Cysteine Oxidative Post-translational Modifications: Emerging Regulation in the Cardiovascular System (original) (raw)
. Author manuscript; available in PMC: 2015 Feb 26.
Abstract
In the cardiovascular system, changes in the oxidative balance can affect many aspects of cellular physiology through redox-signaling. Depending on the magnitude, fluctuations in the cell's production of reactive oxygen and nitrogen species can regulate normal metabolic processes, activate protective mechanisms, or be cytotoxic. Reactive oxygen and nitrogen species can have many effects including the post-translational modification of proteins at critical cysteine (Cys) thiols. A subset can act as redox-switches, which elicit functional effects in response to changes in oxidative state. While the general concepts of redox-signaling have been established, the identity and function of many regulatory switches remains unclear. Characterizing the effects of individual modifications is the key to understanding how the cell interprets oxidative signals under physiological and pathological conditions. Here, we review the various Cys oxidative post-translational modifications (Ox-PTMs) and their ability to function as redox-switches that regulate the cell's response to oxidative stimuli. In addition, we discuss how these modifications have the potential to influence other post-translational modifications' signaling pathways though cross-talk. Finally, we review the growing number of tools being developed to identify and quantify the various Cys Ox-PTMs and how this will advance our understanding of redox-regulation.
Keywords: Cys Ox-PTM, redox-switches, cardiac protection, heart failure, proteomics
Over the last two decades, reactive oxygen/nitrogen species (ROS/RNS) have been found to function as important physiological regulators of intracellular signaling pathways.1,2 The specific effects of ROS/RNS are modulated, in part, through the covalent modification of specific cysteine (Cys) residues found within redox-sensitive proteins. Oxidative post-translational modification (Ox-PTM) of Cys residues is an important mechanism that regulates protein structure and ultimately function. The unique properties of a Cys side chain permit a variety of different Ox-PTMs, which potentially result in diverse regulatory effects.3 In this review, we will focus on the unique aspects of Cys Ox-PTMs that have potential to regulate cell phenotype. We also discuss recent findings examining how each Cys residue can have a different reactivity to the Cys Ox-PTMs, provide examples of how Ox-PTMs can modulate the function of different target proteins, and review a small subset of Cys residues that have been found to act as redox-sensors or switches which may compete for different Ox-PTMs. The latter we speculate contributes to the coordination of the cellular response to oxidative stress. Finally, we discuss the growing number of tools being developed to identify and quantify the various Cys Ox-PTMs and how this will continue to advance our understanding of redox-regulation.
1. Unique characteristics of Cys and its Ox-PTMs
The side chain of a Cys residue contains a terminal thiol (-SH) functional group. The sulfur atom at the core of the thiol is electron rich and its d-orbitals allow for multiple oxidation states.4,5 The availability of different oxidation states permits the formation of a diverse range of Ox-PTMs including S-nitrosylation (also called S-nitrosation, SNO), sulfhydration (SSH), S-glutathionylation (SSG), disulfide bonds (RS-SR), sulfenylation (SOH), sulfinic acid (SO2H) and sulfonic acid (SO3H). As shown in Figure 1, most Cys Ox-PTMs are stimulated by diffusible small molecules and are reversible. They can be reduced back to a free thiol (SH) by the antioxidant defense system or converted to other Ox-PTMs depending on the cell's redox-state. The formation of an individual Ox-PTM depends on many factors including the reactivity of the individual Cys residue, its surrounding environment and the composition of the local redox-environment. A summary of the various Ox-PTMs and the redox-chemistry associated with their formation is presented in Table 1. Most Cys Ox-PTMs are induced by reactive oxygen or nitrogen species molecules (ROS/RNS) that react with the free thiol on a Cys side chain.
Figure 1. Ox-PTMs on cysteine.
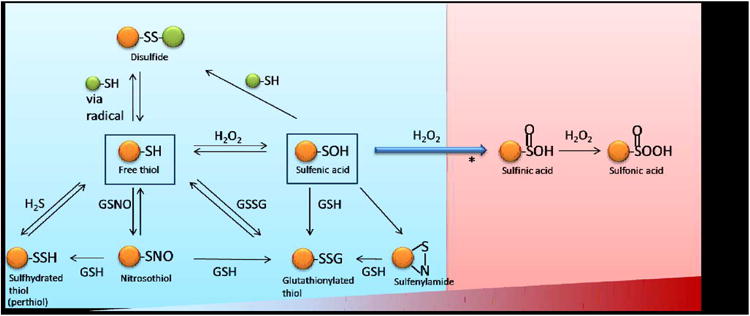
A, Protective mode (Blue), from free thiol, modifications induced by small molecules: sulfhydration, S- nitrosylaion, S-glutahionylation (on bottom) and sulfenylation (in the middle). From sulfenic acid to reversible modifications: disulfide bond formation, sulfenylamide and S-glutathionylation. B, Non-protective/detrimental mode (red), irreversible modification: sulfinic acid and sulfonic acid as ROS level increases (bar on bottom).
*: It has long been regarded this modification is irreversible but recently, there have been examples of enzymatic reduction including sulfiredoxin (Srx), which can reduce sulfinic acid in a subset of Peroxiredoxins (Prxs).23–25
Table 1.
Chemistry and methods of Ox-PTMs on Cys.
| Mediator | PTM | Reactions by mediator (two electro transfer) | Reactions by thiyl radical (single electron transfer) | Methods for detection |
|---|---|---|---|---|
| GSNO N2O3 | Nitrosylation (SNO) | P-SH + RSNO → P-SNO + RSH N2O3 + RSH→P-SNO + HNO | PS• + NO• → P-SNO | -Biotin switch style Biotin-HPDP64 SNO-RAC81 ICAT67 SNOCAP82 CysTMT12 |
| -Organomercury30 | ||||
| H2O2 | Sulfenylation (SOH) | P-SH + H2O2 → P-SOH + H2O | PS• + O2 → PSOO• PSOO• + R SH → RSO• + P-SOH | -Biotin switch style Biotin-HPDP with arsenite.83 OxICAT84,85 |
| -Dimedone chemistry69,70 | ||||
| -Yap1 probe86 | ||||
| H2O2 | Sulfinic acid (SO2H) | P-SOH + H2O2 → P-SO2H + H2O | -Direct detection by MS87 | |
| H2O2 | Sulfonic acid (SO3H) | P-SO2H + H2O2 → P-SO3H + H2O | -Direct detection by MS72,73 | |
| H2S | Sulfhydrylation (SSH) | * | -Biotin switch style17,88 | |
| GSSG | Glutathionylation (SSG) | P-SH + GSSG → P-SSG+GSH | PS• + GS− → PSSG•− PSSG•− + O2 → PSSG + O2•− | -Biotinylated reagent89–91 |
| -Antibody-based assay9,92,93 | ||||
| Disulfide bond formation (RSSR) | P-SOH + P'SH → PSSP' + H2O | PS• + P′-SH− → PSSP′•− PSSP'•− + O2 → PSSP' + O2•- | -2D gel analysis94,95 | |
| -Thiol-Sepharose96,97 |
1a. S-Nitrosylation (SNO)
ROS/RNS can react with a thiol group either by a single or double electron transfer resulting in the formation of an Ox-PTM (Figure 1). Research has mainly focused on reversible Cys Ox-PTMs in cardiovascular biology. Within this research, there is an emerging role for S-nitrosylation (SNO) in cardioprotection.6,7 Seminal papers by Murphy and colleagues6–8 have suggested that SNO-modification is a central phenomenon for cardioprotection. Murphy et al proposed that the SNO-modification of critical thiols during ischemic preconditioning (mild ischemic/oxidative stress) shields them from further oxidative (irreversible) damage during a longer or more severe ischemic insult ensuring that the cell can subsequently regain normal function quickly. This concept is supported by recent work from our group in a canine model of heart failure (HF) in which there is competition among different Ox-PTMs for the same Cys residue (C294) in ATP synthase.9 Excitingly, the actual Ox-PTM occupying this Cys residue differed depending on the phenotype: control vs. HF or when the HF animals were treated with cardiac resynchronization therapy (CRT).10 This suggests that C294 in the ATP synthase alpha subunit may function as a redox-switch.
To determine which Cys residues act as redox-switches requires the identification of residues sensitive to multiple Ox-PTMs. We compiled the first database for all SNO-modified proteins (and the modified site) from the four different proteomic studies on cardiac tissue,8,11,12 endothelial cells12 and smooth muscle cells13 (online Table 1). The SNO-modified proteins were found to be involved in a variety of cellular functions, including energy production. The three methodologies used to capture and identify SNO-modified proteins are discussed later in the review. As shown in Figure 2, there is little overlap in the proteins identified by the various methods although this may, in part, reflect differences in the sample fractionation and sample types. Only 15 proteins are found in common between 2 of the 3 data sets. Of these proteins, 6 are involved in mitochondria/metabolism; the remainder of the group is made up of 1 ion channel and a number of nuclear proteins. In total, 359 SNO-modified Cys were identified which corresponded to 258 non-redundant proteins. This represents less than 15% of 2525 total Cys residues present in these 258 modified proteins (Figure 2A). Of these, the majority of the proteins had only a single SNO-modified Cys (Figure 2B). This suggests that there is a high degree of specificity for the residues that are modified. However, previous attempts to determine a linear amino acid consensus sequence were largely unsuccessful, suggesting other physical characteristics play a role in reactivity. Another interesting aspect of these data sets was the opportunity to assess the subcellular localization of the SNO-modifiable proteins. These proteins were found to be disturbed across many cellular organelles including the mitochondria, nucleus, plasma membrane and cell junction as well as the cytoskeleton (Figure 2C). This indicates a potential for SNO to have a broad effect on the cell. Based on this assumption, we hypothesized that this could be a possible mechanism for a coordinated cellular response to RNS.
Figure 2. Overview of the SNO modified Cys residues.
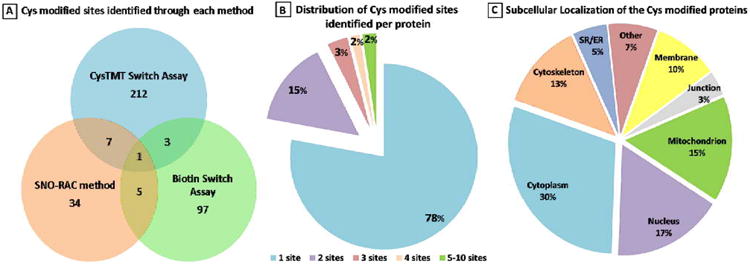
A, The distribution of the 359 modified sites identified based on different experimental protocols is described in the text. B, Distribution of modified sites identified per protein. C, The subcellular localization of modified proteins as determined by annotation in the Uniprot database.
1b. Sulfhydration (SSH)
To date, hydrogen sulfide (H2S) has been proposed mostly to be a physiological vasorelaxant.14,15 Transgenic mice lacking cystathionine gamma-lyase (CSE), an enzyme involved in the synthesis of H2S, were found to be hypertensive.16 Despite its known biological effects, only a small number of target proteins have been identified. Recently, H2S-mediated sulfhydration has been suggested to be an important Cys modification, resulting in the formation of a persulfide bond (R-SSH). Some sulfhydrated proteins have been identified using LC-MS/MS17, although the mechanism for how these modified proteins experience the physiological effects of sulfhydration is not yet clear. Recently, expanding roles for H2S and its target protein were proposed by Tonks et al.18 They suggested that H2S is the key aspect of the endoplasmic reticulum (ER) stress response. H2S produced by CSE in response to ER stress sulfhydrates protein tyrosine phosphatase 1B (PTP1B) and decreases its activity. This results in the inhibition of the PTP1B-mediated dephosphorylation of PERK (protein kinase–like endoplasmic reticulum kinase), thereby facilitating PERK phosphorylation and its activation during the response to ER stress.
1c. S-Glutathionylation (SSG)
S-Glutathionylation is the formation of a reversible R-SSG modification. Glutathionylation forms by a reaction with GSSG or GSNO through either a single electron via thiyl radicals (RS•) or dual electron pathways. An S-nitrosylated thiol is one of the possible intermediates for S-glutathionylation and it can also be formed from sulfenylated thiols or sulfenylamide by reacting with GSH (Figure 1). Some proteins involved in diseases such as cardiovascular diseases, diabetes, lung or neurodegenerative diseases, have been found to be S-glutathionylated.19 Studies for the physiological effects by S-glutathionylation are still being elucidated and one of the studies about ATP synthase is discussed below.
1d. Disulfide bonds (RS-SR)
Disulfide bonds are formed between the thiyl radicals of two independent free thiols that are in close proximity, either within a protein or between proteins (termed an intra- or inter-molecular disulfide bond, respectively). Disulfides can also form through reaction with a sulfenic acid (Figure 1). In this case, the presence of high concentration of ROS will convert SOH groups to thiyl radicals (RS•) which react with other thiolates preferentially to form a disulfide bond.5,20 Disulfide bonds are often involved in protein folding or complex formation and are assumed to induce a static protein conformation.21 Recent work has suggested that some disulfide bonds are dynamic and can confer changes in protein structure and function (see discussion below on ATP synthase).
1e. Sulfenylation (SOH) and irreversible modifications
Sulfenylation is the lowest oxidation state induced by the ROS hydrogen peroxide (H2O2) and superoxide (O2-). It has long been considered as deleterious oxidative damage but over the last 10 years sulfenylation has been emerging as a critical intermediate in redox-signaling.5 Sulfenic acid is reactive, unstable and is believed to be short lived in the cell.22 Because of its reactive nature, sulfenic acid can be converted to other Ox-PTMs, reversible or irreversible modifications depending on the local redox-environment (Figure 1). As a reversible modification, it can react with the abundant tripeptide glutathione (Glu-Cys-Gly), forming S-glutathionylated Cys (RSSG) or react with a nearby available thiol to form a disulfide bond (R-SSR'). Once R-SSG (or R') has formed, it has been suggested that these modifications could protect the host Cys from additional oxidative reactions.5 This is feasible as both R-SSG (and R') are largely reversible by the cell's redox-defense mechanisms.
An alternative path for SOH is to undergo further oxidation directly to the irreversible Ox-PTMs, sulfinic acid (SO2H), and then sulfonic acid (SO3H), both having a higher oxidation state (+2 and +4, respectively). Although there are examples where sulfinic acid modification can be reversed enzymatically,23–25 these Ox-PTMs have been primarily associated with higher levels of ROS/RNS and are largely viewed as irreversible oxidative damage in the cell.
2. Differential reactivity: not all Cys residues are created equally
Not all Cys residues react equally to changes in local redox-state. This diversity in reactivity provides the basis for specificity in ROS/RNS-mediated signaling. As mentioned above, although a given protein may contain numerous Cys amino acids, only a minority of these will have the availability and chemical properties to function as possible target sites for ROS/RNS. Redox-reactivity depends on both known and unknown factors. It is known that the physical availability and pKa of a thiol can impact reactivity of a particular Cys residue. The average pKa of a thiol is around 8.6; however, this can vary from 3.5 to 10 depending on the molecular environment that surrounds the individual thiols.26,27 Thiols act as a nucleophile and become more reactive in their thiolate form (Cys-S-); the lower a thiol's pKa, the more likely it will be deprotonated and reactive. One factor that can lower the pKa is the composition of the neighboring amino acid residues. A thiolate anion is stabilized by positively charged or protonated amino acids but destabilized by negative charges. Wilson and co-workers demonstrated that the oxidative sensitivity of Cys106 in DJ-1 (Parkinson disease protein 7), a protein which protects cells against oxidative stress, is determined by neighboring amino acids' side chains.28 They found that the protonated glutamic acid (E18) reduced pKa of Cys106 from 8.3 to 5.4 by stabilizing the interaction with the thiolate. Despite these influences, no amino acid consensus sequence has been identified for predicting which Cys residues may be targets for redox-regulation. This suggests that pKa, hydrophobicity and primary and secondary structure (including an acid/base pocket near the SNO-reactive Cys)29 may contribute to different degrees toward an individual thiol's reactivity30 rather than to a single unified consensus sequence.12
The consequence of Cys having differences in their pKa is that proteins can vary in their reactivity to Ox-PTMs. Two recent proteomic studies have addressed this point. The first study used a general thiol-reactive iodoacetamide-based probe and identified a large number of Cys-modifiable proteins in three human cell lines.31 The authors reported that general cysteine reactivity predicts that the most highly reactive sites are likely involved in an active site and have greater regulatory potential. In the second study, our group used a novel reagent (CysTMT, described below) to determine the reactivity of Cys specifically for SNO-modifications.12 Endothelial cell lysates exposed to increasing concentrations of NO-donor showed that most of the available Cys residues were not modified. However, the Cys residues that were susceptible to SNO-modification showed a continuum of reactivity. Some Cys residues were very sensitive to SNO-modification while others were less sensitive. Endothelial cells in culture exposed to increasing oxidative stimuli resulted in the modification of more of the insensitive Cys residues. Based on these findings, it is tempting to speculate that differences in thiol-reactivity comprise a coordinated mechanism for the cell to gauge the magnitude of an oxidative stimulus and respond accordingly.
3. Redox-sensors and redox-switches: ATP synthase and alpha actin
Since Cys residues can have different reactivity to oxidation, it is reasonable to hypothesize that each Cys Ox-PTM will have a unique reactivity curve. Thus, for a given ROS/RNS exposure there would be a selective group of Cys containing proteins that may be receptive to different Ox-PTMs. It is this subset of proteins, where various Cys Ox-PTMs compete for a single Cys residue, which could be reactive hubs and act as a cellular redox-sensor capable of monitoring and coordinating the cell's redox-balance. The term redox sensing is commonly used to describe global Cys-dependent control mechanisms while redox signaling refers to a specific signaling process in which a redox element transmits an activation/inhibition signal.32 As such, redox sensors are defined as switches that control the cellular redox homeostasis and redox switches are biochemical components that control the redox state of the reactive Cys in proteins. The concept of Cys redox-switches is emerging as a significant regulatory mechanism in the cardiovascular system and elsewhere.33,34 Modifications of this type have been found to affect cellular signaling, impacting both physiological and pathological pathways.34 In addition, redox compartmentalization can have a dramatic impact on redox switches since different intracellular compartments have different redox characteristics, including protein composition, pH, reducing power, and sources of ROS/RNS generation.35 The following sections review some selected examples of redox-switches involved in cardioprotection, heart failure, ischemia/reperfusion injury and cardiac hypertrophy and specifically focuses on mitochondrial F1FO-ATP synthase and myofibril actin.
3a. ATP synthase
F1FO-ATP synthase (also known as complex V) is located on the mitochondrial inner membrane, chloroplast thylakoid membranes, and bacterial plasma membranes. Its role is to synthesize ATP from ADP and phosphate at the expense of the proton gradient generated by the electron transport chain (ETC) across the mitochondria membrane.36 The F1FO-ATP synthase complex is responsible for the majority of cellular ATP production, and as a result, is very tightly regulated.37 ATP synthase consists of multiple highly conserved core subunits including alpha, beta and gamma, which are located in the F1 catalytic domain and other less conserved subunits which differ between plants and other life forms38–40 (Figure. 3A).
Figure 3. Redox sensor proteins and redox switches.

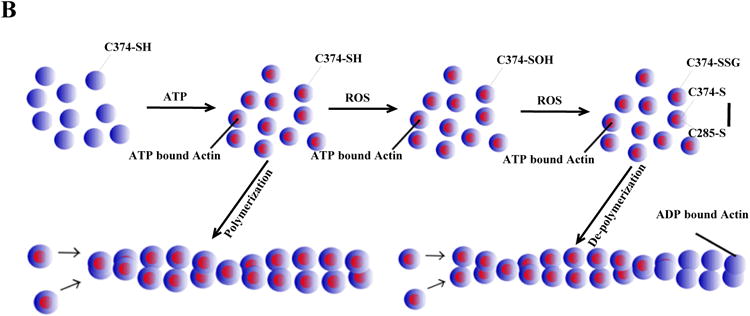
A, ATP synthase α subunit Cys 294 functions as a redox switch. In dyssynchronous heart failure (DHF), Cys294 of the α-subunit are _S_-glutathionylated, or inter-disulfide bonding occurs between Cys294 of alpha-subunits, as well as between Cys294 and Cys103 of the gamma-subunit. Cys cross-linking inhibits its ATP production, leading to mitochondrial dysfunction. Cardiac resynchronization therapy (CRT) increases _S_-nitrosylation of ATP synthase at Cys294 of the alpha-subunit by reverse Cys cross-linking, along with recovered ATPase activity. B, Regulation of actin polymerization through S-glutathionylation of Cys374 of actin.
Redox-regulation of plant chloroplast ATP synthase is well documented.41 It involves the formation and reduction of a disulfide bridge between two Cys residues located in the gamma-subunit. Reduction of the disulfide bond shifts the threshold for ATP synthesis toward lower proton gradients making it easier for the mitochondria to produce ATP.41 Interestingly, the chloroplast redox-sensitive gamma-subunit region is not present in mammalian cells; however, mitochondrial F1FO-ATP synthase complex is still a “hot spot” for Ox-PTMs.
We have directly shown in an animal model of dyssynchronous heart failure (DHF) that the mitochondrial ATP synthase is regulated through the site-specific Ox-PTMs of some of its subunits.9 During heart failure (HF), the ATP synthase alpha subunit forms disulfide bonds between Cys294 on neighboring alpha subunits and to gamma-subunits at Cys103. Furthermore, Cys294 in the alpha-subunit can also be S-glutathionylated and S-nitrosylated. It is noteworthy that so far, only these few sites have indications of regulation by Ox-PTMs and that those sites experience multiple modifications. The formation of disulfide bonds and S-glutathionylation at these regulatory sites is negatively correlated with ATP hydrolytic activity, suggesting these modifications cause profound conformational changes leading to the inactivation of ATP synthase complex. It is interesting to note that during CRT, the only ongoing clinical effective therapy for DHF,10 the disulfide bond at Cys294 has been found to be reversed and replaced by _S_-nitrosylation, which resulted in the recovery of ATPase activity. CRT can increase mitochondrial ATP synthase activity through the reversal of Cys cross-linking of its specific subunits, suggesting that CRT treatment enhances mitochondrial antioxidant defense systems or increases the cellular and mitochondrial reducing status. Reversible Cys Ox-PTMs of ATP synthase subunits are proposed to serve as a protective mechanism to prevent permanent oxidative damage to ATP synthase complex at the expense of decreased ATP production. However, in HF, this could preserve the cellular concentration of ATP, reducing the mitochondrial membrane potential (Δψ), thereby lowering the driving force for Ca2+ uptake into the mitochondrial matrix as suggested by Murphy and colleagues.6 This would ultimately produce a protective phenotype in the heart.
Since Cys294 in the ATP synthase alpha subunit is actively involved in various Ox-PTMs, including intermolecular disulfide bond formation, S-glutathionylation, and S-nitrosylation, each potentially competing with the others for occupancy of this residue, we have suggested that this amino acid functions as a redox-sensor.9 Site-directed mutagenesis analysis confirmed that Cys294 is critical for the functionality of ATP synthase in vitro, and that it plays a critical role in redox-regulation of ATP production.9 The precise mechanism by which these Ox-PTMs form has yet to be determined. Given that Cys294 is located on the surface ATP synthase where it is exposed to the environment and is surrounded by several basic amino acid residues;9,42 it is likely to be deprotonated at physiological pH, making it a good candidate for oxidant attack. With increased oxidative stress in early stages of HF, it is conceivable that Cys294 could be oxidized to a sulfenic acid (although there is no evidence yet) and this initial thiol modification of Cys294 or other post-translational modifications (PTMs)43,44 could cause conformational changes that expose other Cys such as Cys103 of ATP synthase gamma-subunit to subsequent Ox-PTMs. This could result in the formation of an intermolecular disulfide bond or modification by glutathione to form a mixed disulfide bond. Nevertheless, all these modifications cause the reversible inhibition of ATP synthase activity and eventually lead to mitochondrial dysfunction in vivo. CRT treatment, which enhances the cellular antioxidant capacity by increased expression of proteins like peroxiredoxin 3 (Prx3)43, could reverse these dysfunctional disulfide bonds through the increased expression of the Prx/thioredoxin (Trx) pathway. Thus, this single modifiable Cys in ATP synthase appears to act as a redox-sensor of local cellular redox environment and modulate mitochondria ATP production.
3b. Actin
Actin is a highly conserved protein in eukaryotic cells, and its three isoforms, alpha, beta and gamma, share over 90% amino acid sequence homology. All three actin isoforms can participate in the cytoskeleton when monomeric globular G-actin is activated to polymerize into filamentous F-actin upon extracellular stimuli.45 In muscle, filamentous alpha actin is also found in the contractile myofilament proteins where the F-actin thin filament (also comprising tropomyosin and the troponin subunits) interacts with the thick filament which is composed primarily of myosin, the enzyme that hydrolyzes the ATP required to drive muscle contraction.46
Actin polymerization is fundamental to many cellular activities, including motility, cytokinesis, and vesicle traffic.45 There is mounting evidence that the actin system (primarily the beta and gamma isoforms) is one of the most redox-sensitive constituents of the cytoskeleton.47 Recent redox-proteomics studies detected actin as the most prominent protein oxidized in response to exposure of cells to oxidants.48–51 In vitro, treatment of alpha-actin with 5–20 mM H2O2 inhibited actin polymerization.52 In vivo, H2O2 treatment of cells has been found to cause cytoskeletal rearrangements, including F-actin fragmentation and an amassment that correlated with gross cell morphological changes such as membrane blebbing.47
Alpha-actin oxidation was also implicated in human diseases such as HF and ischemia/reperfusion (I/R) injury.53 In human end-stage HF, alpha-actin in the myofilaments was found to be a major target of protein carbonylation and is significantly correlated with both loss of viability and contractile dysfunction.54 Protein carbonylation is an Ox-PTM that occurs to many different amino acid residues (Pro, Arg, Lys and Thr) and is considered to occur during higher levels of oxidative stress.55 Exposure of isolated rat hearts to 30 min global ischemia followed by 60 min reperfusion resulted in 80% increase in alpha-actin carbonyl group which was associated with significant depression of post-ischemia contractile function.56
One of the most studied examples of reversible actin Cys Ox-PTM is the formation of a mixed disulfide between Cys374 and glutathione.48 Several studies indicated that this S-glutathionylation involves the initial oxidative activation of the Cys374 thiol group, likely through a sulfenic acid intermediary, which then reacts with GSH to form the protein-GSH mixed disulfide.50,51 Importantly, Cys374 is conserved among all actin isoforms and the three-dimensional structure reveals that Cys374 is exposed on the surface of the molecule,57 so as to be accessible to solvents and highly mobile. In addition, the surface exposure of Cys374 is not influenced by polymerization into F-actin57 which makes it a good candidate for a redox-switch in the filamentous form, in both the cytoskeleton and myofilament proteins. Impairment of actin S-glutathionylation, either through GSH depletion or expression of the C374A redox-insensitive mutant, greatly affects cell spreading and the formation of stress fibers, leading to inhibition of the disassembly of the actin myosin complex.58 Deglutathionylation of Cys374, likely through glutaredoxin (Grx), leads to an increased rate of actin polymerization.59
Other than S-glutathionylation, actin is also capable of forming disulfide bonds upon external stimuli. During episodes of crisis, like in sickled red blood cells, there is an accumulation of Cys285-Cys374 intra-molecularly disulfide bonded beta-actin, which reduces actin filament dynamics and may help to protect the cell from oxidative stress arising from normal oxidative metabolism and contribute to the cell's general adaptive response to oxidative stress.60 Crystallization of H2O2 treated beta-actin has shown that a Cys374-Cys374 intermolecular disulfide bond can form under such conditions,57 preventing polymerization. Crucially, the reduction of these oxidative Cys modifications likely takes place via the thioredoxin system since thioredoxin1 (Trx1) physically interacts with actin and has an important role in regulation of actin polymerization.51 Given this evidence, it seems actin Cys374 is in a prime position and functions as a redox-switch to help coordinate the response to oxidative stress.
3c. Other examples of proteins modified by Ox-PTMs
The list of proteins involved in cardiovascular diseases that undergo Ox-PTMs in Cys has been increasing over the last several years. For example, Sadoshima and colleagues61,62 have shown the redox-state of HDAC4 plays a role in cardiac hypertrophy, suggesting the molecular mechanism by which Trx1 triggers nuclear translocation of HDAC4 through reduction of Cys residues in HDAC4, thereby inhibiting hypertrophy. HDAC4 at Cys667 and Cys669 are oxidized to form disulfide bonds in response to hypertrophic stimulation and this oxidation on Cys is responsible for cytoplasmic translocation. Trx1, an anti-oxidant protein, forms complex with TBP-2 (thioredoxin binding protein-2) and DnaJb5, a heat shock protein. DnaJb5 is oxidized resulting in a disulfide bond between Cys274 and Cys276 in response to hypertrophic stimulation. Reduction of DnaJb5 by Trx1 through interaction with TBP-2 allows it to associate with HDAC4, whereas the oxidized form of DnaJb5 does not bind to HDAC4. Trx1 can reduce the disulfide bond of Cys667 and Cys669 in HDAC4 in the complex with TBP-2 and DnaJb5. This induces nuclear translocation of HDAC4, thereby attenuating hypertrophy through the suppression of key transcription factors such as NFAT (nuclear factor of activated T cells).
Lovelock and co-workers63 also recently showed a new example of the redox-state of Ox-PTMs in cardiac dysfunction. They demonstrated a significant increase of S-glutathionylation of myosin binding protein C with diastolic dysfunction, further demonstrating the necessity for understanding Ox-PTMs in the cardiovascular system.
4. Experimental tools for the detection of Cys Ox-PTMs
In 2001, Jaffrey and Snyder developed the technique for detection of SNO termed ‘biotin switch assay’.64 Since then, multiple variations of this technique have been presented for the detection of not only S-nitrosylation but also S-glutathionylation, disulfide bonds, sulfhydration and sulfenylation (Table 1). The workflow of the biotin switch assay includes i) blocking all free thiols (non-target), ii) specifically reducing the target Cys Ox-PTM (SNO-modified thiols are reduced by ascorbic acid) and iii) labeling the newly reduced thiol with biotin-HPDP(N-[6-(Biotinamido)hexyl]-3′-(2′-pyridyldithio)-propionamide) which forms a disulfide bond with the available thiol. Since the initial description of the technique, other labeling reagents have been developed including those which alkylate target thiols. Once labeled the SNO-modified proteins can be analyzed by gel-based assay/western blot or be digested and specifically enriched and analyzed by mass spectrometry (MS). This technique has also been modified to detect Cys Ox-PTMs other than SNO. Different reducing reagents are used to specifically reduce the different modifications. Reducing agents such as ascorbate, glutaredoxin and m-arsenite have been used with S-nitrosylation (also for sulfhydration), S-glutathionylation, and sulfenylation, respectively.65,66 Depending on the goals of the study, there are a variety of reagents available, including those for general cellular detection or site identification and relative quantification. The latter has been has been achieved using labeling reagents such as ICAT.67 More recently, we used CysTMT12 to detect and determine individual Cys's reactivity for SNO while Murphy and co-workers68 measured SNO occupancy in the myocardium with the CysTMT.
In the case of sulfenic acid, dimedone chemistry has also been applied to detect sulfenylated proteins.69,70 Dimedone is a cell-permeable small molecule and can trap sulfenic acid specifically by nucleophilic attack. Recently, Carroll and co-workers synthesized dimedone derivative, Dyn-2, which traps sulfenic acids efficiently in cells and can be used to enrich modified proteins using a click-chemistry reaction to an azide bead.71
Unlike SNO or sulfenylation, stable PTMs like sulfinic acid or sulfonic acid can be detected directly by MS.72,73 Recently, Carroll's group74 proposed a new one-step reaction of sulfinic acid in physiological pH with potential utility for the detection of sulfinylated proteins to handle concerns with similar mass shift of sulfinic acid and sulfhydrated species.
5. Future and cross-talk between different signaling systems within the cell
Emerging evidence suggests that Ox-PTMs may work in concert with other PTMs (such as phosphorylation, acetylation, ubiquitination, among others) to determine the ultimate biological outcome and cell phenotype (Figure 4). In this context, it appears that cross-talk between PTMs represent a complex regulatory network with characteristics of a dynamic “code”.75 These networks should be fundamental to normal development and disease pathogenesis.76 In this context, the oxidation of the redox-sensitive Cys must lead to the activation or inactivation of the target enzymes (for example, kinase, Figure 4A) which in turn change the cellular PTM status (e.g. phosphorylation) of their downstream targets. For instance, upon treatment with H2O2, the Iα isoform of the cGMP-dependent protein kinase (PKGIα) forms an intermolecular disulfide bond linking two subunits.77,78 The H2O2-induced protein dimerization directly activates PKG-Iα and leads to the opening of Ca2+-activated K+(BKCa) channel. This participates in the subsequent smooth muscle hyperpolarization and dilation in the human coronary microcirculation.78 Alternatively, it may be possible that other PTMs indirectly regulate the state of Ox-PTM (Figure 4B). For example, Trx1 has been suggested to play a role in denitrosylation. Ma and co-workers have demonstrated that Trx1 can be glycated by lipopolysaccharide and that glycation (not Ox-PTM) inhibited Trx1's antioxidant action.79 The direct effect on S-nitrosylation by glycation still needs to be elucidated but this type of indirect regulation will be promising.
Figure 4. Proposed schemes for indirect (A and B) and direct (C and D) regulation by Ox-PTM of other PTMs and, vice versa.
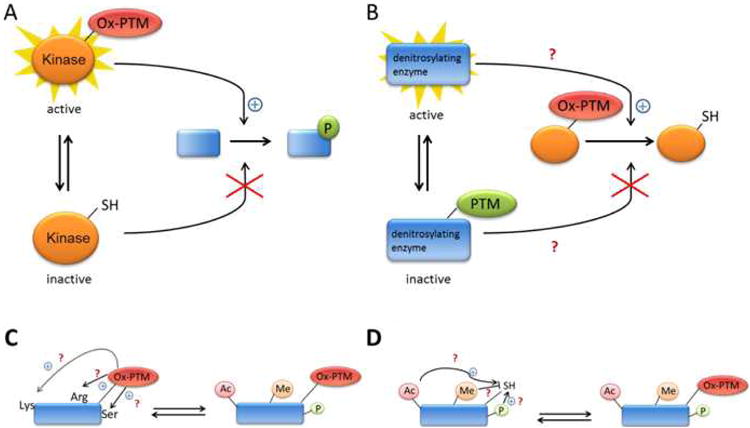
A, Indirect regulation by Ox-PTM of other PTM (ex.phosphorylation).B, Indirect regulation by other PTM (ex.glycation) of Ox-PTM (proposed mechanism). C and D, Direct regulation (cross-talk) between Ox-PTM and other PTMs in the same protein (proposed mechanism).
In addition to the indirect activation effects, it is feasible that Cys Ox-PTMs could enhance or inhibit the addition of another modification (e.g. acetylation, methylation, and phosphorylation) within the same protein to change its activity (cross-talk) (Figure 4C&4D). This is best exemplified by the forkhead transcription factor FoxO4, which plays a major role in the control of cellular proliferation, oxidative stress and apoptosis. FoxO4 has been found to interact with the acetyltransferase p300/CBP via an intermolecular disulfide link mediated through Cys477.80 Formation of the disulfide bond triggers acetylation of FoxO4 and represses transcriptional activity.80 Characterizing these types of interactions will be necessary for understanding how the cell detects and responds to various redox-signals and stresses.
In conclusion, the application of proteomic methods has increased the number of Ox-PTM proteins and the modified Cys residue, at least for some specific modifications. This collective data supports the concept that Ox-PTMs are specific and selective. Furthermore, studies focused on specific protein as well as the occupancy of different Cys Ox-PTMs have increased our understanding of redox regulation. It is also clear that redox regulation is an important component of cardiovascular disease and that it is critical to unravel its complex regulation. In the meantime, the high selectivity inherent of Cys Ox-PTMs will provide novel therapeutic targets for the design of innovative drugs as a means to prevent and manage cardiovascular disease risk.
Supplementary Material
1
Acknowledgments
The authors would like to thank Kate Ferguson for editing the manuscript.
Source of funding: This work was supported by National Heart, Lung, and Blood Institute grants NHLBI-HV-05(2), PO1HL77189-01 and 1RO1HL101235-01 to J.E.V.E. C.I.M. is supported by American Heart Association Pre-Doctoral Fellowship 0815145E
Non-standard Abbreviations and Acronyms
CRT
cardiac resynchronization therapy
Cys
cysteine
CysTMT
Cysteine tandem mass tag
DHF
dyssynchronous heart failure
ETC
electron transport chain
I/R injury
ischemia/reperfusion (I/R) injury
HF
heart failure
H2O2
hydrogen peroxide
LC-MS/MS
liquid chromatography-tandem mass spectrometry
MS
mass spectrometry
Ox-PTM
oxidative post-translational modifications
PTM
post-translational modifications
ROS/RNS
reactive oxygen/nitrogen species
RS-SR
disulfide bonds
SNO
S-nitrosation
SOH
sulfenylation
SO2H
sulfinic acid
SO3H
sulfonic acid
SSH
sulfhydration
SSG
S-glutathionylation
Footnotes
Disclosure: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–28. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 3.Marino SM, Gladyshev VN. Analysis and functional prediction of reactive cysteine residues. J Biol Chem. 2012;287:4419–25. doi: 10.1074/jbc.R111.275578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddie KG, Carroll KS. Expanding the functional diversity of proteins through cysteine oxidation. Curr Opin Chem Biol. 2008;12:746–54. doi: 10.1016/j.cbpa.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 5.Roos G, Messens J. Protein sulfenic acid formation: from cellular damage to redox regulation. Free Radic Biol Med. 2011;51:314–26. doi: 10.1016/j.freeradbiomed.2011.04.031. [DOI] [PubMed] [Google Scholar]
- 6.Sun J, Murphy E. Protein S-nitrosylation and cardioprotection. Circ Res. 2010;106:285–96. doi: 10.1161/CIRCRESAHA.109.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy E, Kohr M, Sun J, Nguyen T, Steenbergen C. S-nitrosylation: a radical way to protect the heart. J Mol Cell Cardiol. 2012;52:568–77. doi: 10.1016/j.yjmcc.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohr MJ, Sun J, Aponte A, Wang G, Gucek M, Murphy E, Steenbergen C. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ Res. 2011;108:418–26. doi: 10.1161/CIRCRESAHA.110.232173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang SB, Foster DB, Rucker J, O'Rourke B, Kass DA, Van Eyk JE. Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circ Res. 2011;109:750–7. doi: 10.1161/CIRCRESAHA.111.246124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kass DA. Pathobiology of cardiac dyssynchrony and resynchronization. Heart Rhythm. 2009;6:1660–5. doi: 10.1016/j.hrthm.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray CI, Kane La, Uhrigshardt H, Wang SB, Van Eyk JE. Site-mapping of in vitro S-nitrosation in cardiac mitochondria: implications for cardioprotection. Mol Cell Proteomics. 2011;10:M110–004721. doi: 10.1074/mcp.M110.004721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murray CI, Uhrigshardt H, O'Meally RN, Cole RN, Van Eyk JE. Identification and quantification of S-nitrosylation by cysteine reactive tandem mass tag switch assay. Mol Cell Proteomics. 2012;11:M111.013441. doi: 10.1074/mcp.M111.013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greco TM, Hodara R, Parastatidis I, Heijnen HFG, Dennehy MK, Liebler DC, Ischiropoulos H. Identification of S-nitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc Natl Acad Sci U S A. 2006;103:7420–5. doi: 10.1073/pnas.0600729103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophysic Res Commun. 1997;237:527–31. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- 15.Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–16. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–90. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krishnan N, Fu C, Pappin DJ, Tonks NK. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal. 2011;4:ra86. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mieyal JJ, Gallogly MM, Qanungo S, Sabens EA, Shelton MD. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid Redox Signal. 2008;10:1941–88. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wardman P, von Sonntag C. Kinetic factors that control the fate of thiyl radicals in cells. Methods Enzymol. 1995;251:31–45. doi: 10.1016/0076-6879(95)51108-3. [DOI] [PubMed] [Google Scholar]
- 21.Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- 22.Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–47. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 23.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–4. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 24.Woo HA, Jeong W, Chang TS, Park KJ, Park SJ, Yang JS, Rhee SG. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J Biol Chem. 2005;280:3125–8. doi: 10.1074/jbc.C400496200. [DOI] [PubMed] [Google Scholar]
- 25.Lowther WT, Haynes AC. Reduction of cysteine sulfinic acid in eukaryotic, typical 2-Cys peroxiredoxins by sulfiredoxin. Antioxid Redox Signal. 2011;15:99–109. doi: 10.1089/ars.2010.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gan ZR, Wells WW. Identification and reactivity of the catalytic site of pig liver thioltransferase. J Biol Chem. 1987;262:6704–7. [PubMed] [Google Scholar]
- 27.Held JM, Gibson BW. Regulatory control or oxidative damage? Proteomic approaches to interrogate the role of cysteine oxidation status in biological processes. Mol Cell Proteomics. 2012;11:R111.013037. doi: 10.1074/mcp.R111.013037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Witt AC, Lakshminarasimhan M, Remington BC, Hasim S, Pozharski E, Wilson MA. Cysteine pKa depression by a protonated glutamic acid in human DJ-1. Biochemistry. 2008;47:7430–7440. doi: 10.1021/bi800282d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marino SM, Gladyshev VN. Structural analysis of cysteine S-nitrosylation: a modified acid-based motif and the emerging role of trans-nitrosylation. J Mol Biol. 2010;395:844–59. doi: 10.1016/j.jmb.2009.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doulias PT, Greene JL, Greco TM, Tenopoulou M, Seeholzer SH, Dunbrack RL, Ischiropoulos H. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc Natl Acad Sci U S A. 2010;107:16958–63. doi: 10.1073/pnas.1008036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–5. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones DP. Redox sensing: orthogonal control in cell cycle and apoptosis signalling. J Intern Med. 2010;268:432–48. doi: 10.1111/j.1365-2796.2010.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tocchetti CG, Stanley BA, Murray CI, Sivakumaran V, Donzelli S, Mancardi D, Pagliaro P, Gao WD, van Eyk J, Kass DA, Wink DA, Paolocci N. Playing with cardiac “redox switches”: the “HNO way” to modulate cardiac function. Antioxid Redox Signal. 2011;14:1687–98. doi: 10.1089/ars.2010.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antelmann H, Helmann JD. Thiol-based redox switches and gene regulation. Antioxid Redox Signal. 2011;14:1049–63. doi: 10.1089/ars.2010.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu Rev Pharmacol Toxicol. 2006;46:215–34. doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- 36.Weber J, Senior AE. ATP synthesis driven by proton transport in F1F0-ATP synthase. FEBS Lett. 2003;545:61–70. doi: 10.1016/s0014-5793(03)00394-6. [DOI] [PubMed] [Google Scholar]
- 37.Capaldi RA, Aggeler R. Mechanism of the F(1)F(0)-type ATP synthase, a biological rotary motor. Trends Biochem Sci. 2002;27:154–60. doi: 10.1016/s0968-0004(01)02051-5. [DOI] [PubMed] [Google Scholar]
- 38.Stock D, Leslie AG, Walker JE. Molecular architecture of the rotary motor in ATP synthase. Science. 1999;286:1700–5. doi: 10.1126/science.286.5445.1700. [DOI] [PubMed] [Google Scholar]
- 39.Kane LA, Van Eyk JE. Post-translational modifications of ATP synthase in the heart: biology and function. J Bioenerg Biomembr. 2009;41:145–50. doi: 10.1007/s10863-009-9218-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balabaskaran Nina P, Dudkina NV, Kane LA, van Eyk JE, Boekema EJ, Mather MW, Vaidya AB. Highly divergent mitochondrial ATP synthase complexes in Tetrahymena thermophila. PLoS Biol. 2010;8:e1000418. doi: 10.1371/journal.pbio.1000418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim Y, Konno H, Sugano Y, Hisabori T. Redox regulation of rotation of the cyanobacterial F1-ATPase containing thiol regulation switch. J Biol Chem. 2011;286:9071–8. doi: 10.1074/jbc.M110.200584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gibbons C, Montgomery MG, Leslie AG, Walker JE. The structure of the central stalk in bovine F(1)-ATPase at 2.4 A resolution. Nat Struct Biol. 2000;7:1055–61. doi: 10.1038/80981. [DOI] [PubMed] [Google Scholar]
- 43.Agnetti G, Kaludercic N, Kane LA, Elliott ST, Guo Y, Chakir K, Samantapudi D, Paolocci N, Tomaselli GF, Kass DA, Van Eyk JE. Modulation of mitochondrial proteome and improved mitochondrial function by biventricular pacing of dyssynchronous failing hearts. Circ Cardiovasc Genet. 2010;3:78–87. doi: 10.1161/CIRCGENETICS.109.871236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kane LA, Youngman MJ, Jensen RE, Van Eyk JE. Phosphorylation of the F(1)F(o) ATP synthase beta subunit: functional and structural consequences assessed in a model system. Circ Res. 2010;106:504–13. doi: 10.1161/CIRCRESAHA.109.214155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dominguez R, Holmes KC. Actin structure and function. Annu Rev Biophys. 2011;40:169–86. doi: 10.1146/annurev-biophys-042910-155359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kobayashi T, Solaro RJ. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu Rev Physiol. 2005;67:39–67. doi: 10.1146/annurev.physiol.67.040403.114025. [DOI] [PubMed] [Google Scholar]
- 47.Dalle-Donne I, Rossi R, Milzani A, Di Simplicio P, Colombo R. The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic Biol Med. 2001;31:1624–32. doi: 10.1016/s0891-5849(01)00749-3. [DOI] [PubMed] [Google Scholar]
- 48.Dalle-Donne I, Giustarini D, Rossi R, Colombo R, Milzani A. Reversible S-glutathionylation of Cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic Biol Med. 2003;34:23–32. doi: 10.1016/s0891-5849(02)01182-6. [DOI] [PubMed] [Google Scholar]
- 49.Dalle-Donne I, Giustarini D, Colombo R, Milzani A, Rossi R. S-glutathionylation in human platelets by a thiol-disulfide exchange-independent mechanism. Free Radic Biol Med. 2005;38:1501–10. doi: 10.1016/j.freeradbiomed.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 50.Johansson M, Lundberg M. Glutathionylation of beta-actin via a cysteinyl sulfenic acid intermediary. BMC Biochem. 2007;8:26. doi: 10.1186/1471-2091-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, Ling S, Zhao D, Sun Q, Li Q, Wu F, Nie J, Qu L, Wang B, Shen X, Bai Y, Li Y, Li Y. Redox regulation of actin by thioredoxin-1 is mediated by the interaction of the proteins via cysteine 62. Antioxid Redox Signal. 2010;13:565–73. doi: 10.1089/ars.2009.2833. [DOI] [PubMed] [Google Scholar]
- 52.DalleDonne I, Milzani A, Colombo R. H2O2-treated actin: assembly and polymer interactions with cross-linking proteins. Biophys J. 1995;69:2710–9. doi: 10.1016/S0006-3495(95)80142-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Canton M, Neverova I, Menabò R, Van Eyk J, Di Lisa F. Evidence of myofibrillar protein oxidation induced by postischemic reperfusion in isolated rat hearts. Am J Physiol Heart Circ Physiol. 2004;286:H870–7. doi: 10.1152/ajpheart.00714.2003. [DOI] [PubMed] [Google Scholar]
- 54.Canton M, Menazza S, Sheeran FL, Polverino de Laureto P, Di Lisa F, Pepe S. Oxidation of myofibrillar proteins in human heart failure. J Am Coll Cardiol. 2011;57:300–9. doi: 10.1016/j.jacc.2010.06.058. [DOI] [PubMed] [Google Scholar]
- 55.Barreiro E, Hussain SNA. Protein carbonylation in skeletal muscles: impact on function. Antioxid Redox Signal. 2010;12:417–29. doi: 10.1089/ars.2009.2808. [DOI] [PubMed] [Google Scholar]
- 56.Powell SR, Gurzenda EM, Wahezi SE. Actin is oxidized during myocardial ischemia. Free Radic Biol Med. 2001;30:1171–6. doi: 10.1016/s0891-5849(01)00514-7. [DOI] [PubMed] [Google Scholar]
- 57.Lassing I, Schmitzberger F, Björnstedt M, Holmgren A, Nordlund P, Schutt CE, Lindberg U. Molecular and structural basis for redox regulation of beta-actin. J Mol Biol. 2007;370:331–48. doi: 10.1016/j.jmb.2007.04.056. [DOI] [PubMed] [Google Scholar]
- 58.Fiaschi T, Cozzi G, Raugei G, Formigli L, Ramponi G, Chiarugi P. Redox regulation of beta-actin during integrin-mediated cell adhesion. J Biol Chem. 2006;281:22983–91. doi: 10.1074/jbc.M603040200. [DOI] [PubMed] [Google Scholar]
- 59.Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal. 2005;7:348–66. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 60.Farah ME, Amberg DC. Conserved actin cysteine residues are oxidative stress sensors that can regulate cell death in yeast. Mol Biol Cell. 2007;18:1359–65. doi: 10.1091/mbc.E06-08-0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, Vatner SF, Sadoshima J. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell. 2008;133:978–93. doi: 10.1016/j.cell.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 62.Oka Sh, Ago I Tetsuro, Kitazono T, Zablocki D, Sadoshima J. The role of redox modulation of class II histone deacetylases in mediating pathological cardiac hypertrophy. J Mol Med (Berl) 2009;87:785–91. doi: 10.1007/s00109-009-0471-2. [DOI] [PubMed] [Google Scholar]
- 63.Lovelock JD, Monasky MM, Jeong EM, Lardin HA, Liu H, Patel BG, Taglieri DM, Gu L, Kumar P, Pokhrel N, Zeng D, Belardinelli L, Sorescu D, Solaro RJ, Dudley SC. Ranolazine improves cardiac diastolic dysfunction through modulation of myofilament calcium sensitivity. Circ Res. 2012;110:841–50. doi: 10.1161/CIRCRESAHA.111.258251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001:l1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 65.Burgoyne JR, Eaton P. Contemporary techniques for detecting and identifying proteins susceptible to reversible thiol oxidation. Biochem Soc Trans. 2011;39:1260–7. doi: 10.1042/BST0391260. [DOI] [PubMed] [Google Scholar]
- 66.Murray CI, Van Eyk JE. Chasing Cysteine Modification: Reflecting the Cellular Redox-Status. Circ Cardiovasc Genet. doi: 10.1161/CIRCGENETICS.111.961425. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–9. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 68.Kohr MJ, Aponte A, Sun J, Gucek M, Steenbergen C, Murphy E. Measurement of S-Nitrosylation Occupancy in the Myocardium With Cysteine-Reactive Tandem Mass Tags. Circ Res. 2012 doi: 10.1161/CIRCRESAHA.112.271320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Charles RL, Schröder E, May G, Free P, Gaffney PRJ, Wait R, Begum S, Heads RJ, Eaton P. Protein sulfenation as a redox sensor: proteomics studies using a novel biotinylated dimedone analogue. Mol Cell Proteomics. 2007;6:1473–84. doi: 10.1074/mcp.M700065-MCP200. [DOI] [PubMed] [Google Scholar]
- 70.Leonard SE, Reddie KG, Carroll KS. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem Biol. 2009;4:783–99. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- 71.Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol. 2012;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kinumi T, Shimomae Y, Arakawa R, Tatsu Y, Shigeri Y, Yumoto N, Niki E. Effective detection of peptides containing cysteine sulfonic acid using matrix-assisted laser desorption/ionization and laser desorption/ionization on porous silicon mass spectrometry. J Mass Spectrom. 2006;41:103–12. doi: 10.1002/jms.973. [DOI] [PubMed] [Google Scholar]
- 73.Medzihradszky KF, Guan S, Maltby DA, Burlingame AL. Sulfopeptide fragmentation in electron-capture and electron-transfer dissociation. J Am Soc Mass Spectrom. 2007;18:1617–24. doi: 10.1016/j.jasms.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 74.Lo Conte M, Carroll KS. Chemoselective ligation of sulfinic acids with aryl-nitroso compounds. Angew Chem Int Ed Engl. 2012;51:6502–5. doi: 10.1002/anie.201201812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 76.Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14:1008–16. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- 77.Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schröder E, Browning DD, Eaton P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317:1393–7. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- 78.Zhang DX, Borbouse L, Gebremedhin D, Mendoza SA, Zinkevich NS, Li R, Gutterman DD. H2O2-induced dilation in human coronary arterioles: role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation. Circ Res. 2012;110:471–80. doi: 10.1161/CIRCRESAHA.111.258871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yuan Y, Jiao X, Lau WB, Wang Y, Christopher TA, Lopez BL, Ramachandrarao SP, Tao L, Ma XL. Thioredoxin glycation: A novel posttranslational modification that inhibits its antioxidant and organ protective actions. Free Radic Biol Med. 2010;49:332–8. doi: 10.1016/j.freeradbiomed.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dansen TB, Smits LMM, van Triest MH, de Keizer PLJ, van Leenen D, Koerkamp MG, Szypowska A, Meppelink A, Brenkman AB, Yodoi J, Holstege FCP, Burgering BMT. Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat Chem Biol. 2009;5:664–72. doi: 10.1038/nchembio.194. [DOI] [PubMed] [Google Scholar]
- 81.Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, Stamler JS. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol. 2009;27:557–9. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Paige JS, Xu G, Stancevic B, Jaffrey SR. Nitrosothiol reactivity profiling identifies S-nitrosylated proteins with unexpected stability. Chemistry & biology. 2008;15:1307–16. doi: 10.1016/j.chembiol.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Burgoyne JR, Eaton P. A rapid approach for the detection, quantification, and discovery of novel sulfenic acid or S-nitrosothiol modified proteins using a biotin-switch method. Methods Enzymol. 2010;473:281–303. doi: 10.1016/S0076-6879(10)73015-9. [DOI] [PubMed] [Google Scholar]
- 84.Sethuraman M, McComb ME, Heibeck T, Costello CE, Cohen RA. Isotope-coded affinity tag approach to identify and quantify oxidant-sensitive protein thiols. Mol Cell Proteomics. 2004;3:273–8. doi: 10.1074/mcp.T300011-MCP200. [DOI] [PubMed] [Google Scholar]
- 85.Sethuraman M, McComb ME, Huang H, Huang S, Heibeck T, Costello CE, Cohen RA. Isotope-coded affinity tag (ICAT) approach to redox proteomics: identification and quantitation of oxidant-sensitive cysteine thiols in complex protein mixtures. J Proteome Res. 2004;3:1228–33. doi: 10.1021/pr049887e. [DOI] [PubMed] [Google Scholar]
- 86.Takanishi CL, Ma LH, Wood MJ. A genetically encoded probe for cysteine sulfenic acid protein modification in vivo. Biochemistry. 2007;46:14725–32. doi: 10.1021/bi701625s. [DOI] [PubMed] [Google Scholar]
- 87.Wagner E, Luche S, Penna L, Chevallet M, Van Dorsselaer A, Leize-Wagner E, Rabilloud T. A method for detection of overoxidation of cysteines: peroxiredoxins are oxidized in vivo at the active-site cysteine during oxidative stress. Biochem J. 2002;366:777–85. doi: 10.1042/BJ20020525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, Kim S, Snyder SH. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol Cell. 2012;45:13–24. doi: 10.1016/j.molcel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sullivan DM, Wehr NB, Fergusson MM, Levine RL, Finkel T. Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry. 2000;39:11121–8. doi: 10.1021/bi0007674. [DOI] [PubMed] [Google Scholar]
- 90.Sullivan DM, Levine RL, Finkel T. Detection and affinity purification of oxidant-sensitive proteins using biotinylated glutathione ethyl ester. Methods Enzymol. 2002;353:101–13. doi: 10.1016/s0076-6879(02)53040-8. [DOI] [PubMed] [Google Scholar]
- 91.Brennan JP, Miller JIA, Fuller W, Wait R, Begum S, Dunn MJ, Eaton P. The utility of N,N-biotinyl glutathione disulfide in the study of protein S-glutathiolation. Mol Cell Proteomics. 2006;5:215–25. doi: 10.1074/mcp.M500212-MCP200. [DOI] [PubMed] [Google Scholar]
- 92.Clavreul N, Bachschmid MM, Hou X, Shi C, Idrizovic A, Ido Y, Pimentel D, Cohen RA. S-glutathiolation of p21ras by peroxynitrite mediates endothelial insulin resistance caused by oxidized low-density lipoprotein. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:2454–61. doi: 10.1161/01.ATV.0000242791.28953.4c. [DOI] [PubMed] [Google Scholar]
- 93.West MB, Hill BG, Xuan YT, Bhatnagar A. Protein glutathiolation by nitric oxide: an intracellular mechanism regulating redox protein modification. FASEB J. 2006;20:1715–7. doi: 10.1096/fj.06-5843fje. [DOI] [PubMed] [Google Scholar]
- 94.Brown JR, Hartley BS. Location of disulphide bridges by diagonal paper electrophoresis. The disulphide bridges of bovine chymotrypsinogen A. Biochem J. 1966;101:214–28. doi: 10.1042/bj1010214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sommer A, Traut RR. Diagonal polyacrylamide-dodecyl sulfate gel electrophoresis for the identification of ribosomal proteins crosslinked with methyl-4-mercaptobutyrimidate. Proc Natl Acad Sci U S A. 1974;71:3946–50. doi: 10.1073/pnas.71.10.3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee K, Lee J, Kim Y, Bae D, Kang KY, Yoon SC, Lim D. Defining the plant disulfide proteome. Electrophoresis. 2004;25:532–41. doi: 10.1002/elps.200305677. [DOI] [PubMed] [Google Scholar]
- 97.Hu W, Tedesco S, McDonagh B, Bárcena JA, Keane C, Sheehan D. Selection of thiol- and disulfide-containing proteins of Escherichia coli on activated thiol-Sepharose. Analytical biochemistry. 2010;398:245–53. doi: 10.1016/j.ab.2009.11.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1