Synaptic Glutamate Release Is Modulated by the Na+-Driven Cl−/HCO3− Exchanger Slc4a8 (original) (raw)
Abstract
On the one hand, neuronal activity can cause changes in pH; on the other hand, changes in pH can modulate neuronal activity. Consequently, the pH of the brain is regulated at various levels. Here we show that steady-state pH and acid extrusion were diminished in cultured hippocampal neurons of mice with a targeted disruption of the Na+-driven Cl−/HCO3− exchanger Slc4a8. Because Slc4a8 was found to predominantly localize to presynaptic nerve endings, we hypothesize that Slc4a8 is a key regulator of presynaptic pH. Supporting this hypothesis, spontaneous glutamate release in the CA1 pyramidal layer was reduced but could be rescued by increasing the intracellular pH. The reduced excitability in vitro correlated with an increased seizure threshold in vivo. Together with the altered kinetics of stimulated synaptic vesicle release, these data suggest that Slc4a8 modulates glutamate release in a pH-dependent manner.
Introduction
The regulation of intra- and extracellular pH is a vital homeostatic function of all cell types. Given that numerous ion channels can be influenced by pH, a role of pH transients in relation to membrane potential and excitability has often been considered. This is of particular relevance in the brain where, on the one hand, changes in intra- and extracellular pH can influence neural activity and, on the other hand, neural activity can elicit rapid changes of pH. As a general rule, it appears that a rise in brain pH is associated with increased neuronal excitability, whereas a fall in pH has been shown to have the opposite effect (Chesler, 2003). In this respect, the mechanisms that generate and regulate changes in pH are of considerable neurobiological and clinical interest.
Because of its paramount importance, intra- and extracellular pH is regulated at various levels. The first line of regulation is the inherent buffering capacity of the intracellular and extracellular space that helps to reduce rapid localized pH shifts (Casey et al., 2010). The most important intracellular buffer is HCO3−, which is generated by hydration of CO2 and subsequent deprotonation of carbonic acid (Cordat and Casey, 2009). This process is sped up by carbonic anhydrases, which thereby help to dissipate local pH gradients (Supuran, 2008). Because of the continuous generation of acid equivalents, together with the transport of ions that alter pH, more dynamic and sustained mechanisms are required to ensure long-term pH homeostasis in neurons. The defense machinery in principle comprises Na+/H+ exchange (Luo and Sun, 2007), the Ca2+/H+-ATPase (Schwiening et al., 1993), monocarboxylate transport (Hertz and Dienel, 2005), passive Cl−/HCO3− exchange (Alper, 2009), Na+-driven Cl−/HCO3− exchange, and Na+-HCO3− cotransport (Boron et al., 2009). The acid regulation machinery may differ among different types of neurons; however, the overlapping expression of pH-relevant transporters and the lack of specific inhibitors have precluded a detailed analysis to date. Indirect studies have suggested that Na+-driven Cl−/HCO3− exchange contributes to acid extrusion in hippocampal neurons (Schwiening and Boron, 1994; Baxter and Church, 1996; Bevensee et al., 1996; Bonnet et al., 2000), but the molecular correlate and its physiological relevance are currently unclear. Both NDCBE/Slc4a8 (encoded by Slc4a8) and NCBE/Slc4a10 (encoded by Slc4a10) have been shown to mediate Na+-driven Cl−/HCO3− exchange (Wang et al., 2000; Grichtchenko et al., 2001; Damkier et al., 2010).
Here, we show that Slc4a8 was enriched in presynaptic glutamatergic nerve endings and, indeed, played an essential role in the regulation of intracellular pH (pHi). Glutamate release was impaired in neurons with a targeted disruption of Slc4a8, but was alleviated by increasing pH. On the systemic level, disruption of Slc4a8 increased the seizure threshold in mice. These findings indicate that pH regulation via Slc4a8 has important consequences for synaptic transmission in both physiological and pathophysiological conditions.
Materials and Methods
All experiments were approved by the responsible local institutions (Behörde für Soziales, Familie, Gesundheit und Verbraucherschutz, Hamburg, Germany, and Landesamt für Lebensmittelsicherheit und Verbraucherschutz, Bad Langensalza, Germany) and complied with the regulations of the National Institutes of Health and those of the Society of Neuroscience (Washington, DC). _Slc4a8_−/− (KO) mice were generated by targeted disruption of Slc4a8. All studies were performed in a mixed 129Sv/C57BL/6 background in the F7 generation. KO animals were born in the expected Mendelian ratio, had a normal life span, and did not show any obvious abnormality (Leviel et al., 2010). KO brains appeared to be unaltered at the structural or histological level (data not shown). If not indicated otherwise, littermates of both genders from heterozygous matings in F7 were used for experiments.
Immunohistochemistry.
For immunohistochemistry, brains of 6- to 8-week-old wild-type (WT) and KO mice were removed after transcardial perfusion and postfixed in 2% paraformaldehyde/0.25% glutaraldehyde in 0.1 m phosphate buffer (PB) for 2 h. The tissue was incubated in 30% sucrose overnight. Free-floating cryosections (50 μm) were stained with our polyclonal rabbit Slc4a8 antibody (Leviel et al., 2010). For 3,3′-diaminobenzidine (DAB) staining, coronal and sagittal brain sections were incubated with a biotinylated anti-rabbit IgG secondary antibody using the Vectastain ABC kit (Vector Laboratories). The peroxidase stain was visualized by 0.05% DAB, 0.04% nickel ammonium sulfate, and 0.03% H2O2 dissolved in 0.01 m PB. For morphological analysis, a standard hematoxylin-eosin staining procedure was used. For immunofluorescence studies on sagittal brain sections, the Slc4a8 signal was amplified with a tyramide signal amplification kit (Invitrogen) (n ≥ 6 from at least two independent experiments). For costainings, the following primary antibodies were used: monoclonal mouse anti-neurofilament 68 (anti-NF68) (1:500, Sigma-Aldrich), monoclonal mouse anti-glutamic acid decarboxylase 67 (anti-GAD67) (1:500, Millipore), polyclonal guinea pig anti-vesicular glutamate transporter 1 (anti-vGLUT1) (1:500, Synaptic Systems), monoclonal mouse anti-vGLUT 2 (1:250, Millipore), monoclonal mouse anti-vGLUT3 (1:5000, Millipore), monoclonal mouse anti-synaptophysin (1:500, Millipore), mouse anti-parvalbumin (1:2000, Swant), monoclonal mouse anti-microtubule-associated protein 2 (anti-MAP2) (1:500, Sigma-Aldrich), monoclonal mouse anti-presynaptic density protein 95 (PSD-95) (1:1000, Abcam), monoclonal mouse anti-glial fibrillary acidic protein (anti-GFAP) (1:500, Millipore), and monoclonal mouse anti-2′,3′-cyclic nucleotide 3′-phosphodiesterase (anti-CNPase) (1:500, Sigma-Aldrich). Alexa Fluor 488- and 555-coupled goat anti-rabbit, goat anti-guinea pig, and goat anti-mouse antibodies (1:500 or 1:1000, Invitrogen) were used as secondary antibodies. Cell nuclei were stained by 4′,6-diamidino-2-phenylindole (DAPI) (1 μg/ml, Sigma-Aldrich). Analysis was performed by confocal microscopy (LSM 510, Zeiss).
To quantify the degree of colocalization, images (n = 12) were taken from the hippocampal CA1 region (stratum radiatum, 225 × 225 μm) of 4 different WT brains. The relative area of colocalization was determined by scatter plot analysis using the colocalization module of AxioVision (Release 4.8.2, Zeiss).
Subcellular fractionation.
Synaptic proteins were prepared as described by Carlin et al. (1980) with minor modifications. Briefly, whole brains of adult mice (WT or KO, 4–8 weeks of age) were homogenized with a Teflon Dounce homogenizer and centrifuged twice (1000 × g, 10 min at 4°C). The crude membrane fraction was pelleted from the supernatants (12,000 × g, 20 min). The cytosolic fraction was harvested by ethanol precipitation of the resulting supernatant and ultracentrifugation (100,000 × g, 1 h at 4°C). The synaptosome fraction was isolated by a discontinuous sucrose gradient (0.85/1/1.2 m). The synaptosomal membrane fraction was prepared by subsequent osmotic lysis and centrifugation (33,000 × g, 30 min at 4°C). Synaptic junction plasma membranes were enriched by a subsequent second sucrose gradient. For the final isolation of the postsynaptic density fraction, two Triton X-100 purification steps were performed.
Immunoblotting.
For immunoblotting, 10 μg of protein (if not otherwise indicated) was separated by reducing SDS-8% polyacrylamide gel and transferred to nylon membranes. Detection was achieved with an enhanced chemiluminescence kit (GE Healthcare). The following primary antibodies were used: rabbit anti-Slc4a8 (1:500), mouse monoclonal anti-PSD-95 (1:1000, Abcam), and mouse monoclonal anti-synaptophysin (1:1000, Millipore). β-Actin (1:40,000, Santa Cruz Biotechnology) served as a loading control.
Freeze-fracture replica immunolabeling.
For freeze-fracture replica immunogold labeling, aliquots of isolated synaptosomes were enclosed between two 0.1 mm copper sandwich profiles. The profiles were rapidly frozen by plunge-freezing in liquid ethane/propane (1:1) cooled by liquid nitrogen. Freeze-fracture was performed in a BAF400T (Leica Microsystems) freeze-fracture unit at −150°C using a double-replica stage. The fractured samples were replicated by perpendicular evaporation of carbon (15–25 nm) as first layer, followed by platinum/carbon (2 nm) shadowing at an angle of 35°. The SDS freeze-fracture replica labeling technique was performed as described previously (Westermann et al., 2005), using specific primary antibodies for Slc4a8 (1:100) and Syntaxin (mouse monoclonal, 1:50, Sigma-Aldrich), followed by gold-conjugated second antibodies goat anti-rabbit (10 nm) and goat anti-mouse (5 nm) (British Biocell International). After immunolabeling, the replicas were fixed with 0.5% glutaraldehyde, washed with distilled water, and finally picked onto Formvar-coated grids. Images were taken as digital pictures in an EM 902 A electron microscope (Zeiss) using a 1 k FastScan CCD camera [Tietz Video and Image Processing Systems (TVIPS) camera and software].
Hippocampal neuron culture.
Mice from homozygous WT and KO matings were killed at postnatal day 0.5–1.5. Brains were removed and hippocampi dissected in ice-cold HBSS (Invitrogen) supplemented with penicillin-streptavidin/HEPES (pH 7.25). Hippocampi were rinsed three times with HBSS and trypsinized with 0.05% trypsin/EDTA (Invitrogen) for 25 min at 37°C. The supernatant was removed and rinsed with HBSS. The tissue was placed in 2 ml of HBSS containing 20 μl of DNase (1 μg/μl in HBSS) and dissociated into single cells by trituration with folded Pasteur pipettes of decreasing diameter. Approximately 50,000 cells were plated onto poly-l-lysine (Sigma-Aldrich)-coated coverslips (ø 18 mm) in plating medium (minimum essential medium, 10% horse serum, 0.6% glucose). Cells were allowed to settle for 10–20 min at 37°C and 5% CO2. Finally, coverslips were transferred to 6 cm culture dishes containing Neurobasal medium (Invitrogen) supplemented with 1 mm l-glutamine, 0.2% horse serum, and B27 supplement. Neurobasal medium supplemented with B27 was added once a week to maintain the initial volume. For pure glial culture, astrocytes were cultured with DMEM (Invitrogen) supplemented with 10% fetal calf serum and penicillin-streptavidin, and split at least 3 times. pH measurements were performed after 21–28 d in culture and FM measurements at day 14 in culture.
Intracellular pH recordings.
The fluorescent indicator 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF) (Rink et al., 1982) was used for noninvasive measurement of pHi in cultured hippocampal neurons. BCECF is a commonly used dual-excitation indicator, which is pH sensitive when excited at 495 nm and nearly pH insensitive at 440 nm (isosbestic point), and thereby allows ratiometric monitoring of pHi independent of photobleaching and dye leakage (Boyarsky et al., 1988). Primary hippocampal cultures were loaded with acetoxymethyl ester of BCECF (Invitrogen) at a final concentration of 1 μm with 0.002% Pluronic F127 in bicarbonate-buffered solution (containing, in mm: 125 NaCl, 2 KCl, 1.2 CaCl2, 1.2 MgSO4, 1.25 NaH2PO4, 10 glucose, and 26 NaHCO3, gassed with 95% O2/5% CO2, pH 7.3) for 5–10 min at 37°C. For recordings, coverslips were mounted onto a perfusion chamber (350 μl volume, Chamlide EC, Live Cell Instrument) and superfused with bicarbonate-buffered solution (32 ± 0.2°C) at a linear flow rate of 1.5–2.0 ml/s corresponding to ∼4 bath changes per minute. A subset of experiments was performed in the nominal absence of bicarbonate and the presence of 30 mm HEPES (pH adjusted to 7.3 with NaOH). Light emission (510–535 nm) upon alternating excitation with 495 and 440 nm was recorded with a 10× objective every 10 s with a CCD camera (AxioCam MRm, Zeiss). The _F_495/_F_440 ratio was converted into pHi values using the nigericin single-point calibration technique (Boyarsky et al., 1988) and nonlinear regression analysis. After defining the region of interest (somata of pyramidal neurons) and 10 min of baseline recording, ratio values were relatively stable, although we still observed a slight alkaline drift in our recordings regardless of the genotype (drift10 min KO, 0.030 ± 0.004 ΔpH/min; WT, 0.031 ± 0.004 ΔpH/min; p = 0.87). Basal pHi was calculated as mean pHi >30 s, 10 min after the beginning of the recording. Cells were acid loaded by the “rebound acidification” technique (Boron and De Weer, 1976) with a 20 mm NH4Cl pulse (bicarbonate-buffered solution with equimolar substitution of Na+ by NH4+), and NH4Cl-induced alkalosis was quantified as pHMaximum-pHBaseline. The rate of pHi recovery (_d_pHi/_d_t) was determined over the first 2 min after peak acidification upon withdrawal of NH4Cl and between pHi 6.7 and 6.8. Data from ≥30 neurons (2–7 cells/experiment) cultured from at least three independent preparations per genotype were averaged. For NH4Cl pulse experiments in the presence of HEPES, ≥9 cells were analyzed from at least two independent preparations per genotype.
FM recordings.
For analysis of presynaptic vesicle release in WT and KO hippocampal neurons, synaptic vesicles were labeled with a fluorescent FM dye. Vesicle exocytosis was investigated upon electrical field stimulation by quantification of fluorescence intensity decline (Gaffield and Betz, 2006).
Coverslips with mixed hippocampal cultures were mounted onto an optical recording chamber supplied with a pair of platinum electrodes for field stimulation (Chamlide EC) at day 14 in culture. During the recording, cells were continuously superfused with bicarbonate-buffered solution (pH 7.3, flow rate 1.1 ml/min at room temperature) supplemented with 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (10 μm, Tocris Bioscience) and (2_R_)-amino-5-phosphonovaleric acid (dl-APV) (50 μm, Sigma-Aldrich) in an optical recording chamber supplied with a pair of platinum electrodes for field stimulation. After 5 min, the cells were exposed to high potassium solution (containing, in mm: 68 NaCl, 60 KCl, 1.2 CaCl2, 1.2 MgSO4, 1.25 NaH2PO4, 10 glucose, and 26 NaHCO3) supplemented with 10 μm FM1-43 (Invitrogen) for 1 min, followed by 1 min of bicarbonate-buffered solution supplemented with 5 μm FM1-43, and washed with bicarbonate-buffered solution for 10 min. Fluorescence was recorded with a 63× water-immersion objective at a frequency of 1 Hz (excitation 480 nm, emission 445 ± 25 nm). Following the baseline recording, 1 ms current pulses were applied for 150 s at a frequency of 10 Hz (corresponding to ∼1500 action potentials). Regions of interest corresponding to nerve terminals (≥25, ø 1.5 μm) were analyzed (6 recordings from three independent preparations per genotype). Bleaching effects were corrected by negative slope analysis. Puncta that did not show a reduction of the corrected fluorescence intensity of >30% after stimulation or that could not be fitted by a monoexponential fit were excluded from analysis of the destaining time constant τ.
Slice preparation for electrophysiological recordings.
Immediately after decapitation, brains from adult mice (WT or KO, 4–8 weeks of age) were removed from the skull and chilled (at ∼4°C) in artificial CSF (aCSF) containing (in mm): 120 NaCl, 3 KCl.5, 1.3 MgSO4, 1.25 NaH2PO4, 2.5 CaCl2, 10 d-glucose, and 25.0 NaHCO3, gassed with 95% O2/5% CO2, pH 7.3. Frontal lobes and cerebellum were removed. Coronal slices were prepared with a vibroslicer (VT 1000S, Leica Instruments) as described previously (Liebmann et al., 2009). Slices (350 μm) were stored at room temperature in aCSF for at least 1 h until use.
Hippocampal field potential recordings.
After a 1 h equilibration period, slices were transferred to an interface recording chamber and perfused with oxygenated aCSF (2–3 ml/min) at 32°C. Bipolar stimulating electrodes with a tip diameter of 100 μm (SNE-200X, Science Products GmbH) were placed onto the Schaffer collateral fibers in the CA1 region. Data of field EPSP (fEPSP) recordings were collected with an extracellular amplifier (EXT-02, NPI Electronic), low-pass filtered at 4 kHz, and digitally stored with a sample frequency of 10 kHz. Data acquisition and analysis of population spike amplitudes were performed using the software, Signal (Cambridge Electronic Design). Upon stimulation (pulse duration 50 μs), fEPSPs were recorded using glass microelectrodes (2–5 MΩ, filled with aCSF) impaled into the pyramidal layer or the stratum radiatum of CA1. The maximal population spike amplitude was determined by gradually increasing the stimulus intensity (0–70 V) for each experiment (interstimulus interval 30 s) until the responses saturated. The relationship between stimulus intensity and the evoked response was fitted by a sigmoid function: R(i) = _R_max/1+exp(i − _i_h), where R(i) is the response at intensity (i), _R_max is the maximal response, and _i_h is the intensity at which half-maximal response was observed.
Following determination of the half-maximal stimulation intensity, paired-pulse stimuli were applied with interstimulus intervals of 15, 20, 30, 50, 80, 120, 180, 280, 430, 650, and 1000 ms to investigate paired-pulse facilitation. Changes of paired-pulse facilitation rather support a presynaptic alteration, because this kind of short-term plasticity depends on a sustained increase in intracellular calcium after a preconditioning pulse (fEPSP1) (Katz and Miledi, 1968; Wu and Saggau, 1994).
Patch-clamp recordings.
Hippocampal slices were placed in a submerged recording chamber mounted on an upright microscope (BX51WI, Olympus). Slices were continuously superfused with gassed aCSF (5% CO2/95% O2, 2–3 ml/min, 32°C, pH 7.2). Patch-clamp recordings were performed under visual control with differential interference contrast and a 40× water-immersion objective. CA1 neurons with a pyramidal-shaped cell body were selected for recordings. The membrane patch under the electrode was ruptured by gentle suction after seal formation (resistance >1 GΩ). Only recordings with an access resistance of <15 MΩ and a resting membrane potential more negative than −55 mV were included in this study. Signals were recorded using a patch-clamp amplifier (Multiclamp 700B, Molecular Devices). Responses were low-pass filtered at 4 kHz and digitized at 20 kHz (Digidata 1440, Molecular Devices). All data were acquired, stored, and analyzed on a PC using pClamp 10.2 and Clampfit 10.2 (Molecular Devices). Patch pipettes with an impedance of 3–4 MΩ were pulled from borosilicate glass (outer diameter 1.5 mm, Science Products GmbH) on a Sutter Instrument micropipette puller (P-97). Miniature EPSCs (mEPSCs) and miniature IPSCs (mIPSCs) were recorded at a holding potential of −70 mV for at least 5 min. Data analysis was performed off-line with the detection threshold levels set to 5 pA for mEPSCs and mIPSCs. mEPSCs were isolated by adding tetrodotoxin (0.5 μm, Tocris Bioscience) and bicuculline methiodide (20 μm, Biomol) to block action potential-induced glutamate release and GABAA receptor-mediated mIPSCs, respectively. dl-APV (30 μm) was added to suppress NMDA currents. The pipette solution contained the following (in mm): 120 CsMeSO4, 17.5 CsCl, 10 HEPES, 5 BAPTA, 2 Mg-ATP, 0.5 Na-GTP, 10 QX-314 [_N_-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide], pH 7.3, adjusted with CsOH.
Recordings of mIPSCs were performed using a CsCl-based intracellular solution (in mm): 122 CsCl, 8 NaCl, 0.2 MgCl2, 10 HEPES, 2 EGTA, 2 Mg-ATP, 0.5 Na-GTP, pH adjusted to 7.3 with CsOH. dl-APV (30 μm), CNQX (10 μm), and tetrodotoxin (0.5 μm) were added to the perfusate. The currents were identified as events when the rise time was faster than the decay time. The following parameters were determined: interevent interval, frequency, rise time, peak amplitude, and τ of decay. The decay of each event was fitted with a monoexponential curve in pClamp. Only residual SDs <0.3 were accepted as a criterion for the quality of the fit. In a subset of mEPSC recordings, the extracellular pH was adjusted to 6.9 or 7.5 using aCSF gassed with carbogen containing 10 or 2.5% CO2, respectively. Alterations of pHi upon application of different CO2 concentrations were confirmed by recordings of pHi in cultured neurons (ΔpHi at 2.5% CO2, +0.26 ± 0.04; at 10% CO2, −0.19 ± 0.05; one-way ANOVA: F = 27.48, p < 0.00001; n = 22/39/17). In a subset of experiments, pHi was alkalinized by substitution of 20 mm NaCl by trimethylamine chloride (TriMA) (Sigma-Aldrich).
To study the regulation of presynaptic release, the open-channel blocker of the NMDA receptor MK-801 maleate (5_H_-dibenzo[a,_d_]cyclohepten-5,10-imine maleate) (40 μm, Tocris Bioscience) was used. CNQX (10 μm) was added to block α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor responses. NMDA receptor-mediated evoked EPSCs were evoked in CA1 pyramidal neurons by electrical stimulation (stimulator model 2100, A-M Systems) of Schaffer collaterals with a frequency of 0.1 Hz. Evoked EPSCs were recorded in response to stimulation with half-maximal stimulus intensity at +40 mV holding potential. MK-801 was added after establishment of a stable baseline. Stimulation was resumed and evoked EPSCs were recorded for an additional 100 stimuli. Pulses were normalized to the first pulse given in the presence of MK-801. Time constants of NMDA receptor blockade were estimated by monoexponential fit and defined as the time point at which amplitudes dropped to 37% of baseline. Furthermore, action potential properties and spike frequency accommodation were recorded under current-clamp conditions. Prolonged current steps (600 ms) were applied from the resting membrane potential in the range of 0–560 pA with a 40 pA increment. Patch pipettes were filled with the following (in mm): 140 K-methane sulfonate, 10 HEPES, 0.1 EGTA, 4 Mg-ATP, and 0.3 Na-GTP, pH 7.3.
Seizure susceptibility.
Seizure threshold was determined as described previously (Jacobs et al., 2008). In adult mice (WT or KO, male, 3–4 months of age), seizures were evoked by administration of the chemical proconvulsants pentylenetetrazol (60 mg/kg body weight, Sigma-Aldrich) or pilocarpine (350 mg/kg body weight, Sigma-Aldrich,) intraperitoneally (dissolved in a total volume of 300 μl of PB). Peripheral cholinergic actions of pilocarpine were blocked by injection of 1 mg/kg body weight methylscopolamine (Sigma-Aldrich) 30 min before the pilocarpine injection. Animals were monitored for 10 min after administration of the proconvulsants. The latency until the first myoclonic jerk (focal seizure), clonic seizure (clamping of the forefeet), or generalized (tonic-clonic) seizures was measured. For hyperthermia-induced epileptic activity, 10-d-old pups of both genotypes of comparable body weight were placed into a chamber with an ambient temperature of 48.0 ± 1.0°C (Schuchmann et al., 2006). The body temperature was monitored with a rectal probe. The latency until loss of postural control due to sustained tonic-clonic generalized epileptic activity (>20 s) was recorded. Experimenters were blinded for the genotypes of mice.
Data analysis.
Data are presented as mean ± SEM. Statistical analysis of two experimental groups was performed using the parametric two-tailed Student's t test. If more than two groups were compared, one-way ANOVA was performed, and differences between subjects were analyzed by a subsequent Newman–Keuls test. In experiments that included repeated measurements, differences between groups were tested by repeated-measures ANOVA. If necessary, two-way ANOVA was applied. Cumulative distributions were tested using the Kolmogorov–Smirnov test. Significance was considered at p values < 0.05.
Results
Slc4a8 localizes to presynaptic glutamatergic nerve endings
DAB staining of brain sections demonstrated that Slc4a8 was broadly expressed in the CNS (Fig. 1A,B). Signals were most prominent in cortical and midbrain structures, whereas large fiber tracts like the corpus callosum were almost spared. In the hippocampus, all layers stained positive, including the stratum lacunosum-moleculare of the CA1 region (Fig. 1C). The specificity of our antibody and the staining procedure was confirmed by the absence of signals on tissue of KO mice (Fig. 1D).
Figure 1.

Neuronal expression of Slc4a8 in mouse brain. A, DAB staining of a coronal WT brain section with an antibody raised against Slc4a8. Scale bar, 1000 μm. B, Slc4a8 staining of a sagittal WT brain section. Scale bar, 1000 μm. C, All hippocampal layers stained positive for Slc4a8. Scale bar, 500 μm. SOr, Stratum oriens; SPyr, stratum pyramidale; SRad, stratum radiatum; SL-M, stratum lacunosum-moleculare. D, The specificity of the antibody was verified by the absence of signals in brain sections of KO mice. Scale bar, 1000 μm. E, The ∼120 kDa band for Slc4a8 was absent in KO brain lysate. Slc4a8 was detected in mixed hippocampal cultures from WT mice, but was not detectable in lysates from pure glia cell cultures.
Slc4a8 was detected in protein lysates of mixed neuron/glia cell cultures, but not of cultured glia cells (Fig. 1E). Accordingly, no overlap of Slc4a8 was noted with the astrocyte marker GFAP or the oligodendrocyte marker CNPase in immunostainings of brain sections (data not shown). The somata of Slc4a8-stained neurons were labeled variably, some displaying a clear plasma membrane-bound signal and some a more intracellular vesicular staining (see also close-up from pyramidal cell layer, Fig. 2H). Slc4a8 did not colocalize with the dendritic marker MAP2 (Fig. 2F). Costaining with the axonal marker NF68 revealed that Slc4a8 localized to axons rather than dendrites (data not shown). Rather, Slc4a8 overlapped more with the presynaptic marker synaptophysin (Fig. 2A,B; relative area of colocalization, 64.1 ± 4.4%) than with the postsynaptic marker PSD-95 (Fig. 2E; relative area of colocalization, 18.9 ± 3.6%; n = 12/12; Student's t test: p = 0.00001). More specifically, Slc4a8 colocalized with glutamate transporters vGLUT1 (Fig. 2C), vGLUT2 (Fig. 2D), and vGLUT3 (data not shown). Overlap between Slc4a8 and GAD67 as a marker of GABAergic presynapses was marginal (Fig. 2G), and Slc4a8 was absent from most parvalbumin-positive inhibitory interneurons in the hippocampus (Fig. 2H). The presynaptic localization of Slc4a8 was further confirmed by fractionation studies from mouse brain lysates. Slc4a8 was increasingly enriched in synaptosomes, synaptosomal membranes, and synaptic junction plasma membranes, but absent from the postsynaptic density (Fig. 2I). Immunogold labeling of freeze-fractured WT and KO synaptosome preparations proved that the Slc4a8 signal was specific (Fig. 2J,K) and that Slc4a8 and Syntaxin—a well established presynaptic protein (Okada et al., 1995)—localized to the same compartment in WT preparations (Fig. 2L).
Figure 2.
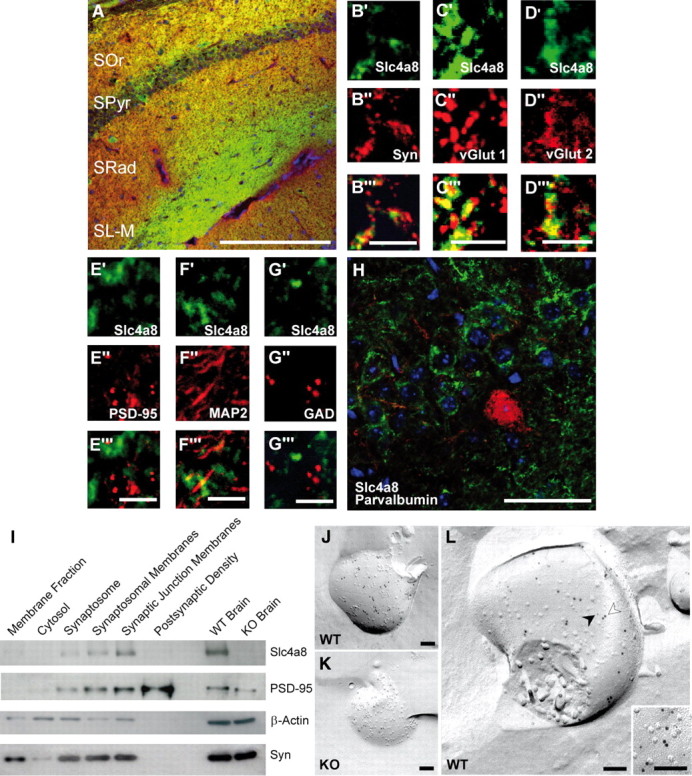
Presynaptic localization of Slc4a8 in the hippocampus. A, Costaining for Slc4a8 (green) and the presynaptic marker synaptophysin (Syn) (red) revealed a significant overlay. Scale bar, 200 μm. SOr, Stratum oriens; SPyr, stratum pyramidale; SRad, stratum radiatum; SL-M, stratum lacunosum-moleculare. B′–B‴, Higher magnification of SRad. Scale bar, 5 μm. C′–C‴, Slc4a8 (green) overlapped with the vesicular glutamate transporter vGLUT1 (red, SRad). Scale bar, 5 μm. D′–D‴, Similar results were obtained for vGLUT2 as shown in a close-up from the SRad. Scale bar, 5 μm. E′–E‴, Slc4a8 and the postsynaptic protein PSD-95 (red, SL-M) (scale bar, 5 μm) did not colocalize. F′–F‴, In agreement, costainings of Slc4a8 (green) and the dendritic marker MAP2 (red, SRad) (scale bar, 5 μm) did not support a dendritic expression. G′–G‴, Costaining of Slc4a8 (green) and GAD (red) revealed that Slc4a8 was not detected in most GABAergic nerve endings (SRad). Scale bar, 5 μm. H, Parvalbumin-positive interneurons (red) in the SPyr of the hippocampus did not express Slc4a8 (green). Scale bar, 50 μm. Nuclei were labeled with DAPI (blue). I, Immunoblot analysis of a density fractionation of WT mouse brain lysates. Slc4a8 was enriched in synaptosomes, synaptosomal membranes, and the synaptic junction plasma membrane fraction, but was not detectable in the postsynaptic density fraction. The postsynaptic protein PSD-95 and the presynaptic marker synaptophysin served as positive controls and β-actin as a loading control. J, Transmission electron microscopy of a freeze-fractured WT synaptosome immunogold labeled for Slc4a8 (large grains, 10 nm). K, The KO control confirmed the specificity of the Slc4a8 antibody. L, Colabeling of Slc4a8 (10 nm, white arrow) and the presynaptic marker syntaxin (small grains, 5 nm, black arrow) of freeze-fractured WT synaptosomes confirmed presynaptic localization of Slc4a8. Inset, Higher magnification. All images show the protoplasmic fracture face of a synaptosome membrane. Scale bars: J–L, 100 nm.
Slc4a8 is important for the regulation of pHi in hippocampal neurons
Because Slc4a8 appeared to be broadly expressed in neurons, and sodium-dependent chloride-bicarbonate exchangers had repeatedly been proposed to be key regulators of neuronal pHi (Russell and Boron, 1976; Schwiening and Boron, 1994; Bevensee et al., 1996; Romero et al., 2000), we investigated the role of Slc4a8 in neuronal pH homeostasis by BCECF fluorescence imaging of cultured hippocampal neurons from KO and WT mice. In bicarbonate-buffered solutions, the resting pHi of KO neurons was significantly lower compared with WT (Fig. 3A,B; pHi KO, 6.88 ± 0.02; WT, 6.97 ± 0.03; one-way ANOVA, F = 7.65, p = 0.0001; Newman–Keuls test, p < 0.05; _n_ = 35/35), whereas it was unchanged in HEPES-buffered and nominally bicarbonate-free solution (pHi KO, 6.75 ± 0.05; WT, 6.78 ± 0.03; Newman–Keuls test, _p_ > 0.05; n = 9/16). To challenge neuronal pHi regulation, cells were alkali loaded with an extracellular NH4Cl pulse. In the presence of bicarbonate, NH4Cl-induced peak alkalosis, compared with baseline (ΔpHMaximum-Basal), was significantly raised in KO neurons compared with WT (Fig. 3C; ΔpHMaximum-Basal KO, 0.50 ± 0.03; WT, 0.33 ± 0.02; one-way ANOVA, F = 30.88, p < 0.0001; Newman–Keuls test, p < 0.001; n = 35/35), whereas rebound acidosis upon withdrawal of NH4Cl compared with baseline (ΔpHBasal-Minimum) was unaltered (Fig. 3C; ΔpHBasal-Minimum KO, 0.22 ± 0.03; WT, 0.17 ± 0.03; one-way ANOVA, F = 0.08, p = 0.97; n = 35/35), although the absolute minimum pHi (pHMinimum) was slightly more acidic in KO neurons (pHMinimum KO, 6.67 ± 0.03; WT, 6.79 ± 0.03; Student's t test, p = 0.005; n = 35/35).
Figure 3.
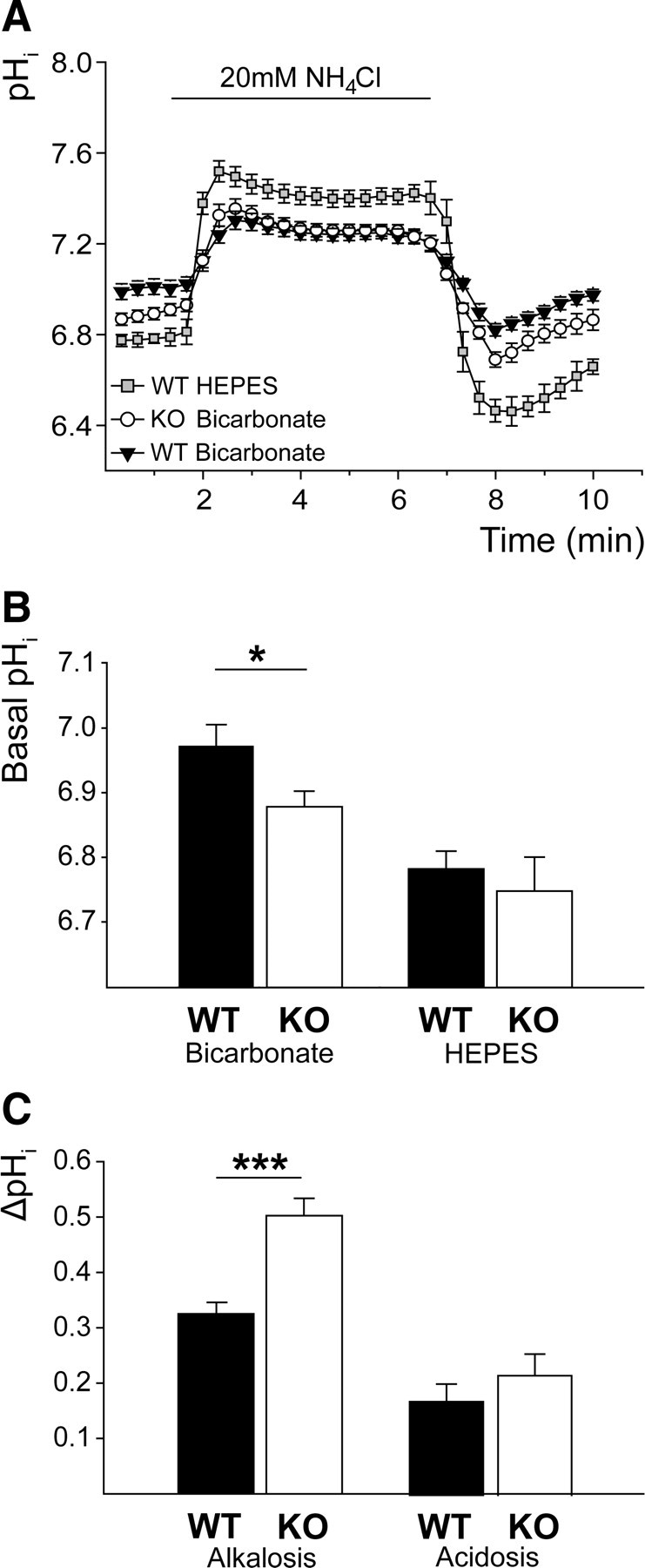
Impaired pH regulation in cultured hippocampal neurons of KO mice. A, Mean pHi of KO and WT neurons challenged with NH4Cl (WTHEPES, n = 9; WTBicarbonate, n = 35; KOBicarbonate, n = 35). B, Steady-state pHi was only reduced in KO neurons in the presence of bicarbonate (bicarbonate, p < 0.05; _n_ = 35/35; HEPES, _p_ > 0.05; n = 9/16). C, Peak alkalosis by NH4Cl was increased in KO neurons (ΔpHMaximum-Basal, p < 0.001; n = 35/35), but net acid load upon withdrawal of NH4Cl did not differ between genotypes (ΔpHBasal-Minimum, p = 0.97; n = 35/35). *p < 0.05; ***p < 0.0005.
Acid extrusion within the first 2 min after the maximal acid load was diminished in the absence of Slc4a8 (ΔpH/min2 min KO, 0.10 ± 0.01; WT, 0.13 ± 0.01; Student's t test, p = 0.019; n = 35/35). To evaluate the net proton fluxes independently of the pHi, we additionally calculated the mean slope for both genotypes between pHi 6.7 and 6.8, which was decreased as well (ΔpH/minpH6.7–6.8 KO, 0.11 ± 0.01; WT, 0.15 ± 0.01; one-way ANOVA, F = 4.24, p = 0.007; Newman–Keuls test, p < 0.01). In the nominal absence of bicarbonate, no significant differences were observed between genotypes in any of the parameters analyzed (NH4Cl-induced alkalosis ΔpHMaximum-Basal: KO, 0.83 ± 0.08; WT, 0.74 ± 0.07; Newman–Keuls test, _p_ > 0.05; acid load ΔpHBasal-Minimum: KO, 0.24 ± 0.06; WT, 0.38 ± 0.07; Newman–Keuls test, p > 0.05; recovery from acid load ΔpH/minpH 6.7–6.8: KO, 0.09 ± 0.01; WT, 0.10 ± 0.01; Newman–Keuls test, p > 0.05; n = 9/16).
These results did not suggest a major compensatory regulation of other acid extruders in KO mice. In accordance, transcript abundance of Slc9a1, Slc4a3, Slc4a4, Slc4a7, and Slc4a10 quantified by real-time PCR did not differ between genotypes (data not shown). In conclusion, acid extrusion via Slc4a8 appears to play an important role for the steady-state pHi and for the control of pHi to an acute acid load.
Altered excitability of CA1 pyramidal cells in the absence of Slc4a8
Because of the localization of Slc4a8 to glutamatergic boutons and its impact on neuronal pH regulation, we measured field potentials in the CA1 region of acute WT and KO slice preparations upon stimulation of Schaffer collaterals. Fiber volley amplitudes analyzed at half-maximal stimulus intensities (WT, 33.6 ± 2.1 V; KO, 35.5 ± 3.3 V; Student's t test, p = 0.65; n = 22/19) did not differ between the genotypes (KO, 0.68 ± 0.06 mV; WT, 0.72 ± 0.08 mV; Student's t test, p = 0.63; n = 22/19). Population spike amplitudes in the pyramidal layer were decreased in KO mice (Fig. 4A; input/output: repeated-measures ANOVA, F = 4.56; p = 0.040; and at half-maximal stimulus intensity: KO, 4.27 ± 0.62 mV; WT, 6.81 ± 0.67 mV; Student's t test, p = 0.008; n = 22/19). In support of a presynaptic defect, paired-pulse facilitation was increased in fEPSP recordings of KO slices (Fig. 4B; repeated-measures ANOVA, F = 5.04, p = 0.03, n = 22/19). Field potential recordings in the stratum radiatum also revealed increased paired-pulse ratios upon stimulation with half-maximal stimulus intensity in KO slices (repeated-measures ANOVA, F = 4.44, p = 0.04; n = 37/41). Passive (input resistance, capacity, resting membrane potential) and active (e.g., action potential height, spike frequency accommodation) membrane properties were unaltered in CA1 pyramidal neurons patched in the current-clamp mode (Table 1; n = 12/12).
Figure 4.
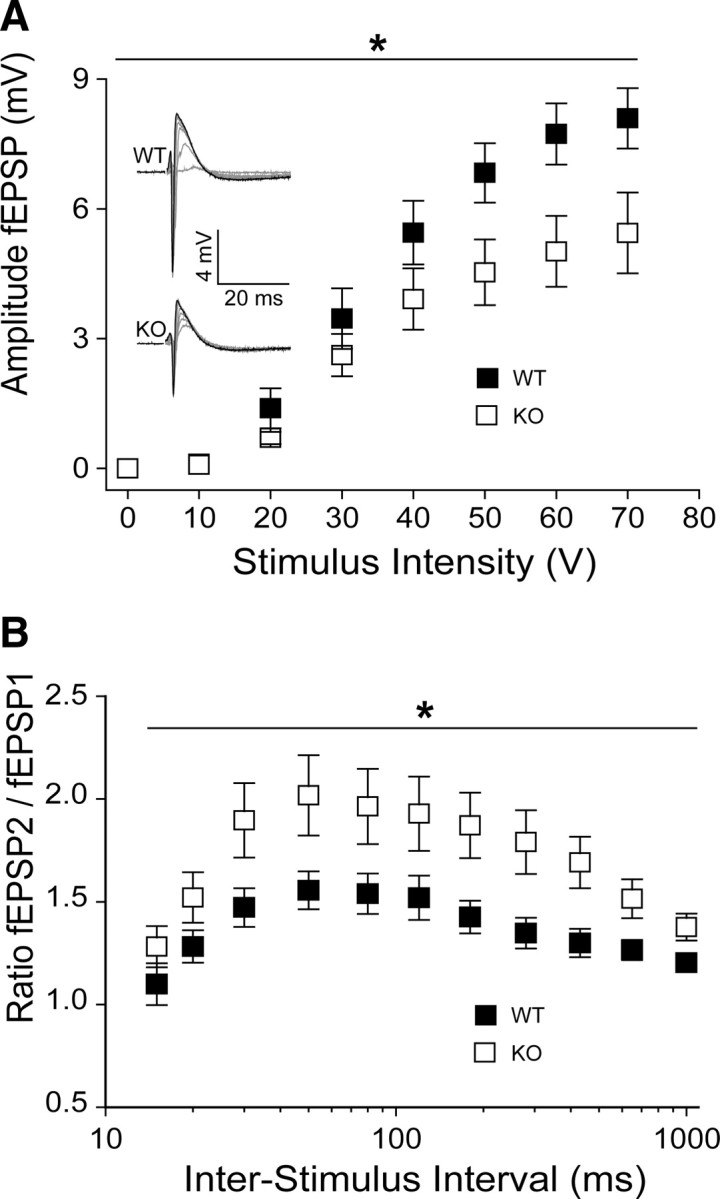
Reduction of network excitability in hippocampal slices from KO mice. A, CA1 population spike amplitudes upon stimulation of Schaffer collaterals were decreased in KO slices (p = 0.04; n = 22/19). Inset, Sample traces of somatic field recordings. Stimulus artifacts were omitted for clarity. B, Paired-pulse facilitation was significantly increased in slices of KO mice at 30–1000 ms interstimulus intervals (p = 0.03; n = 22/19). *p < 0.05.
Table 1.
Passive and active membrane properties in principal neurons of the CA1
| KO (n = 12) | WT (n = 13) | p value | |
|---|---|---|---|
| Capacity (pF) | 19.1 ± 1.7 | 17.1 ± 1.1 | 0.45 |
| Resting membrane potential (mV) | −66.5 ± 1.3 | −64.4 ± 1.0 | 0.26 |
| Input resistance (MΩ) | 62.6 ± 6.1 | 73.5 ± 7.8 | 0.28 |
| Action potential threshold (mV) | −44.7 ± 1.0 | −42.2 ± 2.0 | 0.28 |
| Action potential height (mV) | 97.0 ± 2.9 | 97.1 ± 2.9 | 0.98 |
| Action potential half-width (ms) | 0.7 ± 0.1 | 0.7 ± 0.0 | 0.61 |
| Action potential rise time (ms) | 0.5 ± 0.1 | 0.5 ± 0.1 | 0.95 |
The altered paired-pulse ratio of hippocampal field recordings and the presynaptic localization of Slc4a8 prompted us to examine synaptic transmission more closely. Spontaneous synaptic vesicle release was assessed by recording mEPSCs (Fig. 5A–F) and mIPSCs (Fig. 5G–J) in acute slice preparations from KO and WT mice. While the amplitudes of the mEPSCs did not differ between genotypes (Fig. 5C; KO, 19.493 ± 1.63 pA; WT, 20.49 ± 1.61 pA; one-way ANOVA, F = 1.51, p = 0.21, n = 38/32), the mean frequency was more than halved in pyramidal CA1 neurons of KO mice (Fig. 5D; KO, 0.31 ± 0.03 Hz; WT, 1.11 ± 0.25 Hz; one-way ANOVA, F = 3.71, p = 0.007; Newman–Keuls test, p < 0.01).
Figure 5.
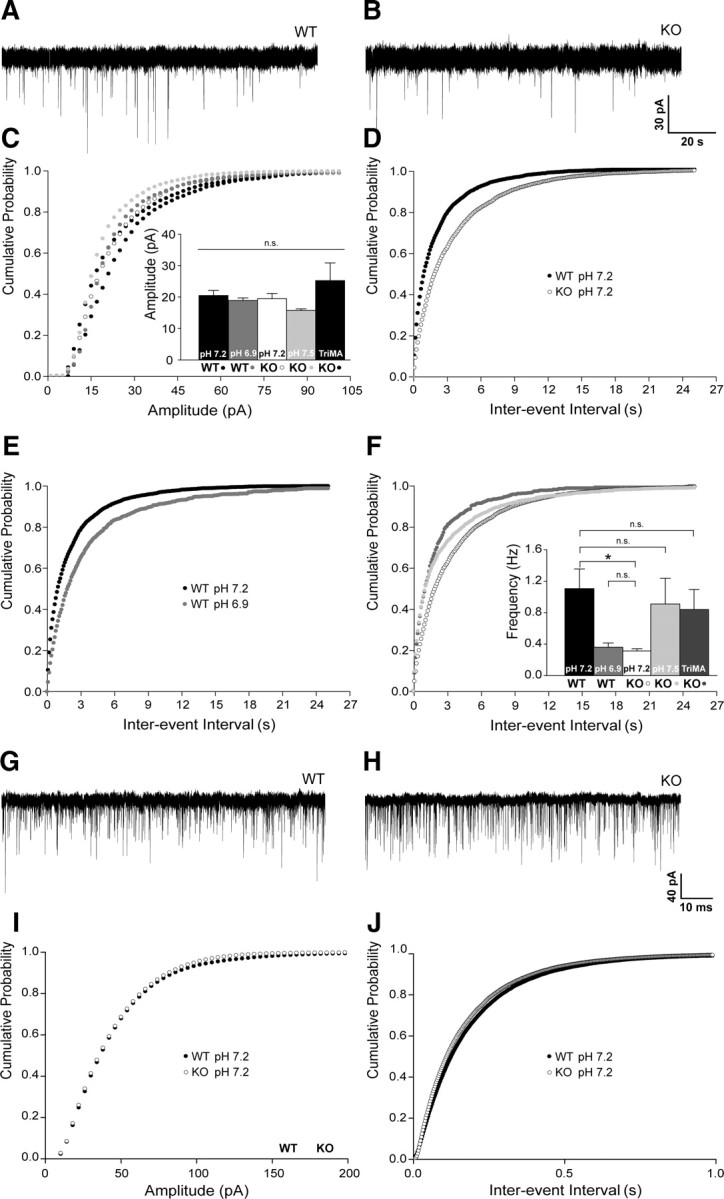
The frequency of mEPSCs but not of mIPSCs is decreased in KO mice in a pH-dependent manner. A, B, Representative traces of mEPSC recordings of WT and KO CA1 pyramidal cells at pHo 7.2. C, Cumulative plots of mEPSC amplitudes at varying pHi did not differ. Inset, Means of mEPSC amplitudes (p = 0.21; n = 32/15/38/14/7). D, Cumulative plots of interevent intervals revealed a shift to longer intervals in KO compared with WT (pHo 7.2, p < 0.001; n = 38/32). E, Shifting the pHo to 6.9 in WT diminished mEPSC frequencies (p < 0.0001; n = 32/15). F, Increasing pHi by decreasing pCO2 (pHo 7.5) or by application of the weak base TriMA raised the mEPSC frequency of KO cells (pHo 7.5, p = 0.03; n = 38/14; TriMA, p = 0.003; n = 38/7) toward WT. Inset, Means of mEPSC frequencies of WT and KO at different pHi. G, H, Representative traces of mIPSC recordings of WT and KO CA1 pyramidal neurons. I, J, Cumulative plots and means of mIPSC amplitude and frequencies did not differ between the genotypes (pHo 7.2, p = 0.78 and p = 0.27; n = 15/16). *p < 0.05.
pH-dependent modulation of mEPSC frequency in WT and KO neurons
To assess whether the electrophysiological changes could be explained by a compromised neuronal pH regulation, we manipulated the pHi by varying pCO2 at a constant extracellular bicarbonate concentration. Under these conditions, altering the pCO2 changed extra- and intracellular pH in the same direction, with a decrease of pCO2 resulting in an increase of pHi and an increase of pCO2 in a decrease of pHi, as demonstrated by previous studies (Lee et al., 1996; Dulla et al., 2005) and confirmed by our pH recordings on cultured neurons. Lowering pH decreased the frequency of mEPSCs in WT to values obtained from KO slices (Fig. 5E; WT pHo 7.2, 1.11 ± 0.25 Hz; WT pHo 6.9, 0.36 ± 0.05 Hz; Kolmogorov–Smirnov test, p = 0.0001; n = 32/15), whereas raising the pH increased the frequency of mEPSCs in KO slices (Fig. 5F; KO pHo 7.2, 0.31 ± 0.03 Hz; KO pHo 7.5, 0.91 ± 0.33 Hz; Kolmogorov–Smirnov test, p = 0.028; n = 38/14). The amplitude was not altered by changing pCO2 (Fig. 5C; KO pHo 7.5, 15.72 ± 0.51 pA; WT pHo 6.9, 18.87 ± 0.84 pA; one-way ANOVA, F = 1.51, p = 0.21, n = 14/15). We additionally recorded mEPSCs in slices from KO mice upon substitution of 20 mm NaCl by TriMA, a membrane-permeant weak base, which raises the pHi without altering the extracellular pHo (Eisner et al., 1989). Again, no significant differences in mEPSC amplitudes were observed (Fig. 5C; KO, 19.49 ± 1.63 pA; KOTriMA, 25.23 ± 5.61 pA; one-way ANOVA, F = 1.51, p = 0.21; n = 38/7), but TriMA raised the mEPSC frequencies in KO slices (KO, 0.34 ± 0.03 Hz; KOTriMA, 0.84 ± 0.25 Hz; Kolmogorov–Smirnov test, p = 0.003; n = 38/7), which did not differ from WT cells under control conditions (Fig. 5F; KOTriMA, 0.84 ± 0.25 Hz; WT, 1.11 ± 0.25 Hz; Kolmogorov–Smirnov test, p = 1.00; n = 7/32).
In accordance with the immunofluorescence data that GAD and Slc4a8 only partially overlap, no differences were noted for mIPSC amplitudes (Fig. 5I; KO pHo 7.2, 38.54 ± 2.39 pA; WT pHo 7.2, 39.43 ± 2.10 pA; Student's t test, p = 0.78; n = 15/16) and frequencies (Fig. 5J; KO pHo 7.2, 7.07 ± 1.02 Hz; WT pHo 7.2, 5.82 ± 0.46 Hz; Student's t test, p = 0.27).
Slc4a8 modulates presynaptic vesicle release
To finally pinpoint the defect of glutamatergic transmission in KO mice to the presynapse, vesicle release upon field stimulation was subsequently quantified by measuring bouton destaining in FM1-43-labeled cultured hippocampal neurons (Fig. 6A,B). No difference was observed in baseline punctual intensity (KO, 476.34 ± 18.02 arbitrary units; WT, 466.25 ± 18.09 arbitrary units; Student's t test, p = 0.69; n = 116/134). However, destaining, and hence synaptic vesicle release, upon repetitive stimulation was significantly diminished in KO cultures after ∼900 action potentials ((_F_0–_F_900)/_F_0); KO, 0.183 ± 0.01; WT, 0.206 ± 0.01; Student's t test, p = 0.04; n = 116/134). Following extensive stimulation with >1500 action potentials, this difference disappeared ((_F_0–_F_1500)/F0); KO, 0.307 ± 0.01; WT, 0.320 ± 0.01; Student's t test, p = 0.31). Fitting single bouton destaining over time monoexponentially allowed the calculation of destaining time constants. Analysis of the cumulative probability of time constants (Fig. 6B) revealed a significantly different distribution between the two groups (Kolmogorov–Smirnov test, p = 0.002), with an increase of mean τ in KO (144.59 ± 9.04 s; n = 54) compared with WT (112.43 ± 7.53 s; Student's t test, p = 0.0079; n = 50) neurons. These findings could be corroborated electrophysiologically: normalized evoked EPSC amplitudes decreased more rapidly per stimulus upon application of the open-channel NMDA receptor blocker MK-801 in WT compared with KO slices (Fig. 6C). The blocking rate of NMDA receptor-mediated EPSCs was significantly delayed in KO slices (54.59 ± 6.32 stimuli; n = 8) compared with WT (27.56 ± 3.64 stimuli; Student's t test, p = 0.005; n = 6), whereas the input/output relationship of evoked NMDA current density did not differ between genotypes at all stimulus intensities tested (repeated-measures ANOVA, F = 1.01, p = 0.32; n = 16/11).
Figure 6.

Impaired release of glutamate vesicles in KO mice. A, Time course of fluorescence destaining following field stimulation of cultured hippocampal WT and KO neurons upon vesicle labeling with FM1-43 (n = 54/50). B, Cumulative plot of the time constants revealed a shift toward a higher τ in KO neurons (p = 0.002; n = 54/50) equivalent with an increased mean time constant. C, Time course of the amplitude decrease of normalized NMDA currents in the presence of MK-801 (n = 8/6). Inset, Sample traces of NMDA receptor-mediated EPSCs recorded from CA1 pyramidal neurons in the presence of MK-801 upon repetitive stimulation of Schaffer collaterals. Mean time constant was increased in CA1 neurons of KO mice in the presence of MK-801. D, Model illustrating the consequences of Slc4a8 disruption on glutamate release. Slc4a8 is involved in presynaptic pH homeostasis. Upon disruption of Slc4a8, the intracellular pH is diminished and glutamate release impaired. Accordingly, fewer postsynaptic glutamate receptors are activated, but the pool of releasable vesicles appears to be unchanged.
Together, these experiments show that deletion of Slc4a8 results in a reduction of the number of released glutamatergic vesicles, although the pool of releasable vesicles appears to be unchanged (Fig. 6D).
In vivo characterization of KO mice
Given the importance of pH for neuronal excitability, we tested the susceptibility of KO mice to proconvulsant substances. Latencies of myoclonic, tonic, and tonic-clonic seizures in response to the intraperitoneal application of pentylenetetrazol (Fig. 7A; KO: 109.23 ± 25.90, 169.2 ± 19.3, and 315.8 ± 29.9 s; WT: 90.73 ± 37.89, 123.2 ± 9.5, and 150.8 ± 14.5 s; repeated-measures ANOVA, F = 21.31, p > 0.0001; n = 22/22) were significantly prolonged in KO mice. Seizure latency after administration of pilocarpine (Fig. 7B; KO, 2709.2 ± 406.8 s; WT, 1530.5 ± 102.4 s; n = 13/13) and ictal activity upon hyperthermia-induced hyperventilation at postnatal day 10 were delayed also (Fig. 7B; KO, 526.3 ± 27.7 s; WT, 431.4 ± 35; n = 11/10; two-way ANOVA: genotype F = 7.53, p = 0.009; interaction F = 5.74, p = 0.02).
Figure 7.

Slc4a8 deletion increases seizure threshold in different in vivo models of epilepsy. A, The latency until onset of pentylenetetrazol-induced myoclonic, tonic, and tonic-clonic seizures was significantly prolonged in KO mice (p < 0.0001; n = 22/22). B, Latency of ictal activity upon pilocarpine injection (p = 0.01; n = 13/13) was prolonged, as was seizure onset in a mouse pup model of hyperthermia-related epileptic activity (n = 11/10). *p < 0.05.
Discussion
Slc4a8 localizes to presynaptic nerve endings
With our Slc4a8 antibody (Leviel et al., 2010), neurons in various regions of the CNS including cerebellum, spinal cord, brainstem, hippocampus, and cortex were stained. This confirms previous data that antibodies generated to detect Slc4a8 labeled hippocampal pyramidal neurons (Damkier et al., 2007; Chen et al., 2008) but not astrocytes (Chen et al., 2008). Our costainings with presynaptic (vGLUT1, -2, -3; synaptophysin; NF68) and postsynaptic (PSD-95; MAP2) markers were in accordance with a mainly presynaptic localization of Slc4a8. This was corroborated by immunoblot analysis of subcellular fractions of mouse brain lysates, Slc4a8 being enriched in synaptic junction membranes compared with synaptosomes and crude membrane preparations, but absent from the postsynaptic density, and by ultrastructural analysis. The closely related Na+-coupled anion exchanger Slc4a10 (Wang et al., 2000; Damkier et al., 2010) has mainly been detected in dendrites, dendritic spines, and postsynaptic membranes including parvalbumin-positive interneurons (Jacobs et al., 2008), whereas Slc4a8 was absent from parvalbumin-positive interneurons, and overlap with GAD was only partial. It has been shown that complex-spike activity in some auditory interneurons results in a pH-dependent negative shift of the glycine reversal potential, and it was suggested that Na+-coupled anion exchange via Slc4a8 may account for the reduction of intracellular chloride (Kim and Trussell, 2009). However, in view of our localization data (Jacobs et al., 2008) and unpublished data from our laboratory, Slc4a10 appears to be the more likely candidate. Similar to Slc4a10, the Na+-HCO3− cotransporter Slc4a7, which also mediates acid extrusion, has been found to be localized to the postsynaptic side (Park et al., 2010), whereas it has been suggested that the Na+/H+ exchanger Slc9a1 is present in presynaptic nerve endings (Jang et al., 2006; Dietrich and Morad, 2010).
Role of Slc4a8 in neuronal pHi regulation
The transport direction of electroneutral transporters like Slc4a8 does not depend on the transmembrane potential. Because of the large Na+ gradient between the extracellular and intracellular compartment, Slc4a8 mediates an inward transport of HCO3−, which is equivalent to acid extrusion from the cells and an increase of the intracellular buffering capacity. In accordance with a reduction of the buffering capacity, NH4Cl-induced peak alkalosis was increased in KO neurons. Because the steady-state pHi was decreased and the recovery from an acid load impaired in the somata of cultured hippocampal neurons of KO mice, Slc4a8 clearly plays an important role in pHi regulation in these neurons. This confirms previous data that rat CA1 hippocampal neurons displayed DIDS-sensitive HCO3−-dependent acid extrusion that required external Na+ as well as internal Cl− (Schwiening and Boron, 1994). Since acid extrusion did not differ between genotypes in HEPES-buffered solutions, a compensatory upregulation of Na+/H+ exchange or other HCO3−-independent mechanisms in KO mice appears unlikely. In addition, Slc9a1, Slc4a3, Slc4a4, Slc4a7, and Slc4a10 transcript abundance was not altered between genotypes (data not shown).
The repeated association and dissociation of intracellular protons with intracellular macromolecules results in a surprisingly low diffusion rate in the cytosol (al-Baldawi and Abercrombie, 1992). Consequently, pH gradients have been described in cells with a polarized expression of pH-relevant transporters (Stewart et al., 1999; Schwiening and Willoughby, 2002). Because of the enrichment of Slc4a8 in presynaptic nerve endings, one may infer that the impact of the disruption of Slc4a8 on pHi regulation may be more pronounced in synaptic boutons compared with the soma. However, to our knowledge, a direct analysis of pHi regulation in central presynaptic boutons has not been possible to date, and the numerous studies dealing with pH regulation in synaptosomes are difficult to interpret as these preparations include both presynaptic and postsynaptic compartments (Bai and Witzmann, 2007). Focal injections of BCECF-AM in combination with slice imaging, as used for measuring calcium transients in small synaptic compartments with the calcium-sensitive dye Fura-2 (Saggau et al., 1999), may pave the way toward adequate pH imaging in small compartments like central presynaptic and postsynaptic terminals. Quite recently, the pH-sensitive properties of YFP were successfully used to analyze the presynaptic pH in large motor endplates (Zhang et al., 2010).
Glutamatergic vesicle release is modulated by Slc4a8
Field recordings in hippocampal slices provided the first indication that Slc4a8 may be relevant for neuronal excitability, as synchronous firing of CA1 pyramidal neurons was strongly reduced upon stimulation of Schaffer collaterals. The increased facilitation by a second pulse, together with the localization of Slc4a8, pointed to a presynaptic defect of the projection of Schaffer collaterals onto pyramidal neurons. Because glutamate uptake into synaptic vesicles is driven by the electrochemical proton gradient across the synaptic vesicle membrane (Maycox et al., 1988), we analyzed the amplitudes of spontaneous mEPSCs as an indirect measure for the quantum of transmitter released per synaptic vesicle. Whereas the normal amplitude distribution indicated that the loading of glutamatergic synaptic vesicles in KO mice was not affected, the drastic decrease of mEPSC frequency revealed that synaptic vesicle release was impaired in the absence of Slc4a8. It has been shown that effects of varying pCO2 on excitatory transmission are apparently caused by changes in pHi (Lee et al., 1996). Thus, supporting our hypothesis that the decrease of frequency could be attributed to a decrease of pHi in presynaptic nerve endings, lowering the pHi by increasing pCO2 in WT slices decreased mEPSC frequency to values as observed in KO mice, whereas the frequency of spontaneous mEPSCs increased with increased pHi in KO slices. Although the optical analysis of synaptic vesicle release could not distinguish between glutamatergic and GABAergic presynapses, which are thought to account for a minor fraction in cultured hippocampal neurons (Walker and Peacock, 1981; Feng et al., 2002), induced synaptic vesicle release upon repetitive field stimulation of cultured hippocampal neurons was significantly decreased over a wide range of the stimulation period. However, the overall synaptic vesicle pool appeared to be unaffected by disruption of Slc4a8, as the difference disappeared after >1500 pulses. The difference between genotypes was more pronounced in the electrophysiological analysis, which was specific for glutamatergic synaptic activity.
How can disruption of Slc4a8 affect neurotransmitter release?
The machinery for the release of synaptic vesicles is initiated by a rise in [Ca2+]i (Schneggenburger and Neher, 2000). However, other ions including Na+ and H+ have also been proposed to play a critical role. As Slc4a8 mediates Na+-coupled anion exchange, its activity increases the [Na+]i concentration, which in turn may increase the [Ca2+]i via the Na+/Ca2+ exchanger (Mulkey and Zucker, 1992; Reuter and Porzig, 1995), whereas Ca2+ extrusion via the ATP-driven Ca2+/H+ exchanger is probably not severely impaired by the reduction in baseline pHi (Schwiening et al., 1993; Trapp et al., 1996). Following this scenario, disruption of Slc4a8 may result in a lower [Ca2+]i and hence a reduction of spontaneous synaptic vesicle release. However, in view of the partial reversibility of the decreased synaptic vesicle release by increasing the pH, a direct pH-related effect may be more likely. Since virtually all proteins depend on pH to maintain their structure and function, multiple effects may add up. For example, the opening and the conductivity of presynaptic voltage-gated calcium channels, which mediate the presynaptic Ca2+ influx, are strongly dependent on both extracellular and intracellular pH (Tombaugh and Somjen, 1997). Neurotransmitter release and synaptic short-term plasticity, as well as spontaneous vesicle release, also depend on calcium release from intracellular stores (Emptage et al., 2001), which is mediated via inositol 1,4,5-trisphosphate and ryanodine receptors, both of which show strong pH dependence (Ma et al., 1988; Tsukioka et al., 1994).
In vivo relevance
Because neuronal activity can result in sustained decreases of pHi (Chesler and Kaila, 1992), the pH-dependent impairment of synaptic vesicle release may be even more relevant in situations with increased neuronal activity (Zhan et al., 1998; Tong and Chesler, 1999), including seizures (Xiong et al., 2000). Whereas a decrease of pH generally reduces neuronal excitability, it is increased by a rise of pH (Aram and Lodge, 1987). Hyperthermia-induced respiratory alkalosis is also considered as a key event in the pathogenesis of febrile seizures. In a rat pup model for febrile seizures, ictal activity was induced by respiratory alkalosis with a threshold of 0.2–0.3 pH units (Schuchmann et al., 2006), and could be stopped by suppressing alkalosis by increasing ambient pCO2. The impaired acid extrusion in neurons of KO mice may thus explain why KO mice have reduced seizure susceptibilities in various paradigms, including the hyperthermia model. Likewise, mice with a targeted disruption of Slc4a10 were protected from seizure-related mortality (Jacobs et al., 2008), whereas the sensitivity to seizure-inducing agents was increased in mice with a disruption of the Na+-independent Cl−/HCO3− exchanger Slc4a3, which imposes an intracellular acid load (Hentschke et al., 2006). Some anticonvulsants, such as acetazolamide, reduce extracellular pH in the brain (Chen and Chesler, 1992), suggesting that acidosis may contribute to their anti-epileptic effects. The increased seizure threshold in KO mice suggests that inhibition of Slc4a8 may be a target for anti-epileptic therapy.
In conclusion, we show that the broadly expressed neuronal Na+-coupled anion exchanger Slc4a8 is enriched in presynaptic glutamatergic nerve terminals. As a key regulator of intracellular pH, Slc4a8 affects glutamatergic synaptic vesicle release in a pH-dependent way. Since consensus phosphorylation sites have been predicted for different members of the gene family (Boron et al., 2009), including Slc4a8 (Grichtchenko et al., 2001), it is tempting to speculate that synaptic strength may be regulated via Slc4a8 in response to various signals. Thereby, Slc4a8 could have important consequences for synaptic transmission in both physiological and pathophysiological conditions.
Footnotes
This study was supported by grants from the Deutsche Forschungsgemeinschaft to C.A.H. We thank Knut Holthoff, Knut Kirmse, Christopher Hennings, Ingo Kurth, and Valentin Stein for helpful discussions. We also thank Mukhran Khundadze for his help, Walter Alt and Jens Schumacher for advice regarding the data analysis, and Antje Hübner for her continuous support.
The authors declare no conflict of interest.
References
- al-Baldawi NF, Abercrombie RF. Cytoplasmic hydrogen ion diffusion coefficient. Biophys J. 1992;61:1470–1479. doi: 10.1016/S0006-3495(92)81953-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alper SL. Molecular physiology and genetics of Na+-independent SLC4 anion exchangers. J Exp Biol. 2009;212:1672–1683. doi: 10.1242/jeb.029454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aram JA, Lodge D. Epileptiform activity induced by alkalosis in rat neocortical slices: block by antagonists of N-methyl-d-aspartate. Neurosci Lett. 1987;83:345–350. doi: 10.1016/0304-3940(87)90112-1. [DOI] [PubMed] [Google Scholar]
- Bai F, Witzmann FA. Synaptosome proteomics. Subcell Biochem. 2007;43:77–98. doi: 10.1007/978-1-4020-5943-8_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter KA, Church J. Characterization of acid extrusion mechanisms in cultured fetal rat hippocampal neurones. J Physiol. 1996;493:457–470. doi: 10.1113/jphysiol.1996.sp021396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevensee MO, Cummins TR, Haddad GG, Boron WF, Boyarsky G. pH regulation in single CA1 neurons acutely isolated from the hippocampi of immature and mature rats. J Physiol. 1996;494:315–328. doi: 10.1113/jphysiol.1996.sp021494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet U, Leniger T, Wiemann M. Alteration of intracellular pH and activity of CA3-pyramidal cells in guinea pig hippocampal slices by inhibition of transmembrane acid extrusion. Brain Res. 2000;872:116–124. doi: 10.1016/s0006-8993(00)02350-7. [DOI] [PubMed] [Google Scholar]
- Boron WF, De Weer P. Intracellular pH transients in squid giant axons caused by CO2, NH3, and metabolic inhibitors. J Gen Physiol. 1976;67:91–112. doi: 10.1085/jgp.67.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boron WF, Chen L, Parker MD. Modular structure of sodium-coupled bicarbonate transporters. J Exp Biol. 2009;212:1697–1706. doi: 10.1242/jeb.028563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyarsky G, Ganz MB, Sterzel RB, Boron WF. pH regulation in single glomerular mesangial cells. I. Acid extrusion in absence and presence of HCO3−. Am J Physiol. 1988;255:C844–C856. doi: 10.1152/ajpcell.1988.255.6.C844. [DOI] [PubMed] [Google Scholar]
- Carlin RK, Grab DJ, Cohen RS, Siekevitz P. Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities. J Cell Biol. 1980;86:831–845. doi: 10.1083/jcb.86.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 2010;11:50–61. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- Chen JC, Chesler M. pH transients evoked by excitatory synaptic transmission are increased by inhibition of extracellular carbonic anhydrase. Proc Natl Acad Sci U S A. 1992;89:7786–7790. doi: 10.1073/pnas.89.16.7786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LM, Kelly ML, Parker MD, Bouyer P, Gill HS, Felie JM, Davis BA, Boron WF. Expression and localization of Na-driven Cl-HCO3− exchanger (SLC4A8) in rodent CNS. Neuroscience. 2008;153:162–174. doi: 10.1016/j.neuroscience.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M. Regulation and modulation of pH in the brain. Physiol Rev. 2003;83:1183–1221. doi: 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- Chesler M, Kaila K. Modulation of pH by neuronal activity. Trends Neurosci. 1992;15:396–402. doi: 10.1016/0166-2236(92)90191-a. [DOI] [PubMed] [Google Scholar]
- Cordat E, Casey JR. Bicarbonate transport in cell physiology and disease. Biochem J. 2009;417:423–439. doi: 10.1042/BJ20081634. [DOI] [PubMed] [Google Scholar]
- Damkier HH, Nielsen S, Praetorius J. Molecular expression of SLC4-derived Na+-dependent anion transporters in selected human tissues. Am J Physiol Regul Integr Comp Physiol. 2007;293:R2136–R2146. doi: 10.1152/ajpregu.00356.2007. [DOI] [PubMed] [Google Scholar]
- Damkier HH, Aalkjaer C, Praetorius J. Na+-dependent HCO3− import by the slc4a10 gene product involves Cl− export. J Biol Chem. 2010;285:26998–27007. doi: 10.1074/jbc.M110.108712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich CJ, Morad M. Synaptic acidification enhances GABAA signaling. J Neurosci. 2010;30:16044–16052. doi: 10.1523/JNEUROSCI.6364-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulla CG, Dobelis P, Pearson T, Frenguelli BG, Staley KJ, Masino SA. Adenosine and ATP link PCO2 to cortical excitability via pH. Neuron. 2005;48:1011–1023. doi: 10.1016/j.neuron.2005.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner DA, Kenning NA, O'Neill SC, Pocock G, Richards CD, Valdeolmillos M. A novel method for absolute calibration of intracellular pH indicators. Pflugers Arch. 1989;413:553–558. doi: 10.1007/BF00594188. [DOI] [PubMed] [Google Scholar]
- Emptage NJ, Reid CA, Fine A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron. 2001;29:197–208. doi: 10.1016/s0896-6273(01)00190-8. [DOI] [PubMed] [Google Scholar]
- Feng J, Chi P, Blanpied TA, Xu Y, Magarinos AM, Ferreira A, Takahashi RH, Kao HT, McEwen BS, Ryan TA, Augustine GJ, Greengard P. Regulation of neurotransmitter release by synapsin III. J Neurosci. 2002;22:4372–4380. doi: 10.1523/JNEUROSCI.22-11-04372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffield MA, Betz WJ. Imaging synaptic vesicle exocytosis and endocytosis with FM dyes. Nat Protoc. 2006;1:2916–2921. doi: 10.1038/nprot.2006.476. [DOI] [PubMed] [Google Scholar]
- Grichtchenko II, Choi I, Zhong X, Bray-Ward P, Russell JM, Boron WF. Cloning, characterization, and chromosomal mapping of a human electroneutral Na+-driven Cl−HCO3 exchanger. J Biol Chem. 2001;276:8358–8363. doi: 10.1074/jbc.C000716200. [DOI] [PubMed] [Google Scholar]
- Hentschke M, Wiemann M, Hentschke S, Kurth I, Hermans-Borgmeyer I, Seidenbecher T, Jentsch TJ, Gal A, Hübner CA. Mice with a targeted disruption of the Cl−/HCO3− exchanger AE3 display a reduced seizure threshold. Mol Cell Biol. 2006;26:182–191. doi: 10.1128/MCB.26.1.182-191.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz L, Dienel GA. Lactate transport and transporters: general principles and functional roles in brain cells. J Neurosci Res. 2005;79:11–18. doi: 10.1002/jnr.20294. [DOI] [PubMed] [Google Scholar]
- Jacobs S, Ruusuvuori E, Sipilä ST, Haapanen A, Damkier HH, Kurth I, Hentschke M, Schweizer M, Rudhard Y, Laatikainen LM, Tyynelä J, Praetorius J, Voipio J, Hübner CA. Mice with targeted Slc4a10 gene disruption have small brain ventricles and show reduced neuronal excitability. Proc Natl Acad Sci U S A. 2008;105:311–316. doi: 10.1073/pnas.0705487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang IS, Brodwick MS, Wang ZM, Jeong HJ, Choi BJ, Akaike N, et al. The Na+/H+ exchanger is a major pH regulator in GABAergic presynaptic nerve terminals synapsing onto rat CA3 pyramidal neurons. J Neurochem. 2006;99:1224–1236. doi: 10.1111/j.1471-4159.2006.04168.x. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Trussell LO. Negative shift in the glycine reversal potential mediated by a Ca2+- and pH-dependent mechanism in interneurons. J Neurosci. 2009;29:11495–11510. doi: 10.1523/JNEUROSCI.1086-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Taira T, Pihlaja P, Ransom BR, Kaila K. Effects of CO2 on excitatory transmission apparently caused by changes in intracellular pH in the rat hippocampal slice. Brain Res. 1996;706:210–216. doi: 10.1016/0006-8993(95)01214-1. [DOI] [PubMed] [Google Scholar]
- Leviel F, Hübner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hatim H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest. 2010;120:1627–1635. doi: 10.1172/JCI40145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebmann L, Karst H, Joëls M. Effects of corticosterone and the beta-agonist isoproterenol on glutamate receptor-mediated synaptic currents in the rat basolateral amygdala. Eur J Neurosci. 2009;30:800–807. doi: 10.1111/j.1460-9568.2009.06882.x. [DOI] [PubMed] [Google Scholar]
- Luo J, Sun D. Physiology and pathophysiology of Na+/H+exchange isoform 1 in the central nervous system. Curr Neurovasc Res. 2007;4:205–215. doi: 10.2174/156720207781387178. [DOI] [PubMed] [Google Scholar]
- Ma J, Fill M, Knudson CM, Campbell KP, Coronado R. Ryanodine receptor of skeletal muscle is a gap junction-type channel. Science. 1988;242:99–102. doi: 10.1126/science.2459777. [DOI] [PubMed] [Google Scholar]
- Maycox PR, Deckwerth T, Hell JW, Jahn R. Glutamate uptake by brain synaptic vesicles. Energy dependence of transport and functional reconstitution in proteoliposomes. J Biol Chem. 1988;263:15423–15428. [PubMed] [Google Scholar]
- Mulkey RM, Zucker RS. Posttetanic potentiation at the crayfish neuromuscular junction is dependent on both intracellular calcium and sodium ion accumulation. J Neurosci. 1992;12:4327–4336. doi: 10.1523/JNEUROSCI.12-11-04327.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995;81:769–780. doi: 10.1016/0092-8674(95)90538-3. [DOI] [PubMed] [Google Scholar]
- Park HJ, Rajbhandari I, Yang HS, Lee S, Cucoranu D, Cooper DS, Klein JD, Sands JM, Choi I. Neuronal expression of sodium/bicarbonate cotransporter NBCn1 (SLC4A7) and its response to chronic metabolic acidosis. Am J Physiol Cell Physiol. 2010;298:C1018–C1028. doi: 10.1152/ajpcell.00492.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter H, Porzig H. Localization and functional significance of the Na+/Ca2+ exchanger in presynaptic boutons of hippocampal cells in culture. Neuron. 1995;15:1077–1084. doi: 10.1016/0896-6273(95)90096-9. [DOI] [PubMed] [Google Scholar]
- Rink TJ, Tsien RY, Pozzan T. Cytoplasmic pH and free Mg2+ in lymphocytes. J Cell Biol. 1982;95:189–196. doi: 10.1083/jcb.95.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero MF, Henry D, Nelson S, Harte PJ, Dillon AK, Sciortino CM. Cloning and characterization of a Na+-driven anion exchanger (NDAE1). A new bicarbonate transporter. J Biol Chem. 2000;275:24552–24559. doi: 10.1074/jbc.M003476200. [DOI] [PubMed] [Google Scholar]
- Russell JM, Boron WF. Role of chloride transport in regulation of intracellular pH. Nature. 1976;264:73–74. doi: 10.1038/264073a0. [DOI] [PubMed] [Google Scholar]
- Saggau P, Gray R, Dani JA. Optical measurements of calcium signals in mammalian presynaptic terminals. Methods Enzymol. 1999;294:3–19. doi: 10.1016/s0076-6879(99)94004-1. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- Schuchmann S, Schmitz D, Rivera C, Vanhatalo S, Salmen B, Mackie K, Sipilä ST, Voipio J, Kaila K. Experimental febrile seizures are precipitated by a hyperthermia-induced respiratory alkalosis. Nat Med. 2006;12:817–823. doi: 10.1038/nm1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiening CJ, Boron WF. Regulation of intracellular pH in pyramidal neurones from the rat hippocampus by Na+-dependent Cl−-HCO3− exchange. J Physiol. 1994;475:59–67. doi: 10.1113/jphysiol.1994.sp020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiening CJ, Willoughby D. Depolarization-induced pH microdomains and their relationship to calcium transients in isolated snail neurones. J Physiol. 2002;538:371–382. doi: 10.1113/jphysiol.2001.013055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiening CJ, Kennedy HJ, Thomas RC. Calcium-hydrogen exchange by the plasma membrane Ca-ATPase of voltage-clamped snail neurons. Proc Biol Sci. 1993;253:285–289. doi: 10.1098/rspb.1993.0115. [DOI] [PubMed] [Google Scholar]
- Stewart AK, Boyd CA, Vaughan-Jones RD. A novel role for carbonic anhydrase: cytoplasmic pH gradient dissipation in mouse small intestinal enterocytes. J Physiol. 1999;516:209–217. doi: 10.1111/j.1469-7793.1999.209aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran CT. Carbonic anhydrases-an overview. Curr Pharm Des. 2008;14:603–614. doi: 10.2174/138161208783877884. [DOI] [PubMed] [Google Scholar]
- Tombaugh GC, Somjen GG. Differential sensitivity to intracellular pH among high- and low-threshold Ca2+ currents in isolated rat CA1 neurons. J Neurophysiol. 1997;77:639–653. doi: 10.1152/jn.1997.77.2.639. [DOI] [PubMed] [Google Scholar]
- Tong CK, Chesler M. Activity-evoked extracellular pH shifts in slices of rat dorsal lateral geniculate nucleus. Brain Res. 1999;815:373–381. doi: 10.1016/s0006-8993(98)01059-2. [DOI] [PubMed] [Google Scholar]
- Trapp S, Lückermann M, Kaila K, Ballanyi K. Acidosis of hippocampal neurones mediated by a plasmalemmal Ca2+/H+ pump. Neuroreport. 1996;7:2000–2004. doi: 10.1097/00001756-199608120-00029. [DOI] [PubMed] [Google Scholar]
- Tsukioka M, Iino M, Endo M. pH dependence of inositol 1,4,5-trisphosphate-induced Ca2+ release in permeabilized smooth muscle cells of the guinea-pig. J Physiol. 1994;475:369–375. doi: 10.1113/jphysiol.1994.sp020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker CR, Peacock JH. Development of GABAergic function of dissociated hippocampal cultures from fetal mice. Brain Res. 1981;254:541–555. doi: 10.1016/0165-3806(81)90022-5. [DOI] [PubMed] [Google Scholar]
- Wang CZ, Yano H, Nagashima K, Seino S. The Na+-driven Cl−/HCO3− exchanger. Cloning, tissue distribution, and functional characterization. J Biol Chem. 2000;275:35486–35490. doi: 10.1074/jbc.C000456200. [DOI] [PubMed] [Google Scholar]
- Westermann M, Steiniger F, Richter W. Belt-like localisation of caveolin in deep caveolae and its re-distribution after cholesterol depletion. Histochem Cell Biol. 2005;123:613–620. doi: 10.1007/s00418-004-0750-5. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Presynaptic calcium is increased during normal synaptic transmission and paired-pulse facilitation, but not in long-term potentiation in area CA1 of hippocampus. J Neurosci. 1994;14:645–654. doi: 10.1523/JNEUROSCI.14-02-00645.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZQ, Saggau P, Stringer JL. Activity-dependent intracellular acidification correlates with the duration of seizure activity. J Neurosci. 2000;20:1290–1296. doi: 10.1523/JNEUROSCI.20-04-01290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan RZ, Fujiwara N, Tanaka E, Shimoji K. Intracellular acidification induced by membrane depolarization in rat hippocampal slices: roles of intracellular Ca2+ and glycolysis. Brain Res. 1998;780:86–94. doi: 10.1016/s0006-8993(97)01149-9. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Nguyen KT, Barrett EF, David G. Vesicular ATPase inserted into the plasma membrane of motor terminals by exocytosis alkalinizes cytosolic pH and facilitates endocytosis. Neuron. 2010;68:1097–1108. doi: 10.1016/j.neuron.2010.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]