Structure of the C-terminal domain of Saccharomyces cerevisiae Nup133, a component of the Nuclear Pore Complex (original) (raw)
. Author manuscript; available in PMC: 2012 May 13.
Published in final edited form as: Proteins. 2011 Mar 1;79(5):1672–1677. doi: 10.1002/prot.22973
Abstract
Nuclear pore complexes (NPCs), responsible for the nucleo-cytoplasmic exchange of proteins and nucleic acids, are dynamic macromolecular assemblies forming an eight-fold symmetric co-axial ring structure. Yeast (Saccharomyces cerevisiae) NPCs are made up of at least 456 polypeptide chains of ~30 distinct sequences. Many of these components (nucleoporins, Nups) share similar structural motifs and form stable subcomplexes. We have determined a high-resolution crystal structure of the C-terminal domain of yeast Nup133 (ScNup133), a component of the heptameric Nup84 subcomplex. Expression tests yielded ScNup133(944-1157) that produced crystals diffracting to 1.9Å resolution.
ScNup133(944-1157) adopts essentially an all α-helical fold, with a short two stranded β-sheet at the C-terminus. The 11 α-helices of ScNup133(944-1157) form a compact fold. In contrast, the previously determined structure of human Nup133(934-1156) bound to a fragment of human Nup107 has its constituent α-helices are arranged in two globular blocks. These differences may reflect structural divergence among homologous nucleoporins.
Keywords: Nuclear Pore Complex, Nup133, structural genomics
INTRODUCTION
Nuclear pore complexes (NPCs) are situated at the interface of the inner and outer nuclear membranes, where they are responsible for transport biomolecules between nucleus and cytoplasm of eukaryotic cells.1-3 Saccharomyces cerevisiae NPCs are ~50MDa assemblies made up of at least 456 polypeptides of ~30 different sequences, called nucleoporins or Nups.4,5 Several of these Nups, sharing similar structural motifs, assemble into stable subcomplexes that are arranged with eight-fold symmetry to form a co-axial ring structure containing a membrane ring, two outer (nuclear and cytoplasmic) rings and two inner rings. One of these subcomplexes in the yeast NPC is the Nup84 complex that is situated within the nuclear and cytoplasmic peripheral rings.5 The Nup84 complex is a heptameric assembly forming a Y-shaped structure composed of Nup84, Nup85, Nup120, Nup133, Nup145C, Sec13, and Seh1.6 The homologous metazoan complex is a nonameric assembly termed the Nup107-160 complex, carrying homologues of all seven yeast Nups plus Nup37 and Nup43 as additional components.7 Nup133 is a ~1150 residue protein containing an N-terminal seven blade β-propeller domain followed by an ~80kDa helical domain.8-10 The C-terminal domain of human Nup133, in particular residues 934 to 1156 [HsNup133(934-1156)], is essential for its binding to Nup107, together forming the stem of the Y-shaped complex. Crystal structures of the HsNup133(934-1156) have been determined in complex with Nup107 (residues 658-925). In this complex, 11 HsNup133(934-1156) helices are arranged as two helical blocks.11,12
We have determined the structure of the S. cerevisiae Nup133 (ScNup133) C-terminal helical domain at 1.9Å resolution, corresponding to residues 944-1157 [ScNup133(944-1157)]. Our structure reveals a compact helical arrangement, differing in several respects from the structure of the homologous HsNup133(934-1156) determined in complex with Nup107. We also report Small Angle X-ray Scattering (SAXS) of ScNup133(944-1157) in solution that is consistent with our compact crystallographically observed structure.
EXPERIMENTAL PROCEDURES
Cloning, expression, and purification of ScNup133(944-1157)
ScNup133 comprising residues 881-1157 was cloned from the genomic DNA of the yeast strain Sc2601D-5 (American Type Culture Collection, USA). The desired construct was PCR amplified using the forward, AAGTACGGTCATGTAGCATGGA and, reverse, CGTATTCTACAGTGTTGGTTTCATAG, primers and subsequently TOPO® (Invitrogen, USA) cloned into pSGX3, a derivative of pET26 b(+) giving rise to proteins with a non-cleavable C-terminal hexa histidine tag. Plasmids were transformed into BL21 (DE3)-Condon+RIL (Invitrogen, USA) cells for overexpression. Expression of selenomethionine labeled proteins was carried out in 3L of HY media at 22 °C with 50 μg/ml of kanamycin and 35 μg/ml of chloramphenicol. Protein expression was induced at O.D.600 ~1.0 by addition of 0.4 mM IPTG. Cells were harvested after 21 hours by centrifugation at 4°C.
For purification, the E. coli cell pellet was resuspended in 30mL of cold buffer containing 20mM Tris HCl pH 8.0, 500mM NaCl, 25mM Imidazole and 0.1% Tween20 and cells were lysed by sonication. Debris was removed by centrifugation at 4°C. The decanted supernatant was applied to a 5 mL HisTrapHP column (GE Health Care, USA) charged with nickel and pre-equilibrated with 20mM Tris HCl pH 8.0, 500mM NaCl, 10% v/v glycerol and 25mM imidazole. The sample was washed with 5 column volumes (CV) of 20mM Tris HCl pH 8.0, 500mM NaCl, 10% (v/v) glycerol and 40mM imidazole and subsequently eluted with 2 CV of same buffer with an imidazole concentration of 250mM. Eluted protein was passed over a 120 mL Superdex 200 column equilibrated with 10mM HEPES pH 7.5, 150mM NaCl, 10% (v/v) glycerol and 5mM DTT (protein storage buffer). SDS-PAGE analysis showed greater than 95% purity and protein fractions corresponding to the symmetric portion of the size exclusion chromatography profile were pooled for concentration using AMICON spin filters. Concentrated protein aliquots were frozen in liquid nitrogen and stored at -80 °C.
Crystallization, data collection and structure determination
ScNup133(944-1157), at a concentration of 17.6 mg/ml, was subjected to crystallization screening with the Classics, Classics II, and PEGs kits (Qiagen, USA) using a Phoenix Liquid Handling Systems (Art Robins, USA) with equal volumes (0.3μL) and reservoir solution in 96-well sitting drop format at 21 °C. Diffracting quality crystals were obtained with 20% PEG3350 in the presence of 200mM potassium thiocyanate (PEGs suite; condition number 62) and were cryo-protected with the addition of ~30% (v/v) glycerol before being flash frozen by immersion in liquid nitrogen. Diffraction data were recorded using the LRL-CAT 31-ID beamline at the Advanced Photon Source (APS) and processed with MOSFLM13 and SCALA (Collaborative Computing Project Number 4, 1994).14 About 800 reflections were randomly selected for cross validation representing an R-free dataset. Structure of ScNup133(944-1157) was determined by the Single-wavelength Anomalous Dispersion (SAD) method, using data collected at a wavelength of 0.97929 Å, corresponding to “Se peak” absorption edge. A single Se atom was located with SHELXD15 and phases were calculated using SHELXE as implemented in HKL2MAP.16 Following numerous rounds of automated and manual model building with ARP/wARP17 and COOT18, respectively, the atomic coordinates were refined using REFMAC519 as implemented in CCP4. The polypeptide backbone of ScNup133 could be traced continuously from residue Lys946 to Tyr1157 in the electron density maps. The final refined atomic model of ScNup133(944-1157) has excellent stereochemistry (Table 1). Structural analyses were carried out using COOT and CCP4 and illustrations were prepared using PyMol (http://pymol.sourceforge.net).
Table 1.
Crystallographic statistics.
| Data Collection | ScNup133(946-1157) |
|---|---|
| PDB code: | 3KFO |
| Space group: | _P_212121 |
| Unit-cell dimensions a, b, c (Å): | a=47.4, b=52.7, c=76.7 |
| Matthew’s coefficient (Å3/Da) | 1.89 |
| Solvent content (%) | 34.45 |
| Resolution (Å): | 31.00-1.90 (2.00-1.90)* |
| Number of unique reflections: | 15683 (2236) |
| Completeness (%): | 99.7 (100.0) |
| Rsymm (%): | 12.1 (41.4) |
| Multiplicity: | 7.7 (7.8) |
| < I/σ(I) >: | 10.6 (4.6) |
| Refinement | |
| Rcryst (%): | 19.1 |
| Rfree (%): | 24.8 |
| RMS deviations from ideal values | |
| Bond length (Å): | 0.019 |
| Bond angles (°) | 1.63 |
| Average B-factors (Å2) | |
| Protein | 23.4 |
| Water molecules | 31.5 |
| Co-solvent (glycerol) | 45.7 |
| Ramachandran Plot33: | |
| MolProbity34 Residues in | |
| Favored region (%): | 99.5 |
| Allowed region (%): | 100.0 |
Small Angle X-ray Scattering (SAXS)
SAXS measurements of ScNup133(944-1157) were carried out on the SIBYLS Beamline 12.3.1 at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory (LBNL). Samples were prepared on a 96-well plate and kept at 10°C until SAXS measurement. An automatic sample delivery system equipped with a Hamilton pipetting robot was used.20 SAXS data was collected on a MAR165 area detector (Rayonix), placed at 1.5 m from the SAXS sample cell. Exposures of 2 sec / 10 sec / 2 sec were made in series for each protein sample maintained at 10 °C. Each of the diffraction images was scaled using the transmitted beam intensity, azimuthally integrated, and averaged to obtain fully processed data in the form of intensity versus q [q = 4πsin(θ)/λ, θ = one-half of the scattering angle; λ = X-ray wavelength]. The buffer profile was obtained in the same manner and subtracted from a protein profile. SAXS profiles of ScNup133(944-1157) were recorded at protein concentrations of 0.5, 1.0, 2.0, 5.0, and 17.6 mg/mL in protein storage buffer. Moreover, mild concentration dependence was removed by extrapolating to zero concentration. We merged the average of the low scattering angle parts (q < 0.15Å-1) of the low concentration profiles (0.5-2.0 mg/mL) and the average of the high scattering angle parts (**_q_** > 0.12Å-1) of the high concentration (5.0-17.6 mg/mL) profiles, to obtain the final, merged experimental SAXS profile. SAXS profiles were calculated with IMP-FoXS21, 22 (http://salilab.org/foxs/) and CRYSOL23, and compared with the merged experimental SAXS profile. A complete structural model of ScNup133(944-1157), including two N-terminal residues (Leu-Arg), C-terminal hexa-histidine tag (Gly-His-His-His-His-His-His), and 16 missing side chains, was generated using its crystal structure with the automatic PSF-based model building option of VMD24 and the automodel function of MODELLER25 (these missing atoms were not included in the final X-ray structure because they appear disordered in the electron density map). Inclusion of the missing atoms improved the fit of the calculated SAXS profiles to our experimental observations (χ2 value improved from 2.95 to 1.25), particularly at higher scattering angles (q > 0.2 Å-1). The shape of ScNup133(946-1157) was calculated from the merged experimental SAXS profile by running DAMMIF26 and GASBOR27 20 times individually, followed by superposition and averaging with DAMAVER.28
RESULTS AND DISCUSSION
Structure of ScNup133(944-1157)
The expected molecular weight of ScNup133 (residues 881 to 1157) is 33,025 Da. The measured molecular weight, using mass spectrometry, of the final purified selenomethionine form of ScNup133 was 25,871 Da (mass error < 0.007%). The difference in mass of -7154 Da was attributed to unintended N-terminal proteolysis during expression and/or purification (the presence of an intact C-terminus with hexa-histidine affinity tag was confirmed by binding the purified ScNup133 to Ni-NTA resin). N-terminal sequencing documented that the truncation occurred at the Thr943-Leu944 peptide bond. The resulting target protein, consisting of residues 944 to 1157 [ScNup133(944-1157)], yielded crystals diffracting to 1.9 Å resolution (orthorhombic _P_212121 space group, one molecule per asymmetric unit, Table 1). Electron density maps phased with a single selenium atom allowed continuous tracing of the ScNup133 polypeptide backbone from Lys946 to Tyr1157.
The overall fold of ScNup133(944-1157) is composed of eleven α-helices with a short two-stranded β-sheet, formed by β2-β3, at the C-terminus [Fig. 1(A)]. Helices α1 to α11 form a compact structure. A four helical bundle (formed by the α3, α4, α5, and α6 helices) at the center of the structure is surrounded by the α1, α2, α9, α10, and α11 helices. A search for structurally similar proteins using DALI29 did not identify any known structures with Z-scores > 10. Highest Z-scores of 5.0 were obtained for PDB (http://www.rcsb.org) codes 3IHV and 3DWL, which correspond to a sugar binding protein SusD homolog (sequence identity = 8%; root mean square deviation = 3.6 Å for 112 Cα atomic pairs) and subunit 5 of the actin-related protein 2/3 complex (sequence identity = 12%; r.m.s.d. = 3.4 Å for 95 Cα atomic pairs), respectively. It is likely that the structural similarities of these two proteins to ScNup133(946-1157) are due to geometrical restraints on the packing of helices rather than to a common ancestor in evolution. The absence of the human Nup133 structure among the top scoring DALI hits came as a surprise, prompting us to measure the SAXS profile of ScNup133(944-1157) to determine whether or not the arrangement of helices observed in our crystal structure is an artifact of crystallization.
Figure 1.
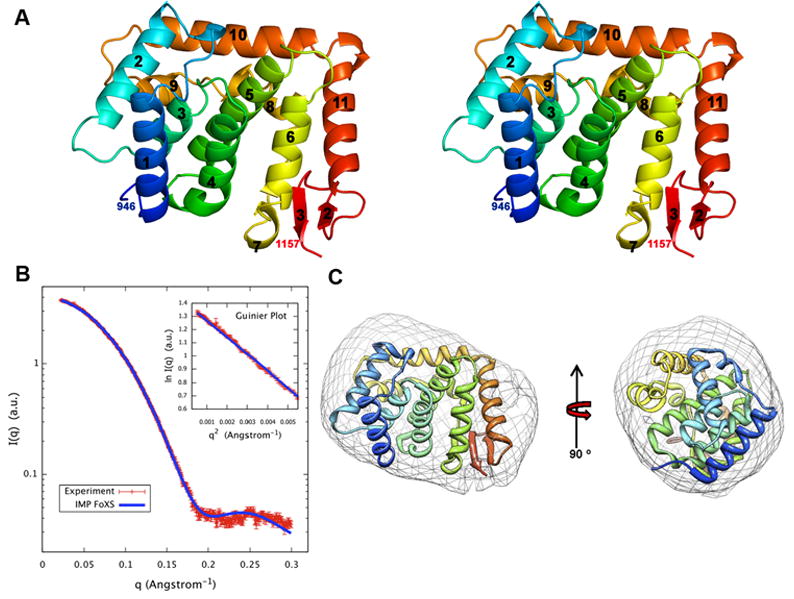
A: Stereo view of a ribbon representation of ScNup133(944-1157). Secondary structure elements were assigned by DSSP.35 The fold is colored blue to red from the N- to C-termini.
B: Comparison of the merged experimental SAXS profile (red) of ScNup133(946-1157) with SAXS profiles computed by IMP-FoXS 31,32 (blue) for the complete model of ScNup133(944-1157). Inset shows the SAXS profiles in the Guinier plot. Dmax of radial distribution function, P(r) is 56.25Å).
C: The shape of ScNup133(944-1157) derived from the experimental SAXS profile, shown in two orthogonal orientations.
The merged experimental SAXS profile closely resembles the SAXS profile calculated from the complete model of ScNup133(944-1157) obtained with the X-ray structure of [Fig. 1(B)]. The measured radius of gyration (Rg) of 18.8 ± 0.5 Å, obtained from AutoRg 30, compares within the experimental errors with the Rg of 18.6 Å for the crystal structure and Rg of 19.3 Å for the complete model. The “_ab initio_” shape reconstructed from the merged experimental SAXS profile with DAMMIF26 [Fig 1(C)] and GASBOR27 (not shown) matches the overall structure of ScNup133(944-1157). Therefore, SAXS analysis indicates that the compact fold of ScNup133 (944-1157) exists in solution and is not an artifact of crystallization.
Comparison of ScNup133(946-1157) with human Nup133
Structures of human Nup133 residues 934 to 1156 [HsNup133(934-1156)] have only been determined in complexes with Nup107 (PDB Codes 3CQC,10 3CQG,10 and 3I4R11). The amino acid sequence identity between the C-terminal domains of Sc and Hs Nup133 is rather low, ~12%. Structure based alignments did not yield complete coverage of the two polypeptide chains. Structure based alignment using Multiprot30 resulted in a r.m.s.d. of 1.6 Å for 88 Cα atomic pairs with sequence identity of ~9%. The r.m.s.d. for structure based alignment using SSM31 is 2.5 Å for 98 Cα atomic pairs with sequence identity of ~8%. In the case of ScNup133 we have lost the putative Nup84 interaction surface due to the proteolysis. Therefore, the first helix α1 of ScNup133 aligns with the α4 of the HsNup133 structure [Fig. 2]. The four helical bundle present at the center of both ScNup133(946-1157) and HuNup133(934-1156) superpose well over each other along with the α11 helix. However, HsNup133(934-1156) lacks counterparts to ScNup133(944-1157) helices α2, α9, and α10; and β-strands. Therefore, two homologous Nups from two distantly related organisms show significant sequence and structural divergence in their C-termini. These differences show that a single example structure from each Nup obtained from just one organism does not represent the range of structures present throughout the Eukaryota among different Nups in different states. Thus, there is a pressing need for more structures of NPC components, from various organisms, both alone and in complexes with their respective binding partners.
Figure 2.
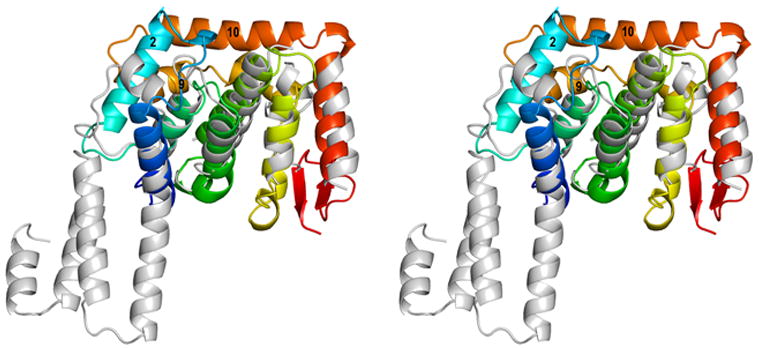
A: Structural superposition of the C-terminal domains of Sc and Hs Nup133. ScNup133(944-1157) is shown as in Fig. 1 (A) and HuNup133(934-1156) is shown in grey.
Acknowledgments
We thank members of the Rout and Sali laboratories for their help and advice, in particular Dina Schneidmann for help with the IMP-FoXS SAXS web server. We are also grateful to Drs. John Tainer and Michal Hammel for help in using the SIBYLS beamline 12.3.1 at ALS. Funding for NYSGXRC was provided by NIH Grant U54 GM074945 (PI: S.K. Burley). Additional funding for this work was provided by NIH R01 GM062427 (MPR), NIH R01 GM083960 (AS), and NIH U54 RR022220 (AS and MPR). Use of the Advanced Photon Source at the Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly and Company, which operates the facility. SSRL operation is funded by the U.S. Department of Energy, Office of Basic Energy Sciences and the SSRL Structural Molecular Biology program by the U.S. Department of Energy, Office of Biological and Environmental Research and the NIH National Center for Research Resources, Biotechnology program. Use of the SIBYLS beamline 12.3.1 at ALS was supported in part by the Office of Science, Office of Biological and Environmental Research, U.S. Department of Energy, under Contract DE-AC02-05CH11231.
Footnotes
Protein Data Bank codes Coordinates and structure factors of ScNup133(944-1157) were deposited to the PDB on October 27, 2009 with accession code 3KFO. The NYSGXRC target identifier for yeast Nup133 is TargetDB (http://targetdb.pdb.org), “NYSGXRC-15133a”. Clone sequences and experimental information are available in PepcDB (http://pepcdb.pdb.org/).
References
- 1.Rout MP, Aitchison JD, Suprapto A, Hjertaas K, Zhao YM, Chait BT. The yeast nuclear pore complex: Composition, architecture, and transport mechanism. J Cell Biol. 2000;148:635–651. doi: 10.1083/jcb.148.4.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D’Angelo MA, Hetzer MW. Structure, dynamics and function of nuclear pore complexes. Trends Cell Biol. 2008;18:456–466. doi: 10.1016/j.tcb.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elizabeth JT, Wente SR. Dynamic nuclear pore complexes: Life on the edge. Cell. 2006;125:1041–1053. doi: 10.1016/j.cell.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 4.Alber F, Dokudovskaya S, Veenhoff LM, Zhang WH, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, Sali A, Rout MP. The molecular architecture of the nuclear pore complex. Nature. 2007;450:695–701. doi: 10.1038/nature06405. [DOI] [PubMed] [Google Scholar]
- 5.Alber F, Dokudovskaya S, Veenhoff LM, Zhang WZ, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, Rout MP, Sali A. Determining the architectures of macromolecular assemblies. Nature. 2007;450:683–694. doi: 10.1038/nature06404. [DOI] [PubMed] [Google Scholar]
- 6.Lutzmann M, Kunze R, Buerer A, Aebi U, Hurt E. Modular assembly of a Y-shaped multiprotein complex from seven nucleoporins. EMBO J. 2002;21:387–397. doi: 10.1093/emboj/21.3.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loiodice I, Alves A, Rabut G, Van Overbeek M, Ellenberg J, Sibarita JB, Doye V. The entire Nup107-160 complex, including three new members, is targeted as one entity to kinetochores in mitosis. Mol Biol Cell. 2004;15:3333–3344. doi: 10.1091/mbc.E03-12-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doye V, Wepf R, Hurt EC. A novel nuclear pore protein Nup133p with distinct roles in poly(A)+ RNA transport and nuclear pore distribution. EMBO J. 2002;21:387–397. doi: 10.1002/j.1460-2075.1994.tb06953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pemberton LF, Rout MP, Blobel G. Disruption of the nucleoporin gene NUP133 results in clustering of nuclear pore complexes. Proc Natl Acad Sci USA. 92:1187–1191. doi: 10.1073/pnas.92.4.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berke IC, Boehmer T, Blobel G, Schwartz TU. Structural and functional analysis of Nup133 domains reveals modeular building blocks of the nuclear pore complex. J Cell Biol. 2004;22:591–597. doi: 10.1083/jcb.200408109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boehmer T, Jeudy S, Berke IC, Schwartz TU. Structural and functional studies of Nup107/Nup133 interaction and its implications for the architecture of the nuclear pore complex. Mol Cell. 2008;30:721–731. doi: 10.1016/j.molcel.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whittle JRR, Schwartz TU. Architectural nucleoporins Nup157/170 and Nup133 are structurally related and discend from a second ancestral element. J Biol Chem. 2009;284:28442–28452. doi: 10.1074/jbc.M109.023580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leslie AGW, Brick P, Wonacott AJ. An improved program package for the measurement of oscillation photographs. CCP4 News. 1986;18:33–39. [Google Scholar]
- 14.Collaborative Computing Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 15.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 16.Pape T, Schneider TR. HKL2MAP: a graphical user interface for phasing with SHELX program. J Appl Cryst. 2004;37:843–844. [Google Scholar]
- 17.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nature Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 18.Emsley P, Cowtan K. COOT: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 19.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the Maximum-Likelihood Method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 20.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, II, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, Yang S, Scott JW, Dillard BD, Adams MWW, Tainer JA. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nature Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forster F, Webb B, Krukenberg KA, Tsuruta H, Agard DA, Sali A. Integration of small-angle X-ray scattering data into structural modeling of proteins and their assemblies. J Mol Biol. 2008;382:1089–1106. doi: 10.1016/j.jmb.2008.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneidman-Duhovny D, Hammel M, Sali A. FoXS: A Web server for Rapid Computation and Fitting of SAXS Profiles. Nucl Acids Res. 2010 doi: 10.1093/nar/gkq461. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Svergun DI, Barberato C, Koch MHJ. CRYSOL - a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J Appl Cryst. 1995;28:768–773. [Google Scholar]
- 24.Humphrey W, Dalke A, Schulten K. VMD - Visual Molecular Dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 25.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 26.Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Cryst. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Svergun DI, Petoukhov MV, Koch MHJ. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Cryst. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure database with DliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PCbased system for small-angle scattering data analysis. J Appl Cryst. 2003;36:1277–1282. [Google Scholar]
- 31.Shatsky M, Nussinov R, Wolfson HJ. A method for simultaneous alignment of multiple protein structures. Proteins: Structure, Function and Bioinformatics. 2004;56:143–156. doi: 10.1002/prot.10628. [DOI] [PubMed] [Google Scholar]
- 32.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 33.Ramakrishnan C, Ramachandran GN. Stereochemical criteria for polypeptide and protein chain conformations. II. Allowed conformations for a pair of peptide units. Biophys J. 1965;5:909–933. doi: 10.1016/S0006-3495(65)86759-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucl Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:1577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]