Emerging Role of Protein-Protein Transnitrosylation in Cell Signaling Pathways (original) (raw)
Abstract
Significance: Protein _S_-nitrosylation, a covalent reaction of a nitric oxide (NO) group with a critical protein thiol (or more properly thiolate anion), mediates an important form of redox-related signaling as well as aberrant signaling in disease states. Recent Advances: A growing literature suggests that over 3000 proteins are _S_-nitrosylated in cell systems. Our laboratory and several others have demonstrated that protein _S_-nitrosylation can regulate protein function by directly inhibiting catalytically active cysteines, by reacting with allosteric sites, or via influencing protein-protein interaction. For example, _S_-nitrosylation of critical cysteine thiols in protein-disulfide isomerase and in parkin alters their activity, thus contributing to protein misfolding in Parkinson's disease. Critical Issues: However, the mechanism by which specific protein _S_-nitrosylation occurs in cell signaling pathways is less well investigated. Interestingly, the recent discovery of protein-protein transnitrosylation reactions (transfer of an NO group from one protein to another) has revealed a unique mechanism whereby NO can _S_-nitrosylate a particular set of protein thiols, and represents a major class of nitrosylating/denitrosylating enzymes in mammalian systems. In this review, we will discuss recent evidence for transnitrosylation reactions between (i) hemoglobin/anion exchanger 1, (ii) thioredoxin/caspase-3, (iii) X-linked inhibitor of apoptosis/caspase-3, (iv) GAPDH-HDAC2/SIRT1/DNA-PK, and (v) Cdk5/dynamin related protein 1 (Drp1). This review also discusses experimental techniques useful in characterizing protein-protein transnitrosylations. Future Directions: Elucidation of additional transnitrosylation cascades will further our understanding of the enzymes that catalyze nitrosation, thereby contributing to NO-mediated signaling pathways. Antioxid. Redox Signal. 18, 239–249.
Introduction
Nitric oxide (NO) is a gaseous free radical that can be produced in many cell types, including muscle cells, endothelial cells, and brain cells (neurons and glia). NO can act as either a physiological (typically cell protective) or pathophysiological (generally toxic) effector, depending on the target signaling pathways affected. Three subtypes of NO synthases (NOSs) are present in mammalian organisms; the two constitutive forms of NOS—neuronal NOS (nNOS) and endothelial NOS (eNOS)—take their names from the cell type in which they were first found, and can be activated by cellular Ca2+. The name of the third subtype, inducible NOS (iNOS), indicates that expression of the enzyme can be induced by acute inflammatory stimuli. For example, activated glial cells in the nervous system can produce neurotoxic amounts of NO via iNOS expression in various neurodegenerative diseases. Initially, the biological action of NO was thought to be mediated principally via guanylate cyclase activation and cyclic guanosine-3′,5′-monophosphate (cGMP) production. However, beginning with our own work and that of our colleagues in this area over the past decade and a half, _S_-nitrosylation, a covalent reaction of an NO group with a reactive cysteine thiol on target proteins, has emerged as the principal mechanism exerting NO bioactivity (20). The formation of _S_-nitrosoproteins [protein-_S_-nitrosothiols (SNOs)] generally regulates protein function either allosterically or by direct modification of an active site cysteine (20, 32). Collaboration between the groups of Stuart Lipton and Jonathan Stamler initially discovered and characterized this biochemical process on _N_-methyl-d-aspartate-type glutamate receptors (NMDARs) in the brain, showing that NO inhibits excessive NMDAR activity _via S_-nitrosylation (32). Currently, ∼3000 proteins have been identified as potential protein-SNOs (56), supporting the notion that NO exerts its major biological activity _via S_-nitrosylation, although the specific function of most of protein-SNOs merits further investigation. Analogous to phosphorylation, Lipton, Stamler, and colleagues coined the term “_S_-nitrosylation,” indicating a biological effect of the chemical reaction of _S_-nitrosation (58). Importantly, _S_-nitrosylation can mediate either the protective or toxic effects of NO, depending on the action of the target protein. Cellular localization and compartmentalization are critical in determining where NO is generated and the identity of specific proteins that are _S_-nitrosylated.
NO generated from NOS efficiently _S_-nitrosylates nearby proteins to produce protein-SNOs. For example, the NMDAR and PSD-95 are substrates of _S_-nitrosylation and colocalize with nNOS (21, 32). In addition to proteins located adjacent to NOS, NO can react to form low-molecular-weight SNOs with cysteine and glutathione (GSH). Such low-molecular-weight SNOs, including _S_-nitrosocysteine (CysNO) and _S_-nitrosoglutathione (GSNO), can then act as NO donors for many cell proteins under physiological conditions depending on their redox potential (20). Until recently, transfer of an NO group from one thiol to another (i.e., transnitrosylation) was thought to occur only between low-molecular-weight SNOs and protein thiols. However, our laboratory as well as others have recently shown that transnitrosylation between cell proteins appears to be an important enzymatic mechanism of nitrosylation and thus protein-SNO production (2, 28, 38, 41, 47, 49, 72). In this review, we explore our current understanding of protein-protein transnitrosylation in NO-based signaling and its significance in biological settings.
Protein _S_-Nitrosylation in Signaling Pathways
NO participates in cell signaling pathways that regulate broad aspects of cellular function, including cell differentiation, normal development, and cell death (9, 20, 40). These effects were thought to be largely achieved by activation of guanylate cyclase to form cGMP, but, as mentioned previously, emerging evidence suggests that a more prominent reaction of NO is _S_-nitrosylation of regulatory protein thiol groups (14, 20, 32). _S_-Nitrosylation is the covalent addition of an NO group to a cysteine thiol/sulfhydryl (RSH or, more properly, thiolate anion, RS−) to form an _S_-nitrosothiol derivative (R-SNO). Such regulatory modifications are broadly found in mammalian, plant, and even in microbial proteins. One mechanism for the specificity of _S_-nitrosylation involves the presence of a motif fostering SNO formation adjacent to the target cysteine. We and our colleagues initially found that a consensus motif of nucleophilic residues (generally an acid and a base) surrounds a critical cysteine, increasing the susceptibility of the sulfhydryl to _S_-nitrosylation (20). Recent studies, involving proteomics, bioinformatics, and structural analyses, have confirmed this initial finding by demonstrating that acidic and/or basic amino acids reside generally within 6 to 8 Å of the _S_-nitrosylated cysteine residue; _S_-nitrosylation can also occur in a facile manner within lipid environments (11, 36, 59). The process of protein-SNO formation is counterbalanced by denitrosylation via enzymes, such as thioredoxin (Trx)/thioredoxin reductase (TrxR), class III alcohol dehydrogenase, protein disulfide isomerase (PDI), intracellular GSH, or other mechanisms (3). Recently, our laboratory and others have reported an important _S_-nitrosylation pathway—protein-protein transnitrosylation (28, 38, 41). Transnitrosylation reactions may propagate many NO-mediated cell signaling pathways via production of either a denitrosylated protein (by donating an NO moiety to a second protein), or a protein-SNO (by accepting an NO group from another protein), or both of these processes. Although accumulating evidence suggests that _S_-nitrosylation is analogous to phosphorylation in regulating the biological activity of many proteins (8, 17, 20, 32, 57, 66, 75), the chemistry of these reactions remains contentious. NO is often a good “leaving group,” allowing further oxidation of thiol to disulfide between neighboring (vicinal) cysteine residues. Alternatively, as NO “leaves” for another reaction partner, the remaining thiol group may react with reactive oxygen species (ROS) to yield sulfenic (-SOH), sulfinic (-SO2H), or sulfonic (-SO3H) acid derivatives on the cysteine residue of the protein (15, 66, 75). _S_-Nitrosylation may also possibly produce a nitroxyl disulfide, in which the NO group is shared by proximate cysteine thiols (22).
Depending on the cellular compartments involved, at low (physiological) levels, NO often mediates cell-protective functions. For example, our group first identified the physiological relevance of _S_-nitrosylation by showing that NO reacts with the NMDAR to downregulate its excessive activity. Under excitotoxic conditions, whereby neuronal cells are damaged by excessive activation of glutamate receptors, _S_-nitrosylation of NMDARs can thus provide neuroprotective affects (32). Specifically, we found that NO can _S_-nitrosylate five different cysteine residues on extracellular domains of NR1 and NR2 subunits of the NMDAR, but cysteine residue #399 (Cys399) on the NR2A subunit mediates ≥90% of NO's effect under ambient conditions (7). From crystal structure models and electrophysiological experiments, we further found that _S_-nitrosylation of Cys399 may induce a conformational change in the receptor protein that enhances glutamate and Zn2+ binding to the receptor. The enhanced binding of glutamate and Zn2+ in turn causes the receptor to desensitize and, consequently, the ion channel to close (33). Interestingly, we also found that the NMDAR becomes more sensitive to inhibition by _S_-nitrosylation when oxygen tension is lowered to the levels observed in normal brain (10–20 torr) as opposed to ambient air (62).
In contrast, high levels of NO are thought to stimulate toxic pathways, depending on source and location of NO. For example, in some cases NO rapidly reacts with superoxide anion, generated from both mitochondrial sources and nonmitochondrial sources (e.g., NADPH oxidase), to form the very toxic product peroxynitrite (ONOO−) (4, 32). Additionally, we and others have also found that excessive (pathophysiological) production of oxidative/nitrosative stress contributes to neuronal cell death through _S_-nitrosylation of a number of targets, including parkin, dynamin related protein 1 (Drp1), PDI, X-linked inhibitor of apoptosis (XIAP), MMP-9, cyclooxygenase-2, N-ethylmaleimide sensitive factor, and GAPDH (15, 17, 23, 32, 63). For instance, recent evidence suggests that the presence of excessive NO-related species may play a significant role in the mechanism of protein misfolding and neuronal damage _via S_-nitrosylation of PDI and parkin. In particular, _S_-nitrosylation inhibits the isomerase and chaperone activities of PDI (66), thus contributing to the accumulation of protein misfolding and subsequent neurotoxicity in models of neurodegenerative disorders. _S_-Nitrosylation of parkin affects its E3 ubiquitin ligase activity, potentially influencing Lewy body formation in Parkinson's disease (8, 75).
Protein-Protein Transnitrosylation
A now classic study of _S_-nitrosylation predicated that the presence of an S_-nitrosylation motif (i.e., an acid-base surrounding a cysteine residue) determines the specificity of this kind of post-translational modification (59). In addition, hydrophobicity in the vicinity of a cysteine residue and low p_Ka may also facilitate _S_-nitrosylation (20). However, these factors may occur in the majority of proteins in vivo, and thus cannot completely account for the selective regulation of signaling pathways by NO. Additional regulation involves compartmentalization of NO reactions, the presence of specific catalytic proteins (e.g., protein nitrosylases, such as hemoglobin (Hb), caspases, and GAPDH, as discussed in more detail below), and possibly the availability of a transition metal to accept an electron from NO.
Recent findings on protein-protein transnitrosylation, whereby an NO moiety is transferred from a protein SNO to a free thiol on another protein, also shed light on the mechanism of NO modulation of specific signaling pathways. Protein-protein transnitrosylation generally takes place when two proteins are directly interacting and possess appropriate redox potentials to allow electron transfer; protein apposition facilitates NO transfer between the two proteins. It is also thought that physical association of the two proteins might result in conformational change, affecting the chemical environment of the critical cysteine residues to facilitate, for example, thiolate anion formation (RS−), which is more susceptible to _S_-nitrosylation and possibly subsequent further oxidation by ROS. When a protein-SNO interacts with a partner protein containing a free thiol, the difference between the redox potentials of the two cysteine residues is a major determinant for NO transfer. Specifically, the protein with the higher redox potential has a tendency to acquire an electron and thus to be reduced (i.e., losing the NO moiety). Accordingly, when a protein with a lower redox potential containing a free thiol surrounded by an appropriate amino acid motif for nitrosylation interacts with a second protein that is _S_-nitrosylated, then transnitrosylation would occur. Given these parameters, only a specific subset of proteins is _S_-nitrosylated, resulting in selective activation/inhibition of particular signaling pathways. Additionally, these findings suggest that not only increased production of NO but also protein-protein interactions may facilitate formation of new protein-SNOs and thereby regulate SNO signaling pathways. Below, we describe recent findings on protein-protein transnitrosylation reactions that affect cell signaling pathways in both health and disease.
Transnitrosylation Between Hb and the Anion Exchanger 1 Mediates NO Release from Erythrocytes
An early important study in this field by Jia et al. revealed that _S_-nitrosylation of Hb is a master regulator of blood pressure in the circulatory system (26). Mammalian Hb contains four subunits (two α chains and two β chains), and each subunit contains a heme group, which includes a central iron molecule. The iron ion in the heme group can bind to oxygen as well as NO to carry these gaseous molecules from the lung to the rest of the body. Interestingly, when NO is present at the heme center, Hb auto-catalyzes _S_-nitrosylation of the Hb β chain at Cys93 to form SNO-Hb, leaving the heme center available for oxygen binding. In oxygen-depleted, peripheral tissues, Hb undergoes a major conformational change from the R to T structure, and SNO-Hb releases the NO group, which after transfer out of red blood cells (RBCs) exerts its effect on blood vessel relaxation. In this manner, blood flow is adjusted for oxygen tension in order to maximize oxygen delivery to tissues.
Protein-protein transnitrosylation has been reported to mediate the extracellular liberation of NO from SNO-Hb. The structural change of Hb under low oxygen levels promotes the binding of SNO-Hb to the cytoplasmic tail of an RBC membrane protein, AE1 (or band 3). AE1 is the most abundant protein on the RBC membrane, and endows RBC membranes with a high permeability to chloride and bicarbonate. The conformational change of Hb is believed to increase the redox potential of Hb-Cys93, inducing an unstable status of SNO-Hb. Pawloski et al. found that, as a consequence of Hb binding to AE1, SNO-Hb transnitrosylates AE1 at the membrane–cytoplasm interface of the RBC (Fig. 1) (47). Subsequently, the NO group detaches from SNO-AE1, is released from the RBC, and diffuses into the smooth muscle cells surrounding blood vessels to exert its vasorelaxant activity. How NO is transferred from SNO-AE1 to the outside of the RBC remains unclear, but the bioactivity of NO may be transferred in the form of a low-molecular-weight SNO, such as GSNO (47). Collectively, this finding was the first report of a physiological protein-protein transnitrosylation, and formed the basis of the notion that other transnitrosylation reactions between two proteins could be important for other cell signal transduction machineries.
FIG. 1.

The pathway exporting NO from erythrocytes requires transnitrosylation of AE1 by SNO-Hb. Hb, which is _S_-nitrosylated at Cys93 (forming SNO-Hb), undergoes conformational change from the R-state to T-state (1), allowing binding to the cytoplasmic tail of AE1 (2). SNO-Hb transnitrosylates AE1, forming SNO-AE1 (3). SNO-AE1 then facilitates export of the NO group out of red blood cells, enabling NO to act as a vasorelaxing factor (4). AE1, anion exchanger 1; Hb, hemoglobin; NO, nitric oxide; SNO, _S_-nitrosothiol.
Transnitrosylation Between Trx and Caspase-3 Regulates Cell Death
Trx or related congeners are known to be present in virtually all organisms throughout the six kingdoms of phylogenetic taxonomy, and generally act as cell-protective factors by preventing oxidative stress (31). With its conserved Cys-Gly-Pro-Cys active site, Trx functions as a major protein disulfide oxidoreductase of the cytosol. For example, the active site cysteines of Trx can donate protons to oxidized peroxiredoxin (Prx), producing an active (reduced) form of Prx that can catabolize hydrogen peroxide. This reaction also results in the formation of a disulfide bond between Cys32 and Cys35 in the Trx active site, thus inactivating Trx. The seleno-flavoprotein TrxR and NADPH then reduce oxidized Trx to regenerate Trx reducing activity.
With its oxidoreductase activity, Trx (and possibly TrxR) can also catalyze denitrosylation of both low-molecular-weight and larger protein SNOs (42, 55, 60). A recent report by Benhar et al. highlighted the denitrosylation activity of Trx under physiologically relevant conditions by showing that Trx denitrosylates caspase-3 (2). Caspase-3 belongs to a cysteine protease family that is important for the execution of apoptotic cell death. Caspase-3 and other caspases are synthesized as inactive zymogens, which possess little or limited protease activity in their pro-state. In response to apoptotic stimuli, upstream caspases (e.g., caspase-8 and -9) cleave and activate caspase-3 to initiate cell death pathways. NO has been reported to constitutively _S_-nitrosylate the pro-form of caspase-3 at its active site cysteine (Cys163); thus, _S_-nitrosylation of caspase-3 is inhibitory. Interestingly, apoptotic stimuli seem to not only activate caspase-3 but also facilitate the selective denitrosylation of the cleaved form of caspase-3, relieving inhibition of the active site cysteine from -SNO (35). While Trx1 can effectively denitrosylate caspase-3 in unstressed conditions, the mitochondrial-specific Trx2 system appears to play a key role in the cell death stimulus-induced denitrosylation of caspase-3 (2). Two biochemical mechanisms for Trx-mediated caspase-3 denitrosylation have been proposed (Fig. 2A). First, the denitrosylation of caspase-3 may proceed through evanescent formation of an intermolecular disulfide bond between Cys32 of Trx2 and Cys163 (the active site cysteine) of caspase-3. Cys35 of Trx subsequently attacks the intermolecular disulfide bridge to generate SNO-free caspase-3 and oxidized Trx (in which case Cys32 and Cys35 form a disulfide bond). An alternative pathway entails transnitrosylation of Trx by caspase-3; SNO-caspase-3 transiently _S_-nitrosylates Cys32 of Trx, thus generating denitrosylated caspase-3 (60). Despite the fact that Trx is an efficient denitrosylase for both the pro-form and cleaved form of caspase-3, selective denitrosylation of the cleaved form of caspase-3 in apoptotic cells had remained uncharacterized. Recent efforts to define the physiological effects of _S_-nitrosylated XIAP, a potent endogenous inhibitor of caspase-3, have suggested that SNO-caspase-3 serves as a nitrosylase for XIAP, resulting in selective reduction of the cleaved (active) form of caspase-3. We discuss transnitrosylation between caspase-3 and XIAP in more detail in a subsequent section.
FIG. 2.

Trx mediates denitrosylation and transnitrosylation of caspase-3. (A) Possible denitrosylation schemes for _S_-nitrosylated caspase-3 by Trx. In reaction 1, Cys32 of Trx attacks SNO-caspase-3, leading to the formation of an intermolecular disulfide between caspase-3 and Trx. This reaction allows denitrosylation of caspase-3. Subsequently, an intramolecular disulfide bridge forms between Cys32 and Cys35 in Trx, releasing the reduced form of caspase-3. In reaction 2, SNO-caspase-3 acts as a nitrosylase, transnitrosylating Trx. This reaction also precipitates denitrosylation of caspase-3. (B) Transfer of an NO group from Trx to caspase-3. After Trx is _S_-nitrosylated at Cys73 (forming SNO-Trx-Cys73), SNO-Trx-Cys73 transnitrosylates Cys163 of caspase-3. This reaction results in the inhibition of caspase-3. Trx, thioredoxin.
Additionally, Trx appears to be _S_-nitrosylated at nonactive site cysteines, including Cys62, 69, and 73 (16, 19, 38, 71, 72). However, a recent report found that stable _S_-nitrosylation of Trx at the nonactive site cysteines can only occur when active site cysteines are oxidized (forming a disulfide bond), probably because oxidized Trx manifests less denitrosylation activity toward itself (72). Under physiological conditions, virtually all cellular Trx proteins are in the reduced (active) form; therefore, _S_-nitrosylation of Trx at nonactive site cysteines may occur only in cells under oxidative stress. Nevertheless, SNO-Trx formation on nonactive site cysteine residues, particularly at Cys73, reportedly possesses transnitrosylation activity toward many cellular targets (72). Among these, caspase-3 is the most well-characterized transnitrosylation target of Trx (Fig. 2B) (38). In this scheme, an NO group transfers from Trx to the active site cysteine of caspase-3, resulting in inhibition of caspase-3 activity. Interestingly, Trx mutants that cannot bind to caspase-3 failed to transnitrosylate caspase-3 (39), supporting the specificity of transnitrosylation between these two proteins.
In summary, Trx seemingly has two diametrically opposed functions: denitrosylation and transnitrosylation of caspase-3. While it is clear that different cysteine residues are responsible for Trx-mediated denitrosylation (Cys32) and transnitrosylation (Cys73) of caspase-3, further studies are warranted to elucidate the mechanism of these seemingly paradoxical effects. For instance, as mentioned previously, transnitrosylatable SNO-Trx-Cys73 may appear only under conditions of oxidative stress. Hence, further work will be necessary to carefully define the relationship of Trx-associated denitrosylation and transnitrosylation activity in cells under severe oxidative/nitrosative stress. Finally, there may be additional transnitrosylation substrates for SNO-Trx-Cys73 (72), but further studies will be required to characterize the physiological roles of these SNO-Trx-Cys73–mediated transnitrosylation substrates.
Caspase-3-Mediated Transnitrosylation of XIAP Regulates Neuronal Cell Death
Inhibitor of apoptosis proteins (IAPs) represent important regulators of apoptosis through their ability to associate with active caspases and repress their catalytic activity (12, 50). In particular, XIAP interacts with active caspases-3/7/9 in the cytosol and is thought to be the most potent endogenous caspase inhibitor among the IAPs. XIAP harbors three copies of the baculovirus IAP repeat (BIR) domain and one RING domain. Characteristic BIR and RING folds contain zinc ions coordinated by histidine and cysteine residues. Biochemical and structural analyses indicate that BIR domains and their flanking sequences bind and inhibit the catalytic activity of apoptotic caspases (13). Additionally, the RING domain of XIAP can act as an E3 ligase, functioning as an ubiquitinase to tag proteins for subsequent degradation by the proteasome. Substrates for the E3 ubiquitin ligase activity of XIAP in vivo include caspases, other IAP proteins, and possibly XIAP itself (34, 52, 61, 67, 74).
Recently, we reported that _S_-nitrosylation of the XIAP-RING domain in a pathophysiologically relevant manner decreases its E3 ubiquitin ligase activity, both in vitro and in intact cells, thereby blocking its ability to inhibit apoptosis by degrading caspases. NMR analysis demonstrated that _S_-nitrosylation of XIAP perturbs the conformation of the RING domain, probably accounting for its suppression of E3 ligase activity. We further found that SNO-XIAP accumulates in neurons during excitotoxic stimulation with NMDA. Moreover, transduction with an _S_-nitrosylation-deficient cysteine mutant of XIAP protected neurons from NMDA-related excitotoxicity, suggesting that SNO-XIAP potentiates cell death under these conditions. Additionally, we observed significant SNO-XIAP formation in brains of patients with Alzheimer's disease (AD), Parkinson's disease, and Huntington's disease, suggesting that this reaction contributes to the etiology of neuronal damage in these disorders (41, 65).
But even more interesting was our finding of the interrelationship between _S_-nitrosylation of caspases and XIAP. Unexpectedly, we found that SNO-caspase-3 transnitrosylated XIAP, thus providing an additional mechanism for proapoptotic signaling. As stated previously, NO has been reported to constitutively _S_-nitrosylate the pro-form of caspase-3. Activation of cell death pathways induces both cleavage and selective denitrosylation of the catalytic cysteine residue of caspase-3, resulting in its activation (35). XIAP can interact only with the cleaved form of caspase-3, not with the pro-form. We observed that the NO group was transferred from caspase-3 to XIAP under pathophysiologically relevant conditions of nitrosative or excitotoxic stress. Since the NO group is transferred from active caspase-3 to XIAP, this transnitrosylation reaction induces (i) denitrosylation of the cleaved form of caspase-3, thus restoring its death-inducing protease activity, and (ii) _S_-nitrosylation of XIAP, to decrease its antiapoptotic activity. Therefore, the consequence of this transnitrosylation is pro-apoptotic for two reasons (Fig. 3). We also found that the pro-form of caspase-3 and the XIAP-D148A mutant, which lack the ability to interact with each other, failed to transnitrosylate one another, indicating that the specific transfer of NO between cleaved caspase-3 and XIAP requires direct protein–protein interaction. Additionally, we found evidence that the transnitrosylation from caspase-3 to XIAP proceeds in vivo by calculating from freshly obtained human postmortem brain the relative redox potential (_ΔEo_′) form the reaction, using a modification of the Nernst equation, as well as the associated Gibbs free energy (_ΔG_o′). This procedure is discussed in a later section.
FIG. 3.

Schematic illustration of the mechanism of SNO-XIAP–mediated apoptotic cell death. (1) Under normal conditions (non-nitrosative conditions), XIAP efficiently binds to and inhibits caspase activity. Additionally, XIAP serves as an E3 ligase that ubiquitinates caspases and thus targets caspases for proteasomal degradation. (2) Under conditions of nitrosative stress, NO inactivates the E3 ligase activity of XIAP _via S_-nitrosylation, thus stabilizing caspases, and sensitizing neurons to apoptotic stimuli, which is mediated by Smac. (3) Caspases are constitutively _S_-nitrosylated, serving as a transnitrosylase for XIAP, producing SNO-XIAP in cells undergoing apoptotic cell death. XIAP, X-linked inhibitor of apoptosis.
Transnitrosylation of Nuclear Proteins by GAPDH Mediates Neuronal Apoptosis
In addition to its well-known role in glycolysis, GAPDH is known to contribute to nuclear signaling pathways that initiate apoptotic cascades (24, 51). Although the sequence encoding GAPDH lacks a nuclear localization signal, translocation of GAPDH from the cytosol to the nucleus occurs in a number of cell systems during apoptosis (24, 51). Hara et al. found a regulated pathway in which NO generated in neurons _S_-nitrosylates the catalytic cysteine of GAPDH (Cys152 for human; Cys150 for mouse and rat), triggering its cytotoxic activity (Fig. 4) (17). _S_-Nitrosylation of GAPDH augments its binding to Siah, which possesses a nuclear localization signal, and thus facilitates translocation of the GAPDH/Siah complex into the nucleus (17). Once in the nucleus, GAPDH stabilizes Siah1, enabling the degradation of nuclear proteins, such as N-CoR, via the ubiquitin E3 ligase activity of Siah1. Hara et al. found that targeting nuclear proteins for degradation by Siah1 can contribute to cell death. Further, SNO-GAPDH influences several other signaling molecules, including p300/CBP. For example, nuclear GAPDH stimulates expression of downstream gene targets of p300/CBP (e.g., the pro-apoptotic p53 gene) to initiate apoptotic cascades (17, 28, 54). Collectively, these studies suggest a potential contribution of SNO-GAPDH-mediated neuronal cell death in the pathogenesis of neurodegenerative diseases.
FIG. 4.

Signaling pathways mediated by S -nitrosylated GAPDH. Toxic levels of NO can _S_-nitrosylate GAPDH in the cytosol. _S_-Nitrosylation of GAPDH promotes interaction between GAPDH and Siah, facilitating nuclear translocation of the GAPDH-Siah1 complex. NO preferentially _S_-nitrosylates GOSPEL to form a SNO-GOSPEL/GAPDH complex when NO levels are relatively low, preventing GAPDH from interacting with Siah1. In the nucleus, the GAPDH-Siah1 complex contributes to neuronal cell injury and death via promoting degradation of nuclear proteins, such as N-CoR, and stimulating expression of genes downstream to p300/CBP, such as p53. Moreover, SNO-GAPDH serves as a nuclear nitrosylase, producing SNO-SIRT1, SNO-HDAC2, and SNO-DNA-PK.
Interestingly, the cytosolic protein GOSPEL, as well as the monoamine oxidase inhibitor R-(-)-deprenyl (deprenyl), manifests neuroprotective effects at least in part by inhibiting the interaction of GAPDH-Siah (18, 53). GOSPEL effectively competes with Siah1 for GAPDH binding, preventing formation of the GAPDH/Siah1 complex. Further, _S_-nitrosylation of GOSPEL at Cys47 enhances the GAPDH–GOSPEL interaction. In the future, it would be interesting to test whether there is transnitrosylation between GAPDH and GOSPEL, as both of these proteins are targets of _S_-nitrosylation and are located in the same complex.
Kornberg et al. also recently reported that GAPDH transnitrosylates several nuclear proteins, including SIRT1, HDAC2, and DNA-PK (28). The specific interaction of GAPDH and SIRT1 facilitates the transnitrosylation reaction because mutations in GAPDH that prevent its binding to SIRT1 abolished transnitrosylation. Kornberg et al. further demonstrated that inhibition of SIRT1 activity in the nucleus is mediated by transnitrosylation from SNO-GAPDH. Consequently, SIRT1 downstream effectors, such as PGC1α, manifest decreased transcriptional activity, suggesting that the SNO-GAPDH-SIRT1 pathway may have physiological relevance not only in neurodegenerative conditions but also in metabolic pathways as well as in normal aging. Additionally, _S_-nitrosylation of HDAC is known to induce chromatin remodeling during neuronal development, promoting dendritic growth and branching via CREB activation (43). Despite the fact that SNO-GAPDH is a well-established mediator of neuronal apoptosis, demonstration of HDAC2 transnitrosylation by GAPDH suggests that SNO-GAPDH can possibly transduce neurotrophic signaling. Therefore, future studies in this area are warranted to elucidate the opposing effects of SNO-GAPDH on neuronal survival via different downstream effectors.
Transnitrosylation of Drp1 by Cdk5 Contributes to Mitochondrial Dysfunction and Synaptic Loss in AD
Cdk5 is a neuronal-specific kinase that is activated by proteins p35 and p25 (30, 64). Cdk5 has been implicated in an array of neuronal functions, including cell survival, axon guidance, neuronal migration, and regulation of synaptic spine density (27, 73, 76). Dysregulation of Cdk5 activity may play a role in several neurodegenerative disorders, including AD, amyotrophic lateral sclerosis, Parkinson's disease, and Huntington's disease (45, 46, 48). We recently demonstrated that aberrant _S_-nitrosylation of Cdk5 at cysteines 83 and 157, which apparently occurs in neurodegenerative disorders but not under normal conditions, results in hyperactivation of the kinase (49). In several neurodegenerative diseases, in particular AD, synaptic damage in the hippocampus and cerebral cortex closely correlates and is therefore thought to underlie cognitive decline. In this context, we showed that _S_-nitrosylation of Cdk5 contributes to amyloid-β (Aβ) peptide-induced synaptic spine loss and that expression of a non-nitrosylatable mutant of Cdk5 reversed these effects. Moreover, we observed significant levels of SNO-Cdk5 in postmortem AD brains but not in normal human brains, suggesting that _S_-nitrosylation of Cdk5 is an aberrant regulatory mechanism of enzyme activity that may contribute to the pathogenesis of AD. However, the mechanism whereby SNO-Cdk5 might injure synapses remained elusive until we realized the relationship of Cdk5 and the mitochondrial fission protein Drp1.
Along these lines, our group recently reported that _S_-nitrosylation of Drp1 at Cys644 mediates Aβ-induced disruption of mitochondrial dynamics and compromise of mitochondrial bioenergetics, consequently contributing to synaptic injury and neuronal damage (6). The mitochondrial fission/fusion machinery proteins are known to maintain mitochondrial integrity and insure that new mitochondria are generated at critical locations. In both familial and sporadic neurodegenerative conditions, abnormal mitochondria regularly appear in the brain as a result of dysfunction in the fission/fusion machinery. Rare genetic mutations in mitochondrial fission/fusion proteins disturb mitochondrial dynamics, causing neurological defects, including certain forms of peripheral neuropathy and optic atrophy (1, 5, 10, 29, 44, 70, 77). Both mitochondrial fusion and fission proteins are widely expressed in human tissues, but since defects preferentially affect nervous tissue, clearly neurons are particularly sensitive to mitochondrial dysfunction.
We found that _S_-nitrosylation of Drp1 causes hyperactivation and excessive mitochondrial fission. Moreover, we observed that Drp1 is _S_-nitrosylated in the brains of virtually all cases of sporadic AD that we examined (6, 68). Emerging evidence suggests that disruption of mitochondrial distribution and bioenergetics in synaptic structures, in part due to abnormal mitochondrial dynamics, can contribute to synaptic loss (69). Consistent with this notion, in response to Aβ, SNO-Drp1 induces excessive mitochondrial fragmentation, producing synaptic damage, an early characteristic feature of AD, and subsequently apoptotic neuronal cell death. Importantly, blockade of Drp1 nitrosylation [using the non-nitrosylatable Drp1(C644A) mutant] prevented Aβ-mediated mitochondrial fission, synaptic loss, and neuronal cell death, suggesting that _S_-nitrosylation of Drp1 contributes to the pathogenesis of AD. Thus, SNO-Drp1 may represent a potential new therapeutic target for protecting neurons and their synapses in AD.
Previously, it was postulated that Cdk5 might activate Drp1 to cause mitochondrial fission, but the pathway remained obscure since direct phosphorylation of Drp1 by Cdk5 was not observed (37). Instead, we found that SNO-Cdk5 potentially contributes to synaptic damage via transfer of the NO group to Drp1 via transnitrosylation. Initially, we found evidence that SNO-Cdk5 could indeed _S_-nitrosylate Drp1 in vitro via transnitrosylation, with consequent formation of SNO-Drp1 (Fig. 5). Next, using a modification of the Nernst equation that we developed in order to predict whether transnitrosylation will indeed proceed in vivo (as described below), we obtained quantitative evidence that transnitrosylation from Cdk5 to Drp1 would favorably proceed in intact cells. These findings suggest that transnitrosylation of Drp1 by SNO-Cdk5 might be involved in the pathway to Aβ-mediated spine loss. Further studies will be required to understand the details of Drp1 transnitrosylation by SNO-Cdk5 in vivo in disease states such as AD. The critical importance of these new findings is that Cdk5 appears to act catalytically by two mechanisms, as a kinase and as a nitrosylase.
FIG. 5.

Schema of SNO-Cdk5 contributing to transnitrosylation of Drp1 and the pathogenesis of AD. In AD, oligomeric Aβ can promote NO production, which results in the formation of SNO-Cdk5. _S_-Nitrosylation of Cdk5 per se activates its activity, thus contributing to neuronal injury. In addition, SNO-Cdk5 transfers an NO group to mitochondrial fission protein Drp1. _S_-Nitrosylated Drp1 manifests excessive fission activity, causing abnormal mitochondrial fragmentation and compromise in mitochondrial bioenergetics, thus impairing synaptic form and function. Aβ, amyloid-β; AD, Alzheimer's disease; Drp1, dynamin related protein 1.
Experimental Approaches for Detection of Protein-Protein Transnitrosylation
The strategy for characterizing protein-protein transnitrosylation involves _S_-nitrosylation assays performed both in vitro and on intact cells (28, 38, 39, 41, 49). Researchers in this field typically employ a biotin-switch assay, a technique pioneered by Jaffrey, Snyder, and colleagues (25), in which _S_-nitrosylated proteins are identified by western blot after substituting SNO(s) with a more stable biotin group via ascorbate-mediated chemical reduction and hence removal of the NO group (Fig. 6A). The in vitro transnitrosylation experiments require two purified proteins that donate or receive an NO group in the transnitrosylation reaction, providing direct evidence for transfer of NO between the two proteins of interest. To determine whether transnitrosylation proceeds, _in vitro_–prepared protein-SNO is mixed with the other purified protein to allow possible transnitrosylation, and subsequently the biotin-switch assay (or another _S_-nitrosylation assay) is used to detect _S_-nitrosylation of the second protein. To demonstrate the specific transfer of NO between the two proteins, one must determine the interaction site of the proteins (if not known) and mutate that amino acid(s) to prevent physical protein–protein interaction. When physical contact is impaired, transnitrosylation should be abolished.
FIG. 6.
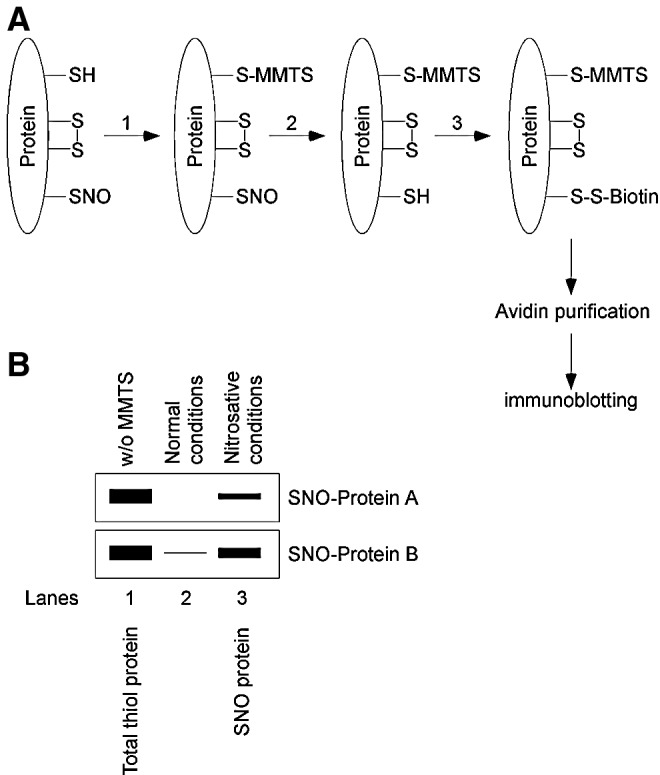
Biotin-switch technique for the calculation of the relative redox potential and the associated change in Gibbs free energy. (A) Overview of the biotin-switch assay. The biotin-switch assay consists of three steps: (1) blocking of free thiol groups by MMTS, (2) conversion of SNOs to free thiols by ascorbate, and (3) labeling newly formed free thiols (formerly _S_-nitrosylated) by biotin-HPDP. Biotinylated proteins are purified by avidin-agarose, and proteins are eluted and analyzed by SDS-PAGE/immunoblotting. (B) Schematic gel image for calculation of standard redox potentials and Gibbs free energy. Cell or tissue lysates are subjected to the biotin-switch assay. MMTS is used to block free thiols during the assay for _S_-nitrosylated protein. “Total thiol protein” represents the relative amount of total protein obtained by the biotin-switch assay performed in the absence of MMTS. “SNO-protein” represents the relative amount of _S_-nitrosylated protein obtained by the biotin-switch assay. Band intensities in lanes 1 and 3 are measured for calculating the standard redox potential using a variant of the Nernst equation and the Gibbs free energy (see text for further details). MMTS, _S_-methyl methanethiosulfonate.
To ascertain whether a transnitrosylation also takes place in cells (i.e., in vivo), one must perform experiments involving overexpression or knockdown of the transnitrosylase, that is, the NO donor protein mediating the transnitrosylation reaction. Although it does not provide direct evidence of transnitrosylation, alteration in the expression of the transnitrosylase should influence _S_-nitrosylation of substrate proteins.
Further, in order to obtain chemical support for transnitrosylation in intact cells, we have recently adapted the Nernst equation to calculate the relative redox potential (_ΔEo_′) and the associated change in Gibbs free energy (_ΔG_o′) associated with a particular transnitrosylation reaction (41, 49). Here, we assume that an NO group is transferred from _S_-nitrosylated protein A (proteinASNO) to the reduced form of protein B (proteinBred), resulting in the formation of reduced Protein A (proteinAred) and _S_-nitrosylated protein B (proteinBSNO).
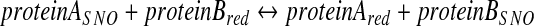 |
(1) |
|---|
To obtain _ΔEo_′ and _ΔG_o′ of the reaction, we expose cells to nitrosative stress, and conduct the biotin-switch assay. Band intensities on the western blots are then analyzed by densitometry; we prefer to use the LICOR Odyssey system for this kind of measurement, due to its wide linear dynamic range. Importantly, in this assay, we include a sample that did not receive the free thiol blocking agent [_S_-methyl methanethiosulfonate (MMTS)] (lane 1 in Fig. 6B), allowing us to observe total protein in the biotin-switch assay [i.e., both the reduced and _S_-nitrosylated (oxidized) forms] and thus to compare that value to the oxidized (_S_-nitrosylated) form. We then acquire the relative ratio of the reduced form (proteinAred or proteinBred) by subtracting the amount of _S_-nitrosylated protein (proteinASNO or proteinBSNO in lane 3 of Fig. 6B) from the total protein (lane 1 of Fig. 6B). Next, these values are used in the following equations (Eqs. 2 and 3, below, which are variants of the Nernst equation) to determine the difference in the standard redox potentials between protein A and protein B, or what we designate as the relative redox potential for the transnitrosylation reaction. Note that this calculation assumes that the transnitrosylation reaction had reached equilibrium when the samples were obtained.
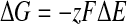 |
(2) |
|---|
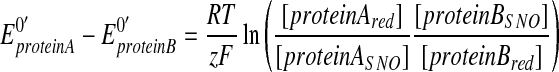 |
(3) |
|---|
Additionally, using these values, we employ the following equation (Eq. 4) to calculate the change in the Gibbs free energy of the transnitrosylation reaction.
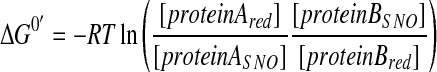 |
(4) |
|---|
When the value of _ΔE_o′ is positive and _ΔG_o′ is negative, this indicates a net energy gain for the transfer of NO from protein A to protein B, suggesting that the transnitrosylation from protein A to B would readily occur in intact cells. For instance, consistent with our finding of transnitrosylation from mature caspase-3 to XIAP, caspase-3 has a higher redox potential than XIAP, producing a positive _ΔE_o′ and a negative value for _ΔG_o′ in this reaction (41).
Conclusions
Both physiological and pathophysiological production of NO triggers many signaling pathways _via S_-nitrosylation of specific protein thiols. _S_-Nitrosylation can either inhibit or activate protein function, depending on the exact site of reaction. For instance, excessively generated NO in the central nervous system is believed to initiate protein misfolding and mitochondrial dysfunction by reacting with specific proteins, thus contributing to neuronal damage and synaptic loss in neurodegenerative diseases.
Recently, we have entered into a new era of _S_-nitrosylation research that will unveil additional NO signaling pathways mediated by the transfer of the NO group between two or more proteins (i.e., protein-protein transnitrosylation). In this review, we have described molecular mechanisms involving transnitrosylation of AE1 by Hb, Trx by caspase-3 (as well as caspase-3 by Trx), XIAP by caspase-3, nuclear proteins (e.g., SITR1) by GAPDH, and Drp1 by Cdk5. These findings have important implications for new drug discovery efforts targeting aberrant transnitrosylation pathways, and suggest that elucidation of additional transnitrosylation pathways should help us attain a more complete understanding of the molecular processes involved in NO-mediated signaling pathways. Importantly, this repertoire of transnitrosylation reactions provides evidence for multiple nitrosylases, that is, enzymes that catalyze _S_-nitrosylation, as each NO donor protein is acting as a nitrosylase in these reactions. Coupled with the fact that several denitrosylating enzymes have recently been described, including the Trx system, class III alcohol deyhdrogenase, and GSNO reductase, the enzymatic machinery involved in reactions of the NO group with cysteine thiol is now becoming more clear.
Abbreviations Used
Aβ
amyloid-β
AD
Alzheimer's disease
AE1
anion exchanger 1
BIR
baculovirus IAP repeat
cGMP
cyclic guanosine-3′,5′-monophosphate
CysNO
_S_-nitrosocysteine
Drp1
dynamin related protein 1
eNOS
endothelial nitric oxide synthase
GSH
glutathione
GSNO
_S_-nitrosoglutathione
Hb
hemoglobin
IAP
inhibitor of apoptosis protein
iNOS
inducible nitric oxide synthase
MMTS
_S_-methyl methanethiosulfonate
NMDAR
_N_-methyl-d-aspartate-type glutamate receptor
nNOS
neuronal nitric oxide synthase
NO
nitric oxide
NOS
nitric oxide synthase
PDI
protein disulfide isomerase
Prx
peroxiredoxin
RBC
red blood cell
ROS
reactive oxygen species
SNO
_S_-nitrosothiol
Trx
thioredoxin
TrxR
Trx reductase
XIAP
X-linked inhibitor of apoptosis
Acknowledgments
This work was supported in part by a grant from the Michael J. Fox Foundation (to T.N. and S.A.L); NIH grants P01 ES016738, P01 HD29587, P30 NS057096, R01 EY05477, and R01 EY09024, the American Parkinson's Disease Association, San Diego Chapter; and an Ellison Senior Scholars Award in Aging (to S.A.L.).
References
- 1.Alexander C. Votruba M. Pesch UE. Thiselton DL. Mayer S. Moore A. Rodriguez M. Kellner U. Leo-Kottler B. Auburger G. Bhattacharya SS. Wissinger B. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 2.Benhar M. Forrester MT. Hess DT. Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benhar M. Forrester MT. Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol. 2009;10:721–732. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 4.Brennan AM. Suh SW. Won SJ. Narasimhan P. Kauppinen TM. Lee H. Edling Y. Chan PH. Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cartoni R. Martinou JC. Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp Neurol. 2009;218:268–273. doi: 10.1016/j.expneurol.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Cho DH. Nakamura T. Fang J. Cieplak P. Godzik A. Gu Z. Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi YB. Tenneti L. Le DA. Ortiz J. Bai G. Chen HS. Lipton SA. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat Neurosci. 2000;3:15–21. doi: 10.1038/71090. [DOI] [PubMed] [Google Scholar]
- 8.Chung KK. Thomas B. Li X. Pletnikova O. Troncoso JC. Marsh L. Dawson VL. Dawson TM. S-nitrosylation of parkin regulates ubiquitination and compromises Parkin's protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 9.Dawson VL. Dawson TM. London ED. Bredt DS. Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci U S A. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delettre C. Lenaers G. Griffoin JM. Gigarel N. Lorenzo C. Belenguer P. Pelloquin L. Grosgeorge J. Turc-Carel C. Perret E. Astarie-Dequeker C. Lasquellec L. Arnaud B. Ducommun B. Kaplan J. Hamel CP. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 11.Doulias PT. Greene JL. Greco TM. Tenopoulou M. Seeholzer SH. Dunbrack RL. Ischiropoulos H. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc Natl Acad Sci U S A. 2010;107:16958–16963. doi: 10.1073/pnas.1008036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eckelman BP. Salvesen GS. Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuentes-Prior P. Salvesen GS. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J. 2004;384:201–232. doi: 10.1042/BJ20041142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garthwaite J. Charles SL. Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature. 1988;336:385–388. doi: 10.1038/336385a0. [DOI] [PubMed] [Google Scholar]
- 15.Gu Z. Kaul M. Yan B. Kridel SJ. Cui J. Strongin A. Smith JW. Liddington RC. Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- 16.Haendeler J. Hoffmann J. Tischler V. Berk BC. Zeiher AM. Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat Cell Biol. 2002;4:743–749. doi: 10.1038/ncb851. [DOI] [PubMed] [Google Scholar]
- 17.Hara MR. Agrawal N. Kim SF. Cascio MB. Fujimuro M. Ozeki Y. Takahashi M. Cheah JH. Tankou SK. Hester LD. Ferris CD. Hayward SD. Snyder SH. Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 18.Hara MR. Thomas B. Cascio MB. Bae BI. Hester LD. Dawson VL. Dawson TM. Sawa A. Snyder SH. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc Natl Acad Sci U S A. 2006;103:3887–3889. doi: 10.1073/pnas.0511321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hashemy SI. Holmgren A. Regulation of the catalytic activity and structure of human thioredoxin 1 via oxidation and S-nitrosylation of cysteine residues. J Biol Chem. 2008;283:21890–21898. doi: 10.1074/jbc.M801047200. [DOI] [PubMed] [Google Scholar]
- 20.Hess DT. Matsumoto A. Kim SO. Marshall HE. Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 21.Ho GP. Selvakumar B. Mukai J. Hester LD. Wang Y. Gogos JA. Snyder SH. S-nitrosylation and S-palmitoylation reciprocally regulate synaptic targeting of PSD-95. Neuron. 2011;71:131–141. doi: 10.1016/j.neuron.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houk KN. Hietbrink BN. Bartberger MD. McCarren PR. Choi BY. Voyksner RD. Stamler JS. Toone EJ. Nitroxyl disulfides, novel intermediates in transnitrosation reactions. J Am Chem Soc. 2003;125:6972–6976. doi: 10.1021/ja029655l. [DOI] [PubMed] [Google Scholar]
- 23.Huang Y. Man HY. Sekine-Aizawa Y. Han Y. Juluri K. Luo H. Cheah J. Lowenstein C. Huganir RL. Snyder SH. S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron. 2005;46:533–540. doi: 10.1016/j.neuron.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 24.Ishitani R. Chuang DM. Glyceraldehyde-3-phosphate dehydrogenase antisense oligodeoxynucleotides protect against cytosine arabinonucleoside-induced apoptosis in cultured cerebellar neurons. Proc Natl Acad Sci U S A. 1996;93:9937–9941. doi: 10.1073/pnas.93.18.9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaffrey SR. Erdjument-Bromage H. Ferris CD. Tempst P. Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 26.Jia L. Bonaventura C. Bonaventura J. Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380:221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 27.Kim Y. Sung JY. Ceglia I. Lee KW. Ahn JH. Halford JM. Kim AM. Kwak SP. Park JB. Ho Ryu S. Schenck A. Bardoni B. Scott JD. Nairn AC. Greengard P. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature. 2006;442:814–817. doi: 10.1038/nature04976. [DOI] [PubMed] [Google Scholar]
- 28.Kornberg MD. Sen N. Hara MR. Juluri KR. Nguyen JV. Snowman AM. Law L. Hester LD. Snyder SH. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol. 2010;12:1094–1100. doi: 10.1038/ncb2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawson VH. Graham BV. Flanigan KM. Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology. 2005;65:197–204. doi: 10.1212/01.wnl.0000168898.76071.70. [DOI] [PubMed] [Google Scholar]
- 30.Lew J. Huang QQ. Qi Z. Winkfein RJ. Aebersold R. Hunt T. Wang JH. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–426. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- 31.Lillig CH. Holmgren A. Thioredoxin and related molecules—from biology to health and disease. Antioxid Redox Signal. 2007;9:25–47. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 32.Lipton SA. Choi YB. Pan ZH. Lei SZ. Chen HS. Sucher NJ. Loscalzo J. Singel DJ. Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 33.Lipton SA. Choi YB. Takahashi H. Zhang D. Li W. Godzik A. Bankston LA. Cysteine regulation of protein function—as exemplified by NMDA-receptor modulation. Trends Neurosci. 2002;25:474–480. doi: 10.1016/s0166-2236(02)02245-2. [DOI] [PubMed] [Google Scholar]
- 34.MacFarlane M. Merrison W. Bratton SB. Cohen GM. Proteasome-mediated degradation of Smac during apoptosis: XIAP promotes Smac ubiquitination in vitro. J Biol Chem. 2002;277:36611–36616. doi: 10.1074/jbc.M200317200. [DOI] [PubMed] [Google Scholar]
- 35.Mannick JB. Hausladen A. Liu L. Hess DT. Zeng M. Miao QX. Kane LS. Gow AJ. Stamler JS. Fas-induced caspase denitrosylation. Science. 1999;284:651–654. doi: 10.1126/science.284.5414.651. [DOI] [PubMed] [Google Scholar]
- 36.Marino SM. Gladyshev VN. Structural analysis of cysteine S-nitrosylation: a modified acid-based motif and the emerging role of trans-nitrosylation. J Mol Biol. 2010;395:844–859. doi: 10.1016/j.jmb.2009.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meuer K. Suppanz IE. Lingor P. Planchamp V. Goricke B. Fichtner L. Braus GH. Dietz GP. Jakobs S. Bahr M. Weishaupt JH. Cyclin-dependent kinase 5 is an upstream regulator of mitochondrial fission during neuronal apoptosis. Cell Death Differ. 2007;14:651–661. doi: 10.1038/sj.cdd.4402087. [DOI] [PubMed] [Google Scholar]
- 38.Mitchell DA. Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol. 2005;1:154–158. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- 39.Mitchell DA. Morton SU. Fernhoff NB. Marletta MA. Thioredoxin is required for S-nitrosation of procaspase-3 and the inhibition of apoptosis in Jurkat cells. Proc Natl Acad Sci U S A. 2007;104:11609–11614. doi: 10.1073/pnas.0704898104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamura T. Lipton SA. Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ. 2011;18:1478–1486. doi: 10.1038/cdd.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakamura T. Wang L. Wong CC. Scott FL. Eckelman BP. Han X. Tzitzilonis C. Meng F. Gu Z. Holland EA. Clemente AT. Okamoto S. Salvesen GS. Riek R. Yates JR., 3rd Lipton SA. Transnitrosylation of XIAP regulates caspase-dependent neuronal cell death. Mol Cell. 2010;39:184–195. doi: 10.1016/j.molcel.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nikitovic D. Holmgren A. S-nitrosoglutathione is cleaved by the thioredoxin system with liberation of glutathione and redox regulating nitric oxide. J Biol Chem. 1996;271:19180–19185. doi: 10.1074/jbc.271.32.19180. [DOI] [PubMed] [Google Scholar]
- 43.Nott A. Watson PM. Robinson JD. Crepaldi L. Riccio A. S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature. 2008;455:411–415. doi: 10.1038/nature07238. [DOI] [PubMed] [Google Scholar]
- 44.Olichon A. Guillou E. Delettre C. Landes T. Arnaune-Pelloquin L. Emorine LJ. Mils V. Daloyau M. Hamel C. Amati-Bonneau P. Bonneau D. Reynier P. Lenaers G. Belenguer P. Mitochondrial dynamics and disease, OPA1. Biochim Biophys Acta. 2006;1763:500–509. doi: 10.1016/j.bbamcr.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 45.Paoletti P. Vila I. Rife M. Lizcano JM. Alberch J. Gines S. Dopaminergic and glutamatergic signaling crosstalk in Huntington's disease neurodegeneration: the role of p25/cyclin-dependent kinase 5. J Neurosci. 2008;28:10090–10101. doi: 10.1523/JNEUROSCI.3237-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patrick GN. Zukerberg L. Nikolic M. de la Monte S. Dikkes P. Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 47.Pawloski JR. Hess DT. Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409:622–626. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- 48.Qu D. Rashidian J. Mount MP. Aleyasin H. Parsanejad M. Lira A. Haque E. Zhang Y. Callaghan S. Daigle M. Rousseaux MW. Slack RS. Albert PR. Vincent I. Woulfe JM. Park DS. Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson's disease. Neuron. 2007;55:37–52. doi: 10.1016/j.neuron.2007.05.033. [DOI] [PubMed] [Google Scholar]
- 49.Qu J. Nakamura T. Cao G. Holland EA. McKercher SR. Lipton SA. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by β-amyloid peptide. Proc Natl Acad Sci U S A. 2011;108:14330–14335. doi: 10.1073/pnas.1105172108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salvesen GS. Duckett CS. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 51.Sawa A. Khan AA. Hester LD. Snyder SH. Glyceraldehyde-3-phosphate dehydrogenase: nuclear translocation participates in neuronal and nonneuronal cell death. Proc Natl Acad Sci U S A. 1997;94:11669–11674. doi: 10.1073/pnas.94.21.11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schile AJ. Garcia-Fernandez M. Steller H. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev. 2008;22:2256–2266. doi: 10.1101/gad.1663108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sen N. Hara MR. Ahmad AS. Cascio MB. Kamiya A. Ehmsen JT. Agrawal N. Hester L. Dore S. Snyder SH. Sawa A. GOSPEL: a neuroprotective protein that binds to GAPDH upon S-nitrosylation. Neuron. 2009;63:81–91. doi: 10.1016/j.neuron.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sen N. Hara MR. Kornberg MD. Cascio MB. Bae BI. Shahani N. Thomas B. Dawson TM. Dawson VL. Snyder SH. Sawa A. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat Cell Biol. 2008;10:866–873. doi: 10.1038/ncb1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sengupta R. Ryter SW. Zuckerbraun BS. Tzeng E. Billiar TR. Stoyanovsky DA. Thioredoxin catalyzes the denitrosation of low-molecular mass and protein S-nitrosothiols. Biochemistry. 2007;46:8472–8483. doi: 10.1021/bi700449x. [DOI] [PubMed] [Google Scholar]
- 56.Seth D. Stamler JS. The SNO-proteome: causation and classifications. Curr Opin Chem Biol. 2011;15:129–136. doi: 10.1016/j.cbpa.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stamler JS. Lamas S. Fang FC. Nitrosylation. The prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 58.Stamler JS. Simon DI. Osborne JA. Mullins ME. Jaraki O. Michel T. Singel DJ. Loscalzo J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A. 1992;89:444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stamler JS. Toone EJ. Lipton SA. Sucher NJ. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 60.Stoyanovsky DA. Tyurina YY. Tyurin VA. Anand D. Mandavia DN. Gius D. Ivanova J. Pitt B. Billiar TR. Kagan VE. Thioredoxin and lipoic acid catalyze the denitrosation of low molecular weight and protein S-nitrosothiols. J Am Chem Soc. 2005;127:15815–15823. doi: 10.1021/ja0529135. [DOI] [PubMed] [Google Scholar]
- 61.Suzuki Y. Nakabayashi Y. Takahashi R. Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc Natl Acad Sci U S A. 2001;98:8662–8667. doi: 10.1073/pnas.161506698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takahashi H. Shin Y. Cho SJ. Zago WM. Nakamura T. Gu Z. Ma Y. Furukawa H. Liddington R. Zhang D. Tong G. Chen HS. Lipton SA. Hypoxia enhances S-nitrosylation-mediated NMDA receptor inhibition via a thiol oxygen sensor motif. Neuron. 2007;53:53–64. doi: 10.1016/j.neuron.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tian J. Kim SF. Hester L. Snyder SH. S-nitrosylation/activation of COX-2 mediates NMDA neurotoxicity. Proc Natl Acad Sci U S A. 2008;105:10537–10540. doi: 10.1073/pnas.0804852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsai LH. Delalle I. Caviness VS., Jr. Chae T. Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- 65.Tsang AH. Lee YI. Ko HS. Savitt JM. Pletnikova O. Troncoso JC. Dawson VL. Dawson TM. Chung KK. S-nitrosylation of XIAP compromises neuronal survival in Parkinson's disease. Proc Natl Acad Sci U S A. 2009;106:4900–4905. doi: 10.1073/pnas.0810595106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Uehara T. Nakamura T. Yao D. Shi ZQ. Gu Z. Ma Y. Masliah E. Nomura Y. Lipton SA. S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 67.Vaux DL. Silke J. IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol. 2005;6:287–297. doi: 10.1038/nrm1621. [DOI] [PubMed] [Google Scholar]
- 68.Wang X. Su B. Lee HG. Li X. Perry G. Smith MA. Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang X. Su B. Zheng L. Perry G. Smith MA. Zhu X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J Neurochem. 2009;109(Suppl 1):153–159. doi: 10.1111/j.1471-4159.2009.05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Waterham HR. Koster J. van Roermund CW. Mooyer PA. Wanders RJ. Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- 71.Weichsel A. Brailey JL. Montfort WR. Buried S-nitrosocysteine revealed in crystal structures of human thioredoxin. Biochemistry. 2007;46:1219–1227. doi: 10.1021/bi061878r. [DOI] [PubMed] [Google Scholar]
- 72.Wu C. Liu T. Chen W. Oka S. Fu C. Jain MR. Parrott AM. Baykal AT. Sadoshima J. Li H. Redox regulatory mechanism of transnitrosylation by thioredoxin. Mol Cell Proteomics. 2010;9:2262–2275. doi: 10.1074/mcp.M110.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xie Z. Sanada K. Samuels BA. Shih H. Tsai LH. Serine 732 phosphorylation of FAK by Cdk5 is important for microtubule organization, nuclear movement, and neuronal migration. Cell. 2003;114:469–482. doi: 10.1016/s0092-8674(03)00605-6. [DOI] [PubMed] [Google Scholar]
- 74.Yang Y. Fang S. Jensen JP. Weissman AM. Ashwell JD. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 2000;288:874–877. doi: 10.1126/science.288.5467.874. [DOI] [PubMed] [Google Scholar]
- 75.Yao D. Gu Z. Nakamura T. Shi ZQ. Ma Y. Gaston B. Palmer LA. Rockenstein EM. Zhang Z. Masliah E. Uehara T. Lipton SA. Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci U S A. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang P. Yu PC. Tsang AH. Chen Y. Fu AK. Fu WY. Chung KK. Ip NY. S-nitrosylation of cyclin-dependent kinase 5 (cdk5) regulates its kinase activity and dendrite growth during neuronal development. J Neurosci. 2010;30:14366–14370. doi: 10.1523/JNEUROSCI.3899-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zuchner S. Mersiyanova IV. Muglia M. Bissar-Tadmouri N. Rochelle J. Dadali EL. Zappia M. Nelis E. Patitucci A. Senderek J. Parman Y. Evgrafov O. Jonghe PD. Takahashi Y. Tsuji S. Pericak-Vance MA. Quattrone A. Battaloglu E. Polyakov AV. Timmerman V. Schroder JM. Vance JM. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]