/+;LSL-KrasG12D mice did not develop SPT-like tumours, or PanINs or PDAC. Instead, they developed a distinct ductal tumour similar to rare intraductal tubular tumours (ITTs) observed in humans104. Therefore, although PDAC shows concomitant KRAS activity and β-catenin accumulation, simultaneous activation of these two pathways in the developing pancreas seems incompatible with specification of the PanIN–PDAC ductal lineage.
The unexpected lack of synergy between β-catenin and KRAS in PDAC initiation suggests that developmental signalling pathways must be tuned to appropriate levels at key time points during transformation to specify the PanIN–PDAC lineage. Recently, the discovery that pancreatitis, a potent risk factor for PDAC in humans109–111, accelerates PanIN and PDAC development in mice expressing mutant KRAS in the exocrine compartment has led to insights into the role of β-catenin in this process. This discovery has also prompted interesting findings regarding which cells in the pancreas have the capacity to be reprogrammed into PanIN–PDAC lesions.
Acinar plasticity and the PanIN–PDAC lineage
Unlike other gastrointestinal organs, such as the intestine and colon, turnover of the pancreatic parenchyma occurs at a very slow rate (for example, as shown by rare incorporation of bromodeoxyuridine into the DNA of healthy, adult mouse pancreas cells112). However, the pancreas has potent regenerative capacity. Investigations of pancreatic regeneration following various types of pancreatic damage (such as chemically induced pancreatitis113 and partial pancreatectomy114) have yielded extensive knowledge regarding the source of cells that repopulate damaged pancreatic compartments and the inherent plasticity of adult pancreatic lineages.
In humans, chronic pancreatitis is a potent risk factor for PDAC109–111. Recently, several studies have used different versions of KRAS-driven mouse PDAC models to evaluate whether pancreatitis is functionally relevant to PDAC initiation and progression. Not only have these studies shown that pancreatitis accelerates PanIN and PDAC development, suggesting that tissue damage and inflammation cooperate with KRAS signalling to drive the disease, they have also indicated that KRAS substantially alters pancreatic regeneration and plasticity. The first study to directly investigate the effect of pancreatitis on PDAC development used a mouse model that allowed doxycycline-induced, temporal expression of constitutively active, mutant KRAS (KrasG12V) exclusively in the acinar and centroacinar compartment24. like models in which KRAS is induced by Cre under control of the Pdx1 or p48 promoters, activation of KRAS in these mice during embryogenesis or soon after birth resulted in the development of frequent PanIN lesions and some PDACs. Although the presence of acinar markers in human and mouse PanINs had been noted115, this result provided direct evidence for a non-ductal source for the PanIN–PDAC sequence of events. Interestingly, these mice were refractory to PanIN–PDAC development when mutant KRAS was activated 60 days after birth. Therefore, the authors investigated whether persistent damage in the form of chronic pancreatitis could provide a permissive environment for the development of PanIN–PDAC. They induced pancreatitis through long-term treatment with caerulein, a cholecystokinin analogue that stimulates precocious activation of acinar cell digestive enzymes, resulting in pancreatic auto-digestion and cellular damage that is associated with inflammation. In response to chronic pancreatitis, mice in which KRAS was activated in adulthood developed PanINs and PDAC at a high frequency. Thus, this study functionally linked pancreatitis and, therefore, tissue damage to the initiation and progression of PDAC.
Since this study, other groups have verified its key findings, namely that KRAS can reprogram acinar cells into the ductal PanIN–PDAC lineage, and that pancreatitis potently accelerates the initiation and progression of KRAS-driven PanIN–PDAC. Using other inducible Cre lines, adult acinar cells have been shown to be sensitive to spontaneous ductal reprogramming into PanINs by KRASG12D in the absence of pancreatitis25,26. Although the mechanisms remain unclear, these results suggest that different KRAS mutations (in this case KRASG12D versus KRASG12v) that both render the protein constitutively active may exert distinct biological and biochemical effects on exocrine cells, potentially due to differences in the levels of Ras signalling. Indeed, there is emerging evidence that acinar to ductal reprogramming into the PanIN–PDAC lineage depends on breaching a crucial KRAS activity threshold (FIG. 4). Ji and colleagues28 showed that levels of active, GTP-bound KRAS increase between cells derived from non-transformed pancreas expressing KRASG12D and cells derived from KRASG12D-driven PDAC. enforcing KRAS activity in acini at levels which mirror those observed in PDAC resulted in acinar to ductal metaplasia that was reminiscent of chronic pancreatitis in young mice. This was followed by the development of PanINs and PDAC as animals aged28. In support of the need for a crucial KRAS activity threshold to initiate ductal reprogramming, Siveke and colleagues116 showed that combining overexpression of TGFα, which can activate KRAS downstream of the epidermal growth factor receptor, with mutant KRAS dramatically accelerates the elimination of normal acini and the development of PanINs and intraductal papillary mucinous neoplasm (IPMN). Currently, which KRAS effectors are important for reprogramming acini into the PanIN–PDAC lineage is unclear; however, MAPK activity29 and Akt signalling117 (downstream of Ras-activated PI3K) have been associated with acinar to ductal plasticity.
Figure 4. Crucial temporal thresholds of developmental signalling pathways and KRAS activity allow pancreatic epithelial neoplasia — pancreatic ductal adenocarcinoma initiation and progression.

KRAS activity above a crucial threshold can drive differentiated pancreatic cells (acinar cells, for example) into a de-differentiated, ductal state that persists in pancreatic epithelial neoplasia (PanIN) and pancreatic ductal adenocarcinoma (PDAC). For these de-differentiated ductal cells to become PanINs, β-catenin signalling must be maintained below a crucial low level. However, once the PanIN state is established, β-catenin signalling is reactivated in parallel with increasing expression of Hedgehog (Hh) ligand that activates target genes in stromal cells of the developing desmoplastic response. Gli activity in the developing tumour epithelium emerges independently of autocrine stimulation. Although Gli activity is probably active in PanINs, its role in the progression from PanIN to PDAC is unknown. Finally, Gli activity is probably important for PDAC maintenance. Figure is modified, with permission, from REF. 128 © (2000) American Association of Cancer Research.
Other studies have verified the ability of pancreatitis to provide a permissive environment for specifying PDAC precursors, demonstrating that acute pancreatitis potently accelerates PanIN development, and in some cases PDAC, in mice in which mutant KRAS is expressed in the exocrine compartment29,118. These studies, alongside that of Guerra and colleagues24, suggest that tissue damage is a permissive environment for the development of PDAC precursors and altering pancreatic regeneration may play a parallel part with KRAS activity in initiating ductal reprogramming into the PanIN–PDAC lineage. In wild-type mice, acinar cells rapidly regenerate following severe caerulein-induced pancreatitis112,113,119. However, acinar cells undergo substantial morphological and molecular changes immediately following injury. During this regenerative phase, acinar cells transiently reactivate elements of embryonic pancreatic development113, frequently assume duct-like morphology112 and express cytokeratin 19, a marker of ductal differentiation29. However, it is important to note that in wild-type animals this dedifferentiated, ductal state does not become fixed and cells resume acinar differentiation. This limited acinar to acinar plasticity has been noted in response to other insults that activate acinar regeneration such as partial pancreatectomy114.Therefore, in the absence of aberrant activation of some signalling pathways, fixed acinar to ductal reprogramming (or acinar to ductal metaplasia (ADM)) is a tightly restricted differentiation fate. It can occur in wild-type acini in vivo, but seems to require persistent damage. Strobel and colleagues120 noted that mice with 7 to 10 weeks of chronic caerulein-induced pancreatitis developed mucinous metaplastic lesions (MMLs) that had some characteristics of early PanINs (and newly defined pancreatic duct glands121). Although most MMLs were derived from ductal or centroacinar cells, only a small percentage (~5%) were the result of ADM. Desai and colleagues114 also noted ADM in mice following pancreatic ductal ligation, a process characterized by severe acinar apoptosis and prolonged acinar loss. However, ADM can readily be induced through activation of other signalling pathways, such as epidermal growth factor (EGF)122 and Notch signalling123,124, by exposing acini to matrix metalloproteinase 7 (MMP7)124 and, as recently appreciated, activation of mutant KRAS25. Furthermore, enforced expression of PDX1 in exocrine cells during development induces ADM125. Therefore, some factors associated with acinar regeneration (such as Notch signalling and PDX1 expression) can drive acinar to ductal reprogramming when inappropriately activated, suggesting that developmental signalling pathways must be tightly regulated to control acinar plasticity.
Taken together, these data raise the possibility that developmental signalling pathways must be specifically tuned for KRAS to reprogram acini into ductal PanINs. Our recent work has compared the regenerative response to acute pancreatitis of normal acini versus acini expressing mutant KRAS and found that this is the case with β-catenin signalling29. Although acini expressing mutant KRAS assume a de-differentiated, ductal state similar to wild-type acini in response to acute pancreatitis, their ability to regenerate the acinar state is blocked. Instead, acini expressing mutant KRAS persistently express duct markers and reactivated elements of embryonic development, and rapidly give rise to PanIN lesions. As such persistently active, regeneration-associated elements of pancreatic development are characteristics of PanINs, these data suggest that assuming a de-differentiated state may be a rate-limiting step in KRAS-driven PDAC development. Other mouse models that combine mutant KRAS with mutations in genes involved in maintaining acinar differentiation support this potentially obligate role for de-differentiation. For example, inhibiting muscle, intestine and stomach 1 (MIST1) function, a transcription factor expressed in acinar cells, results in ADM that expresses markers characteristic of the de-differentiated acini found following acute pancreatitis and during KRAS-driven ductal reprogramming and PanIN formation126. Indeed, combining _Mist1_-knockout mice with mutant KRAS significantly accelerates the development of acinar-derived PanINs27, supporting a role for de-differentiation as a component of KRAS-driven PDAC initiation.
Furthermore, β-catenin signalling seems to be a crucial difference between transiently de-differentiated, regenerating acini and acini undergoing persistently de-differentiated ductal reprogramming and PanIN formation29. In wild-type mice, β-catenin signalling is activated during acinar regeneration following caerulein-induced pancreatitis. However, β-catenin signalling is blocked at an equivalent time point in mice expressing mutant KRAS, representing an early stage of ductal reprogramming. Challenging adult acinar cells expressing both mutant KRAS and stabilized β-catenin with caerulein inhibits PanIN formation, and instead results in abnormal duct structures that frequently display nuclear accumulation of β-catenin26. Taken together, it seems that the ability of mutant KRAS to exploit the de-differentiated acinar state to drive the usually restricted, ductal PanIN lineage is sensitive to the molecular activity of developmental signalling pathways and specifically involves a low threshold of β-catenin signalling (FIG. 4).
Therefore, β-catenin seems to have opposing roles during PanIN initiation and progression to PDAC. These studies suggest that mutations in classical β-catenin pathway modulators are mainly absent in PDAC because they may block the ability of KRAS to initiate cells into a progenitor-like lineage capable of being driven into PDAC. However, as discussed above,β-catenin accumulation and signalling occur in PanIN lesions and support PDAC maintenance. These data suggest that β-catenin signalling is tuned, similar to the temporal regulation of developmental signalling pathways required during normal organ specification, in an ordered fashion to allow KRAS to specify the PanIN–PDAC lineage. The exact mechanisms that block β-catenin signalling during the reprogramming of acini into the PanIN lineage and enable its reactivation during PanIN progression are currently being investigated. Insights from recent studies suggest that differing β-catenin levels may be achieved because of the reactivation of other developmental signalling pathways. Siveke and colleagues119 have shown that Notch receptor activation inhibits β-catenin activity in acinar cells in culture and during regeneration following acute pancreatitis. Also, De la O and colleagues26 showed that transgenic NOTCH1 activation significantly accelerated ADM–PanIN. This may indicate that Notch signalling (which is activated in response to acute pancreatitis) may help to maintain a permissive β-catenin signalling threshold during KRAS-driven ductal reprogramming. It is likely that the roles of Notch in PanIN initiation, PDA progression and control of β-catenin signalling will prove to be complex. For example, Hanlon and colleagues have recently demonstrated that elimination of Notch1 in Pdx1-Cre;LSL-KrasG12D mice accelerates PanIN and PDAC development127. However, these mice showed neither gross changes in the activation of Notch signalling targets nor an increase in β-catenin accumulation. Therefore, the interactions between Notch and β-catenin in PanIN development and PDAC progression may depend on input from other signalling cascades. For example, Gli signalling activated non-canonically in PDAC epithelium96 and TGF-β signalling have been shown to support β-catenin signalling in PDAC cells131. These pathways, along with other novel regulators such as ATDC, may help drive β-catenin signalling in PDAC after the PanIN–PDAC lineage has been established.
Conclusion
The advent of KRAS-driven models has generated much knowledge about how PDAC is initiated and how the disease progresses. both Hh and β-catenin signalling clearly affect PDAC development and maintenance. The next challenge is to understand how to exploit these pathways for therapeutic success. Crucial to this effort will be determining the molecular basis of how each pathway is activated, through both known mechanisms and discovering new regulators. Also, it is important to understand how the pathways interact and to compare and contrast these interactions in the tumour epithelium and the microenvironment. Finally, efforts must be made to determine which Hh and β-catenin targets are ‘mission critical’ for maintaining proliferation, viability and differentiation, and to develop efficient methods (pharmacological and otherwise) to block these crucial signalling nodes. As these important goals are achieved, they open a window of opportunity to twist the developmental biology of PDAC away from malignancy.
At a glance.
- Mutations in KRAS are nearly universal in human pancreatic ductal adenocarcinoma (PDAC). Mouse models in which mutant KRAS is targeted to the pancreas reveal that KRAS signalling is sufficient to reprogram pancreatic cells into duct-like lineages capable of progressing through preneoplastic lesions and, ultimately, PDAC in stages that are reminiscent of human disease.
- The latency, differentiation and type of preneoplastic lesion observed in KRAS-driven PDAC mouse models is sensitive to tumour suppressor loss, suggesting that PDAC evolution is dependent on sequential tuning of signalling pathways.
- PDAC is characterized by frequent deregulation of embryonic signalling pathways, including Hedgehog (Hh) and Wnt–β-catenin signalling. Recent evidence points to temporal and spatial control of these pathways in PDAC development and maintenance.
- PDAC cells frequently display aberrant Hh ligand expression. Recent studies suggest that classical ligand-dependent signalling is activated in cells in the tumour microenvironment, supporting tumour maintenance in a paracrine fashion, but not in the tumour epithelium. However, Hh signalling at the level of Gli transcriptional factors is active in the tumour epithelium, dictated by non-canonical regulators of the pathway. Both paracrine ligand activity and epithelial Gli signalling seem to independently support KRAS-driven PDAC evolution in mouse models.
- Wnt–β-catenin signalling is frequently activated in PDAC and contributes to tumour cell proliferation and biology. Genetic models that allow Wnt–β-catenin deregulation reveal that this pathway can transform pancreatic cells but is insufficient to drive PDAC initiation.
- Mouse models have revealed that the ability of KRAS to reprogram cells into a duct-like fate that can give rise to PDAC is sensitive to cell differentiation and levels of KRAS signalling. Temporal regulation of embryonic signalling pathways seems to play a part in preneoplastic reprogramming, as shown by a requirement for control of Wnt–β-catenin signalling during KRAS-driven de-differentiation of acinar cells into PDAC precursor lesions.
Acknowledgements
The authors thank R. Vanderlaan and A. Folias for critical reading of the manuscript and stimulating discussions. Work in the M.H. laboratory on pancreatic cancer is supported by grants from the National Institutes of Health (NIH) (CA112537) and American Association for Cancer Research Pancreatic Cancer Network (PanCAN). S.C.W. is supported by the NIH under the Ruth L. Kirschstein National Research Service Award F32 from the National Cancer Institute and the American College of Surgeons Resident Research Scholarship.
Footnotes
Competing interests statement The authors declare no competing financial interests.
References
- 1.Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–1249. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 2.Corcoran RB, Scott MP. A mouse model for medulloblastoma and basal cell nevus syndrome. J. Neurooncol. 2001;53:307–318. doi: 10.1023/a:1012260318979. [DOI] [PubMed] [Google Scholar]
- 3.Romer JT, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1+/−p53−/− mice. Cancer Cell. 2004;6:229–240. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 4.Taketo MM, Edelmann W. Mouse models of colon cancer. Gastroenterology. 2009;136:780–798. doi: 10.1053/j.gastro.2008.12.049. [DOI] [PubMed] [Google Scholar]
- 5.Habbe N, Langer P, Sina-Frey M, Bartsch DK. Familial pancreatic cancer syndromes. Endocrinol. Metab. Clin. North Am. 2006;35:417–430. doi: 10.1016/j.ecl.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 6.Wescott MP, Rustgi AK. Pancreatic cancer: translating lessons from mouse models and hereditary syndromes. Cancer Prev. Res. 2008;1:503–506. doi: 10.1158/1940-6207.CAPR-08-0195. [DOI] [PubMed] [Google Scholar]
- 7.Almoguera C, et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 8.Caldas C, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nature Genet. 1994;8:27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
- 9.Ruggeri B, et al. Human pancreatic carcinomas and cell lines reveal frequent and multiple alterations in the p53 and Rb-1 tumor-suppressor genes. Oncogene. 1992;7:1503–1511. [PubMed] [Google Scholar]
- 10.Scarpa A, et al. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am. J. Pathol. 1993;142:1534–1543. [PMC free article] [PubMed] [Google Scholar]
- 11.Hahn SA, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez-Viciana P, et al. Cancer targets in the Ras pathway. Cold Spring Harb. Symp. Quant. Biol. 2005;70:461–467. doi: 10.1101/sqb.2005.70.044. [DOI] [PubMed] [Google Scholar]
- 13.Jones S, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hruban RH, Maitra A, Kern SE, Goggins M. Precursors to pancreatic cancer. Gastroenterol. Clin. North Am. 2007;36:831–849. vi. doi: 10.1016/j.gtc.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hruban RH, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am. J. Surg. Pathol. 2004;28:977–987. doi: 10.1097/01.pas.0000126675.59108.80. [DOI] [PubMed] [Google Scholar]
- 16.Feldmann G, Beaty R, Hruban RH, Maitra A. Molecular genetics of pancreatic intraepithelial neoplasia. J. Hepatobiliary Pancreat. Surg. 2007;14:224–232. doi: 10.1007/s00534-006-1166-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lohr M, Kloppel G, Maisonneuve P, Lowenfels AB, Luttges J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. 2005;7:17–23. doi: 10.1593/neo.04445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maitra A, et al. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod. Pathol. 2003;16:902–912. doi: 10.1097/01.MP.0000086072.56290.FB. [DOI] [PubMed] [Google Scholar]
- 19.Wilentz RE, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60:2002–2006. [PubMed] [Google Scholar]
- 20.Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. 2003;63:2016–2019. [PubMed] [Google Scholar]
- 21.Brembeck FH, et al. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003;63:2005–2009. [PubMed] [Google Scholar]
- 22.Tuveson DA, et al. Endogenous oncogenic K-rasG12D stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 23.Hingorani SR, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. The first example of a conditional _Kras_-driven PDAC mouse model that recapitulates the progression observed in humans.
- 24.Guerra C, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. The first direct functional demonstration that PDAC can arise from non-ductal cells. This paper also established a functional link between pancreatitis and PDAC initiation and progression.
- 25.Habbe N, et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc. Natl Acad. Sci. USA. 2008;105:18913–18918. doi: 10.1073/pnas.0810097105. This study provided evidence that mutant Kras is sufficient to reprogram acini into the PanIN lineage in the absence of tissue damage.
- 26.De La O J, et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl Acad. Sci. USA. 2008;105:18907–18912. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi G, et al. Loss of the acinar-restricted transcription factor Mist1 accelerates Kras-induced pancreatic intraepithelial neoplasia. Gastroenterology. 2009;136:1368–1378. doi: 10.1053/j.gastro.2008.12.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji B, et al. Ras activity levels control the development of pancreatic diseases. Gastroenterology. 2009;137:1072–1082. e6. doi: 10.1053/j.gastro.2009.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris JP, 4th, Cano DA, Sekine S, Wang SC, Hebrok M. β-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J. Clin. Invest. 2010;120:508–520. doi: 10.1172/JCI40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gidekel Friedlander SY, et al. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16:379–389. doi: 10.1016/j.ccr.2009.09.027. This study provided evidence that mutant Kras combined with chronic pancreatitis can drive endocrine cells into the PanIN–PDAC lineage.
- 31.Aguirre AJ, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bardeesy N, et al. Both p16Ink4a and the p19Arf–p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl Acad. Sci. USA. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hingorani SR, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. Mutant p53 drives both PDAC progression and the development of metastasis and genomic instability, which are hallmarks of the human disease.
- 34.Bardeesy N, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006;20:3130–3146. doi: 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ijichi H, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-β signaling in cooperation with active Kras expression. Genes Dev. 2006;20:3147–3160. doi: 10.1101/gad.1475506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Izeradjene K, et al. KrasG12D and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell. 2007;11:229–243. doi: 10.1016/j.ccr.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 37.Kojima K, et al. Inactivation of Smad4 accelerates KrasG12D-mediated pancreatic neoplasia. Cancer Res. 2007;67:8121–8130. doi: 10.1158/0008-5472.CAN-06-4167. [DOI] [PubMed] [Google Scholar]
- 38.Vincent DF, et al. Inactivation of TIF1γ cooperates with Kras to induce cystic tumors of the pancreas. PLoS Genet. 2009;5:e1000575. doi: 10.1371/journal.pgen.1000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quint E, et al. Bone patterning is altered in the regenerating zebrafish caudal fin after ectopic expression of sonic hedgehog and bmp2b or exposure to cyclopamine. Proc. Natl Acad. Sci. USA. 2002;99:8713–8718. doi: 10.1073/pnas.122571799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karhadkar SS, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 41.Stecca B, Ruiz i Altaba A. Brain as a paradigm of organ growth: Hedgehog–Gli signaling in neural stem cells and brain tumors. J. Neurobiol. 2005;64:476–490. doi: 10.1002/neu.20160. [DOI] [PubMed] [Google Scholar]
- 42.Liu S, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–6071. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li N, et al. Reciprocal intraepithelial interactions between TP63 and hedgehog signaling regulate quiescence and activation of progenitor elaboration by mammary stem cells. Stem Cells. 2008;26:1253–1264. doi: 10.1634/stemcells.2007-0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hooper JE, Scott MP. Communicating with Hedgehogs. Nature Rev. Mol. Cell Biol. 2005;6:306–317. doi: 10.1038/nrm1622. [DOI] [PubMed] [Google Scholar]
- 45.Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev. Cell. 2008;15:801–812. doi: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006;26:3365–3377. doi: 10.1128/MCB.26.9.3365-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423–434. doi: 10.1016/s0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- 48.Kenney AM, Rowitch DH. Sonic hedgehog promotes G1 cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol. Cell. Biol. 2000;20:9055–9067. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mill P, et al. Shh controls epithelial proliferation via independent pathways that converge on N-Myc. Dev. Cell. 2005;9:293–303. doi: 10.1016/j.devcel.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 50.Regl G, et al. Activation of the BCL2 promoter in response to Hedgehog/GLI signal transduction is predominantly mediated by GLI2. Cancer Res. 2004;64:7724–7731. doi: 10.1158/0008-5472.CAN-04-1085. [DOI] [PubMed] [Google Scholar]
- 51.Teh MT, et al. FOXM1 is a downstream target of Gli1 in basal cell carcinomas. Cancer Res. 2002;62:4773–4780. [PubMed] [Google Scholar]
- 52.Brancaccio A, et al. Requirement of the forkhead gene Foxe1, a target of sonic hedgehog signaling, in hair follicle morphogenesis. Hum. Mol. Genet. 2004;13:2595–2606. doi: 10.1093/hmg/ddh292. [DOI] [PubMed] [Google Scholar]
- 53.Rubin LL, de Sauvage FJ. Targeting the Hedgehog pathway in cancer. Nature Rev. Drug Discov. 2006;5:1026–1033. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]
- 54.Hahn H, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 55.Xie J, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 56.Raffel C, et al. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997;57:842–845. [PubMed] [Google Scholar]
- 57.Thayer SP, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–856. doi: 10.1038/nature02009. Hh ligands are frequently overexpressed in human PDAC, and enforced Hh ligand expression during pancreatic development can lead to elements of PDAC initiation.
- 58.Feldmann G, et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol. Cancer Ther. 2008;7:2725–2735. doi: 10.1158/1535-7163.MCT-08-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feldmann G, et al. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut. 2008;57:1420–1430. doi: 10.1136/gut.2007.148189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feldmann G, et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007;67:2187–2196. doi: 10.1158/0008-5472.CAN-06-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Olive KP, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. SMO inhibition increases accessibility of chemotherapeutic agents and decreases the fibroblast compartment in a mouse model of PDAC.
- 62.Yauch RL, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406–410. doi: 10.1038/nature07275. PDAC cells do not respond to Hh ligand or SMO inhibition. This study provides evidence that Hh ligand activates the signalling cascade in cells in the tumour microenvironment, providing paracrine tumour support.
- 63.Tian H, et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl Acad. Sci. USA. 2009;106:4254–4259. doi: 10.1073/pnas.0813203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nature Rev. Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 65.Olumi AF, et al. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–5011. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Orimo A, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 67.Bachem MG, et al. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology. 2005;128:907–921. doi: 10.1053/j.gastro.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 68.Hwang RF, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vonlaufen A, et al. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 2008;68:2085–2093. doi: 10.1158/0008-5472.CAN-07-2477. [DOI] [PubMed] [Google Scholar]
- 70.Bailey JM, Mohr AM, Hollingsworth MA. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene. 2009;28:3513–3525. doi: 10.1038/onc.2009.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bailey JM, et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008;14:5995–6004. doi: 10.1158/1078-0432.CCR-08-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Menke A, et al. Down-regulation of E-cadherin gene expression by collagen type I and type III in pancreatic cancer cell lines. Cancer Res. 2001;61:3508–3517. [PubMed] [Google Scholar]
- 73.Armstrong T, et al. Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2004;10:7427–7437. doi: 10.1158/1078-0432.CCR-03-0825. [DOI] [PubMed] [Google Scholar]
- 74.Shintani Y, Hollingsworth MA, Wheelock MJ, Johnson KR. Collagen I promotes metastasis in pancreatic cancer by activating c-Jun NH2-terminal kinase 1 and up-regulating N-cadherin expression. Cancer Res. 2006;66:11745–11753. doi: 10.1158/0008-5472.CAN-06-2322. [DOI] [PubMed] [Google Scholar]
- 75.Koenig A, Mueller C, Hasel C, Adler G, Menke A. Collagen type I induces disruption of E-cadherin-mediated cell–cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res. 2006;66:4662–4671. doi: 10.1158/0008-5472.CAN-05-2804. [DOI] [PubMed] [Google Scholar]
- 76.Nakamura K, et al. Hedgehog promotes neovascularization in pancreatic cancers by regulating Ang-1 and IGF-1 expression in bone-marrow derived pro-angiogenic cells. PLoS ONE. 2010;5:e8824. doi: 10.1371/journal.pone.0008824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Burris HA, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 78.Tempero M, et al. Randomized Phase II comparison of dose-intense gemcitabine: thirty-minute infusion and fixed dose rate infusion in patients with pancreatic adenocarcinoma. J. Clin. Oncol. 2003;21:3402–3408. doi: 10.1200/JCO.2003.09.140. [DOI] [PubMed] [Google Scholar]
- 79.Tsuda N, et al. Synthetic microRNA designed to target glioma-associated antigen 1 transcription factor inhibits division and induces late apoptosis in pancreatic tumor cells. Clin. Cancer Res. 2006;12:6557–6564. doi: 10.1158/1078-0432.CCR-06-0588. [DOI] [PubMed] [Google Scholar]
- 80.Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl Acad. Sci. USA. 2007;104:8455–8460. doi: 10.1073/pnas.0609699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nolan-Stevaux O, et al. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23:24–36. doi: 10.1101/gad.1753809. KRAS-driven PDAC develops in the absence of smoothened expression. However, aberrant Hh ligand expression and epithelial Gli signalling remain present, suggesting that they are uncoupled during PDAC development.
- 82.Ji Z, Mei FC, Xie J, Cheng X. Oncogenic KRAS activates Hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007;282:14048–14055. doi: 10.1074/jbc.M611089200. [DOI] [PubMed] [Google Scholar]
- 83.Dennler S, et al. Induction of sonic hedgehog mediators by transforming growth factor-β: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007;67:6981–6986. doi: 10.1158/0008-5472.CAN-07-0491. [DOI] [PubMed] [Google Scholar]
- 84.Apelqvist A, Ahlgren U, Edlund H. Sonic hedgehog directs specialised mesoderm differentiation in the intestine and pancreas. Curr. Biol. 1997;7:801–804. doi: 10.1016/s0960-9822(06)00340-x. [DOI] [PubMed] [Google Scholar]
- 85.Pasca di Magliano M, et al. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 2006;20:3161–3173. doi: 10.1101/gad.1470806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clevers H. Wnt/β-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 87.MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schwartz AL, et al. Phenylmethimazole decreases Toll-like receptor 3 and noncanonical Wnt5a expression in pancreatic cancer and melanoma together with tumor cell growth and migration. Clin. Cancer Res. 2009;15:4114–4122. doi: 10.1158/1078-0432.CCR-09-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pilarsky C, et al. Activation of Wnt signalling in stroma from pancreatic cancer identified by gene expression profiling. J. Cell. Mol. Med. 2008;12:2823–2835. doi: 10.1111/j.1582-4934.2008.00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531–7537. doi: 10.1038/sj.onc.1210059. [DOI] [PubMed] [Google Scholar]
- 91.Seymour AB, et al. Allelotype of pancreatic adenocarcinoma. Cancer Res. 1994;54:2761–2764. [PubMed] [Google Scholar]
- 92.Gerdes B, et al. Analysis of β-catenin gene mutations in pancreatic tumors. Digestion. 1999;60:544–548. doi: 10.1159/000007704. [DOI] [PubMed] [Google Scholar]
- 93.Abraham SC, et al. Solid-pseudopapillary tumors of the pancreas are genetically distinct from pancreatic ductal adenocarcinomas and almost always harbor β-catenin mutations. Am. J. Pathol. 2002;160:1361–1369. doi: 10.1016/s0002-9440(10)62563-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Al-Aynati MM, Radulovich N, Riddell RH, Tsao MS. Epithelial-cadherin and β-catenin expression changes in pancreatic intraepithelial neoplasia. Clin. Cancer Res. 2004;10:1235–1240. doi: 10.1158/1078-0432.ccr-03-0087. [DOI] [PubMed] [Google Scholar]
- 95.Zeng G, et al. Aberrant Wnt/β-catenin signaling in pancreatic adenocarcinoma. Neoplasia. 2006;8:279–289. doi: 10.1593/neo.05607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pasca di Magliano M, et al. Common activation of canonical Wnt signaling in pancreatic adenocarcinoma. PLoS ONE. 2007;2:e1155. doi: 10.1371/journal.pone.0001155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang L, et al. Oncogenic function of ATDC in pancreatic cancer through Wnt pathway activation and β-catenin stabilization. Cancer Cell. 2009;15:207–219. doi: 10.1016/j.ccr.2009.01.018. Cell autonomous ATDC supports β-catenin accumulation and signalling during PDAC development and impacts tumour cell proliferation and transformed characteristics.
- 98.Froeling FE, et al. Organotypic culture model of pancreatic cancer demonstrates that stromal cells modulate E-cadherin, β-catenin, and Ezrin expression in tumor cells. Am. J. Pathol. 2009;175:636–648. doi: 10.2353/ajpath.2009.090131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nawroth R, et al. Extracellular sulfatases, elements of the Wnt signaling pathway, positively regulate growth and tumorigenicity of human pancreatic cancer cells. PLoS ONE. 2007;2:e392. doi: 10.1371/journal.pone.0000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Takahashi N, et al. Dickkopf-1 is overexpressed in human pancreatic ductal adenocarcinoma cells and is involved in invasive growth. Int. J. Cancer. 2009;126:1611–1620. doi: 10.1002/ijc.24865. [DOI] [PubMed] [Google Scholar]
- 101.Kemler R, et al. Stabilization of β-catenin in the mouse zygote leads to premature epithelial–mesenchymal transition in the epiblast. Development. 2004;131:5817–5824. doi: 10.1242/dev.01458. [DOI] [PubMed] [Google Scholar]
- 102.Heiser PW, Lau J, Taketo MM, Herrera PL, Hebrok M. Stabilization of β-catenin impacts pancreas growth. Development. 2006;133:2023–2032. doi: 10.1242/dev.02366. [DOI] [PubMed] [Google Scholar]
- 103.Strom A, et al. Unique mechanisms of growth regulation and tumor suppression upon Apc inactivation in the pancreas. Development. 2007;134:2719–2725. doi: 10.1242/dev.02875. [DOI] [PubMed] [Google Scholar]
- 104.Heiser PW, et al. Stabilization of β-catenin induces pancreas tumor formation. Gastroenterology. 2008;135:1288–1300. doi: 10.1053/j.gastro.2008.06.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nishimori I, et al. Non-cystic solid-pseudopapillary tumor of the pancreas showing nuclear accumulation and activating gene mutation of β-catenin. Pathol. Int. 2006;56:707–711. doi: 10.1111/j.1440-1827.2006.02034.x. [DOI] [PubMed] [Google Scholar]
- 106.Janssen KP, et al. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology. 2006;131:1096–1109. doi: 10.1053/j.gastro.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 107.Phelps RA, et al. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell. 2009;137:623–634. doi: 10.1016/j.cell.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sansom OJ, et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc. Natl Acad. Sci. USA. 2006;103:14122–14127. doi: 10.1073/pnas.0604130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lowenfels AB, et al. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N. Engl. J. Med. 1993;328:1433–1437. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- 110.Malka D, et al. Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut. 2002;51:849–852. doi: 10.1136/gut.51.6.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hassan MM, et al. Risk factors for pancreatic cancer: case–control study. Am. J. Gastroenterol. 2007;102:2696–2707. doi: 10.1111/j.1572-0241.2007.01510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fendrich V, et al. Hedgehog signaling is required for effective regeneration of exocrine pancreas. Gastroenterology. 2008;135:621–631. doi: 10.1053/j.gastro.2008.04.011. This study demonstrated that acinar cells regenerate from pre-existing acinar cells through ductal intermediates following chemically induced pancreatitis.
- 113.Jensen JN, et al. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology. 2005;128:728–741. doi: 10.1053/j.gastro.2004.12.008. This study provided evidence that embryonic signalling pathways are reactivated during acinar regeneration following chemically induced pancreatitis.
- 114.Desai BM, et al. Preexisting pancreatic acinar cells contribute to acinar cell, but not islet β cell, regeneration. J. Clin. Invest. 2007;117:971–977. doi: 10.1172/JCI29988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhu L, Shi G, Schmidt CM, Hruban RH, Konieczny SF. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am. J. Pathol. 2007;171:263–273. doi: 10.2353/ajpath.2007.061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Siveke JT, et al. Concomitant pancreatic activation of KrasG12D and Tgfa results in cystic papillary neoplasms reminiscent of human IPMN. Cancer Cell. 2007;12:266–279. doi: 10.1016/j.ccr.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 117.Elghazi L, et al. Regulation of pancreas plasticity and malignant transformation by Akt signaling. Gastroenterology. 2009;136:1091–1103. doi: 10.1053/j.gastro.2008.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Carriere C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem. Biophys. Res. Commun. 2009;382:561–565. doi: 10.1016/j.bbrc.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Siveke JT, et al. Notch signaling is required for exocrine regeneration after acute pancreatitis. Gastroenterology. 2008;134:544–555. doi: 10.1053/j.gastro.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 120.Strobel O, et al. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology. 2007;133:1999–2009. doi: 10.1053/j.gastro.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Strobel O, et al. Pancreatic duct glands are distinct ductal compartments that react to chronic injury and mediate Shh-induced metaplasia. Gastroenterology. 138:1166–1177. doi: 10.1053/j.gastro.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Means AL, et al. Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development. 2005;132:3767–3776. doi: 10.1242/dev.01925. [DOI] [PubMed] [Google Scholar]
- 123.Miyamoto Y, et al. Notch mediates TGF α-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3:565–576. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 124.Sawey ET, Johnson JA, Crawford HC. Matrix metalloproteinase 7 controls pancreatic acinar cell transdifferentiation by activating the Notch signaling pathway. Proc. Natl Acad. Sci. USA. 2007;104:19327–19332. doi: 10.1073/pnas.0705953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Miyatsuka T, et al. Persistent expression of PDX-1 in the pancreas causes acinar-to-ductal metaplasia through Stat3 activation. Genes Dev. 2006;20:1435–1440. doi: 10.1101/gad.1412806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhu L, et al. Inhibition of Mist1 homodimer formation induces pancreatic acinar-to-ductal metaplasia. Mol. Cell. Biol. 2004;24:2673–2681. doi: 10.1128/MCB.24.7.2673-2681.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hanlon L, et al. Notch1 functions as a tumor suppressor in a model of K-ras-induced pancreatic ductal adenocarcinoma. Cancer Res. 2010;70:4280–4286. doi: 10.1158/0008-5472.CAN-09-4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin. Cancer Res. 2000;6:2969–2972. [PubMed] [Google Scholar]
- 129.Carrière C, Seeley ES, Goetze T, Longnecker DS, Korc M. The Nestin progenitor lineage is the compartment of origin for pancreatic intraepithelial neoplasia. Proc. Natl Acad. Sci. USA. 2007;104:4437–4442. doi: 10.1073/pnas.0701117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mao J, et al. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006;66:10171–10178. doi: 10.1158/0008-5472.CAN-06-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Romero D, Iglesias M, Vary CP, Quintanilla M. Functional blockade of Smad4 leads to a decrease in β-catenin levels and signaling activity in human pancreatic carcinoma cells. Carcinogenesis. 2008;29:1070–1076. doi: 10.1093/carcin/bgn054. [DOI] [PubMed] [Google Scholar]