Hyperpolarization Induces a Long-Term Increase in the Spontaneous Firing Rate of Cerebellar Golgi Cells (original) (raw)
Abstract
Golgi cells (GoCs) are inhibitory interneurons that influence the cerebellar cortical response to sensory input by regulating the excitability of the granule cell layer. While GoC inhibition is essential for normal motor coordination, little is known about the circuit dynamics that govern the activity of these cells. In particular, although GoC spontaneous spiking influences the extent of inhibition and gain throughout the granule cell layer, it is not known whether this spontaneous activity can be modulated in a long-term manner. Here we describe a form of long-term plasticity that regulates the spontaneous firing rate of GoCs in the rat cerebellar cortex. We find that membrane hyperpolarization, either by mGluR2 activation of potassium channels, or by somatic current injection, induces a long-lasting increase in GoC spontaneous firing. This spike rate plasticity appears to result from a strong reduction in the spike after hyperpolarization. Pharmacological manipulations suggest the involvement of calcium-calmodulin-dependent kinase II and calcium-activated potassium channels in mediating these firing rate increases. As a consequence of this plasticity, GoC spontaneous spiking is selectively enhanced, but the gain of evoked spiking is unaffected. Hence, this plasticity is well suited for selectively regulating the tonic output of GoCs rather than their sensory-evoked responses.
Introduction
Mossy fibers provide the main source of sensory input to the cerebellar cortex by exciting granule cells, Golgi cells (GoCs), and other interneurons in the granule cell layer (Eccles et al., 1967; Ito, 2006). In turn, GoCs provide the sole source of inhibition onto granule cells and can hence regulate activation of both the granule cell layer and ultimately the entire cerebellar cortical circuitry (Eccles et al., 1964).
Cerebellar GoCs characteristically fire spontaneous action potentials (APs) at frequencies between 1 and 20 Hz (Forti et al., 2006; Ruigrok et al., 2011). This produces a continuous barrage of fast inhibitory currents onto both granule cells and other GoCs. The GABA released by GoCs also activates extrasynaptic high-affinity α6 GABAA receptors on granule cells to produce a tonic inhibitory current (Brickley et al., 1996; Hamann et al., 2002). Recent evidence suggests that this tonic current accounts for the majority of granule cell inhibitory charge in vivo and plays a crucial role in regulating sensory-evoked responses (Duguid et al., 2012). In addition to tonically inhibiting granule cells, GoC spiking can inhibit release from mossy fibers by activating presynaptic GABAB receptors (Mitchell and Silver, 2000). In these ways, the firing rate of GoCs critically determines the ongoing excitability of the granule cell layer.
The rate of GoC firing is also likely important at the behavioral level, as manipulations that alter GoC firing can lead to ataxia in rodents (Watanabe et al., 1998; Cheron et al., 2009). Such severe behavioral consequences could arise from an overall change in the level of GoC inhibition provided to the granule cell layer. It is also possible that the tendency of GoCs to fire synchronously as a population depends on the frequency of GoC spontaneous firing (Gibson et al., 2005; Dugue et al., 2009; Vervaeke et al., 2010). Thus, modulating the tonic rate of GoC firing could provide an important mechanism for regulating activity throughout the granule cell layer.
In this study, we demonstrate that brief hyperpolarization produces a robust long-term increase in the spontaneous firing rate of cerebellar GoCs. This modulation likely involves calcium-calmodulin-dependent kinase II (CaMKII) as well as BK-type calcium-activated potassium channels, and is similar to a form of spike rate plasticity termed firing rate potentiation (FRP), which has been described previously in the vestibular nucleus (Nelson et al., 2003, 2005). Hence, these results suggest that modulation of calcium-activated potassium channels by CaMKII could represent a more widespread mechanism for spontaneously active cells to adjust their activity. However, we find that BK-type channel modulation cannot explain all of the observed plasticity in GoCs. Moreover, we find a strong frequency dependence of the plasticity, with lower initial spike rates allowing larger changes in spiking. Finally, in contrast with cells in the vestibular nucleus, GoC FRP selectively regulates spontaneous spiking without altering the gain of evoked spiking. These data hence suggest that FRP in GoCs is specifically tailored to modulate their tonic inhibitory output rather than their evoked activity.
Materials and Methods
Slices and recordings.
Acute sagittal slices (250 μm thick) were prepared from the cerebellar vermis of postnatal day 17 (P17) to P23 Sprague Dawley rats of either sex. Slices were cut and stored as described previously (Hull and Regehr, 2012). Visually guided whole-cell recordings were obtained with patch pipettes (3–6 MΩ) pulled from borosilicate capillary glass (World Precision Instruments). Electrophysiological recordings were performed at 31°C-33°C.
Spontaneous IPSCs (sIPSCs) were recorded at 0 mV using an internal pipette solution containing the following (in mm): 140 Cs-gluconate, 15 HEPES, 0.5 EGTA, 2 TEA-Cl, 2 MgATP, 0.3 NaGTP, 10 phosphocreatine-tris2, 2 QX 314-Cl, pH 7.2. Current-clamp recordings were performed with an internal solution containing the following (in mm): 150 K-gluconate, 3 KCl, 10 HEPES, 0.5 EGTA, 3 MgATP, 0.5 GTP, 5 phosphocreatine-tris2, and 5 phosphocreatine-Na2, pH 7.2.
For FRP experiments, it was critical to avoid exposing cells to internal solution before patching because this produced a prolonged depolarization block of spiking and greatly reduced the amount of subsequent FRP (likely by preinducing plasticity when spiking was blocked). Hence, pipettes were not positioned near the cell before patching, and positive pressure was not applied until immediately before approaching to form a seal. To check that cells were not in depolarization block, we recorded in cell-attached mode before break-in and then confirmed after break-in that the spike rate was within 1 Hz of the cell-attached spike rate.
No current was injected to regulate GoC spiking before or after induction of FRP. Current-clamp experiments were performed in synaptic blockers of AMPA receptors (NBQX, 5 μm), NMDA receptors (R-CPP, 2.5 μm), GABAA receptors (picrotoxin, 20 μm), and GABAB receptors (CGP55845, 1 μm). Voltage-clamp experiments used the same drugs, except without picrotoxin. For incubation experiments, slices were placed in the recording chamber and perfused with the appropriate drug for ∼30 min before patching. Drugs were purchased from Sigma-Aldrich, Tocris, or Abcam.
Electrophysiological data were acquired using a Multiclamp 700B amplifier (Molecular Devices), digitized at 20 kHz with either a National Instruments USB-6229 or ITC-18 board, and filtered at 2 kHz. Acquisition was controlled with custom software written in IgorPro (Matthew Xu-Friedman, State University of New York at Buffalo).
Analysis.
Linear fits were used to describe input/output relationships. Mean AP waveforms were generated by averaging 5 s of spikes per condition in each experiment, and then by averaging these mean waveforms across experiments. Action potentials were aligned based on the time of the peak derivative. Changes in the AP waveforms were determined by subtraction of the average waveforms for each individual cell per condition, and then by averaging these subtracted waveforms across cells. As we focused on the repolarization of the waveform, difference traces were plotted with a baseline of 0. As is apparent from the superimposed plots of waveforms, differences before the action potential were very small. These differences in waveforms were quantified 2 ms after the peak of the AP (ΔVm2ms) to provide a sensitive measure of spike broadening. Changes in the spike after-hyperpolarization (AHP) were quantified at ΔVm20ms, a time where the AHP is near maximal. Data are reported as mean ± SEM, and statistical analysis was performed using a two-tailed Student's t test.
Results
Our recent study of the inhibitory connection between GoCs provided a clue that GoC spontaneous activity can be modulated (Hull and Regehr, 2012). We took advantage of the fact that GoCs are the only inhibitory cerebellar interneuron that expresses mGluR2 (Ohishi et al., 1994). Consistent with the known role of mGluR2 in opening potassium channels (Watanabe and Nakanishi, 2003), we found that activating these receptors with 2_R_,4_R_-4-aminopyrrolidine-2,4-dicarboxylate (APDC) reduced evoked inhibition onto other GoCs. Here we find that APDC (2 μm) also reduces the frequency of sIPSCs onto other GoCs (65 ± 8% reduction, n = 6; Fig. 1A). Surprisingly, however, when APDC was washed out of the slice, there was an immediate increase in the rate of sIPSCs onto GoCs (46 ± 27% mean increase; Fig. 1A). Based on our previous finding that GoCs provide the majority of GABAergic inhibition onto other GoCs, this observation led us to hypothesize that transient APDC application could increase the rate of spontaneous GoC firing.
Figure 1.

Transient activation of GoC mGluR2 receptors increases spontaneous inhibition by producing a long-term increase in tonic spiking. A, Left, recording configuration for all experiments. Right, Voltage-clamp recording of sIPSCs at 0 mV (top). Example recording shows that the mGluR2 agonist APDC reduces the sIPSC rate (middle), and upon washout the sIPSC rate is elevated (bottom). B, Top, Current-clamp recording reveals that APDC eliminates GoC spontaneous spiking, and spike rate is elevated upon washout. Bottom, Summary time course showing the effect of transient mGluR2 activation on the GoC spike rate (n = 8). C, Average AP waveform in control (top, black) and 15 min after APDC (top, gray) reveals a reduction in the AHP (n = 8). Bottom, Subtracted AP waveforms illustrating the mean difference (±SEM) quantified 20 ms after the AP peak (ΔVm20ms).
To test this hypothesis, we recorded GoC spontaneous spiking before, during, and after APDC application (2 μm for ∼3 min; Fig. 1B). In these experiments, APDC hyperpolarized GoCs to an average membrane potential of −61 ± 2 mV (n = 8). Upon washout of APDC, there was an immediate increase in the rate of GoC spiking. GoCs spike rates then continued to increase for ∼10 min before stabilizing at a rate that was >2-fold faster than it had been before APDC (148.7 ± 37% increase, 5.2 ± 1.2–11.6 ± 1.4 Hz, n = 8). This FRP lasted for the duration of our recordings.
To investigate possible cellular mechanisms underlying this plasticity, we compared the AP waveforms in control conditions with those at least 15 min after APDC application (Fig. 1C). By averaging mean AP waveforms across all experiments in control and post-APDC conditions, we observed a consistent decrease in the magnitude of the GoC AHP without any change in the spike width. Subtraction of the mean waveforms (ΔVm) revealed a decrease in the AHP after washout of APDC, as measured 20 ms after the AP peak (ΔVm20ms = 4.9 ± 0.4 mV).
To determine whether mGluR2 activation produced FRP simply by hyperpolarizing GoCs and transiently suppressing their spontaneous firing, or whether a downstream cascade initiated by mGluR2 activation was required, we performed additional current-clamp experiments. Negative current injections for 3 min were used to suppress spontaneous firing by hyperpolarizing GoCs to ∼−65 mV (−64 ± 1 mV, n = 20; Fig. 2A). After hyperpolarization, there was an immediate increase in GoC spike rates. The spike rates then continued to increase for 10–15 min before stabilizing at nearly double the initial control rate (84 ± 14% increase, 4.3 ± 0.5–7.9 ± 0.8 Hz, n = 20). At the same time, we observed a consistent decrease in the magnitude of the GoC AHP that closely mirrored the time course of spike rate increases. There was a consistent decrease in the AHP 15 min after hyperpolarization (ΔVm20ms = 2.9 ± 0.3 mV; Fig. 2B). These experiments indicate that a brief suppression of GoC firing is sufficient to induce FRP, and mGluR2 activation is not required.
Figure 2.
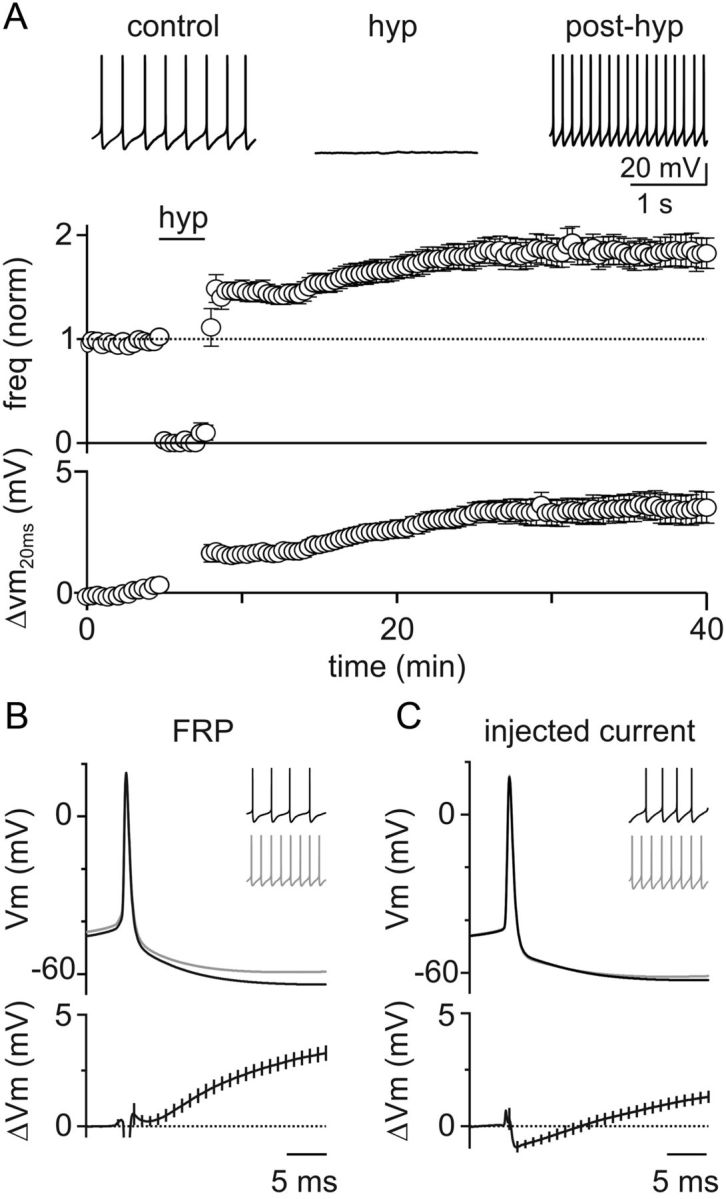
Transient hyperpolarization induces a long-term increase in GoC spiking and a corresponding decrease in spike AHPs. A, Top, Example experiment showing that a 3 min negative current injection of −50 pA hyperpolarizes the GoC membrane to ∼−65 mV and suppresses spiking. An increase in GoC spiking occurred after hyperpolarization. Bottom, Summary of all experiments (n = 20) where GoCs were hyperpolarized with current injection shows a long-term increase in spontaneous spiking and a decrease in AHP amplitudes. B, Mean AP waveform before (top, black) and after (top, gray) hyperpolarization, and difference waveforms (bottom) reveal a reduction in AHPs (n = 20). Inset, 1 s of spiking from an example cell before and after induction of FRP with a 3 min current injection of −75 pA. C, Current injection was used to adjust GoC spike to the average mean rates observed before and after hyperpolarization in A. Averaged APs at low frequency (4.4 ± 0.1 Hz, top black), high frequency (7.7 ± 0.1 Hz, top gray), and the mean difference trace (bottom, black) reveal little change in the waveforms at low and high frequency. Inset, 1 s of spiking from an example cell before and after a 7 pA current injection.
The reduction in spike AHP after FRP induction could reflect modulation of an ion channel, or it could simply be a consequence of increased GoC spike rates. To discriminate between these possibilities, we modulated GoC spike rates with positive current injections under control conditions and measured the resulting AP waveform changes (Fig. 2C). In these experiments, we matched the absolute spike rates to the averages obtained in FRP experiments (4.4 ± 0.1 Hz and 7.7 ± 0.1 Hz, n = 11). The waveform changes associated with differences in firing frequency were significantly smaller than those associated with FRP (ΔVm20ms = 1.0 ± 0.3 mV, p = 0.0003). These data suggest that FRP may involve modulation of the currents underlying the spike AHP.
To investigate the cellular mechanism underlying this AHP change, we began by testing the involvement of CaMKII, which has been implicated in a similar form of spike rate plasticity in the vestibular nucleus (Nelson et al., 2005). In the vestibular nucleus, membrane hyperpolarization reduces CaMKII activity by lowering AP-evoked calcium influx, which in turn leads to a long-term reduction in the overall BK channel conductance by reducing the open probability of individual channels (van Welie and du Lac, 2011). This mechanism decreases the AHP of vestibular nucleus cells and produces a long-term increase in their spontaneous firing rate (Nelson et al., 2003). If this mechanism also controls GoC FRP, then inhibiting CaMKII should mimic FRP by increasing spontaneous firing rates, and should also occlude subsequent induction of FRP by hyperpolarization.
In GoCs, we found that the CaMKII inhibitor KN-62 (10 μm) increased spike rates to a similar degree as the induction of FRP with hyperpolarization (65 ± 19% increase, n = 7; Fig. 3A). This increase in spike rate was also associated with a decrease in the GoC AHP, similar to what was observed during FRP (ΔVm20ms = 3.1 ± 0.5 mV, p = 0.74). Moreover, incubation of slices in KN-62 largely occluded hyperpolarization-evoked FRP (frequency = 19 ± 8% of control, n = 6; Fig. 3B) and produced a much smaller change in the AHP after hyperpolarization compared with controls (ΔVm20ms = 1.3 ± 0.5 mV, p = 0.012). These data suggest that GoC FRP involves a CaMKII-dependent long-term modification of an ion channel involved in the GoC AHP.
Figure 3.

FRP involves CaMKII and BK-type potassium channels. Current-clamp experiments were performed in which the spontaneous firing of GoCs and the GoC spike waveform was monitored. A, C, E, The wash-in effect of the CaMKII inhibitor KN-62 (A, n = 7), the SK-type potassium channel antagonist apamin (C, n = 7), and the BK-type potassium channel inhibitor paxilline (E, n = 10) was assessed. B, D, F, The effect of a 3 min somatic hyperpolarization was also assessed in slices incubated in the presence of KN-62 (B, n = 6), apamin (D, n = 10), and paxilline (F, n = 17).
To identify the target of CaMKII modulation, we focused on GoC conductances known to regulate spontaneous firing and contribute to their AHP. We therefore tested the role of small and large conductance calcium-activated potassium channels (SK and BK channels, respectively), both of which are present in GoCs (Forti et al., 2006). Application of the selective SK channel antagonist apamin (150 nm) significantly increased spontaneous GoC firing (192 ± 50% increase, n = 7) and greatly reduced the GoC AHP (ΔVm20ms = 6.9 ± 1.5 mV; Fig. 3C). Many GoCs that were incubated in apamin entered a sustained depolarized state and were unable to fire normal rhythmic action potentials (6 of 16 cells). In GoCs that fired spontaneously in the presence of apamin, hyperpolarization was still able to evoke FRP (76 ± 36% increase, n = 10; Fig. 3D), suggesting that the bulk of FRP is not a consequence of SK channel modulation. Changes in the AHP after hyperpolarization, however, were smaller than in controls (ΔVm20ms = 1.6 ± 0.5 mV, p = 0.036; Fig. 3D). These data suggest that, after the dramatic decrease in the GoC AHP when SK channels are blocked, additional small changes in the AHP can significantly change the overall spike rate.
We also examined the involvement of BK channels in FRP. The selective BK-channel antagonist paxilline (10 μm) increased GoC spike rates >2-fold (145 ± 38% increase, n = 10; Fig. 3E) and greatly reduced the GoC AHP (ΔVm20ms = 4.0 ± 0.6 mV). In addition, paxilline also broadened the APs (control ΔVm2ms = 0.3 ± 0.2 mV, paxilline ΔVm2ms = 6.8 ± 1.3 mV, p = 0.0001). Paxilline incubation, however, largely occluded FRP (24 ± 13% increase, n = 17; Fig. 3F), and there was very little change in the GoC AHP after hyperpolarization compared with controls (ΔVm20ms = 1.3 ± 0.4 mV, p = 0.0025).
It was not always possible to test the contribution of different channel types to FRP using pharmacology. For example, the role of Ih in FRP could not be assessed because the selective Ih blocker ZD7288 disrupted firing and led to unstable, depolarized membrane potentials along with prolonged membrane oscillations after FRP induction. We also tried to determine whether simultaneous blockade of SK and BK channels led to a more complete suppression of FRP. However, coapplication of paxilline and apamin had such a profound effect on membrane repolarization that most GoCs went into a depolarized state and could not fire action potentials. These findings indicate that stable spontaneous activity in GoCs involves multiple channels, and removing more than one channel type critically destabilizes their pacemaking ability. Moreover, these findings place practical limitations on the use of pharmacological approaches to study the role of ion channels in FRP.
Our findings suggest that the mechanism of FRP in GoCs has many similarities to the FRP previously described in vestibular nucleus neurons. These similarities include a comparable decrease in the AHP, insensitivity to SK channel blockade, and occlusion of FRP by BK channel blockade. However, in GoCs, we find that paxilline both reduces the spike AHP and broadens the spikes. These data suggest that BK channels with both fast and slow kinetics are present in GoCs and that the contributions of BK channels to GoC firing might be complex.
To clarify the role of BK channels in GoC FRP, we performed additional experiments. First, we sought to resolve the differential effect of paxilline and FRP on the GoC action potential by applying paxilline after FRP induction. In contrast with experiments where paxilline was applied without inducing FRP, we found that paxilline had a very small effect on the AHP after FRP induction (ΔVm20ms = 1.8 ± 0.4 mV, p = 0.0061). Furthermore, paxilline did not further increase GoC firing rates above FRP levels (FRP = 170 ± 42% of control, paxilline = 167 ± 41% of control, n = 7; Fig. 4A). However, paxilline still had a large effect on the width of APs (ΔVm2ms = 5.0 ± 1.2 mV). This selective effect on AP width was quite different from the effect of paxilline application before inducing FRP, which both broadened APs and reduced the spike AHP (Fig. 4B). These experiments reveal that the slow component of the AHP modulated by FRP is much smaller than the total paxilline-sensitive component of the AHP (Fig. 4B, arrow). In addition, they suggest that paxilline blocks BK channel subtypes with both fast and slow kinetics (Brenner et al., 2005), whereas FRP involves a selective modulation of a subset of slow BK channels active during the AHP.
Figure 4.

GoC FRP involves a subset of BK-type potassium channels. A, Left, Paxilline was applied after the induction of FRP (n = 7). Right, FRP reduced the spike AHP (blue), whereas paxilline selectively broadened the AP (red). B, Comparison of the difference waveforms for paxilline applied without FRP induction (Fig. 3, black) and after FRP induction (red) reveals that paxilline has a much larger effect on the AHP (later time points) when applied without inducing FRP. The paxilline-sensitive component of FRP is small and late in the AHP (double arrow). C, Iberiotoxin wash-in elevates the spike rate of GoCs and selectively decreases the spike AHP. D, Iberiotoxin incubation does not occlude FRP, and the spike AHP is still significantly reduced after induction of plasticity.
The finding that BK channels can contribute rapidly to the width of APs and more slowly to spike AHPs is consistent with the behavior of these channels in other cell types. Previous studies have established that the presence or absence of the β4 subunit can influence the kinetics of BK channels (Brenner et al., 2005). The peptide inhibitor iberiotoxin is thought to target a subset of BK channels lacking the β4 subunit (Meera et al., 1997) and is thus a promising tool for identifying the subpopulation of BK channels that specifically contribute to FRP. In the vestibular nucleus, iberiotoxin selectively affects the slow AHP and completely occludes FRP (Nelson et al., 2003).
To segregate the contribution of distinct subtypes of BK channels to GoC FRP, we tested the effects of iberiotoxin on GoC spiking and plasticity. Iberiotoxin (150 nm) increased the spike rate of GoCs (124 ± 36% increase, n = 7; Fig. 4C) and reduced the GoC spike AHP (ΔVm20ms = 2.5 ± 0.3 mV) without broadening the AP waveform (ΔVm2ms = 0.3 ± 0.2 mV). However, when slices were incubated in iberiotoxin, membrane hyperpolarization still produced robust FRP (FRP = 111 ± 22% of control, n = 8; Fig. 4D) and a decrease in the spike AHP (ΔVm20ms = 3.3 ± 0.4 mV). It is surprising that FRP remained intact in the presence of iberiotoxin given that (1) paxilline blocked FRP, (2) iberiotoxin selectively decreased the AHP without affecting the spike width, and (3) iberiotoxin completely occludes FRP in the vestibular nucleus (Nelson et al., 2003).
These surprising differences between BK channel antagonists led us to reexamine our FRP experiments and test whether any other recording parameter might provide further insight to our pharmacological manipulations. Hence, we tested the role of initial spike rate in all our experiments on an individual cell basis (Fig. 5). We first examined the firing rate of GoCs after each pharmacological drug wash-in as a function of initial spike rate (Fig. 5A). For wash-ins of each drug, this analysis revealed a strong correlation, with lower initial spike rates favoring larger rate enhancements. At initial spike rates >5 Hz, there was little or no increase in spiking in for drug wash-ins.
Figure 5.

The magnitude of GoC FRP depends on initial firing rate. A, In all drug wash-ins, the increase in firing rate was greatest for cells with low initial firing rates. Little or no increase in firing rate was observed for initial firing rates >5 Hz. B, The magnitude of FRP was also dependent on initial firing rates, with lower firing rates producing the largest FRP. However, FRP was present at initial rates >5 Hz. C, For most drug conditions, FRP was also dependent on initial firing rate, and little or no FRP was evident for initial firing rates >5 Hz. D, Summary of FRP magnitude for cells with initial firing rates <5 Hz. Whereas FRP was still occluded by KN62, the effect of paxilline was reduced in cells with lower initial firing rates.
These data led us to question whether FRP might have a similar frequency dependence. For FRP induced with both hyperpolarization and APDC, we also found that lower initial spike rates led to larger FRP (Fig. 5B). In these experiments, however, even spike rates >5 Hz consistently led to an ∼50% enhancement of spiking (FRP, >5 Hz initial spike rate: 52 ± 8% of control, n = 9). These data suggest that some of the differences we observed between pharmacological conditions may involve variability in the initial spike rates. Therefore, we next examined the magnitude of FRP in different drug conditions as a function of initial spike rates (Fig. 5C).
For most drug conditions, we observed larger FRP after lower initial spike rates (Fig. 5C). In addition, we observed little or no enhancement of spiking in any drug condition where initial spike rates were >5 Hz. Interestingly, whereas apamin and iberiotoxin increased spike rates to a similar extent as paxilline in our wash-in experiments (Figs. 3 and 4), we found very few cells with spike rates >5 Hz in apamin or iberiotoxin after incubation of slices in these drugs. These data may suggest some type of compensation that allows spike rates to return to near control levels after extended periods of SK or β4-lacking BK channel blockade.
To determine whether variability in the initial spike rates could affect our interpretation of FRP experiments in various drug conditions, we reanalyzed our FRP data for all cells with initial spike rates <5 Hz (Fig. 5D). FRP induced by hyperpolarization (control) was increased in cells with initial spike rates <5 Hz (control FRP = 127 ± 17%, n = 20). However, KN62 still largely occluded FRP in this restricted population (FRP = 21 ± 9% of control, n = 5). In contrast, paxilline had less impact on FRP for cells with low initial firing rates (FRP = 77 ± 28% of control, n = 5). As in the total population, apamin and iberiotoxin had no significant effect on FRP (apamin FRP = 130 ± 41% of control, n = 5, p = 0.925; iberiotoxin FRP = 123 ± 28% of control, n = 7, p = 0.930). Although FRP in paxilline was still somewhat reduced relative to control cells that had initial spike rates <5 Hz, these data suggest that BK channel modulation cannot account for the all of the FRP in GoCs.
To further explore the conditions required for inducing FRP in GoCs, we tested the duration of hyperpolarization necessary for plasticity (Fig. 6). Spike rates were stable in the absence of hyperpolarization (1 ± 20% of control, n = 8; Fig. 6A), or after 5 s of hyperpolarization (8 ± 23% of control, n = 6). In contrast, hyperpolarizing for 20 s induced robust FRP (84 ± 45% increase, n = 10). This plasticity was slightly less, but not significantly different from, FRP induced by 1 min (97 ± 19% increase, n = 15, p = 0.766) or 3 min of hyperpolarization (112 ± 17% increase, n = 22, p = 0.472; Fig. 6B). Hence, a hyperpolarization of just 20 s is sufficient to induce large increases in GoC spiking.
Figure 6.

Time dependence of FRP induction. A, GoC spike rates were recorded after hyperpolarizations of 0 (n = 8), 5 (n = 6), 20 (n = 10), 60 (n = 15), and 180 (n = 22) seconds. Although 5 s hyperpolarizations did not increase spike rates, longer durations each produced a nearly twofold increase in spiking. Data points represent 20 s intervals, resulting in a non-0 spiking value for 5 s of hyperpolarization B, Summary of GoC spike rates as a function of hyperpolarization time.
To explore the consequences of FRP on GoC function, we tested their responses to injected current (F/I curves) before and after induction of FRP (Fig. 7). In the vestibular nucleus, FRP prominently increases the slope of F/I curves (Nelson et al., 2003), which is thought to be important for setting the gain of the vestibulo-ocular reflex at the behavioral level. In GoCs, however, we find that FRP produces only an offset change in the F/I relationship (control intercept = 1.67 ± 1.10 Hz, FRP intercept = 5.03 ± 1.43 Hz, n = 10), without altering the slopes of the F/I curves (control slope = 0.078 ± 0.006 Hz/pA, FRP slope = 0.077 ± 0.007 Hz/pA, p = 0.91). Hence, FRP produces the largest differences in firing rate in response to the smallest current injections, and there is essentially no change in the firing evoked by large current injections. These data suggest that FRP will play a primary role in regulating the tonic output of GoCs but will have little influence on their sensory-evoked output.
Figure 7.
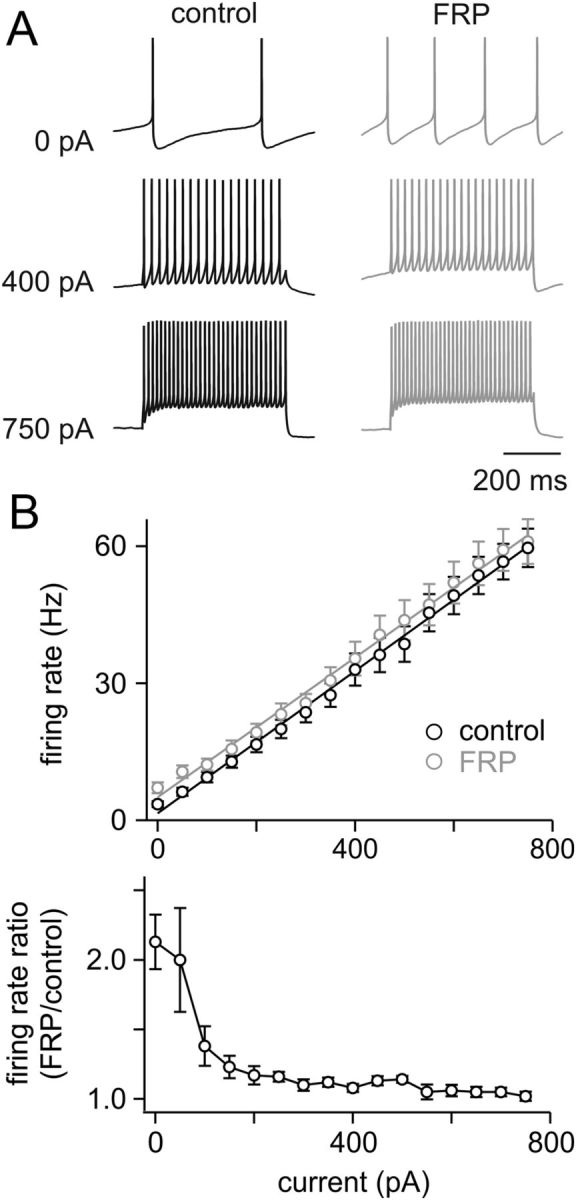
FRP alters the offset but not the gain of GoC input/output transformations. A, Example experiment showing the responses to injected current for a GoC before (black) and 15 min after (gray) inducing FRP with a 3 min hyperpolarization. Although FRP doubles the spontaneous spike rate, it has little influence on responses to injected current. B, Top, Summary of input/output relationships for injected current before and after inducing FRP with hyperpolarization. Lines represent linear fits to the averaged data (n = 10). The mean slope before hyperpolarization was 0.078 ± 0.006 Hz/pA, and 15 min after hyperpolarization was 0.077 ± 0.007 Hz/pA, indicating that FRP does not change the gain of transmission. Bottom, Ratio of the firing rate evoked by a current injection (after inducing FRP)/(before inducing FRP) reveals that the largest increase in spiking after induction of FRP occurs with no injected current, and this difference falls off rapidly with increasing current steps.
Discussion
Here we have shown that a brief hyperpolarization leads to a robust long-term increase in the spontaneous firing rate of cerebellar GoCs. Previously, FRP had only been observed in the vestibular nucleus (Nelson et al., 2003, 2005). Our findings therefore suggest that this form of plasticity may be a more widespread means of regulating spontaneously active neurons. Despite sharing many similarities, however, the underlying mechanism of FRP and its influence on input/output transformations are markedly different in cerebellar GoCs. Here, we find that iberiotoxin-sensitive BK-type channels do not underlie plasticity as they do in the vestibular nucleus. Furthermore, GoC FRP selectively increases spontaneous firing rates without altering the gain of evoked transmission. Thus, FRP in GoCs is specialized to preferentially enhance tonic spiking while minimally altering evoked spiking.
Mechanism of FRP
The basic mechanism of FRP in GoCs shares some similarities to what has been described in neurons of the vestibular nucleus (Nelson et al., 2003, 2005). In both instances, FRP decreases the amplitude of spike AHPs, and these changes are largely occluded by inhibiting CaMKII. It therefore seems likely that FRP in GoCs also results from a decrease in CaMKII activation during hyperpolarization when AP-evoked calcium levels are reduced. However, in contrast with the vestibular nucleus, iberiotoxin-sensitive BK-type channels do not appear to contribute to FRP in GoCs, and blocking paxilline-sensitive BK channels can only partially occlude plasticity. Hence, our data suggest that in GoCs not all FRP is accounted for by a long-term reduction in BK channel activation and that plasticity in these cells may be more complicated. It seems likely that GoC FRP results from the CaMKII-dependent regulation of multiple channel types, which could include a subset of BK channels involved in the spike AHP or possibly the specific calcium channels that regulate their opening.
We find that FRP can be induced by hyperpolarizations as short as 20 s. This induction time course is consistent with the observation that CaMKII is only active for tens of seconds after depolarization-induced calcium influx (Lee et al., 2009). Furthermore, this short period of hyperpolarization may allow multiple types of GoC inputs to produce firing rate enhancement, depending on how this plasticity operates in vivo.
One possibility is that this plasticity acts as a homeostatic mechanism to keep GoC firing within an optimal range by elevating activity in response to a tonic inhibitory input or by compensating for variability in potassium channel expression. This possibility seems to agree with our finding that GoC FRP is highly frequency dependent and can increase lower spike rates to a much greater extent than rates >5 Hz.
Another possibility, which is not mutually exclusive, is that GoC FRP is preferentially engaged by an acute inhibitory input and serves to dynamically regulate spiking. This could include a GABAergic or glycinergic synaptic input, mGluR2 activation, or possibly a neuromodulatory input. Although GoCs do not receive the same type of powerful synaptic inhibition that vestibular nucleus cells receive from Purkinje cell synapses, they do receive inhibition from multiple other GoCs. This inhibition may be sufficient to suppress spiking in vivo on a time scale of 20 s. Indeed, previous in vivo work has shown that the predominant response of GoCs to brief, single tactile stimuli is a depression of firing, which lasts a few hundred milliseconds (Holtzman et al., 2006; Xu and Edgley, 2010). Although the mechanism of this inhibition is not known, it seems reasonable that repetitive stimuli could lead to a depression of firing that lasts for tens of seconds and would be appropriate for activating GoC FRP. Future studies will be required to clarify the physiological conditions necessary for inducing FRP in GoCs.
Functional consequences of FRP
It is remarkable that the effect on input/output transformations is so different in GoCs compared with neurons in the vestibular nucleus despite sharing a similar mechanism. Although both cell types exhibit a prominent increase in spontaneous activity, only the vestibular neurons have an increase in the gain of transmission (Nelson et al., 2003). This gain change is well suited to their physiological role in regulating the gain of the vestibulo-ocular reflex (Bagnall et al., 2008). GoCs, however, do not appear to be involved in such a computation, and FRP accordingly produces no change in their gain of transmission. The differences between these two cell types illustrate the importance of the entire complement of ion channels in a neuron for determining the outcome of modulating a particular channel.
The observation that FRP preferentially affects spontaneous firing in GoCs suggests that this plasticity is tailored for modulating the level of tonic GABA release without dramatically changing the levels of fast, sensory-evoked inhibition they produce. Previous studies suggest that such an increase in tonic GoC spiking will significantly decrease the response of granule cells to mossy fiber inputs by activating α6-containing GABA receptors on granule cells (Hamann et al., 2002; Mitchell and Silver, 2003; Chadderton et al., 2004; Crowley et al., 2009), and possibly by increasing presynaptic inhibition of mossy fibers boutons (Mitchell and Silver, 2000). Recent in vivo work suggests that modulating tonic granule cell inhibition could play an important role in regulating the signal-to-noise ratio of sensory-evoked transmission from mossy fibers (Duguid et al., 2012). GoC FRP may thus provide a mechanism to achieve this type of modulation and, hence, regulate sensory processing in the cerebellar cortex.
Footnotes
This work was supported by National Institutes of Health Grant R37 NS032405 to W.G.R.
The authors declare no competing financial interests.
References
- Bagnall MW, McElvain LE, Faulstich M, du Lac S. Frequency-independent synaptic transmission supports a linear vestibular behavior. Neuron. 2008;60:343–352. doi: 10.1016/j.neuron.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL, Aldrich RW. BK channel β4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci. 2005;8:1752–1759. doi: 10.1038/nn1573. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Cull-Candy SG, Farrant M. Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J Physiol. 1996;497:753–759. doi: 10.1113/jphysiol.1996.sp021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Häusser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Cheron G, Sausbier M, Sausbier U, Neuhuber W, Ruth P, Dan B, Servais L. BK channels control cerebellar Purkinje and Golgi cell rhythmicity in vivo. PloS One. 2009;4:e7991. doi: 10.1371/journal.pone.0007991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley JJ, Fioravante D, Regehr WG. Dynamics of fast and slow inhibition from cerebellar Golgi cells allow flexible control of synaptic integration. Neuron. 2009;63:843–853. doi: 10.1016/j.neuron.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugué GP, Brunel N, Hakim V, Schwartz E, Chat M, Lévesque M, Courtemanche R, Léna C, Dieudonné S. Electrical coupling mediates tunable low-frequency oscillations and resonance in the cerebellar Golgi cell network. Neuron. 2009;61:126–139. doi: 10.1016/j.neuron.2008.11.028. [DOI] [PubMed] [Google Scholar]
- Duguid I, Branco T, London M, Chadderton P, Häusser M. Tonic inhibition enhances fidelity of sensory information transmission in the cerebellar cortex. J Neurosci. 2012;32:11132–11143. doi: 10.1523/JNEUROSCI.0460-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles J, Llinas R, Sasaki K. Golgi cell inhibition in the cerebellar cortex. Nature. 1964;204:1265–1266. doi: 10.1038/2041265a0. [DOI] [PubMed] [Google Scholar]
- Eccles J, Ito M, Szentagothai J. The cerebellum as a neuronal machine. Berlin: Springer; 1967. [Google Scholar]
- Forti L, Cesana E, Mapelli J, D'Angelo E. Ionic mechanisms of autorhythmic firing in rat cerebellar Golgi cells. J Physiol. 2006;574:711–729. doi: 10.1113/jphysiol.2006.110858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Beierlein M, Connors BW. Functional properties of electrical synapses between inhibitory interneurons of neocortical layer 4. J Neurophysiol. 2005;93:467–480. doi: 10.1152/jn.00520.2004. [DOI] [PubMed] [Google Scholar]
- Hamann M, Rossi DJ, Attwell D. Tonic and spillover inhibition of granule cells control information flow through cerebellar cortex. Neuron. 2002;33:625–633. doi: 10.1016/s0896-6273(02)00593-7. [DOI] [PubMed] [Google Scholar]
- Holtzman T, Rajapaksa T, Mostofi A, Edgley SA. Different responses of rat cerebellar Purkinje cells and Golgi cells evoked by widespread convergent sensory inputs. J Physiol. 2006;574:491–507. doi: 10.1113/jphysiol.2006.108282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull C, Regehr WG. Identification of an inhibitory circuit that regulates cerebellar Golgi cell activity. Neuron. 2012;73:149–158. doi: 10.1016/j.neuron.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. Cerebellar circuitry as a neuronal machine. Prog Neurobiol. 2006;78:272–303. doi: 10.1016/j.pneurobio.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meera P, Wallner M, Song M, Toro L. Large conductance voltage- and calcium-dependent K+ channel, a distinct member of voltage-dependent ion channels with seven N-terminal transmembrane segments (S0–S6), an extracellular N terminus, and an intracellular (S9–S10) C terminus. Proc Natl Acad Sci U S A. 1997;94:14066–14071. doi: 10.1073/pnas.94.25.14066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. GABA spillover from single inhibitory axons suppresses low-frequency excitatory transmission at the cerebellar glomerulus. J Neurosci. 2000;20:8651–8658. doi: 10.1523/JNEUROSCI.20-23-08651.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Shunting inhibition modulates neuronal gain during synaptic excitation. Neuron. 2003;38:433–445. doi: 10.1016/s0896-6273(03)00200-9. [DOI] [PubMed] [Google Scholar]
- Nelson AB, Krispel CM, Sekirnjak C, du Lac S. Long-lasting increases in intrinsic excitability triggered by inhibition. Neuron. 2003;40:609–620. doi: 10.1016/s0896-6273(03)00641-x. [DOI] [PubMed] [Google Scholar]
- Nelson AB, Gittis AH, du Lac S. Decreases in CaMKII activity trigger persistent potentiation of intrinsic excitability in spontaneously firing vestibular nucleus neurons. Neuron. 2005;46:623–631. doi: 10.1016/j.neuron.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Ogawa-Meguro R, Shigemoto R, Kaneko T, Nakanishi S, Mizuno N. Immunohistochemical localization of metabotropic glutamate receptors, mGluR2 and mGluR3, in rat cerebellar cortex. Neuron. 1994;13:55–66. doi: 10.1016/0896-6273(94)90459-6. [DOI] [PubMed] [Google Scholar]
- Ruigrok TJ, Hensbroek RA, Simpson JI. Spontaneous activity signatures of morphologically identified interneurons in the vestibulocerebellum. J Neurosci. 2011;31:712–724. doi: 10.1523/JNEUROSCI.1959-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Welie I, du Lac S. Bidirectional control of BK channel open probability by CaMKII and PKC in medial vestibular nucleus neurons. J Neurophysiol. 2011;105:1651–1659. doi: 10.1152/jn.00058.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervaeke K, Lorincz A, Gleeson P, Farinella M, Nusser Z, Silver RA. Rapid desynchronization of an electrically coupled interneuron network with sparse excitatory synaptic input. Neuron. 2010;67:435–451. doi: 10.1016/j.neuron.2010.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe D, Nakanishi S. mGluR2 postsynaptically senses granule cell inputs at Golgi cell synapses. Neuron. 2003;39:821–829. doi: 10.1016/s0896-6273(03)00530-0. [DOI] [PubMed] [Google Scholar]
- Watanabe D, Inokawa H, Hashimoto K, Suzuki N, Kano M, Shigemoto R, Hirano T, Toyama K, Kaneko S, Yokoi M, Moriyoshi K, Suzuki M, Kobayashi K, Nagatsu T, Kreitman RJ, Pastan I, Nakanishi S. Ablation of cerebellar Golgi cells disrupts synaptic integration involving GABA inhibition and NMDA receptor activation in motor coordination. Cell. 1998;95:17–27. doi: 10.1016/s0092-8674(00)81779-1. [DOI] [PubMed] [Google Scholar]
- Xu W, Edgley SA. Cerebellar Golgi cells in the rat receive convergent peripheral inputs via a lateral reticular nucleus relay. Eur J Neurosci. 2010;32:591–597. doi: 10.1111/j.1460-9568.2010.07307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]