Why do bacteria use so many enzymes to scavenge hydrogen peroxide? (original) (raw)
. Author manuscript; available in PMC: 2013 Sep 15.
Published in final edited form as: Arch Biochem Biophys. 2012 May 16;525(2):145–160. doi: 10.1016/j.abb.2012.04.014
Abstract
Hydrogen peroxide (H2O2) is continuously formed by the autoxidation of redox enzymes in aerobic cells, and it also enters from the environment, where it can be generated both by chemical processes and by the deliberate actions of competing organisms. Because H2O2 is acutely toxic, bacteria elaborate scavenging enzymes to keep its intracellular concentration at nanomolar levels. Mutants that lack such enzymes grow poorly, suffer from high rates of mutagenesis, or even die. In order to understand how bacteria cope with oxidative stress, it is important to identify the key enzymes involved in H2O2 degradation. Catalases and NADH peroxidase (Ahp) are primary scavengers in many bacteria, and their activities and physiological impacts have been unambiguously demonstrated through phenotypic analysis and through direct measurements of H2O2 clearance in vivo. Yet a wide variety of additional enzymes have been proposed to serve similar roles: thiol peroxidase, bacterioferritin comigratory protein, glutathione peroxidase, cytochrome c peroxidase, and rubrerythrins. Each of these enzymes can degrade H2O2 in vitro, but their contributions in vivo remain unclear. In this review we examine the genetic, genomic, regulatory, and biochemical evidence that each of these is a bona fide scavenger of H2O2 in the cell. We also consider possible reasons that bacteria might require multiple enzymes to catalyze this process, including differences in substrate specificity, compartmentalization, cofactor requirements, kinetic optima, and enzyme stability. It is hoped that the resolution of these issues will lead to an understanding of stress resistance that is more accurate and perceptive.
Keywords: Thiol peroxidase, catalase, alkyl hydroperoxide reducatse, bacterioferritin comigratory protein, cytochrome c peroxidase, rubrerythrin
In 1900 Oscar Loew reported that higher organisms invariably contain a protein that degrades hydrogen peroxide (H2O2) to oxygen and water [1]:
At the time few proteins were known that could catalyze reactions, and so he named his new enzyme “catalase.” Catalase was later found in many bacteria as well. However, so were H2O2-degrading proteins of another class: peroxidases, which are defined by their ability to reduce rather than disproportionate H2O2:
Peroxidases can differ in their physiological electron donors (i.e., RH2), which may be glutathione, thioredoxins, NAD(P)H, or cytochrome c. Some proteins with peroxidase activity can also oxidize a range of non-physiological electron donors, including dyes; in a few cases these assays have allowed peroxidases to be detected even while their physiological electron donors remain unknown. The abundance of scavenging enzymes can perplex the physiologist who wants to understand the contributions that they make to cell fitness. Consider, for example, the model bacterium Escherichia coli, which expresses at least nine enzymes that have been proposed to be catalases or peroxidases. The obvious issue arises: Why so many? This review seeks to address this question.
The threat of hydrogen peroxide
Virtually every organism—animal, plant, or microbe—exhibits progressive indicators of toxicity when it is exposed to high concentrations of oxygen. The loss of fitness manifests as high mutation rates, growth defects, and ultimately death. Molecular oxygen itself cannot directly react with most biomolecules [2], and so early workers quickly recognized that the direct toxin might be a partially reduced form of oxygen that is formed inside the cell. The progressive addition of single electrons to oxygen generates superoxide (O2−·), H2O2, the hydroxyl radical (HO·), and water (H2O) (Fig.1). Molecular oxygen can adventitiously steal electrons from the dihydroflavin cofactors of a wide range of reduced redox enzymes in vitro; the higher the oxygen concentration, the more rapid the reaction [3–6]. A mixture of superoxide and hydrogen peroxide is formed, and the occurrence of such reactions in vivo would validate the presence of superoxide dismutases and reductases on the one hand, and catalases and peroxidases on the other. Measurements in Escherichia coli confirm that H2O2 is formed inside well-fed aerobic cells at a constant rate of 10–15 μM/second [7] (Fig. 2A). Consistent with the enzymatic data, the rate increases in proportion to oxygen concentration, which likely explains the toxicity of hyperoxia.
Fig. 1.
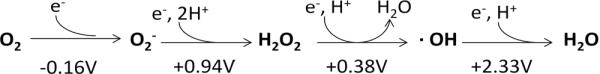
Stepwise univalent reduction of oxygen to superoxide, hydrogen peroxide, the hydroxyl radical, and water. Standard potentials are indicated for pH 7; the oxygen/superoxide potential is designated for 1 M molecular oxygen (not 1 atmosphere).
Fig. 2.


A requirement for endogenous scavengers of H2O2. A. E. coli mutants that lack scavenging enzymes continuously release H2O2 into the extracellular medium. Growing cells were suspended at time zero into fresh glucose/amino-acids medium, and the concentration of extracellular H2O2 was determined at intervals using a peroxidase assay [10]. Medium was gassed either with air or with pure oxygen, as indicated. The rate of intracellular formation can be deduced to be 10–15 μM/sec in air-saturated medium. B. The same mutants are unable to grow in simple aerobic glucose medium. At time zero the strains were switched from anaerobic to aerobic media, and biomass was monitored by measurements of optical density. The mutant strain lacks both catalases and NADH peroxidase (Kat− Ahp−); it does not exhibit any defect under anaerobic conditions.
These doses are enough to require the presence of scavenging systems: bacterial mutants that lack either superoxide or H2O2-scavenging enzymes exhibit growth defects (Fig. 2B), and the phenotypes become more severe when the level of environmental oxygen concentration is raised [8–11]. In H2O2-stressed cells, even 1 μM H2O2 efficiently oxidizes the pool of loose ferrous iron that is not incorporated in proteins, generating hydroxyl radicals that subsequently damage DNA [12]. This reaction explains the mutagenic effect of oxygen:
| H2O2+Fe2+→OH−+OH.+Fe3+(the Fenton reaction) | (3) |
|---|
Through analogous reactions H2O2 oxidizes and destroys the solvent-exposed iron-sulfur clusters of dehydratases such as aconitase, thereby blocking key pathways requisite for energy production and biosynthesis [13]. H2O2 also inactivates non-redox enzymes that employ a single ferrous iron atom as a substrate-activating cofactor [14], (Anjem and Imlay, in press). This basic catalytic strategy is used by enzymes that drive diverse reactions, from epimerizations to dehydrogenations, and therefore a wide variety of metabolic pathways are potentially vulnerable to H2O2 stress.
In vitro data reveal that the rate constants for the reaction of H2O2 with these iron centers can range from 1000–50,000 M−1 s−1. Such high values suggest that even sub-micromolar H2O2 can inactivate such enzymes within minutes—and indeed that is what is observed in bacterial mutants that cannot degrade H2O2. Thus the challenge for aerobic organisms is to keep H2O2 concentrations below these levels. They do so by expressing high titers of extremely active peroxidases and catalases. The levels of these scavenger enzymes in E. coli are so high that, despite the rapidity of endogenous H2O2 formation, the steady-state level of H2O2 is restricted to about 20 nM [15].
Flavoprotein autoxidation is not the only source of these oxidants; on occasion reactive oxygen species are generated by extracellular redox chemistry (Fig. 3). At anoxic/oxic interfaces, reduced metals and sulfur species encounter oxygenated waters, and the subsequent oxidation reactions generate environmental O2−· and H2O2 with which local bacteria must contend. The same oxidants are formed when UV/visible radiation illuminates extracellular chromophores, including pigments that are released by decaying vegetation. O2−· is a charged species and cannot cross bacterial membranes at physiological pH, but H2O2 penetrates lipid bilayers with a permeability coefficient similar to that of water [15, 16]. When environmental H2O2 concentrations exceed 0.2 μM, in fact, the rate of H2O2 influx into E. coli exceeds the rate of its endogenous formation and drives the steady-state intracellular concentration upwards [15]. Similar effects are expected when bacteria are exposed to the H2O2 that is excreted by lactic acid bacteria, which themselves eschew iron enzymes and, thus being relatively immune to H2O2, can use it to poison their competitors [17]. More famously, amoebae, plants, and mammals all douse invading bacteria with H2O2 as a way of suppressing infection [18–20]. Once captured in phagosomes, bacteria are exposed to ca. 5–10 μM H2O2, a concentration that is expected to elevate intracellular H2O2 levels by more than an order of magnitude [21]. A variety of bacteria and plants also release aromatic redox-cycling compounds; after infusion into bacteria, these compounds catalyze rapid electron transfer from flavoproteins to oxygen, thereby generating crippling doses of superoxide and H2O2 [22, 23]. The common theme is that the microbial world is a battlefield, and O2−·2 and H2O2 are weapons of choice.
Fig. 3.

Exogenous sources of H2O2 include H2O2-excreting microbes such as lactic acid bacteria, the NADPH oxidase responses of plants and macrophages, photochemically driven redox reactions, and chemical thiol/metal oxidations that occur at oxic-anoxic interfaces. Endogenous H2O2 is constantly formed by the adventitious oxidations of flavoenzymes. The H2O2 damages DNA through the Fenton reaction. It also disables dehydratases that contain iron-sulfur clusters and non-redox mononuclear enzymes that contain iron.
Scavenging enzymes are key defenses against these assaults, and when the rates of H2O2 formation or entry are high, the basal levels of defenses are inadequate. For this reason virtually all organisms respond to an infusion of H2O2 by raising the rate of synthesis of catalases, peroxidases, or both. These adaptive responses have been most thoroughly defined in bacteria and, among eukarya, in yeasts [24]. The OxyR protein is a transcription factor found in many bacteria, including the model bacterium E. coli. OxyR is largely inactive until H2O2 oxidizes a key cysteine residue, triggering the formation of a disulfide bond that locks the protein into an active conformation [25, 26]. The oxidized OxyR then directly binds to the promoter regions of members of the regulon, including genes encoding catalases and peroxidases, and recruits RNA polymerase. Other members of the E. coli regulon include an iron-storage protein that suppresses DNA damage by sequestering unincorporated iron [27–30], an iron-sulfur-cluster assembly system that repairs damaged iron-sulfur clusters [31–33], and a manganese importer that provides an alternative metal to mononuclear iron enzymes [14, 34, 35]. The OxyR protein is quickly oxidized by 0.1 μM H2O2 [15, 36], ensuring that the regulon is activated when intracellular H2O2 concentrations rise from the basal 20 nM level and begin to approach the 400 nM level at which disabilities become apparent.
Other bacteria use the PerR repressor as an alternative H2O2 sensor in place of OxyR [37, 38]. PerR customarily binds ferrous iron, and this metallated form blocks transcription of defensive enzymes; when H2O2 sweeps into the cell, the Fenton reaction leads to oxidation of the iron and the concerted oxidation of a histidine ligand, presumably irreversibly inactivating the protein. The transcription of regulon members ensues. The PerR regulon has been defined in detail in Bacillus subtilis; it again includes a peroxidase (Ahp), catalase (KatA), and iron-sequestering protein (MrgA), but an apparent zinc-import system stands in place of the manganese importer that is induced in E. coli [39].
Why so many scavengers of H2O2? General considerations
At first blush it is surprising and puzzling to discover that organisms encode so many enzymes that can degrade H2O2. In eukaryotes—and higher organisms especially—the presence of multiple scavengers might be ascribed to isoform-dependent compartmentalization within organelles or to tissue-specific expression patterns. This argument clearly does not hold for bacteria, thereby forcing us to look more closely. In the pages that follow we will see that this puzzle has not yet been solved. In considering each of the proposed scavenging enzymes, we will need to evaluate two things: whether its physiological job is indeed to degrade H2O2; and, if so, what special role the enzyme plays that the other scavengers do not fulfill.
The degradation of H2O2 can be achieved to some extent by any protein that displays exposed iron or thiol moieties, and so the demonstration of such an activity in vitro is not sufficient to conclude that the enzyme serves this purpose in vivo. How does one evaluate each case? The first argument arises from catalytic efficiency. As we will see, authentic catalases and peroxidases are extremely efficient, reacting with H2O2 at rates that exceed by orders of magnitude the reaction rates of typical iron chelates or cysteine residues. Regulation comprises a second piece of evidence: for example, the strong induction of the Ahp peroxidase in response to H2O2 stress connoted a role in H2O2 degradation rather than (as originally believed) in the scavenging of only organic hydroperoxides. Third, the genomic context of a structural gene sometimes implies its physiological purpose, since scavenging enzymes are often encoded by loci adjacent to those of other defensive enzymes, sometimes including the OxyR or PerR transcription factors themselves. Fourth, in some cases the absence of a proposed scavenging enzyme results in a characteristic sensitivity to H2O2. Such is not always the case, however, as diagnostic phenotypes are often obscured by the compensatory induction of other defensive systems. And finally, in some happy examples the presence or absence of a particular enzyme measurably affects the rate at which a specially constructed strain clears H2O2 from the growth medium.
We will see that uncontestable cases for authentic scavenging have been made for only a few enzymes: catalases and NADH peroxidase (Ahp). The evidence for other peroxidases ranges from compelling to dubious (Table 1).
Table 1.
Summary of biochemical and physiological evidences supporting H2O2 scavenging role of various enzymes.
| Enzymes | Biochemical evidence | Physiological evidences | |||
|---|---|---|---|---|---|
| Degrades H2O2 | H2O2 scavenging in growth medium | Impact on H2O2 toxicity | Expression under H2O2 stress | Growth phenotype (aeration) | |
| Monofunctional catalase | [56, 216] | [10] | Observed on treatment with mM doses of H2O2 | Induced | Masked by Ahp in most cases |
| Bifunctional catalase | [56–58] | [10] | Observed on treatment with mM doses of H2O2 | Induced | Masked by Ahp in most cases |
| Manganese catalase | [82, 83, 87, 91] | [92] | [92] | None | [217] |
| Alkyl hydroperoxide reductase | [104, 113] | [10] | [10, 137] | Induced | [10] |
| Bacterioferritin comigratory protein | [117, 118] | None | None | None | None |
| Thiol peroxidase | [128, 129, 133] | None | [136, 137] | [218] | None |
| Glutathione peroxidase | [152, 153] | None | None | None | None |
| Organic hydroperoxide reductase | None | None | None | None | None |
| Cytochrome c peroxidase | [175–182, 192–194] | 196 | [181] | None | [196] |
| Rubrerythrin | [205–208] | None | [209] | [218] | [219] |
| Reverse rubrerythrin | [204, 205] | None | [205] | [212–214, 218] | None |
Still, even by rigorous standards, E. coli expresses at least three authentic scavengers, two catalases plus the Ahp peroxidase—enough to pose the question of what circumstances might require more than a single scavenger. In the discussion that follows we will consider this issue for each candidate enzyme, but in general several possibilities have been considered (Table 2). First, the kinetic behaviors of catalases and peroxidases are distinct in ways that suit them to distinct doses of H2O2. Catalases operate through a disproportionation cycle that includes a reactive intermediate; this species is potentially problematic for cells, which might in general favor the use of peroxidases at low concentrations of H2O2. The latter enzymes, however, typically have a reductive step that is slow. They also are ultimately limited by the capacity of the cell to provide electrons. Both of these features cause peroxidases to be saturated by higher levels of H2O2, whereas catalases turn over at progressively higher rates.
Table 2.
Why multiple H2O2 scavenging enzymes? Some possible answers.
| Requirement (or not) for electron donors: e.g., peroxidases vs. catalases. |
|---|
| Distinct optima for H2O2 concentration: e.g., Ahp vs. catalase. |
| Reliance on the availability of different metals: e.g., heme-catalases vs. manganocatalases. |
| Compartmentalization: e.g., cytoplasmic Ahp vs. periplasmic cytochrome c peroxidase. |
| Stability of catalytic intermediate: e.g., Ahp vs. heme-catalase. |
| Substrate specificity: organic hydroperoxide reductase vs. Ahp. |
| Identity of the peroxidatic electron donor: e.g., Ahp vs. rubrerythrin. |
Other factors may also pertain. Catalases rely on iron or manganese, and some peroxidases depend on selenium; therefore when bacteria enter environments that lack one of these metals, it stands to reason that such enzymes might fail and a metal-independent enzyme might become necessary. Peroxidases can differ in the identities of their electron donors, and the ability of cells to provide various donors may depend upon metabolic circumstances, particularly if severe H2O2 stress or cell starvation disables some branches of metabolism. Some scavenging enzymes may be compartmentalized in the periplasm, a location that might enable them to degrade exogenous H2O2 before enters the cytoplasm, which houses all the known targets of damage. And some enzymes might trade catalytic efficiency for stability—an important issue in bacteria, which routinely experience protracted periods of starvation that preclude protein biosynthesis as a strategy to replace unstable enzymes.
Each of these reasons has been proposed to explain the particular role of one scavenging enzyme or another. As we will see, most of these ideas have not yet been well-tested by experiment.
Heme catalases
Most bacteria contain catalases, although streptococci, enterococci and leuconostocs do not. The nature of their cofactor distinguishes catalases into heme and non-heme (or manganese) catalases. All heme catalases catalyze the typical dispropornation reaction, while some exhibit an additional peroxidatic activity. Catalases with only catalatic activity are called monofunctional catalases, and those with both catalatic and peroxidatic activities are referred to as bifunctional catalases or catalase-peroxidases.
Monofunctional catalases are the earliest known bacterial catalases [40, 41] and are virtually ubiquitous among both aerobic and anaerobic bacteria. Bifunctional catalases are less abundant; as of November 2011, 435 catalase-peroxidase sequences were deposited in the peroxibase data set [42]. Crystal structures have been determined for the monofunctional catalases of Proteus mirabilis [43], E. coli [44, 45], Micrococcus lysodeikticus [46], Pseudomonas syringae [47], and Helicobacter pylori [48]. Catalase/peroxidase structures are available for the enzymes from Burkholderia pseudomallei [49], Mycobacterium tuberculosis [50], and Synechococcus PCC 7942 [51], and for the C-terminal domain of the E. coli enzyme [52]. These two classes differ in their overall 3D structures but share a characteristic deep burial of heme, which can be accessed by through a narrow channel [53]. The narrowness of the channel blocks the entry of molecules larger than H2O2 and thus provides substrate specificity. Thus catalases do not degrade organic hydroperoxides.
For all heme catalases the catalatic reaction requires two consecutive encounters with molecules of H2O2. In the first step, Fe3+-heme reduces H2O2, releasing water and forming a radical intermediate, compound I (Por˙+-Fe4+=O). Two electrons are removed from the heme in this process, one from the Fe3+ atom, which becomes Fe4+, and the second from the porphyrin ring of the heme, which acquires cation radical character:
| Enzyme(Por−Fe3+)+H2O2→Compound I(Por•+−Fe4+=O)+H2O | (4) |
|---|
In the second step, compound I oxidizes a second molecule of H2O2 and the enzyme returns to the original Fe3+ state:
| Compound I(Por•+−Fe4+=O)+H2O2→Enzyme(Por−Fe3+)+H2O+O2 | (5) |
|---|
Catalase is an exceptional enzyme in that its population inside a cell will, in principle, consist of an equimolar mixture of enzymes in the resting and highly oxidizing (compound I) states. The reactivity of the latter form is potentially problematic. Many monofunctional catalases bind NADPH, apparently providing a mechanism to discharge the porphyrin/ferryl radicals when H2O2 is scant [54]. The failure to do so is believed to enable the compound I species to abstract electrons from nearby polypeptide, ultimately leading to enzyme inactivation. However, the NADPH-free enzyme remains active when H2O2 is provided as a physiological reductant for compound I. Not all monofunctional catalases bind NADPH, however, leaving open the question of how these enzymes cope with the compound I species.
Catalase-peroxidases also exhibit a low-level peroxidatic activity: exogenous electron donors can reduce the compound I species in two sequential one electron steps back to the Por-Fe3+ state:
Compound I(Por•+−Fe4+=O)AH→Compound II(Por−Fe4+−OH)+A(ox)
Compound II(Por−Fe4+−OH)+AH→Enzyme(Por−Fe3+)+H2O+A(ox)
This activity is low (ca. 1% the kcat of the catalase activity) and thus seems unlikely to be an important contributor to H2O2 degradation. However, the activity could conceivably provide a mechanism that discharges the compound I species when H2O2 levels decline. Access to the heme site is provided by a channel that is distinct from the path of H2O2 entry. A wide range of biological and artificial reductants can be used, and so the physiological electron donor (if any) remains unknown.
The apparent Km and kcat values of monofunctional catalases are in the range of 40–600 mM and 54000–83300 sec-1 [55], whereas those of characterized catalase-peroxidases are 3.7–8 mM and 3500–6000 sec-1, respectively [56–58]. Thus the catalytic efficiencies (kcat / Km) of both catalases are in the range of ~106 M-1 sec-1; this is the pertinent parameter, as physiological concentrations of H2O2 are in the low-micromolar range, far below the KM of either enzyme. One infers then that the two enzymes are equally proficient in degrading H2O2, and one must look elsewhere to understand why nature maintains two distinct enzyme families.
Physiological role
When bacteria are exposed to millimolar doses of H2O2, they require catalase to degrade it—mutants cannot do so. This observation initially led to the incorrect conclusion that catalases are the primary scavengers of H2O2, with peroxidases playing only a secondary role. What we have learned more recently, however, is that peroxidases are often the primary scavengers when the dose of H2O2 is in the low-micromolar range [10]—the range that is most commonly found in nature. Catalase activity predominates only at higher doses, when peroxidases are saturated due to the slowness of electron delivery and/or inactivation by over-oxidation. (See below.) Thus, in phenotypic studies catalase mutants often exhibit no apparent defects unless millimolar levels of H2O2 are imposed; at lower doses the mutants behave like wild-type cells. Catalases may also be the primary scavengers when bacteria are starved for carbon sources and thereby cannot provide reductants to their peroxidases. Indeed, catalase is commonly induced when cells enter stationary phase. A special catalase is expressed in Bacilli spores, which are metabolically inert, presumably for the same reason [59].
Many bacteria contain more than one catalase isozyme. Bifunctional catalases seem to be the preferred enzyme during exponential growth, and they are usually the enzymes that are induced when the OxyR or PerR systems detect environmental H2O2 [60–65]. In contrast, monofunctional enzymes are induced upon entry into stationary phase by stationary-phase-specific sigma factors [59, 66]. At this point the rationale for this arrangement is a matter of conjecture. Perhaps the bifunctional catalases rely on cellular reductants to revive them from the compound I state when H2O2 levels dwindle; because these reductants are less available in non-growing cells, the monofunctional enzymes are induced in their place. Perhaps monofunctional enzymes are more structurally stable and persist during the long periods of stasis that bacteria must periodically endure.
Aside from the spore example, the requirement for multiple isozymes typically does not arise from compartmentalization, as catalase is usually a cytoplasmic protein. However, data suggest that bifunctional catalases are secreted to the periplasms of some pathogenic bacteria, including enterohaemorrhagic E. coli O157: H7 [67], Brucella abortus [68], Pseudomonas syringae [69], Legionella pneumophila [70], Vibrio fischeri [71], and Helicobacter pylori [72]. The N-terminal amino acid sequencing of KatP of E. coli O157:H7 and KatA of B. abortus indicated the presence of a signal peptide sequence, whereas in other cases signal peptide sequences were absent. Interestingly, these bacteria typically also contain cytoplasmic catalases. Such dual localization potentially offers an advantage to bacteria that must survive phagosomal environments: the presence of scavenging systems in both periplasm and cytoplasm would comprise a two-stage system that might protect cytoplasmic targets from exogenous H2O2 more effectively than would a similar amount of catalase located entirely within the cytoplasm. The arrangement could also provide a communal benefit to bacteria that live within clonal biofilms. At this point the physiological significance of the periplasmic catalase in the pathogenic bacteria has not been demonstrated. More than one hundred years after their discovery, the precise roles of catalases remain unclear.
The role of bifunctional catalase in isoniazid action
The standard treatment for tuberculosis involves a multidrug regime consisting of isoniazid (INH), ethambutol, rifampin and pyrazinamide. Of these, INH is quite effective against slow growing mycobacterial species and targets the mycolic acid biosynthetic pathway of bacteria [73–75]. Mycolic acids are long chain α-alkyl β-hydroxy fatty acids and are an integral component of mycobacterial cell wall.
In recent years INH-resistant Mtb strains have been isolated from patients with increasing frequency, and in at least half the cases the resistance arises from mutations in katG [76]. This finding has been recently explained. INH is administered into patients as a prodrug, which is activated by KatG inside mycobacteria. The compound I form of KatG removes the hydrazine group from INH and forms a isonicotinyl radical, which is later added to the NAD+ to generate isonicotinyl-NAD radical [77]. Interestingly, studies of mutant enzyme have suggested that the peroxidase activity of KatG may not be necessary for drug activation. The activated form of INH, INNAD, acts as an NADH analogue to inhibit enoyl-ACP reductase, an enzyme in the mycolic acid biosynthetic pathway [78, 79]. The observation lends credence to the notion that Compound I may act indiscriminately as cellular oxidant—which might justify the fact that it is maintained at a relatively low titer, ceding the responsibility for H2O2 clearance to peroxidases, until H2O2 levels rise to the point at which peroxidases might be overwhelmed.
Manganese catalase
In a survey of pediococci, lactobacilli, leuconostocs, streptococci, and aerococci, Whittenbury observed that some bacteria exhibit an azide- and cyanide-resistant catalase activity [80]. This catalase was purified from the Pediococcus that had been grown in the absence of any heme source [81] and later from Lactobacillus plantarum [82]. Biochemical characterization and atomic absorption spectroscopy showed that the protein contained manganese atoms instead of heme as a cofactor [82, 83]. The absence of heme led to the initial use of the name “pseudocatalase” [82]; however, it soon became clear that the catalase activity of the protein is neither artifactual nor adventitious.
Manganese catalases from Lactobacillus plantarum [82], Thermus theromophilus [84], Theromoleophilum album [83], Thermus sp. [85], Salmonella enterica [86] and an archeaon, Pyrobaculum caldifontis [87] have been isolated and characterized, and crystal structure data are available for the L. plantarum and T. thermophilus enzymes [88, 89]. Each subunit contains two manganese atoms [90]. After the fashion of monofunctional heme catalases, access to the dimanganese active site is through a narrow channel that likely provides specificity to a small substrate like H2O2.
The reaction of manganese catalase, like that of heme catalases, occurs in two steps:
2Mn2++H2O2+2H+→2Mn3++2H2O
The kinetic constants of manganese catalases purified from L. plantarum, T. album, T. thermophilus and P. calidifontis have been determined: the Km values for H2O2 range between 15–170 mM, which resembles those of heme catalases, while the kcat/KM values are in the range of 2–10 × 105 M−1 s−1, which is an order of magnitude lower [82, 83, 87, 91].
The authenticity of manganese catalase as a functional scavenger of H2O2 was first indicated by an inverse correlation between enzyme content and the accumulation of H2O2 in aerobic varieties of L. plantarum [82]. The deletion of _katN_—the gene encoding manganese catalase—from Salmonella enterica serovar Typhimurium had no effect on survival of H2O2 treated cells [92]; however, these bacteria contain both heme-catalases and peroxidases, and it is likely that the activities of these enzymes masked the impact of manganese catalase. Overexpression of katN, however, protected the rpoS mutants, which fail to induce the stationary-phase heme catalase and which otherwise showed sensitivity to H2O2 [92]. These data affirm the biochemical activity of manganese catalase in vivo, but they do not explain its particular role.
Manganese catalases are not as widespread as the heme-catalases, with genes found thus far in only 100 bacteria (Peroxibase database, November, 2011) [42]. The isozyme is exclusively found in bacteria, and it is especially common among cyanobacteria. In a naturally manganese-rich bacterium, L. plantarum, high manganese catalase activity was only observed in a medium devoid of any heme source [93], suggesting that its raison d'etre is to serve as a catalase when the more-efficient heme-dependent enzymes cannot be activated. In S. enterica, enzyme expression is induced by the stationary-phase sigma factor[92], presumably as an alternative to the stationary-phase (KatE) heme catalase. Interestingly, manganese importers are strongly induced in enteric bacteria when iron levels fall, due to deactivation of the Fur:Fe transcription repressor. Through this mechanism manganese is imported to activate manganese-specific isozymes of enzymes whose housekeeping form requires iron. The iron-dependent ribonucleotide reductase (NrdAB) is replaced by a manganese enzyme (NrdEF), while the iron-dependent superoxide dismutase (SodB) is replaced by the manganese isozyme (SodA); both of the manganese enzymes, like the manganese transporter itself, are normally repressed by Fur [34, 94–96]. One might speculate that katN is regulated in an analogous manner—which would validate the notion that it is the low-iron catalase.
Thiol peroxidases (peroxiredoxins)
Peroxidases fall into two categories: thiol-based peroxidases and non-thiol peroxidases. The latter are also known as peroxiredoxins. All peroxiredoxins contain a conserved peroxidatic cysteine that reacts with H2O2 or organic hydroperoxides and forms a cysteine sulfenic acid. The sulfenic acid residue subsequently reacts with another thiol (intra- or intermolecularly) and forms a disulfide before getting reduced back to free thiol. Variations in the mechanism by which thiols are regenerated and in the architecture of the protein allow peroxiredoxins to be further classified into four groups: alkylhydroperoxide reductases (AhpCF), thiol peroxidases (Tpx), bacterioferritin comigratory protein (BCP), and glutathione peroxidase (Gpx). These groups will each be considered in turn in the sections that follow.
The recognition of thiol chemistry in H2O2 scavenging came quite late. A eukaryotic non-heme H2O2 scavenging enzyme, glutathione peroxidase, was discovered in 1957 but did not get much attention until the 1970s. In 1988 a thiol-specific antioxidant, TSA, was first identified in yeast as an enzyme that could protect glutamine synthetase from metal-catalyzed oxidation during dithiothreitol/Fe2+/O2 exposure [97]. TSA functioned in cell extracts only because it was able to employ dithiothreitol as an artificial electron donor—a harbinger of the uncertainty that is attached to many of the peroxidases that have been identified through biochemical approaches. A year later a thiol peroxidase, AhpC, was isolated from Salmonella typhimurium and was characterized as a member of the OxyR H2O2-stress regulon [98]. Identifications of other peroxiredoxins have followed; these enzymes appear to be more prevalent than catalase, being expressed in almost all bacteria. The few exceptions include Streptococcus pneumoniae, Rhodobacter capsulatus, Rhodobacter sphaeroides, Borrelia spp., and Bartonella spp.
The reaction of H2O2 with free cysteine has a rate constant of 2 M−1 s−1 at neutral pH [99], whereas a cysteine-based peroxidase reacts with H2O2 with a rate constant of ~108 M−1 s−1 [100]. A minor component of the ~106 fold difference in reactivity arises from local polypeptide context, which ensures deprotonation of the active-site thiol to the reactive thiolate form. Still, deprotonated free cysteine reacts with H2O2 at a rate of only 20 M−1 s−1. The mechanism by which peroxidases promote thiol reactivity remains fundamentally unclear. In other enzymes whose reactivity depends upon the nucleophilicity of an active site thiol that residue exhibits reaction rates with H2O2 no higher than 102 M−1 s−1 [101]. Therefore the basis of the high reaction rates of thiol peroxidases remains a key mechanistic question.
Alkylhydroperoxide reductase (Ahp)
The alkylhydroperoxide reductase system consists of two cytoplasmic proteins (Fig. 4). AhpC and AhpF were first discovered in Salmonella typhimurium [102], but homologues are found throughout the aerobic and anaerobic biota. AhpC contains two conserved cysteines, Cys46 and Cys165 (numbering based on AhpC from S. typhimurium). Cys46 is present in the N-terminal region and lies in a VCP motif, which also remains conserved in AhpC and other proposed thiol-based peroxidases. Mutation of Cys46 to serine completely abolishes the peroxidase activity, whereas C165S mutation retains ~60% activity, suggesting that Cys46 is the peroxidatic cysteine [103]. Cys46 has been shown by NBD-Cl assay to form a cysteine sulfenic acid on reaction with peroxide substrate [104]. The sulfenic acid formation causes an unfolding of ~10 residues in the loop region of active site and brings Cys46 closer to the Cys 165 of the other subunit in the head-to-tail dimer of AhpC, permitting the formation of an intermolecular disulfide bond [105, 106]. Disulfide bond formation was confirmed by the migration pattern of oxidized AhpC on a non-reducing SDS-PAGE [104]. A conserved arginine residue (Arg132 in S. typhimurium) lies nearby in the three-dimensional structure and is proposed to stabilize the thiolate anion of Cys46; mutation of the arginine codon abolishes activity [107].
Fig. 4.

The catalytic cycle of alkylhydroperoxide reductase (Ahp).
AhpF is a flavoprotein with NADH:disulfide oxidoreductase activity; it restores the disulfide in AhpC to its reduced form [102, 103]. In almost all cases ahpF lies adjacent to ahpC on the chromosome and is co-transcribed. AhpF probably evolved from thioredoxin reductase or a common ancestor, and of course it functionally resembles them in reducing disulfide bonds. It contains three domains: an FAD-binding domain, an NADH-binding domain with a thiol/disulfide redox active center, and a thioredoxin-like domain [108]. Electron movement within AhpF occurs from NADH to FAD, from FAD to a C345/C348 thiol/disulfide center, and from those thiols to the N-terminal redox-active center (C129/C132) [109]. Mutational analysis of the N-terminal cysteines has demonstrated that electron transfer from AhpF to AhpC occurs via disulfide exchange reactions between AhpF C129/C132 and AhpC C46/C165.
In bacteria such as H. pylori and M. tuberculosis, AhpC is present but its reducing partner, AhpF is absent [110, 111]. The H. pylori AhpC is reduced by a thioredoxin/thioredoxin reductase system [111], whereas AhpD reduces AhpC in mycobacterial species [112].
The physical arrangement of AhpC and AhpF was unexpected. Reduced AhpC assembles in vitro as a head-to-tail homodimer, but upon oxidation it regroups into a decameric [(α2)5] complex. AhpF—which is less abundant in the cell—must then diffuse to AhpC and reduce it during each turnover event. This arrangement raises important questions about the rate-limiting step in vivo, and on a practical level it posed hurdles to workers who sought to measure the catalytic efficiency of the system in vitro. Purified AhpC exhibited a Km of 1.4 μM for H2O2 and a catalytic efficiency (kcat/Km) of 3.7 × 107 M−1 s−1 [113]. That value indicates that AhpC is the equal, if not the superior, of catalase when physiological levels of H2O2 pertain. While AhpC can also react with various organic hydroperoxides, it does so with a much higher KM (~240 and 110 μM, respectively. for t-butyl hydroperoxide and cumene hydroperoxide) and a kcat/Km that is two orders of magnitude lower than that for H2O2. Thus the kinetic data suggest that H2O2 per se is the physiological substrate of AhpC. Indeed, some bacteria that possess AhpCF also express a distinct peroxidase (Ohr) that operates most efficiently on organic hydroperoxide species. The significance of this finding lies in the fact that AhpCF system was originally discovered as an organic hydroperoxide scavenger in S. typhimurium, which led to its initial naming as an alkylhydroperoxide reductase [102]. Thus its name is a misnomer.
Physiological role
The presence of AhpC has been documented in ~3000 bacterial species. Its role in scavenging H2O2 became apparent when it was shown that both ahpCF and katE/katG must be deleted to abolish H2O2 scavenging in E. coli (Fig. 5). Further, the fact that Ahp is the primary scavenger was evidenced by the fact that in its absence, the OxyR regulon is induced by endogenous H2O2 [10]. In contrast, OxyR remains off in katG katE mutants. Kinetic studies showed that whole cells containing AhpC were able to rapidly degrade low doses of H2O2 (< 20 μM), but at H2O2 doses above this value the Ahp system was saturated—whereas catalase activity was not saturated even at millimolar concentrations of H2O2 (Fig. 6). Therefore, AhpC can be proposed to act as H2O2 scavenger at low concentrations of H2O2 whereas catalase can be effective at high doses of H2O2.
Fig. 5.

H2O2 decomposition by aerobic E. coli depends upon catalase and alkylhydroperoxide reductase (Ahp). H2O2 (1.5 μM) was added to a dilute suspension of cells at time zero, and the amount of remaining extracellular H2O2 was subsequently determined at intervals. Note that for the wild-type strain and the single mutants, the rate-limiting step in H2O2 degradation was diffusion into the cell; consequently, the rates of H2O2 clearance from the medium were equivalent. Measurements were made as described [10].
Fig. 6.


Decomposition of exogenous H2O2 by E. coli Ahp+ Kat− mutants (solid line) or Ahp− Kat+ mutants (smooth line). Alkylhydroperoxide reductase (Ahp) is effective at low H2O2 concentrations, but catalase is the more effective enzyme at high concentrations. Note the difference in scale between panels A and B. Measurements were made as discussed in Fig. 5.
The fact that AhpC and AhpF associate and dissociate during every catalytic cycle may introduce a slow step that is responsible for the apparent saturation of the enzyme, and so at first blush this arrangement seems counterproductive. Further consideration suggests otherwise. Aerobically growing E. coli generates internal H2O2 at a rate of about 10 μM/sec [10]. The AhpC concentration itself is about 10 μM—which means that each AhpC subunit needs to react only about once per second. This turnover rate is very low and probably is not limited by the AhpF binding and dissociation steps. Yet the steady-state concentration of intracellular H2O2 depends upon its lifetime—that is, upon the concentration and reactivity of AhpC. Thus the threat of oxidative damage places great evolutionary selection upon the first step of the AhpC reaction, but very little pressure upon the AhpF-driven recycling process. Thus AhpF can be relatively low in concentration, with a few molecules servicing an abundance of AhpC [10].
In the initial studies, the reactivity of AhpC with H2O2 was overlooked because the millimolar concentrations of H2O2 that were used in those experiments immediately inactivated the enzyme. The inactivation apparently occurs when H2O2 further oxidizes the nascent sulfenic acid before it can condense with the resolving cysteine; the consequence is a sulfinic (RSOOH) species that cannot be reduced. This event may not be accidental: if it occurs in vivo—which is not yet established—the effect would be to turn off an NADH-exhausting enzyme whenever H2O2 concentrations become very high. Under such circumstances, peroxidase action may be futile, and catalase may be the preferred path of scavenging, since at high doses of H2O2 this enzyme is kinetically superior to Ahp anyway, and it does not deplete a key cell energy source.
Membrane permeability and the effectiveness of H2O2 scavengers
Because Ahp and catalase were identified as the primary scavenging enzymes of E. coli, it is appropriate to pause and consider a basic question that was considered for this bacterium: Do these scavengers work? In the 1980s workers were perplexed to realize that because H2O2 crosses membranes relatively quickly, the basal catalase activity of E. coli is too low to prevent the equilibration of H2O2 across the cell membrane. That is, if catalase were the sole scavenger of H2O2, then when cells are confronted with external H2O2, it will flow so quickly across membranes that catalase will be unable to keep up, and the intracellular concentration will rise to virtually match that outside the cell. Catalase, they inferred, serves only as a sink that ultimately depletes H2O2 from the environment by mass action [114]. This idea was startling, as it suggested that catalase was “communal” defense that altruistically served the entire microbial community by steadily degrading all the H2O2 in the local habitat. This arrangement might make sense for clonal biofilms, but it was hard to understand why open-sea bacteria, for instance, would attempt this, or why such a system would be selected in bacteria (like E. coli) that live as minority organisms in a planktonic population.
However, knowing now that E. coli catalase is the primary scavenger only when H2O2 is abundant—a situation which triggers catalase induction to ten-fold higher levels—then one must reconsider. Both calculations and experiments (Fig. 5) show that under inducing circumstances the catalatic activity becomes high enough to outstrip H2O2 entry into the cell, so that the intracellular H2O2 concentration is held lower than that in the external environment [15]. Thus catalase serves to protect the specific cell that contains it. Furthermore, at low concentrations of H2O2, the same can be said of Ahp, even without induction. Calculations show that when E. coli enters a habitat containing micromolar H2O2, Ahp drives the intracellular H2O2 concentration at least four-fold lower than that outside the cell. A proportionately larger gradient is expected when Ahp is induced. In sum, despite the ability of H2O2 to penetrate membranes, these scavenging activities are sufficiently high to substantially shield the cytoplasm from external stress. This fact neatly fits the observation that all the H2O2-sensitive iron-sulfur dehydratases and mononuclear iron enzymes are compartmentalized in the cytoplasm, rather than in the unprotected periplasm.
Bacterioferritin comigratory protein (BCP)
Other peroxidases have also been identified, but our understanding is much less complete. BCP is a model for a set of proteins that exhibit thiol-dependent peroxidase activities in vitro but whose in vivo function is much less clear. BCP was originally discovered in E. coli as a protein that migrates with bacterioferritin on SDS gels [115, 116]. The primary sequence suggested a resemblance to AhpC, and biochemical assays confirmed its ability to react with H2O2. BCP homologues are present in both Gram-positive and –negative bacteria.
The N-terminal cysteine (Cys48 in Xanthomonas campestris BCP) acts as the peroxidatic thiolate, and in BCPs with two conserved cysteines Cys84 is a resolving cysteine that enables the formation of a disulfide bond. The loss of either residue eliminates activity [117, 118]. Crystal structures of the reduced and oxidized proteins are available for the BCPs of X. campestris [118] and Aeropyrum pernix (2CX4 released in 2005 by the RIKEN Structural genomics/Proteomics Initiative).
Unlike for Ahp, a dedicated mechanism for BCP reduction has not been identified. Catalytic turnover with H2O2 and organic hydroperoxides has been achieved using both biological (thioredoxin and glutathione/glutaredoxin) and artificial (DTT) electron donors [118–121]. The thioredoxin system has been proposed to be the most likely source of electrons in vivo (Fig. 7), but the absence of a clear phenotype has not allowed this proposal to be tested.
Fig. 7.

Proposed electron transfer pathway of the thiol-based peroxidases: bacterioferritin comigratory protein (BCP), thiol peroxidase (Tpx), and glutathione peroxidase (Gpx). Prx in the figure is a general representation of all three thiol based peroxidases. Trx and TrxR represents thioredoxin and thioredoxin reductase, respectively.
So is BCP an authentic scavenger of peroxides? Although BCP is well-expressed in exponentially growing E. coli, strains that lack Ahp and catalases do not degrade H2O2 at a significant rate, suggesting that BCP must have some other function. Recombinant E. coli BCP reacts with t-butylhydroperoxide (KM = 40 μM) and linoleic hydroperoxide (KM = 10 μM) as well as with H2O2 (_K_m = 50 μM), raising the prospect that the reduction of an organic hydroperoxide might be its actual role [122]. It should be noted, however, that while lipid peroxidation is well-established in eukaryotic organisms, biological sources of organic hydroperoxides are not known in bacteria.
Neither phenotypic nor regulatory studies shed much light. The bcp mutants of Helicobacter pylori, Campylobacter jejuni, and Porphyromonas gingivalis showed eventual impairment of cell viability upon aeration as well as sensitivity to cumene hydroperoxide, but they did not display sensitivity to H2O2 [119, 120, 123, 124]. The latter result should perhaps be viewed with caution, since excess H2O2 can able to overwhelm and inactivate Ahp, an authentic peroxidase, thus obviating any phenotype. In these studies Ahp exerted a much larger effect than BCP upon markers of oxidative stress, including protein carbonylation, intracellular free iron, and 8-oxoG DNA lesions.
The expression of bcp increased when Desulfovibrio vulgaris Hildenborough and E. coli were aerated [122, 125]. However, there is no evidence of a response to H2O2 or other oxidants; it is not regulated by either OxyR or PerR. The genomic context varies among bacterial species and does not provide insight into the function of the protein [124, 126]. Thus in the absence of a compelling phenotype or regulatory pattern, the physiological role of BCP remains mysterious.
Thiol Peroxidase (Tpx)
Thiol peroxidase was discovered as a factor in Saccharomyces cerevisiae cell lysates that could protect glutamine synthetase from H2O2 [127]. This activity depended upon a thiol-reducing agent, and in the early literature Tpx was denoted thiol-specific antioxidant (TSA) [97]. Subsequently, Cha et al. isolated and characterized a homologous protein from periplasmic fractions of E. coli [128]. The E. coli enzyme was initially designated as p20 and later as thiol peroxidase.
Like Ahp, thiol peroxidase contains a conserved cysteine residue in its N-terminal region; however, they are not homologs at the sequence level. Thiol peroxidases from various bacteria show conservation of cysteine 61 and cysteine 95 (numbering based on E. coli thiol peroxidase), and upon exposure to H2O2 these residues form an intramolecular disulfide bond [129]. Mutations in Cys61 eliminate its peroxidatic activity [129, 130]. Crystal structures have been solved in both reduced and oxidized state [131, 132].
The physiological reductant of Tpx is unknown. In vitro peroxidase activity of Tpx with H2O2, t-butylhydroperoxide, cumene hydoperoxide, and linoleic acid hydroperoxide has been demonstrated using dithiothreitol as an electron donor [133]. In a thioredoxin/thioredoxin reductase system [129], the kcat/KM values were determined to be 4 × 106 M−1 and 7.7 × 106 M−1 sec−1 for H2O2 and cumene hydroperoxide, respectively [129], favoring the notion that it might serve to scavenge organic hydroperoxides. Indeed, the peroxidatic cysteine sits at the bottom of a cleft that is lined with hydrophobic residues [132]. These kcat/KM numbers are much lower than those cited for Ahp (107–108 M−1 s−1), but a valid comparison awaits an experimental system in which the reductive step is not rate-limiting.
The cellular localization of thiol peroxidases has been uncertain. Because Tpx was first isolated from E. coli after osmotic shock, it seemed likely to be a periplasmic protein [128], and as such it might appear to have a function that was discrete from that of the cytoplasmic Ahp system. However, thiol peroxidases from E. coli and other bacteria do not exhibit N-terminal signal sequences when analyzed by SignalP 4.0 software, and other workers subsequently identified E. coli thiol peroxidase in cytoplasmic fractions [120, 134]. The presence of tpx homologs in Gram-positive bacteria further supports its cytoplasmic localization.
E. coli, Campylobacter jejuni, Helicobacter pylori and Enterococcus faecalis tpx mutants did not show any phenotype under aerobic growth conditions [120, 135–137], while a modest sensitivity to organic hydroperoxides was observed in anaerobic cultures of E. coli. The H. pylori and E. faecalis tpx mutants were sensitive to exogenous H2O2 and cumene hydroperoxide [136, 137]. These experiments were conducted with doses of oxidants that exceed natural environmental doses by orders of magnitude, so the relevance of these phenotypes is uncertain.
Thiol peroxidase concentration is modestly (2–3-fold) induced by aeration in E. coli [138, 139]. There is no evidence of control by reactive oxygen species. In E. faecalis, tpx expression is not induced by H2O2 treatment [137]; however, during H2O2 stress, expression was ~20 fold downregulated in a mutant lacking the oxidative stress regulator, HypR, suggesting that tpx expression is high even in the absence of H2O2 stress and is positively regulated by HypR. Purified HypR bound the tpx promoter in a gel-shift assay.
In sum, the state of knowledge of Tpx function is similar to that of BCP: enzyme activity can be demonstrated in vitro, but even modest phenotypes can be elicited in vivo only under unnaturally stressful conditions. Neither the expression pattern nor genomic context implies its physiological role. Finally, Ahp and/or catalase are the predominant scavengers of organic peroxide and H2O2 itself, which leaves room for specialization only in a particular circumstance when the other enzymes are non-functional or with a particular substrate that has not yet been identified.
Glutathione peroxidase (Gpx)
Glutathione peroxidase was first isolated in 1957 from lysates of human erythrocytes; it comprised an activity that employed glutathione to prevent the oxidation of hemoglobin [140]. This effect arises from its ability to reduce hydroperoxides to H2O (or the corresponding alcohols), using glutathione as an electron donor in the majority of cases:
Oxidized glutathione is then recycled by glutathione reductase. Gpx occurs in most tissues from higher organisms, and it is also prevalent in eukaryotic microbes, including yeasts. In these organisms it appears to be the primary scavenger that operates at low concentrations of H2O2; in this respect it is functionally analogous to the prokaryotic Ahp. Its distinguishing characteristic is that it features selenocysteine, rather than cysteine, as the peroxidatic residue [141]. Selenium has similar redox potential and electronegativity as sulfur; however, the rate constant of Gpx with H2O2 is ~ 5 orders of magnitude higher with selenocysteine than cysteine. The basis of the superior reactivity of selenocysteine is not completely clear. Their reduction potentials and electronegativity are similar, although selenium has slightly higher nucleophilicity [142] and lower pKa than does sulfur [143, 144].
Glutathione peroxidase orthologues have been predicted in bacteria based on their sequence homology to the eukaryotic enzymes (Fig. 8). Multiple sequence alignments show extensive homology and conservation of Cys37, Gln70, and Trp145 (numbers based on E. coli glutathione peroxidase homolog, BtuE), which constitute the catalytic triad [145]. However, the bacterial enzymes contain sulfur instead of selenium, a feature that is rare but not unknown among the eukaryotic Gpxs [146–149].
Fig. 8.

Multiple sequence alignment showing conservation of glutathione peroxidase (Gpx) active-site residues (yellow) within various bacteria (Ec: E. coli; Se: Salmonella enteric serovar Typhimurium; Pa: Pseudomonas aeruginosa; Bs: Bacillus subtilis 168; Nm: Neisseria meningitides; Sy: Synechocystis sp.; Bj: Bradyrhizobium japonicum; Rs: Rhodobacter sphaeroides; Bf: Bacteroides fragilis) and human glutathione peroxidase I (Hs). The presence of selenocysteine in place of cysteine in human Gpx 1 has been marked by an arrow. Other conserved residues are highlighted in green.
Is the bacterial enzyme an authentic peroxidase? Peroxidase activity has been demonstrated in vitro in the case of E. coli BtuE [150] with H2O2, t-butyl hydroperoxide, cumene hydroperoxide and linoleic acid hydroperoxide as functional substrates. The enzyme exhibited activity with thioredoxin as well as glutathione, a trait that has been observed with some eukaryotic glutathione peroxidases and that might explain the presence of the enzyme in bacteria that lack the glutathione biosynthetic pathway [151]. The kcat/Km for H2O2 in case of eukaryotic glutathione peroxidases is 106−107 M−1 s−1 [151], but that of the bacterial homologues has not been reported.
Deletion of homologs from Neisseria meningitides and E. coli does not create sensitivity to H2O2, although the mutants are more sensitive to paraquat and tellurite, which both have H2O2 stress among their sequelae [152, 153]. The E. coli gene (btuE) is not regulated like a H2O2 scavenger: unlike catalase and Ahp, it is not induced during H2O2 stress [152, 153]. More strikingly, even overproduced BtuE does not confer detectable H2O2-scavenging capacity upon a strain that lacks these other enzymes (unpublished data). Its genomic context is not conserved and does not imply its function [154, 155]. So, in broad strokes, the bacterial Gpx homologs resemble Bcp and Tpx in that they evidence peroxide-scavenging activity in vitro while standard indicators do not suggest that this is their proper function in vivo.
Organic hydroperoxide resistance protein (Ohr)
Organic hydroperoxides (ROOH) can be produced either by the lipoxygenase- or cyclooxygenase-catalyzed oxidation of linoleic acid and arachidnoic acid or by reactions between polyunsaturated fatty acids and free radicals [156]. The subsequent reaction of ROOH with intracellular iron can trigger radical chain reactions that are thousands of events long, generating patches of lipid peroxides that disrupt membrane function and produce collateral damage to proteins and nucleic acids [157, 158]. For this reason eukaryotic cells express peroxidases that specifically degrade lipid peroxides to unreactive alcohols.
It is not immediately clear that bacteria require analogous enzymes, since bacterial synthesize only saturated or monounsaturated fatty acids, neither of which are vulnerable to peroxidation chains. However, many bacteria will take up exogenous polyunsaturated fatty acids if they are presented in the culture medium [159], which suggests that bacteria growing among eukaryotic organisms may incorporate peroxidizable lipids into their cell membranes. Indeed, polyunsaturated lipids were observed in marine bacteria that had been recovered from the intestinal tract of deep-sea subsurface fishes [160].
Thus lipid hydroperoxides seem to be the organic peroxide species most likely to be problematic for bacteria—and a resistance mechanism against them has been found. The _o_rganic _h_ydroperoxide _r_esistance (Ohr) protein [161] exhibits peroxidatic activity that is much more substantial against model organic peroxides than against H2O2 itself [162]. The enzyme was first identified when a Xanthomonas campestris plasmid library was screened for genes imparting resistance to t-butyl hydroperoxides in an E. coliahpCF mutant [161]. Subsequent studies demonstrated that the ohr mutants of X. campestris, A. tumefaciens and P. aeruginosa show normal resistance to H2O2 but are unusually sensitive to t-butyl hydroperoxide and cumene hydroperoxide [161, 163, 164]. Thus the genetic evidence strongly supports the idea that Ohr is the primary scavenger of organic peroxides in these organisms. Peroxidatic activity was demonstrated in vitro using dithiothreitol as an electron donor.
Sequence alignments indicate the conservation of Cys60 and Cys125 (numbering based on X. campestris Ohr). Cys60 has been proposed to be the peroxidatic cysteine. Crystal structures of the dimeric protein indicate that the cysteine residues sit in a cleft at the dimer interface [165–167]; the enzyme structure is compatible with a preference for substrates with hydrophobic side chains. Indeed, the KM values for t-butylhydroperoxide and cumene hydroperoxide were in the low-micromolar range, whereas those for H2O2 were far higher [168, 169].
Coprecipitation experiments found an interaction of X. fastidiosa Ohr with lipoic acid. On this basis it seemed possible that the physiological donor of reducing equivalents might actually be a lipoylated protein such as the dihydrolipoamide dehydrogenase (Lpd) subunits of the α-ketoglutarate dehydrogenase and pyruvate dehydrogenase complexes. Recombinant Lpd from X. fastidiosa supported the NADH dependent peroxidase activity of Ohr but not of AhpC. Lpd is a flavoprotein disulfide reductase with an NADH binding domain. Based on these studies, a physiological electron transfer pathway has been proposed (Fig. 9).
Fig. 9.

Proposed electron-transfer pathway for reduction of organic hydroperoxides by organic hydroperoxide reductase (Ohr). LpdA represents the lipoylated disulfide-based enzymes that reduce oxidized Ohr.
The expression level of ohr increased ~15 fold upon treatment of X. campestris with t-butyl hydroperoxide [161] and of P. aeruginosa, A. tumefaciens, and Sinorhizobium meliloti with cumene hydroperoxide [163, 164, 170]. This effect depends on a transcription factor, OhrR, that is encoded immediately adjacent to ohr [171, 172]. OhrR does not respond to H2O2. Conversely, the same bacteria express H2O2-inducible Ahp and/or catalases, underscoring the apparent specificity of Ohr for organic peroxides.
Finally, X. campestris and A. tumefaciensohr mutants degrade cumene hydroperoxides in lab media at only 15–20% the rate of wild-type cells [163, 173]. Thus the case that Ohr is a dedicated scavenger of organic hydroperoxides is convincing. What remains is to identify the organic species that is its natural substrate.
Non-thiol peroxidases
Cytochrome c peroxidase
Cytochrome c peroxidases (CCPs) are heme enzymes that catalyze the two-electron reduction of H2O2 to water by accepting electrons from a soluble cytochrome c. The first CCP was discovered in baker's yeast [174]; like other eukaryotic CCPs, it is a single-heme enzyme that is localized in the mitochondrial intermembrane space. The first bacterial CCP that was reported and characterized was found in Pseudomonas aeruginosa [175, 176]; homologs were later studied in other bacteria, namely Rhodobacter capsulatus [177], Paracoccus pantotrophus [178], Nitrosomonas europaea [179], Methylococcus capsulatus [180], Neisseria gonorrhoea [181], and Campylobacter jejuni [182]. These bacterial enzymes are found in the periplasm—the functional equivalent of the mitochondrial intermembrane space—but they are not structural homologs of the eukaryotic enzymes, as bacterial CCPs are diheme enzymes. These CCPs are well distributed among Gram-negative bacteria and are absent from Gram-positive bacteria (Peroxibase), which is in line with their periplasmic localization.
Structural features and mechanism
Crystallization data of CCPs from P. aeruginosa, N. europaea, P. pantotrophus, P. denitrificans, P. stutzeri and Geobacter sulfurreducens are available [183–186]. CCP exists as a homodimer. The monomers consist of two domains, each of which binds a c-type heme [179, 183, 187–191]. The resting enzyme has a mixed valence, with Fe2+ at the high-potential heme (HP-heme) and Fe3+ at the low-potential heme (LP-heme) (Fig. 10). H2O2 binds and oxidizes the LP-heme site; simultaneous electron transfer from the HP-heme creates an LP-heme ferryl (Fe[IV]=O) species but avoids the porphyrin cation radical that is potentially problematic for catalases. The oxidized enzyme is then apparently reduced by consecutive electron transfers from reduced cytochrome c, the first transfer generating Fe3+ heme at both HP and LP positions, and the second regenerating the initial mixed-valence enzyme with Fe2+ HP-heme.
Fig. 10.

Mechanism of H2O2 degradation by cytochrome c peroxidase. The active form of enzyme (Fe2+/Fe3+) reduces H2O2 and forms a Fe4+=O species at the LP-heme site. Reduction occurs through two consecutive deliveries of electrons from cytochome c to the HP-heme site.
Measured KM values for H2O2 are in the range of 25–50 μM [192–194]. Following the argument presented previously for Ahp, one might expect that the physiologically important kinetic step in the turnover of CCP is the oxidation part of the reaction cycle, whereas the reduction stage may only be functionally limiting when H2O2 levels are high (when peroxidases generally saturate). Unfortunately, because it is difficult to establish the mixed-valence form of the enzyme in vitro without continuous turnover of the enzyme, it is not easy to isolate and quantify the rate of the oxidation step. The overall turnover numbers, which might provide a lower limit, have been reported to be orders of magnitude lower than those of Ahp and catalase—perhaps because the measurements were obtained using heterologous cytochrome c proteins. Thus the kinetic data alone do not yet provide a sufficient argument that peroxidase activity is the bona fide function of the enzyme.
Cytochrome c proteins are central electron carriers in many electron transport chains, but it is not self-evident that the same cytochrome c that delivers electrons to cytochrome c oxidases also delivers them to CCP. Inhibitors of the bc1 complex did not slow H2O2 reduction by P. denitrificans, indicating either that this was not the route of electron delivey to CCP or that CCP was not the predominant scavenger under the conditions of the experiment [195]. CCP-encoding genes do not sit adjacent to those of prospective partners. Recently, a small monoheme cytochrome c, ScyA, was indicated to be the redox partner of the Shewanella oneidensis CCP: abolition of either CCP or ScyA resulted in ~70% loss of H2O2 scavenging activity [196]. The mutants grew somewhat more slowly than did their wild-type parents, but only under aerobic conditions. To date these are the only phenotypic data for CCP-deficient mutants, and they are broadly consistent with the proposal that CCP relieves oxidative stress.
However, a dilemma arises in considering E. coli and Salmonella typhimurium. They both possess CCPs as well as a cytochrome c biogenesis mechanism and two cytochrome c proteins. However, the latter proteins appear to be dedicated to the delivery of electrons to periplasmic nitrate and nitrite reductases; their structural genes are co-transcribed and are co-regulated. It is not clear then how a peroxidase would fit into the mix, as this electron source would seemingly only be available during periods of nitrate respiration. An alternative is that something other than a cytochrome c might be its physiological electron donor. To compound matters, studies determined that CCP transcription in E. coli, N. gonorrhoea, P. denitrificans, P. stutzeri, and P. aeruginosa is driven by the oxygen-sensitive Fnr transcription factor—meaning that CCP is synthesized only in anaerobic habitats [181, 197–200]. This observation suggests an alternative role for CCP: that in the absence of oxygen CCP enables H2O2 to serve as an alternative respiratory electron acceptor. If so, the fundamental function of CCP is not simply to scavenge H2O2. In any case, E. coli mutants that lack catalase and Ahp exhibit very little residual ability to scavenge H2O2 (Fig. 5), supporting the idea that CCP does not play this role in aerobic cells. The E. coli structural gene is also positively regulated by the OxyR protein, albeit in anaerobic media that presumably lack the H2O2 that normally activates this transcription factor [199]. This arrangement seems to imply some connection to H2O2. In sum, CCP is another protein that displays peroxidase activity in vitro but whose physiological function remains to be shown. At best, its pattern of synthesis indicates that its function is more specialized than service as a general scavenger of periplasmic H2O2.
Rubrerythrin
Spirillum volutans is a microaerophilic bacterium that lacks catalase and is quickly killed by micromolar levels of H2O2. Alban and Krieg selected an H2O2-resistant mutant that had acquired the ability to degrade H2O2 [201]. Mutant extracts exhibited high levels of an NADH-dependent peroxidase activity; proteomic analysis identified rubrerythrin as one protein that the mutant expressed in unusually high levels. This protein had been encountered previously [202] but without knowledge of its biochemical activity. It is broadly distributed among anaerobic and microaerophilic bacteria and archaea.
Rubrerythrins are defined by their two distinctive metal centers: a rubredoxin-type iron center [Fe(Cys)4] and a discrete di-iron site with a μ-oxo bridge [203] (Fig. 11). H2O2 concertedly oxidizes the two ferrous iron atoms at the di-iron site and is thereby reduced to water, thus constituting a peroxidase reaction [204–208]. The KM for H2O2 of this activity is as low as 2 μM, and in vitro systems (below) have achieved turnover numbers as high as 5000 min−1. Completion of the catalytic cycle requires movement of two consecutive electrons from the rubredoxin site to the (now) di-ferric site. In turn, the oxidized rubredoxin site probably is then reduced in vivo by an authentic rubredoxin protein, as its structural gene is sometimes located in an operon with that of the rubrerythrin. A complete peroxidase system has been reconstituted in vivo using NADH:rubredoxin oxidoreductase, rubredoxin, and rubrerythrin [205, 208].
Fig. 11.


Rubrerythrins. A. Domain organization. B. Scheme of electron delivery to recycle rubrerythrin (Rbr). NROR: NADH:rubredoxin oxidoreductase.
It is not surprising that anaerobic bacteria would contain antioxidant enzymes: anaerobes almost invariably express superoxide dismutases and/or reductases and catalase and/or Ahp, presumably to forestall damage upon the occasional exposure to oxygen. Such enzymes are preemptively expressed even during stringent anaerobiosis; were this not the case, aeration would damage metabolic enzymes to the point that defensive systems could not be synthesized. However, given the presence of other scavenging enzymes, it is not self-evident that another peroxidase is needed; one must again consider the possibility that a peroxidase activity might be merely adventitious. Notably, the high NADH peroxidase activity of the S. volutans mutant was not definitively demonstrated to arise from the overexpressed rubrerythrin.
The rubrerythrin of Porphyromonas gingivalis is induced upon oxygenation, and rubrerythrin mutants exhibit sensitivity to both H2O2 and oxygen [209]. Further, “reverse rubrerythrins”—enzymes in which the positions of the rubredoxin and di-iron sites are reversed from that of the conventional enzyme (Fig. 11)—are encoded in Clostridium acetobutylicum and Desulfovibrio vulgaris adjacent to an apparent PerR transcription factor [210, 211]. While the conventional rubrerythrins in Pyrococcus furiosus and C. acetobutylicum are not induced by H2O2 treatment, the reverse rubrerythrins are [212–214], and in C. acetobutylicum this induction appears to be controlled by the PerR protein [215]. Strikingly, perR null mutants exhibit some growth upon aeration—a feature that even the wild-type cell does not share—which confirms that the PerR regulon effectively defends the cell against oxidative stress. These data do not prove that the contribution of the reverse rubrerythrin is to scavenge H2O2, but they certainly align with that idea.
Prospects
Two overarching themes run through this accounting of scavenging enzymes. First, only catalase and Ahp have been rigorously demonstrated to be authentic scavengers in vivo. For the others definitive evidence—in the form of phenotypes or measurements of H2O2 degradation—has been hard to come by. Perhaps this failure reflects the second theme: cells typically possess multiple scavenging systems, so that the absence of one is hidden by the presence of another. Efforts are underway to genetically peel back these multiple layers so that the contribution of each can be appraised in isolation from the others. If and when the true scavengers are identified, the next step will be to probe the raison d'etre of each. In this review we have considered a number of possible reasons why a single enzyme might not suffice: the need for multiple forms of compartmentalization, substrate specificity, metal requirements, stability, and electron demand. The irony is that 112 years after catalase was first reported, we are still struggling to understand the basic roles of these enzymes.
Highlights
- Cells require scavenging enzymes to degrade both endogenous and exogenous hydrogen peroxide.
- Bacteria contain multiple H2O2 scavenging enzymes, which can be distinguished by variation in expression pattern, cofactor requirements, kinetic properties, substrate specificity, compartmentalization, and stability.
- Catalase and Ahp are the only scavenging enzymes whose activities have been clearly demonstrated in vivo.
- The activities of other potential peroxidases have been verified in vitro, but their roles in vivo remain uncertain.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Loew O. Science. 1900;11:701–702. doi: 10.1126/science.11.279.701. [DOI] [PubMed] [Google Scholar]
- [2].Naqui A, Chance B. Ann Rev Biochem. 1986;55:137–166. doi: 10.1146/annurev.bi.55.070186.001033. [DOI] [PubMed] [Google Scholar]
- [3].Massey V, Strickland S, Mayhew SG, Howell LG, Engel PC, Matthews RG, Schuman M, Sullivan PA. Biochem. Biophys. Res. Commun. 1969;36:891–897. doi: 10.1016/0006-291x(69)90287-3. [DOI] [PubMed] [Google Scholar]
- [4].Messner KR, Imlay JA. J. Biol. Chem. 1999;274:10119–10128. doi: 10.1074/jbc.274.15.10119. [DOI] [PubMed] [Google Scholar]
- [5].Messner KR, Imlay JA. J. Biol. Chem. 2002;277:42563–42571. doi: 10.1074/jbc.M204958200. [DOI] [PubMed] [Google Scholar]
- [6].Korshunov S, Imlay JA. Mol Microbiol. 2010;75:1389–1401. doi: 10.1111/j.1365-2958.2010.07059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Seaver LC, Imlay JA. J. Biol. Chem. 2004;279:48742–48750. doi: 10.1074/jbc.M408754200. [DOI] [PubMed] [Google Scholar]
- [8].Carlioz A, Touati D. EMBO J. 1986;5:623–630. doi: 10.1002/j.1460-2075.1986.tb04256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Farr SB, D'Ari R, Touati D. Proc Natl Acad Sci USA. 1986;83:8268–8272. doi: 10.1073/pnas.83.21.8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Seaver LC, Imlay JA. J Bacteriol. 2001;183:7173–7181. doi: 10.1128/JB.183.24.7173-7181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hebrard M, Viala JP, Meresse S, Barras F, Aussel L. J Bacteriol. 2009;191:4605–4614. doi: 10.1128/JB.00144-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Park S, You X, Imlay JA. Proc Natl Acad Sci USA. 2005;102:9317–9322. doi: 10.1073/pnas.0502051102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jang S, Imlay JA. J Biol Chem. 2007;282:929–937. doi: 10.1074/jbc.M607646200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sobota JM, Imlay JA. Proc. Natl. Acad. Sci. USA. 2011;108:5402–5407. doi: 10.1073/pnas.1100410108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Seaver LC, Imlay JA. J Bacteriol. 2001;183:7182–7189. doi: 10.1128/JB.183.24.7182-7189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Winterbourn CC, Hampton MB, Livesey JH, Kettle AJ. J. Biol. Chem. 2006;281:39860–39869. doi: 10.1074/jbc.M605898200. [DOI] [PubMed] [Google Scholar]
- [17].Pericone CD, Overweg K, Hermans PW, Weiser JN. Infect. Immun. 2000;68:3990–3997. doi: 10.1128/iai.68.7.3990-3997.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bedard K, Lardy B, Krause K-H. Biochemie. 2007;89:1107–1112. doi: 10.1016/j.biochi.2007.01.012. [DOI] [PubMed] [Google Scholar]
- [19].Mehdy MC. Plant Physiol. 1994;105:467–472. doi: 10.1104/pp.105.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Glass GA, DeLisle DM, DeTogni P, Gabig TG, Magee BH, Markert M, Babior BM. J Biol Chem. 1986;261:13247–13251. [PubMed] [Google Scholar]
- [21].Imlay JA. In: EcoSal--Escherichia coli and Salmonella: Cellular and Molecular Biology. Curtiss BA,R III, JB K, PD K, FC N, N. T, JM S, CL S, U. D, editors. ASM Press; Washington, D.C.: 2009. http://www.ecosal.org. [Google Scholar]
- [22].Inbaraj JJ, Chignell CF. Chem Res Toxicol. 2004;17:55–62. doi: 10.1021/tx034132s. [DOI] [PubMed] [Google Scholar]
- [23].Turner JM, Messenger AJ. Adv Microb Physiol. 1986;27:211–275. doi: 10.1016/s0065-2911(08)60306-9. [DOI] [PubMed] [Google Scholar]
- [24].Carmel-Harel O, Storz G. Annu Rev Microbiol. 2000;54:439–461. doi: 10.1146/annurev.micro.54.1.439. [DOI] [PubMed] [Google Scholar]
- [25].Zheng L, Cash VL, Flint DH, Dean DR. J. Biol. Chem. 1998;273:13264–13272. doi: 10.1074/jbc.273.21.13264. [DOI] [PubMed] [Google Scholar]
- [26].Lee C, Lee SM, Mukhopadhyay P, Kim SJ, Lee SC, Ahn WS, Yu MH, Storz G, Ryu SE. Nat. Struct. Mol. Biol. 2004;11:1179–1185. doi: 10.1038/nsmb856. [DOI] [PubMed] [Google Scholar]
- [27].Grant RA, Filman DJ, Finkel SE, Kolter R, Hogle JM. Nat Struct Biol. 1998;5:294–303. doi: 10.1038/nsb0498-294. [DOI] [PubMed] [Google Scholar]
- [28].Ilari A, Ceci P, Ferrari D, Rossi G, Chiancone E. J Biol Chem. 2002;277:37619–37623. doi: 10.1074/jbc.M206186200. [DOI] [PubMed] [Google Scholar]
- [29].Zhao G, Ceci P, Ilari A, Giangiacomo L, Laue TM, Chiancone E, Chasteen ND. J Biol Chem. 2002;277:27689–27696. doi: 10.1074/jbc.M202094200. [DOI] [PubMed] [Google Scholar]
- [30].Park S, Imlay JA. J. Bacteriol. 2003;185:1942–1950. doi: 10.1128/JB.185.6.1942-1950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jang S, Imlay JA. Mol Microbiol. 2010;78:1448–1467. doi: 10.1111/j.1365-2958.2010.07418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee JH, Yeo WS, Roe JH. Mol. Microbiol. 2004;51:1745–1755. doi: 10.1111/j.1365-2958.2003.03946.x. [DOI] [PubMed] [Google Scholar]
- [33].Outten FW, Djaman O, Storz G. Mol Microbiol. 2004;52:861–872. doi: 10.1111/j.1365-2958.2004.04025.x. [DOI] [PubMed] [Google Scholar]
- [34].Kehres DG, Janakiraman A, Slauch JM, Maguire ME. J Bacteriol. 2002;184:3151–3158. doi: 10.1128/JB.184.12.3151-3158.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Anjem A, Varghese S, Imlay JA. Mol Microbiol. 2009;72:844–858. doi: 10.1111/j.1365-2958.2009.06699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Aslund F, Zheng M, Beckwith J, Storz G. Proc Natl Acad Sci U S A. 1999;96:6161–6165. doi: 10.1073/pnas.96.11.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Herbig AF, Helmann JD. Mol. Microbiol. 2001;41:849–859. doi: 10.1046/j.1365-2958.2001.02543.x. [DOI] [PubMed] [Google Scholar]
- [38].Lee JW, Helmann JD. Nature. 2006;440:363–367. doi: 10.1038/nature04537. [DOI] [PubMed] [Google Scholar]
- [39].Gaballa A, Helmann JD. Mol Microbiol. 2002;45:997–1005. doi: 10.1046/j.1365-2958.2002.03068.x. [DOI] [PubMed] [Google Scholar]
- [40].Herbert D, Pinsent J. Biochem J. 1948;43:193–202. doi: 10.1042/bj0430193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Clayton RK. Biochim Biophys Acta. 1959;36:40–47. doi: 10.1016/0006-3002(59)90067-8. [DOI] [PubMed] [Google Scholar]
- [42].Passardi F, Theiler G, Zamocky M, Cosio C, Rouhier N, Teixera F, Margis-Pinheiro M, Ioannidis V, Penel C, Falquet L, Dunand C. Phytochemistry. 2007;68:1605–1611. doi: 10.1016/j.phytochem.2007.04.005. [DOI] [PubMed] [Google Scholar]
- [43].Gouet P, Jouve HM, Dideberg O. J Mol Biol. 1995;249:933–954. doi: 10.1006/jmbi.1995.0350. [DOI] [PubMed] [Google Scholar]
- [44].Bravo J, Verdaguer N, Tormo J, Betzel C, Switala J, Loewen PC, Fita I. Structure. 1995;3:491–502. doi: 10.1016/s0969-2126(01)00182-4. [DOI] [PubMed] [Google Scholar]
- [45].Bravo J, Mate MJ, Schneider T, Switala J, Wilson K, Loewen PC, Fita I. Proteins. 1999;34:155–166. doi: 10.1002/(sici)1097-0134(19990201)34:2<155::aid-prot1>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- [46].Murshudov GN, Melik-Adamyan WR, Grebenko AI, Barynin VV, Vagin AA, Vainshtein BK, Dauter Z, Wilson KS. FEBS Lett. 1992;312:127–131. doi: 10.1016/0014-5793(92)80919-8. [DOI] [PubMed] [Google Scholar]
- [47].Carpena X, Perez R, Ochoa WF, Verdaguer N, Klotz MG, Switala J, Melik-Adamyan W, Fita I, Loewen PC. Acta crystallographica. Section D, Biological crystallography. 2001;57:1184–1186. doi: 10.1107/s0907444901009817. [DOI] [PubMed] [Google Scholar]
- [48].Loewen PC, Carpena X, Rovira C, Ivancich A, Perez-Luque R, Haas R, Odenbreit S, Nicholls P, Fita I. Biochemistry. 2004;43:3089–3103. doi: 10.1021/bi035663i. [DOI] [PubMed] [Google Scholar]
- [49].Carpena X, Loprasert S, Mongkolsuk S, Switala J, Loewen PC, Fita I. J Mol Biol. 2003;327:475–489. doi: 10.1016/s0022-2836(03)00122-0. [DOI] [PubMed] [Google Scholar]
- [50].Bertrand T, Eady NA, Jones JN, Jesmin, Nagy JM, Jamart-Gregoire B, Raven EL, Brown KA. J Biol Chem. 2004;279:38991–38999. doi: 10.1074/jbc.M402382200. [DOI] [PubMed] [Google Scholar]
- [51].Wada K, Tada T, Nakamura Y, Kinoshita T, Tamoi M, Shigeoka S, Nishimura K. Acta crystallographica. Section D, Biological crystallography. 2002;58:157–159. doi: 10.1107/s0907444901017735. [DOI] [PubMed] [Google Scholar]
- [52].Carpena X, Melik-Adamyan W, Loewen PC, Fita I. Acta crystallographica. Section D, Biological crystallography. 2004;60:1824–1832. doi: 10.1107/S0907444904020621. [DOI] [PubMed] [Google Scholar]
- [53].Amara P, Andreoletti P, Jouve HM, Field MJ. Protein Sci. 2001;10:1927–1935. doi: 10.1110/ps.14201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hillar A, Nicholls P. FEBS Lett. 1992;314:179–182. doi: 10.1016/0014-5793(92)80969-n. [DOI] [PubMed] [Google Scholar]
- [55].Chelikani P, Fita I, Loewen PC. Cell Mol Life Sci. 2004;61:192–208. doi: 10.1007/s00018-003-3206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hillar A, Peters B, Pauls R, Loboda A, Zhang H, Mauk AG, Loewen PC. Biochemistry. 2000;59:5868–5875. doi: 10.1021/bi0000059. [DOI] [PubMed] [Google Scholar]
- [57].Regelsberger G, Jakopitsch C, Engleder M, Ruker F, Peschek GA, Obinger C. Biochemistry. 1999;38:10480–10488. doi: 10.1021/bi990886n. [DOI] [PubMed] [Google Scholar]
- [58].Singh R, Wiseman B, Deemagarn T, Jha V, Switala J, Loewen PC. Arch Biochem Biophys. 2008;471:207–214. doi: 10.1016/j.abb.2007.12.008. [DOI] [PubMed] [Google Scholar]
- [59].Loewen PC, Switala J. Biochem Cell Biol. 1988;66:707–714. doi: 10.1139/o88-081. [DOI] [PubMed] [Google Scholar]
- [60].Christman MF, Storz G, Ames BN. Proc. Natl. Acad. Sci. USA. 1989;86:3484–3488. doi: 10.1073/pnas.86.10.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Bsat N, Herbig A, Casillas-Martinez L, Setlow P, Helmann JD. Mol Microbiol. 1998;29:189–198. doi: 10.1046/j.1365-2958.1998.00921.x. [DOI] [PubMed] [Google Scholar]
- [62].Bol DK, Yasbin RE. J Bacteriol. 1994;176:6744–6748. doi: 10.1128/jb.176.21.6744-6748.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hahn JS, Oh SY, Chater KF, Cho YH, Roe JH. J Biol Chem. 2000;275:38254–38260. doi: 10.1074/jbc.M006079200. [DOI] [PubMed] [Google Scholar]
- [64].Zou P, Borovok I, Ortiz de Orue Lucana D, Muller D, Schrempf H. Microbiology. 1999;145(Pt 3):549–559. doi: 10.1099/13500872-145-3-549. [DOI] [PubMed] [Google Scholar]
- [65].LeBlanc JJ, Brassinga AK, Ewann F, Davidson RJ, Hoffman PS. J Bacteriol. 2008;190:3444–3455. doi: 10.1128/JB.00141-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Engelmann S, Lindner C, Hecker M. J Bacteriol. 1995;177:5598–5605. doi: 10.1128/jb.177.19.5598-5605.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Brunder W, Schmidt H, Karch H. Microbiology. 1996;142(Pt 11):3305–3315. doi: 10.1099/13500872-142-11-3305. [DOI] [PubMed] [Google Scholar]
- [68].Sha Z, Stabel TJ, Mayfield JE. J Bacteriol. 1994;176:7375–7377. doi: 10.1128/jb.176.23.7375-7377.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Klotz MG, Hutcheson SW. Appl Environ Microbiol. 1992;58:2468–2473. doi: 10.1128/aem.58.8.2468-2473.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Amemura-Maekawa J, Mishima-Abe S, Kura F, Takahashi T, Watanabe H. FEMS Microbiol Lett. 1999;176:339–344. doi: 10.1111/j.1574-6968.1999.tb13681.x. [DOI] [PubMed] [Google Scholar]
- [71].Visick KL, Ruby EG. J Bacteriol. 1998;180:2087–2092. doi: 10.1128/jb.180.8.2087-2092.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Harris AG, Hazell SL. FEMS Microbiol Lett. 2003;229:283–289. doi: 10.1016/S0378-1097(03)00850-4. [DOI] [PubMed] [Google Scholar]
- [73].Pansy F, Stander H, Donovick R. Am Rev Tuberc. 1952;65:761–764. [PubMed] [Google Scholar]
- [74].Winder FG, Collins P, Rooney SA. Biochem J. 1970;117:27P. doi: 10.1042/bj1170027pa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Takayama K, Wang L, David HL. Antimicrob Agents Chemother. 1972;2:29–35. doi: 10.1128/aac.2.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Zhang Y, Heym B, Allen B, Young D, Cole S. Nature. 1992;358:591–593. doi: 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
- [77].Wiseman B, Carpena X, Feliz M, Donald LJ, Pons M, Fita I, Loewen PC. J Biol Chem. 2010;285:26662–26673. doi: 10.1074/jbc.M110.139428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Larsen MH, Vilcheze C, Kremer L, Besra GS, Parsons L, Salfinger M, Heifets L, Hazbon MH, Alland D, Sacchettini JC, Jacobs WR., Jr. Mol Microbiol. 2002;46:453–466. doi: 10.1046/j.1365-2958.2002.03162.x. [DOI] [PubMed] [Google Scholar]
- [79].Vilcheze C, Jacobs WR., Jr. Annu Rev Microbiol. 2007;61:35–50. doi: 10.1146/annurev.micro.61.111606.122346. [DOI] [PubMed] [Google Scholar]
- [80].Whittenbury R. J Gen Microbiol. 1964;35:13–26. doi: 10.1099/00221287-35-1-13. [DOI] [PubMed] [Google Scholar]
- [81].Johnston MA, Delwiche EA. J Bacteriol. 1965;90:347–351. doi: 10.1128/jb.90.2.347-351.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kono Y, Fridovich I. J Biol Chem. 1983;258:6015–6019. [PubMed] [Google Scholar]
- [83].Allgood GS, Perry JJ. J Bacrteriol. 1986;168:563–567. doi: 10.1128/jb.168.2.563-567.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Barynin VV, Grebenko AI. Dokl. Akad. Nauk. SSSR. 1986;286:461–464. [Google Scholar]
- [85].Kagawa M, Murakoshi N, Nishikawa Y, Matsumoto G, Kurata Y, Mizobata T, Kawata Y, Nagai J. Arch Biochem Biophys. 1999;362:346–355. doi: 10.1006/abbi.1998.1041. [DOI] [PubMed] [Google Scholar]
- [86].Robbe-Saule V, Coynault C, Ibanez-Ruiz M, Hermant D, Norel F. Mol Microbiol. 2001;39:1533–1545. doi: 10.1046/j.1365-2958.2001.02340.x. [DOI] [PubMed] [Google Scholar]
- [87].Amo T, Atomi H, Imanaka T. J Bacteriol. 2002;184:3305–3312. doi: 10.1128/JB.184.12.3305-3312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Barynin VV, Whittaker MM, Antonyuk SV, Lamzin VS, Harrison PM, Artymiuk PJ, Whittaker JW. Structure. 2001;9:725–738. doi: 10.1016/s0969-2126(01)00628-1. [DOI] [PubMed] [Google Scholar]
- [89].Antonyuk SV, Barynin VV. Crystallogr. Reports. 2000;45:105–116. [Google Scholar]
- [90].Beyer WF, Jr., Fridovich I. Biochemistry. 1985;24:6460–6467. doi: 10.1021/bi00344a023. [DOI] [PubMed] [Google Scholar]
- [91].Shank M, Barynin V, Dismukes GC. Biochemistry. 1994;33:15433–15436. doi: 10.1021/bi00255a025. [DOI] [PubMed] [Google Scholar]
- [92].Robbe-Saule V, Coynault C, Ibanez-Ruiz M, Hermant D, Norel F. Mol. Microbiol. 2001;39:1533–1545. doi: 10.1046/j.1365-2958.2001.02340.x. [DOI] [PubMed] [Google Scholar]
- [93].Kono Y, Fridovich I. J Bacteriol. 1983;155:742–746. doi: 10.1128/jb.155.2.742-746.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Martin JE, Imlay JA. Mol Microbiol. 2011;80:319–334. doi: 10.1111/j.1365-2958.2011.07593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Patzer SI, Hantke K. J Bacteriol. 2001;183:4806–4813. doi: 10.1128/JB.183.16.4806-4813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Tardat B, Touati D. Mol Microbiol. 1991;5:455–465. doi: 10.1111/j.1365-2958.1991.tb02129.x. [DOI] [PubMed] [Google Scholar]
- [97].Kim K, Kim IH, Lee KY, Rhee SG, Stadtman ER. J Biol Chem. 1988;263:4704–4711. [PubMed] [Google Scholar]
- [98].Tartaglia LA, Storz G, Ames BN. J Mol Biol. 1989;210:709–719. doi: 10.1016/0022-2836(89)90104-6. [DOI] [PubMed] [Google Scholar]
- [99].Winterbourn CC, Metodiewa D. Free Rad. Biol. Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- [100].Peskin AV, Low FM, Paton LN, Maghzal GJ, Hampton MB, Winterbourn CC. J. Biol. Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- [101].Imlay JA. Annu Rev Biochem. 2008;77:755–776. doi: 10.1146/annurev.biochem.77.061606.161055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Jacobson FS, Morgan RW, Christman MF, Ames BN. JBC. 1989;264:1488–1496. [PubMed] [Google Scholar]
- [103].Poole LB, Ellis HR. Biochemistry. 1996;35:56–64. doi: 10.1021/bi951887s. [DOI] [PubMed] [Google Scholar]
- [104].Ellis HR, Poole LB. Biochemistry. 1997;36:13349–13356. doi: 10.1021/bi9713658. [DOI] [PubMed] [Google Scholar]
- [105].Poole LB. Biochemistry. 1996;35:65–75. doi: 10.1021/bi951888k. [DOI] [PubMed] [Google Scholar]
- [106].Wood ZA, Poole LB, Hantgan RR, Karplus PA. Biochemistry. 2002;41:5493–5504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- [107].Choi HJ, Kang SW, Yang CH, Rhee SG, Ryu SE. Nat Struct Biol. 1998;5:400–406. doi: 10.1038/nsb0598-400. [DOI] [PubMed] [Google Scholar]
- [108].Bieger B, Essen L-O. J Mol Biol. 2001;307:1–8. doi: 10.1006/jmbi.2000.4441. [DOI] [PubMed] [Google Scholar]
- [109].Li Calzi M, Poole LB. Biochemistry. 1997;36:13357–13364. doi: 10.1021/bi9713660. [DOI] [PubMed] [Google Scholar]
- [110].Deretic V, Philipp W, Dhandayuthapani S, Mudd MH, Curcic R, Garbe T, Heym B, Via LE, Cole ST. Mol Microbiol. 1995;17:889–900. doi: 10.1111/j.1365-2958.1995.mmi_17050889.x. [DOI] [PubMed] [Google Scholar]
- [111].Baker LM, Raudonikiene A, Hoffman PS, Poole LB. J. Bacteriol. 2001;183:1961–1973. doi: 10.1128/JB.183.6.1961-1973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Bryk R, Griffin P, Nathan C. Nature. 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- [113].Parsonage D, Karplus PA, Poole LB. Proc Natl Acad Sci U S A. 2008;105:8209–8214. doi: 10.1073/pnas.0708308105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Ma M, Eaton JW. Proc Natl Acad Sci U S A. 1992;89:7924–7928. doi: 10.1073/pnas.89.17.7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Neidhardt FC, Vaughn V, Phillips TA, Bloch PL. Microbiol Rev. 1983;47:231–284. doi: 10.1128/mr.47.2.231-284.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Andrews SC, Harrison PM, Guest JR. J Gen Micro. 1991;137:361–367. doi: 10.1099/00221287-137-2-361. [DOI] [PubMed] [Google Scholar]
- [117].Clarke DJ, Mackay CL, Campopiano DJ, Langridge-Smith P, Brown AR. Biochemistry. 2009;48:3904–3914. doi: 10.1021/bi900189e. [DOI] [PubMed] [Google Scholar]
- [118].Liao SJ, Yang CY, Chin KH, Wang AH, Chou SH. J Mol Biol. 2009;390:951–966. doi: 10.1016/j.jmb.2009.05.030. [DOI] [PubMed] [Google Scholar]
- [119].Wang G, Olczak AA, Walton JP, Maier RJ. Infect Immun. 2005;73:378–384. doi: 10.1128/IAI.73.1.378-384.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Atack JM, Harvey P, jone MA, Kelly DJ. J Bacrteriol. 2008;190:5279–5290. doi: 10.1128/JB.00100-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Clarke DJ, Ortega XP, Mackay CL, Valvano MA, Govan JR, Campopiano DJ, Langridge-Smith P, Brown AR. Biochemistry. 2010;49:1319–1330. doi: 10.1021/bi901703m. [DOI] [PubMed] [Google Scholar]
- [122].Jeong W, Cha MK, Kim IH. J Biol Chem. 2000;275:2924–2930. doi: 10.1074/jbc.275.4.2924. [DOI] [PubMed] [Google Scholar]
- [123].Wang G, Hong Y, Johnson MK, Maier RJ. Biochim Biophys Acta. 2006;1760:1596–1603. doi: 10.1016/j.bbagen.2006.05.005. [DOI] [PubMed] [Google Scholar]
- [124].Johnson NA, McKenzie RM, Fletcher HM. Mol Oral Microbiol. 2011;26:62–77. doi: 10.1111/j.2041-1014.2010.00596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Fournier M, Aubert C, Dermoun Z, Durand MC, Moinier D, Dolla A. Biochimie. 2006;88:85–94. doi: 10.1016/j.biochi.2005.06.012. [DOI] [PubMed] [Google Scholar]
- [126].Ghrist AC, Stauffer GV. J Bacteriol. 1998;180:1803–1807. doi: 10.1128/jb.180.7.1803-1807.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Kim K, Rhee SG, Stadtman ER. JBC. 1985;260:15394–15397. [PubMed] [Google Scholar]
- [128].Cha MK, Kim HK, Kim IH. J Biol Chem. 1995;270:28635–28641. doi: 10.1074/jbc.270.48.28635. [DOI] [PubMed] [Google Scholar]
- [129].Baker LMS, Poole LB. J Biol Chem. 2003;278:9203–9211. doi: 10.1074/jbc.M209888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Zhou Y, Wan XY, Wang HL, Yan ZY, Hou YD, Jin DY. Biochem Biophys Res Commun. 1997;233:848–852. doi: 10.1006/bbrc.1997.6564. [DOI] [PubMed] [Google Scholar]
- [131].Choi J, Choi S, Cha MK, Kim IH, Shin W. J Biol Chem. 2003;278:49478–49486. doi: 10.1074/jbc.M309015200. [DOI] [PubMed] [Google Scholar]
- [132].Hall A, Sankaran B, Poole LB, Karplus PA. J Mol Biol. 2009;393:867–881. doi: 10.1016/j.jmb.2009.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Cha MK, Kim W, Lim CJ, Kim K, Kim IH. J. Biol. Chem. 2004;279:8769–8778. doi: 10.1074/jbc.M312388200. [DOI] [PubMed] [Google Scholar]
- [134].Tao K. FEMS Microbiol Lett. 2008;289:41–45. doi: 10.1111/j.1574-6968.2008.01372.x. [DOI] [PubMed] [Google Scholar]
- [135].Cha MK, Kim WC, Lim CJ, Kim K, Kim IH. J Biol Chem. 2004;279:8769–8778. doi: 10.1074/jbc.M312388200. [DOI] [PubMed] [Google Scholar]
- [136].Comtois SL, Gidley MD, Kelly DJ. Microbiology. 2003;149:121–129. doi: 10.1099/mic.0.25896-0. [DOI] [PubMed] [Google Scholar]
- [137].La Carbona S, Sauvageot N, Giard JC, Benachour A, Posteraro B, Auffray Y, Sanguinetti M, Hartke A. Mol Microbiol. 2007;66:1148–1163. doi: 10.1111/j.1365-2958.2007.05987.x. [DOI] [PubMed] [Google Scholar]
- [138].Kim HK, Kim SJ, Lee JW, Lee JW, Cha MK, Kim IH. Biochem Biophys Res Commun. 1996;221:641–646. doi: 10.1006/bbrc.1996.0649. [DOI] [PubMed] [Google Scholar]
- [139].Kim SJ, Han YH, Kim IH, Kim HK. IUBMB Life. 1999;48:215–218. doi: 10.1080/713803496. [DOI] [PubMed] [Google Scholar]
- [140].Mills GC. J Biol Chem. 1957;229:189–197. [PubMed] [Google Scholar]
- [141].Flohe L, Gunzler WA, Schock HH. FEBS Lett. 1973;32:132–134. doi: 10.1016/0014-5793(73)80755-0. [DOI] [PubMed] [Google Scholar]
- [142].Kang SI, Spears CP. J Pharm Sci. 1990;79:57–62. doi: 10.1002/jps.2600790114. [DOI] [PubMed] [Google Scholar]
- [143].Brandt W, Wessjoham LA. Chembiochem. 2005;6:386–394. doi: 10.1002/cbic.200400276. [DOI] [PubMed] [Google Scholar]
- [144].Gromer S, Wessjoham LA, Eubel J, Brandt W. ChemBioChem. 2006;7:1649–1652. doi: 10.1002/cbic.200600080. [DOI] [PubMed] [Google Scholar]
- [145].Maiorino M, Aumann KD, Brigelius-Flohe R, Doria D, van den Heuvel J, McCarthy J, Roveri A, Ursini F, Flohe L. Biol Chem Hoppe Seyler. 1995;376:651–660. doi: 10.1515/bchm3.1995.376.11.651. [DOI] [PubMed] [Google Scholar]
- [146].Criqui MC, Jamet E, Parmentier Y, Marbach J, Durr A, Fleck J. Plant Mol Biol. 1992;18:623–627. doi: 10.1007/BF00040684. [DOI] [PubMed] [Google Scholar]
- [147].Holland D, Ben-Hayyim G, Faltin Z, Camoin L, Strosberg AD, Eshdat Y. Plant Mol Biol. 1993;21:923–927. doi: 10.1007/BF00027124. [DOI] [PubMed] [Google Scholar]
- [148].Herbette S, Roeckel-Drevet P, Drevet JR. FEBS J. 2007;274:2163–2180. doi: 10.1111/j.1742-4658.2007.05774.x. [DOI] [PubMed] [Google Scholar]
- [149].Inoue Y, Matsuda T, Sugiyama K, Izawa S, Kimura A. J Biol Chem. 1999;274:27002–27009. doi: 10.1074/jbc.274.38.27002. [DOI] [PubMed] [Google Scholar]
- [150].Arenas FA, Diaz WA, Leal CA, Perez-Donoso JM, Imlay JA, Vasquez CC. Biochem Biophys Res Commun. 2010;398:690–694. doi: 10.1016/j.bbrc.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Ohdate T, Kita K, Inoue Y. FEMS Yeast Res. 2010;10:787–790. doi: 10.1111/j.1567-1364.2010.00651.x. [DOI] [PubMed] [Google Scholar]
- [152].Moore TD, Sparling PF. J Bacteriol. 1996;178:4301–4305. doi: 10.1128/jb.178.14.4301-4305.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Arenas FA, Covarrubias PC, Sandoval JM, Perez-Donoso JM, Imlay JA, Vasquez CC. PLoS ONE. 2011;6:e15979. doi: 10.1371/journal.pone.0015979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Friedrich MJ, de Veaux LC, Kadner RJ. J Bacteriol. 1986;167:928–934. doi: 10.1128/jb.167.3.928-934.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].de Veaux LC, Clevenson DS, Bradbeer C, Kadner RJ. J Bacteriol. 1986;167:920–927. doi: 10.1128/jb.167.3.920-927.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Halliwell B, Gutteridge JM. Free radicals in biology and medicine. Clarendon Press; Oxford: 1999. [Google Scholar]
- [157].Akaike T, Sato K, Ijiri S, Miyamoto Y, Kohno M, Ando M, Maeda H. Arch Biochem Biophys. 1992;294:55–63. doi: 10.1016/0003-9861(92)90136-k. [DOI] [PubMed] [Google Scholar]
- [158].Halliwell B, Gutteridge JM. Free radicals in biology and medicine. Clarendon Press; Oxford: 1989. [Google Scholar]
- [159].Watanabe K, Ishikawa C, Inoue H, Cenhua D, Yazawa K, Kondo K. J Am Oil Chem Soc. 1994;71:325–330. [Google Scholar]
- [160].Yano Y, Nakayama A, Yoshida K. Appl Environ Microbiol. 1997;63:2572–2577. doi: 10.1128/aem.63.7.2572-2577.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Mongkolsuk S, Praituan W, Loprasert S, Fuangthong M, Chamnongpol S. J Bacteriol. 1998;180:2636–2643. doi: 10.1128/jb.180.10.2636-2643.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Cussiol JR, Alves SV, de Oliveira MA, Netto LE. J Biol Chem. 2003;278:11570–11578. doi: 10.1074/jbc.M300252200. [DOI] [PubMed] [Google Scholar]
- [163].Chuchue T, Tanboon W, Prapagdee B, Dubbs JM, Vattanaviboon P, Mongkolsuk S. J Bacteriol. 2006;188:842–851. doi: 10.1128/JB.188.3.842-851.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Ochsner UA, Hassett DJ, Vasil ML. J Bacteriol. 2001;183:773–778. doi: 10.1128/JB.183.2.773-778.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Meunier-Jamin C, Kapp U, Leonard GA, McSweeney S. J Biol Chem. 2004;279:25830–25837. doi: 10.1074/jbc.M312983200. [DOI] [PubMed] [Google Scholar]
- [166].Oliveira MA, Guimaraes BG, Cussiol JR, Medrano FJ, Gozzo FC, Netto LE. J Mol Biol. 2006;359:433–445. doi: 10.1016/j.jmb.2006.03.054. [DOI] [PubMed] [Google Scholar]
- [167].de Oliveira MA, Netto LE, Medrano FJ, Barbosa JA, Alves SV, Cussiol JR, Guimaraes BG. Acta crystallographica. Section D, Biological crystallography. 2004;60:337–339. doi: 10.1107/S0907444903026799. [DOI] [PubMed] [Google Scholar]
- [168].Cussiol JR, Alegria TG, Szweda LI, Netto LE. J Biol Chem. 2010;285:21943–21950. doi: 10.1074/jbc.M110.117283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Lesniak J, Barton WA, Nikolov DB. Embo J. 2002;21:6649–6659. doi: 10.1093/emboj/cdf670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Fontenelle C, Blanco C, Arrieta M, Dufour V, Trautwetter A. BMC Microbiol. 2011;11:100. doi: 10.1186/1471-2180-11-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Fuangthong M, Atichartpongkul S, Mongkolsuk S, Helmann JD. J Bacteriol. 2001;183:4134–4141. doi: 10.1128/JB.183.14.4134-4141.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Sukchawalit R, Loprasert S, Atichartpongkul S, Mongkolsuk S. J Bacteriol. 2001;183:4405–4412. doi: 10.1128/JB.183.15.4405-4412.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Vattanaviboon P, Whangsuk W, Panmanee W, Klomsiri C, Dharmsthiti S, Mongkolsuk S. Biochem Biophys Res Commun. 2002;299:177–182. doi: 10.1016/s0006-291x(02)02602-5. [DOI] [PubMed] [Google Scholar]
- [174].Altschul AM, Abrams R, Hogness TR. J Biol Chem. 1940;136:777–793. [Google Scholar]
- [175].Ellfolk N, Soininen R. Acta Chem Scand. 1970;24:2126–2136. doi: 10.3891/acta.chem.scand.24-2126. [DOI] [PubMed] [Google Scholar]
- [176].Ronnberg M, Ellfolk N. Biochim Biophys Acta. 1978;504:60–66. doi: 10.1016/0005-2728(78)90006-3. [DOI] [PubMed] [Google Scholar]
- [177].Hochman A, Figueredo A, Wall JD. J Bacteriol. 1992;174:3386–3391. doi: 10.1128/jb.174.10.3386-3391.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Goodhew CF, Wilson IB, Hunter DJ, Pettigrew GW. Biochem J. 1990;271:707–712. doi: 10.1042/bj2710707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Arciero D, Hooper A. J Biol Chem. 1994;269:11878–11886. [PubMed] [Google Scholar]
- [180].Villalain J, Moura I, Liu MC, Payne WJ, LeGall J, Xavier AV, Moura JJ. Eur J Biochem. 1984;141:305–312. doi: 10.1111/j.1432-1033.1984.tb08192.x. [DOI] [PubMed] [Google Scholar]
- [181].Turner S, Reid E, Smith H, Cole J. Biochem J. 2003;373:865–873. doi: 10.1042/BJ20030088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Bingham-Ramos LK, Hendrixson DR. Infec Immun. 2008;76:1105–1114. doi: 10.1128/IAI.01430-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Fulop V, Ridout CJ, Greenwood C, Hajdu J. Structure. 1995;3:1225–1233. doi: 10.1016/s0969-2126(01)00258-1. [DOI] [PubMed] [Google Scholar]
- [184].Shimizu H, Schuller DJ, Lanzilotta WN, Sundaramoorthy M, Arciero DM, Hooper AB, Poulos TL. Biochemistry. 2001;40:13483–13490. doi: 10.1021/bi011481h. [DOI] [PubMed] [Google Scholar]
- [185].Echalier A, Goodhew CF, Pettigrew GW, Fulop V. Structure. 2006;14:107–117. doi: 10.1016/j.str.2005.09.011. [DOI] [PubMed] [Google Scholar]
- [186].Echalier A, Goodhew CF, Pettigrew GW, Fulop V. Acta crystallographica. Section D, Biological crystallography. 2004;60:331–333. doi: 10.1107/S0907444903026519. [DOI] [PubMed] [Google Scholar]
- [187].Gilmour R, Goodhew CF, Pettigrew GW, Prazeres S, Moura JJ, Moura I. Biochem J. 1994;300(Pt 3):907–914. doi: 10.1042/bj3000907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Zahn JA, Arciero DM, Hooper AB, Coats JR, DiSpirito AA. Arch Microbiol. 1997;168:362–372. doi: 10.1007/s002030050510. [DOI] [PubMed] [Google Scholar]
- [189].Alves T, Besson S, Duarte LC, Pettigrew GW, Girio FMF, Devreese B, Vandenberghe I, Beeumen JV, Fauque G, Moura I. Biochim Biophys Acta. 1999;1434:248–259. doi: 10.1016/s0167-4838(99)00188-0. [DOI] [PubMed] [Google Scholar]
- [190].De Smet L, Pettigrew GW, Van Beeumen JJ. Eur J Biochem. 2001;268:6559–6568. doi: 10.1046/j.0014-2956.2001.02610.x. [DOI] [PubMed] [Google Scholar]
- [191].Ronnberg M, Kalkkinen N, Ellfolk N. FEBS Lett. 1989;250:175–178. doi: 10.1016/0014-5793(89)80714-8. [DOI] [PubMed] [Google Scholar]
- [192].Becker CF, Watmough NJ, Elliott SJ. Biochemistry. 2009;48:87–95. doi: 10.1021/bi801699m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Timoteo CG, Tavares P, Goodhew CF, Duarte LC, Jumel K, Girio FM, Harding S, Pettigrew GW, Moura I. J Biol Inorg Chem. 2003;8:29–37. doi: 10.1007/s00775-002-0382-y. [DOI] [PubMed] [Google Scholar]
- [194].Bradley AL, Chobot SE, Arciero DM, Hooper AB, Elliott SJ. J Biol Chem. 2004;279:13297–13300. doi: 10.1074/jbc.C400026200. [DOI] [PubMed] [Google Scholar]
- [195].Koutny M, Kriz L, Kucera I, Pluhacek I. Biochim Biophys Acta. 1999;1410:71–76. doi: 10.1016/s0005-2728(98)00176-5. [DOI] [PubMed] [Google Scholar]
- [196].Schutz B, Seidel J, Sturm G, Einsle O, Gescher J. Appl Environ Microbiol. 2011;77:6172–6180. doi: 10.1128/AEM.00606-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Van Spanning RJ, De Boer AP, Reijnders WN, Westerhoff HV, Stouthamer AH, Van Der Oost J. Mol Microbiol. 1997;23:893–907. doi: 10.1046/j.1365-2958.1997.2801638.x. [DOI] [PubMed] [Google Scholar]
- [198].Vollack KU, Hartig E, Korner H, Zumft WG. Mol Microbiol. 1999;31:1681–1694. doi: 10.1046/j.1365-2958.1999.01302.x. [DOI] [PubMed] [Google Scholar]
- [199].Partridge JD, Poole RK, Green J. Microbiology. 2007;153:1499–1507. doi: 10.1099/mic.0.2006/004838-0. [DOI] [PubMed] [Google Scholar]
- [200].Zimmermann A, Reimmann C, Galimand M, Haas D. Mol Microbiol. 1991;5:1483–1490. doi: 10.1111/j.1365-2958.1991.tb00794.x. [DOI] [PubMed] [Google Scholar]
- [201].Alban PS, Popham DL, Rippere KE, Krieg NR. J Appl. Microbiol. 1998;85:875–882. doi: 10.1046/j.1365-2672.1998.00602.x. [DOI] [PubMed] [Google Scholar]
- [202].Bruschi M, Le Gall J. Biochim Biophys Acta. 1972;263:279–282. [PubMed] [Google Scholar]
- [203].LeGall J, Prickril BC, Moura I, Xavier AV, Moura JJ, Huynh BH. Biochemistry. 1988;27:1636–1642. doi: 10.1021/bi00405a037. [DOI] [PubMed] [Google Scholar]
- [204].Kawasaki S, Ono M, Watamura Y, Sakai Y, Satoh T, Arai T, Satoh J, Niimura Y. FEBS Lett. 2007;581:2460–2464. doi: 10.1016/j.febslet.2007.04.050. [DOI] [PubMed] [Google Scholar]
- [205].Riebe O, Fischer RJ, Wampler DA, Kurtz DM, Jr., Bahl H. Microbiology. 2009;155:16–24. doi: 10.1099/mic.0.022756-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Coulter ED, Emerson JP, Kurtz J, D M, Cabelli DE. J. Amer. Chem. Soc. 2000;122:11555–11556. [Google Scholar]
- [207].Coulter ED, Shenvi NV, Kurtz DM., Jr. Biochem Biophys Res Commun. 1999;255:317–323. doi: 10.1006/bbrc.1999.0197. [DOI] [PubMed] [Google Scholar]
- [208].Zhao W, Ye Z, Zhao J. Mol Microbiol. 2007;66:1219–1230. doi: 10.1111/j.1365-2958.2007.05994.x. [DOI] [PubMed] [Google Scholar]
- [209].Sztukowska M, Bugno M, Potempa J, Travis J, Kurtz DM., Jr. Mol Microbiol. 2002;44:479–488. doi: 10.1046/j.1365-2958.2002.02892.x. [DOI] [PubMed] [Google Scholar]
- [210].Bahl H, Muller H, Behrens S, Joseph H, Narberhaus F. FEMS Microbiol Rev. 1995;17:341–348. doi: 10.1111/j.1574-6976.1995.tb00217.x. [DOI] [PubMed] [Google Scholar]
- [211].Lumppio HL, Shenvi NV, Garg RP, Summers AO, Kurtz DM., Jr. J Bacteriol. 1997;179:4607–4615. doi: 10.1128/jb.179.14.4607-4615.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].May A, Hillmann F, Riebe O, Fischer RJ, Bahl H. FEMS Microbiol. Lett. 2004;238:249–254. doi: 10.1016/j.femsle.2004.07.042. [DOI] [PubMed] [Google Scholar]
- [213].Kawasaki S, Ishikura J, Watamura Y, Niimura Y. FEBS Lett. 2004;571:21–25. doi: 10.1016/j.febslet.2004.06.047. [DOI] [PubMed] [Google Scholar]
- [214].Hillmann F, Fischer RJ, Bahl H. Arch Microbiol. 2006;185:270–276. doi: 10.1007/s00203-006-0091-y. [DOI] [PubMed] [Google Scholar]
- [215].Hillmann F, Fischer RJ, Saint-Prix F, Girbal L, Bahl H. Mol Microbiol. 2008;68:848–860. doi: 10.1111/j.1365-2958.2008.06192.x. [DOI] [PubMed] [Google Scholar]
- [216].Switala J, Loewen PC. Arch Biochem Biophys. 2002;401:145–154. doi: 10.1016/S0003-9861(02)00049-8. [DOI] [PubMed] [Google Scholar]
- [217].Kono Y, Fridovich I. J Biol Chem. 1983;258:13646–13648. [PubMed] [Google Scholar]
- [218].Brioukhanov AL, Durand MC, Dolla A, Aubert C. FEMS Microbiol Lett. 2010;310:175–181. doi: 10.1111/j.1574-6968.2010.02061.x. [DOI] [PubMed] [Google Scholar]
- [219].Lumppio HL, Shenvi NV, Summars AO, Voordouw G, D M Kurtz J. J. Bacteriol. 2001;183:101–108. doi: 10.1128/JB.183.1.101-108.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]