From human genome to cancer genome: The first decade (original) (raw)
Abstract
The realization that cancer progression required the participation of cellular genes provided one of several key rationales, in 1986, for embarking on the human genome project. Only with a reference genome sequence could the full spectrum of somatic changes leading to cancer be understood. Since its completion in 2003, the human reference genome sequence has fulfilled its promise as a foundational tool to illuminate the pathogenesis of cancer. Herein, we review the key historical milestones in cancer genomics since the completion of the genome, and some of the novel discoveries that are shaping our current understanding of cancer.
The 40 years' “war on cancer”
The U.S. declaration of war on cancer, in 1971, was the product of heady optimism following the remarkable technological feat of landing on the moon. Recombinant DNA cloning was still in its infancy and not yet a match for the task of unraveling the full complexity of the human cell. Nevertheless, the proof that mutation of normal genes could cause cancer, a fact we take for granted today, led Dulbecco to realize as early as 1985 the value of obtaining the sequence of all the human chromosomes as the foundation for understanding cancer (Dulbecco 1986). The contemporaneous discussion about generating the complete human sequence represented an astonishing leap since, at the time, sequencing a single gene of about 1 kilobase was worthy of a PhD dissertation, and the human genome was 3 million times larger. The repeat structure of the human genome was well characterized and also viewed by some to be an insurmountable obstacle to such an endeavor and grounds for early resistance (Robertson 1986) to the project. Despite these obstacles, an international effort led to the completion of the human reference genome in 2003 (International Human Genome Sequencing Consortium 2004), and with a finished human genome reference in hand, it became possible to test in earnest the value of genomic approaches to decipher the genetic changes that lead to cancer. The data that have emerged since 2003 overwhelmingly support the value of this vision and have changed the way cancer is researched and understood.
During the period of the sequencing of the human genome (1990–2003) (http://www.ornl.gov/sci/techresources/Human_Genome/project/about.shtml), cancer researchers continued to accumulate knowledge of the basic mechanisms of cancer, and using a variety of clever cloning strategies, with steadily improving sequencing capabilities, identified the majority of the most potent oncogenes and tumor suppressors. An inventory of the genes associated with cancer yielded 291 cancer genes based on mutation data available in the literature: ∼1% of the coding sequence (Futreal et al. 2004). It was noted that 90% of these genes were somatically mutated, 20% germline mutated, and 10% could be found in both categories. The division between germline and somatic genes is a mysterious dichotomy that remains unexplained in the most current inventory (http://www.sanger.ac.uk/genetics/CGP/Census/). The most common form of variation in the 2004 inventory was translocation leading to the production of oncogenic fusion proteins. Until 2004, no one had studied more than a handful of genes at any one time in a single patient.
That was the state of cancer genomic research at the threshold of the “genomic era” of cancer research: an era heralded by the availability of the high-quality reference genome, and the dramatic explosion of DNA sequence data fueled by the introduction of inexpensive massively parallel sequencing instruments. This year is the 10th anniversary of the completion of that remarkable milestone in science―the completion of the reference human genome. At this juncture, we recapitulate some of the key findings and challenges that have emerged from the sequence analysis of the cancer genome.
The armamentarium
With base-level resolution of the human reference genome in hand, cancer researchers turned to the large-scale study of mutation, with the promise of generating the entire catalog of mutations peculiar to a given disease as well as to a single patient. Figure 1 tracks the development of some of the key technologies, resources, and milestones in the development of the present-day armamentarium of cancer mutation discovery. Massively parallel sequencing was introduced by Roche 454 and Illumina in 2004–2006 and soon demonstrated the feasibility of sequencing complete normal and tumor genomes of exemplar human subjects on both platforms (Ley et al. 2008; Wheeler et al. 2008). At the time, it appeared that the application of whole-genome sequencing to routine research and clinical diagnosis might be on the horizon. Although the use of whole-genome sequence (WGS) is far from routine today, the results generated so far are lending insight into the potential of WGS for diagnostic, prognostic, and therapeutic improvement in the treatment of cancer patients.
Figure 1.

Major events in a decade of cancer genomics. (Dark blue) Major advances in massively parallel sequencing platforms and targeted enrichment technologies; (black) major large-scale projects designed to catalog genomic variations of normal human individuals; (red) cancer genomics. (dbSNP) Database of single nucleotide polymorphism; (HapMap) haplotype map of the human genome; (ENCODE) Encyclopedia of DNA Elements; (COSMIC) Catalog of Somatic Mutations in Cancer; (TCGA) The Cancer Genome Atlas; (GA) genome analyzer; (CRC) colorectal carcinoma; (WES) whole-exome sequencing; (ICGC) International Cancer Genome Consortium; (TSP) tumor sequencing project; (AML) acute myeloid leukemia; (WGS) whole-genome sequencing; (OSCC) ovarian small cell carcinoma.
Using PCR and dye-terminator sequencing, Vogelstein and colleagues amplified and sequenced each coding exon of 18,000 genes, defined by the human genome sequence, in 11 each of breast and colorectal tumors (Wood et al. 2007). This brute force whole-exome sequencing (WES) approach afforded for the first time a comprehensive view of the mutation profile of each patient, which, when summed across patients, revealed the “cancer genes” for the patients in the given cohort. In one stroke, the mutation profile, composed of recurrently mutated genes, plus a collection of one-off mutations belonging to pathways and processes known to be involved in tumorigenesis, were revealed for a cancer. The fact that the most frequently mutated genes they observed, APC, TP53, and KRAS for colon cancer and TP53 for breast cancer, recapitulated what was already known, validated the approach and paved the way for expanded application of genome-scale sequencing.
The introduction of DNA sequence enrichment technologies from NimbleGen and Agilent (Albert et al. 2007; Gnirke et al. 2009) enabled WES on large scales. WES has additional advantages over WGS in that the average depth of coverage is about fivefold greater, and the cost of sequencing, data processing and storage are all much less. Given the relative tractability of interpreting variation in the coding sequence compared to intergenic or intronic mutations, the period between 2004 and 2013 has seen a profusion of tumor types analyzed in large cohorts (100–500 patients), mainly by WES (see http://www.sanger.ac.uk/genetics/CGP/cosmic/papers/ for a comprehensive listing). WGS for a variety of tumors has also been reported and, in spite of the smaller numbers of patients, has led to surprising insights into cancer biology, based largely on analysis of structural variation in tumor genomes. Using WGS, genetic alterations observed in the DNA of the cancer cell span six orders of magnitude, from single-base point mutations to chromosome-scale amplification, using different modes of sequence analysis (see Chin et al. 2011) available today.
With these tools in hand, The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/), the Cancer Genome Project (http://www.sanger.ac.uk/genetics/CGP/), the International Cancer Genome Consortium (ICGC) (Hudson et al. 2010), Therapeutically Applicable Research to Generate Effective Treatments (http://target.cancer.gov/), and other privately funded large-scale projects (Downing et al. 2012) began in earnest to systematically catalog all the mutations in a wide variety of adult and pediatric cancers (see Garraway and Lander 2013 for a recent tally of large-scale projects).
WGS and WES sequencing have been augmented by cDNA sequencing (referred to as RNA-seq) to explore alterations to the transcriptome. RNA-seq provides not only gene expression levels, but also aberrant splicing, chimeric gene fusion transcripts characteristic of cancer cells and expressed somatic mutations (Bainbridge et al. 2006; Dong et al. 2009; Maher et al. 2009; Shah et al. 2009; Berger et al. 2010; Tuch et al. 2010; Wang et al. 2012). Analysis of chromatin modification is in its infancy as applied to the cancer cell, but the recent reporting of the ENCODE Project Consortium's genome-wide results (The ENCODE Project Consortium 2012) may provide the tools and technologies to enable new approaches. The technology behind DNA sequencing is improving rapidly in accuracy, cost reduction, and speed, making advances in cancer biology and clinical testing, all based on analysis of the primary sequence of the tumor genome, an essential strategy in the war on cancer. However, the coordinated acquisition and integrated interpretation of all this data has been possible because of a reference genome for comparison. What have we learned so far?
Mutation frequencies
By patient
The median frequency of point mutation varies over more than three orders of magnitude across human tumors; within a given tumor type, the variation in frequency is about one order of magnitude (Fig. 2A). The variation in mutation frequency is a function of the number of somatic cell divisions prior to initiation of the tumor, the exposure to environmental mutagens―notably UV radiation and tobacco leaf by-products―and in some cancers, altered fidelity of the tumor DNA replication system. At the low end of the range are pediatric cancers, followed by adult leukemia and adult solid tumors. Tumors that exceed 10 coding mutations per megabase pair (Mbp) are often found deficient in mismatch repair, either through mutation or epigenetic silencing of MLH1 or one of the other mismatch repair enzymes. Tumors with coding mutation frequencies of 100 per Mbp or greater are mutated in the exonuclease domain of POLE, one of two DNA replicative enzymes of the cells (The Cancer Genome Atlas Research Network 2013). These patterns may have important implications for clinical testing in that colorectal patients with high rates of mutation due to mismatch repair (Walther et al. 2009) or replicative dysfunction tend to have improved survival compared to their lower mutation rate counterparts for the same tumor type. At the other end of the scale, many pediatric patients have so few coding mutations that DNA sequencing sheds less light on the etiology or prognosis of their disease. For the broad range of adult cancers with intermediate rates, mutation discovery is becoming increasingly important in subclassifying disease for prognosis and treatment (e.g., Patel et al. 2012).
Figure 2.

Frequencies of somatic mutations in cancer patients. All data represents primary tumors. Only nonsilent mutations (missense, nonsense, frameshift, and splice site) were counted. (A) Overall frequencies of somatic mutations. Each black dot represents a tumor. The light blue shaded group indicates pediatric tumors, and the deeper blue shaded group indicates adult tumors. Red horizontal lines within each cluster of points indicate median value of the mutation frequency of each tumor type. (ALL) Acute lymphoblastic leukemia; (AML) acute myeloid leukemia; (C) carcinoma; (GCT) germ cell tumor; (CRC) colorectal carcinoma; (MSI) microsatellite instability; (MSS) microsatellite stable; (POLE) patients with somatic mutation in the nuclease (proofreading) domain of the POLE gene. The outlier in the low-grade glioma patient with >100 mutations per Mb is also _POLE_-mutated. (B) Frequency classification of tumors. The pie charts divide the patients into three groups based on frequency of nonsilent mutation: 0 detectable somatic mutations, less than 30, and greater than or equal to 30 for selected representative tumor types (30 mutations represent a frequency of 1 per Mbp in A). The nested histograms below the pie charts show the percentage of patients with no significantly mutated genes (SMG, calculated by MutSig, q ≤ 0.1), no cancer census genes (CGC), or no mutations at all. The sequencing data for all the pediatric tumors, CRC, and hepatocellular carcinoma were generated at the Human Genome Sequencing Center at Baylor College of Medicine. The sequencing data for all other adult tumors were from the TCGA Genome Data Analysis Center (https://confluence.broadinstitute.org/display/GDAC/Home). Pediatric AML, ALL, and Wilm's Tumor data were obtained from the TARGET project (http://www.targetproject.net/).
By gene
The aforementioned cancer gene census (Futreal et al. 2004) has given way to COSMIC, the Catalogue of Somatic Mutations in Cancer (Forbes et al. 2011). The most recent inventory of cancer mutations in COSMIC (http://www.sanger.ac.uk/genetics/CGP/Census/) chronicles nearly 800,000 somatic events. Essentially, every gene is mutated at least once, reflecting the explosion of data available to researchers.
Every tumor type has a characteristic collection of frequently mutated genes―as exemplified by colorectal cancer, shown in Figure 3—which are mutated significantly more often than expected, given the tumor's background mutation rate (see Fig. 2A). Higher-than-expected mutation frequency is prima facie evidence of the important role the mutated genes play in the given cancer. The pattern established in the first completed exome screening in colorectal and breast cancers (Wood et al. 2007) and repeated in every cancer since, is that one to three genes are mutated in >20% of the tumors. A shoulder on the distribution may display several more genes mutated in 10%–20% of the tumors, and then infrequently mutated genes form a long tail to ever-decreasing mutation frequency. Adding new low-frequency genes to the significantly mutated gene list requires sequencing patients in greater numbers. Large-scale projects such as TCGA and the ICGC projects aim to sequence 500 patients in each tumor type surveyed, with the expectation of collecting a sizeable fraction of the genes mutated in the 3%–5% range. Whereas the most potent cancer genes, often referred to as cancer “drivers,” are small in number, the tail of genes mutated at <20% is long and is characteristic of a given tumor or tumor subtype (see Fig. 3A,B).
Figure 3.

Significantly mutated genes and principal cancer pathways deregulated by somatic mutations in human colorectal carcinoma. Patients are divided into two groups based on mutation rate. All genes shown are significantly mutated with a false discovery rate of less than 0.1. (A) Profile determined from 193 patients with chromosome instable, low mutation rate, disease (see Fig. 2A, CRC MSS). (B) Profile determined from 29 microsatellite instable CRC plus 7 _POLE_-mutated patients (see Fig. 2A, CRC MSI and CRC MSS POLE). (C) Principal cancer pathways deregulated by somatic mutation in CRC. Alterations are defined by somatic mutations, homozygous deletions, high-level focal amplifications, and, in some cases, by significant up- or down-regulation of gene expression (black up-triangle). All genes from Figure 3 except MLK4, GPC6, and EDNRB can be placed in one of the four pathways shown here. WNT signaling is disrupted by one or more mutations in 93% of patients; TGFbeta signaling is disrupted in 26% of all patients with a low mutation rate and in 94% of patients, and RTK/RAS/PI3K signaling is disrupted in over 80% of patients. (Red) Activated genes; (blue) inactivated genes. Deep red or blue are genes on the significantly mutated list from panels A and B. Lighter shaded genes are not mutated significantly in this cohort but contribute to pathway disruption in some patients. Panels A and B adapted from Figure 1, and panel C from Figure 4, of The Cancer Genome Atlas Research Network (2012a).
The genes on the tail of the frequency distribution have great importance for the understanding of tumor biology. First, they may exhibit redundancy of mutation in a given signaling pathway in a single patient, which contributes to our understanding of how the pathway is utilized by the cancer cell. These features are again illustrated by colorectal cancer (Fig. 3C). APC is the primary driver of WNT signaling in this cancer; however, 10 other genes, altered in 1%–15% of the patients, also appear to participate in activation of this pathway, sometimes even in the context of APC mutations. That one of the key outputs of the WNT pathway is up-regulation of MYC, this mutation pattern suggests ever increasing selection for activity of the MYC transcription program within the tumor cell, a prediction borne out by transcriptome analysis of these patients' tumors (The Cancer Genome Atlas Research Network 2012a). Second, weaker drivers have often revealed entirely new pathways or processes, as discussed below.
There are also important clinical implications for the low-frequency recurrent cancer genes. The tail of the mutation profile is often found to be populated with genes that are strong drivers in other cancers and for which therapies have already been designed or are in the pipeline (Taylor et al. 2012). This has given rise to the notion that it might be better to classify cancers based on mutation profile for developing treatment strategies rather than histopathology―a radical departure from well-established clinical standards. On the other hand, some patient's cancers have no obvious mutations in any known cancer genes, so mutational screening alone appears to have limited utility in such tumors (see Fig. 2B). Every tumor population studied so far appears to have a few percent of patients with no obvious cancer genes mutated, necessitating alternative diagnostic approaches. The probability of finding no mutated cancer genes increases with decreasing mutation frequency in the tumor.
The low-frequency genes reveal added complexity for the application of therapies targeted to a specific gene in a pathway in that they might have one or more downstream genes already mutated. The well-known EGFR inhibitors erlotinib (Tarceva) and gefitinib (Iressa) fail in the context of mutation in KRAS in nonsmall cell lung cancers (Pao et al. 2005). Redundant mutation in key pathways is an increasingly common feature of cancer genomes (Fig. 3C), revealed by comprehensive exome or genome sequencing.
When DNA sequencing is augmented by genomic approaches such as copy number analysis, RNA expression, or epigenetic profiling, evidence for aberrant silencing or activation of cancer genes adds to the list of genes relevant to each cancer. From the perspective of translating the rapidly accumulating knowledge of mutations to the bedside, the question arises as to how many genes clinicians will have to pay attention to for each cancer type and for any given patient. A fundamental question is whether the set of cancer loci that can participate in the maintenance and survival of the cancer cell is bounded, or will we one day have to monitor the entire genome to effect a cure? Studies with patient populations in the range of 100 to 500 patients (see Garraway and Lander 2013) integrating results of several different platforms suggest that the number of genes involved in any given tumor is small. Mutation profiles like that shown in Figure 3 identify 15–20 genes mutated. Data from copy number analysis add another roughly 20 genes from recurrent focal amplifications or deletions. Aberrant epigenetic and expression profiles reinforce the somatic mutation data and may add a few more genes. Based on results so far, it seems likely that the cast of genes involved in any single cancer type will be in the neighborhood of 50–100, as opposed to 500–1000.
Novel high-frequency cancer genes
Many important new cancer genes, mutated in 20% or more of the patients with common cancers, were absent from the 2004 cancer gene census (Futreal et al. 2004). Among the genes emerging from large-scale studies, the most impressive advances have provided insights into the role of chromatin remodeling in tumorigenesis. Isocitrate dehydrogenase 1 and 2, IDH1 and IDH2, were surprising additions to the list of cancer drivers emerging from glioblastoma multiforme (GBM) among the first whole-exome screens reported (Parsons et al. 2008). Both enzymes normally convert isocitrate to α-ketoglutarate (α-KG), a cofactor for α-KG dioxygenases, including TET family DNA demethylases, KDM-family histone demethylases, and many other proteins (Yang et al. 2012). Mutated IDH1/2 both produce 2-oxyglutarate, a structural analog of α-KG but potent inhibitor of α-KG-dependent enzymes, the methyltransferases involved in DNA and chromatin methylation. These inhibitors, therefore, result in aberrant epigenetic modification as well as the potential for deregulation of many other cellular pathways. The DNA demethylase, DNMT3A, is mutated in 22% of AML patients, suggesting a significant role for transcriptional regulation through epigenetic modification of DNA in the pathogenesis of this cancer (Ley et al. 2010). Mutations in this gene are clinically important in AML patients since they are associated with poor survival. Since its initial discovery, DNMT3A is now recognized as a less frequent, although important, contributor to transformation in nearly all myeloid neoplasms (Ley et al. 2010).
PBRM1, discovered in 41% of clear-cell renal carcinomas, was the first member of the large SWI/SNF chromatin remodeling complex to be found mutated to high levels in any cancer (Varela et al. 2011). Since then, mutations in many of the other SWI/SNF components have been steadily accumulating. Disruption in the normal function of SWI/SNF in over half the cancer patients is inferred for renal, ovarian, hepatocellular, gastric, and other cancers (Shain and Pollack 2013).
BAP1 carries an enzymatic function that deubiquitinates histone H2A as well as other proteins involved in chromatin remodeling. It exhibits a high frequency (23%) of inactivating mutations in mesothelioma (Bott et al. 2011) and in uveal melanomas, where 84% of patients at high risk for metastasis (Harbour et al. 2010) harbor mutations. It is also mutated in 15% of clear-cell renal cell carcinomas, ccRCC, wherein it is anti-correlated with the more frequent PBRM1 mutations, noted above (Peña-Llopis et al. 2012). Inactivating mutation in BAP1 defines a molecular subclass of high-grade aggressive tumors, which led the authors to develop a clinical immunohistochemical assay for the presence or absence of BAP1. Anti-correlation in the mutations of two genes usually indicates they are active in the same pathway. In this case, differing RNA expression profiles associated with mutations in the two genes suggested this was not the case. Instead, the authors concluded that it is disadvantageous to the tumor to have both genes mutated and that the two genes likely affect different epigenetic programs.
This list of new high-frequency genes, added to the cancer gene compendium as a direct result of the human reference genome and next-generation sequencing, reveals the importance of epigenetic regulation for tumorigenesis.
Novel low-frequency cancer genes
At the other end of the mutation profile are cancer genes contributing to 10% or fewer tumors of each cancer type. For most adult solid tumors, they constitute a greater number of mutated genes, filling in details of key pathways and adding interesting insight into the mechanisms of cancer. Perhaps the most interesting ones among the novel genes are those involved in RNA processing functions. Discovered first in myelodysplastic syndrome, U2AF1, U2AF2, SF3B1, and SRSF2 (Yoshida et al. 2011), involved in splice acceptor recognition in the RNA splicing machinery, were mutated cumulatively in over 50% of patients. Subsequently, mutations were found in these and other splicing components in other myeloid neoplasms (Wang et al. 2011; Makishima et al. 2012). Functional testing of the commonly recurrent mutation sites in U2AF1 demonstrated that they promote exon skipping in in vitro assays (Graubert et al. 2012). U2AF1 was recently reported in lung adenocarcinoma at the same frequency as seen in AML (Imielinski et al. 2012). Additional RNA binding proteins―RBM10 in lung (Imielinski et al. 2012) and DDX3X in CML (Wang et al. 2011)―hint at additional contributions to cancer in RNA processing pathways.
Whole-genome sequencing and structural variation
WGS sequencing is a generalized route to reliable discovery and interpretation of mutation in noncoding regions of the genome. This fact was driven home dramatically in the recent reports of somatic base substitution at one of only two positions in the promoter of TERT in over 70% of melanoma patients (Horn et al. 2013; Huang et al. 2013). Although recurrent mutations in noncoding DNA had been reported previously (Chapman et al. 2011), they were clustered within predefined conserved regions rather than recurrent at the same site and, moreover, were without functional follow-up. Both TERT promoter mutations created novel ETS transcription factor binding sites, and the mutations were demonstrated to up-regulate TERT transcription in in vitro luciferase reporter assays (Huang et al. 2013). This astonishing rate of mutation within and between tumors may turn out to be more prevalent than mutation of TP53, the most frequently mutated gene across all cancers to date.
Large-scale translocations, deletions, and inversions leading to oncogenic fusion proteins or promoter activation of an oncogene seldom involve the coding sequence directly and are therefore not visible in WES data; although there has been limited success in capturing known fusion junctions in cohorts expected to harbor them when the target can be restricted to a small interval (Lipson et al. 2012). WGS analyses have dramatically increased our knowledge of the number of translocations and deletions giving rise to gene fusions, so the list of these events is still growing. Among the 487 genes in the current cancer gene census (http://www.sanger.ac.uk/genetics/CGP/Census/), 326 are observed in chimeric protein fusions generated through translocation. Like point mutations, most translocations observed in most individual tumors seem to be random and do not lead to apparent functional changes in oncogenes or tumor suppressors. However, across populations of AML (The Cancer Genome Atlas Research Network 2013), lung adenocarcinoma (Imielinski et al. 2012), and lung squamous cell carcinoma (The Cancer Genome Atlas Research Network 2012b), nonfunctional fusions involving tumor suppressors contribute significantly to the overall mutation spectrum in these cancers.
Analysis of paired reads from WGS data led to the discovery of chromothripsis (Stephens et al. 2011), the result of a catastrophic shattering of one or more chromosomes followed by misjoining of the scrambled fragments upon repair, and kataegis, the localized bursts of point mutations often correlated with somatic structural variation. The mechanisms for neither of these processes is clearly understood, although it is interesting to note that chromothripsis-like events have been observed in normal human germline DNA, at translocation break points, and are associated with congenital deformities and mental retardation (Kloosterman et al. 2011; Chiang et al. 2012). Cancer cells are apparently utilizing a mechanism for recovering from catastrophic error that is available in normal cells, suggesting the possibility that the extremes of chromothripsis seen in some tumors have accumulated over several cell divisions rather than in a single event. In any case, the association of extensive rearrangement with key tumor suppressors and oncogenes in some patients suggests that chromothripsis may sometimes mediate tumorigenesis and associates with clinical outcome (Magrangeas et al. 2011; Molenaar et al. 2012; Hirsch et al. 2013; Malhotra et al. 2013).
Genetic heterogeneity of tumors
Although the existence of intra-tumor phenotypic heterogeneity has been recognized from the early days of experimental cancer research, the intra-tumor genetic heterogeneity has not been well demonstrated until recently with the significant advances of high-throughput next-generation sequencing technologies. In the past two years, researchers have characterized in detail the population structures and evolutionary paths in a variety of primary and metastatic cancers, including childhood acute lymphoblastic leukemia (Anderson et al. 2011) and secondary acute myeloid leukemia (Walter et al. 2012), human pancreatic cancers (Campbell et al. 2010; Yachida et al. 2010), breast cancers (Geyer et al. 2010; Navin et al. 2010, 2011), renal clear-cell carcinomas (Gerlinger et al. 2012), and metastatic medulloblastoma (Wu et al. 2012). Investigations have been done using both the spatially separated cancer samples (Gerlinger et al. 2012) and the regionally separated cancer samples (Navin et al. 2010; Yachida et al. 2010). Genetically distinguishable subpopulations in a mixture of cancer cells were well demonstrated by single-nucleus and single-cell sequencing (Navin et al. 2011; Hou et al. 2012; Xu et al. 2012). Overwhelming evidence shown at the DNA level and the RNA level has revealed the coexistence of genetically divergent clonal subpopulations within tumors. Intra-tumor heterogeneity has been suggested as a common feature in both solid tumors and hematological malignancies.
Recent studies have evaluated the clonal relationships among primary and metastatic cancers (Campbell et al. 2010; Yachida et al. 2010; Navin et al. 2011; Gerlinger et al. 2012). These studies showed that seeding metastases require few, if any, additional driver mutations beyond those found in the primary tumors. Continuous acquisition of mutation and clonal expansion during the development of a tumor results in an assemblage of subclones that can be represented by a branching evolutionary tree. The metastatic cells seem to emerge from small, seemingly arbitrary subclones in the original tumor. Surprisingly, different regions of a single tumor may harbor different mutations in the same driver gene, suggesting a process of convergent evolution in the development of the tumor (Gerlinger et al. 2012). The implications for clinical treatment of tumors are daunting since this suggests that genomic analysis from needle biopsies may yield a biased view of the genetic landscape of the tumor (Gerlinger and Swanton 2010; Anderson et al. 2011; Sequist et al. 2011). Yap et al. (2012) suggest that the genetic heterogeneity observed within most tumors, combined with the polygenic nature of drug resistance, may hamper both the discovery of predictive biomarkers and the development of anti-tumor molecular therapeutics.
Challenges for the future
These exciting insights have only come in the past several years as the technologies involved in sequence capture and sequencing have matured (Fig. 1), enabling sequencing centers and consortia to more effectively exploit the utility of the reference genome. Here, we have focused on novel discoveries. The wealth of information accumulated so far has illuminated the roles of all cancer genes in the biology and pathogenesis of the disease, but there is much more to come, along with new challenges.
There now exist data sets from hundreds of cancer sequencing projects for which most of the focus of the studies from those projects was on the components we know best how to analyze. Thus, our analysis is biased toward variants whose function we readily understand―the low-hanging fruit―even if the sequence data itself is unbiased. Although much progress has been made, there is still considerable potential for discovery in existing data; however, the downstream functional analyses are overwhelmed.
First, very few of the mutations receive functional validation in in vitro and in vivo studies. New mutations in known cancer genes are not necessarily functional in a given patient's disease, whereas the ones that are functional can add important new insights into the mechanism of pathogenesis in play, exemplified in the case of novel mutations in the extracellular domain of ERBB2 (Greulich et al. 2012). ERBB2 signaling is most commonly activated by amplification of the gene. These mutations suggested novel mechanisms of activation of ERBB2 signaling in lung cancer.
Second, the beneficial value of functional follow-up in mouse models is recognized but still expensive and time consuming, although new approaches with genetically engineered mice may help (Heyer et al. 2010).
Third, the tail of the mutation profiles (Fig. 3) grows, as study cohorts become larger, adding many novel significantly mutated genes to the cancer inventory. Computational approaches that predict functional consequences of amino acid substitution have advanced significantly as more mutational data has accumulated (Radivojac et al. 2013), but further improvements are required. Knowing which mutations are likely to be deleterious will be extremely helpful in culling the list of mutations in cancer genes; though this may still be insufficient. Many of the proteins have multiple functions, some of which are unknown. So, functional testing will always be required. This is creating a bottleneck in discovery, which must be met with increasingly rapid and high-throughput methods for functional validation (Chin et al. 2011).
The prediction of the functional consequences of chromosomal-scale structural variation can be challenging. When individual events disrupt gene structures, the interpretation is simplified. In most cases, however, genomic changes are difficult to interpret. Distinguishing functional from passenger mutations in the noncoding portions of the genome is an open frontier. The completion of ENCODE, which has identified myriad functional elements in DNA, brings an enormous resource to bear on the challenge of interpreting the functional role of variants outside coding sequences. Methods to make functional predictions are not yet in hand, although approaches that parallel coding sequence strategies are a starting point (Chapman et al. 2011) and leave a formidable challenge for functional follow-up.
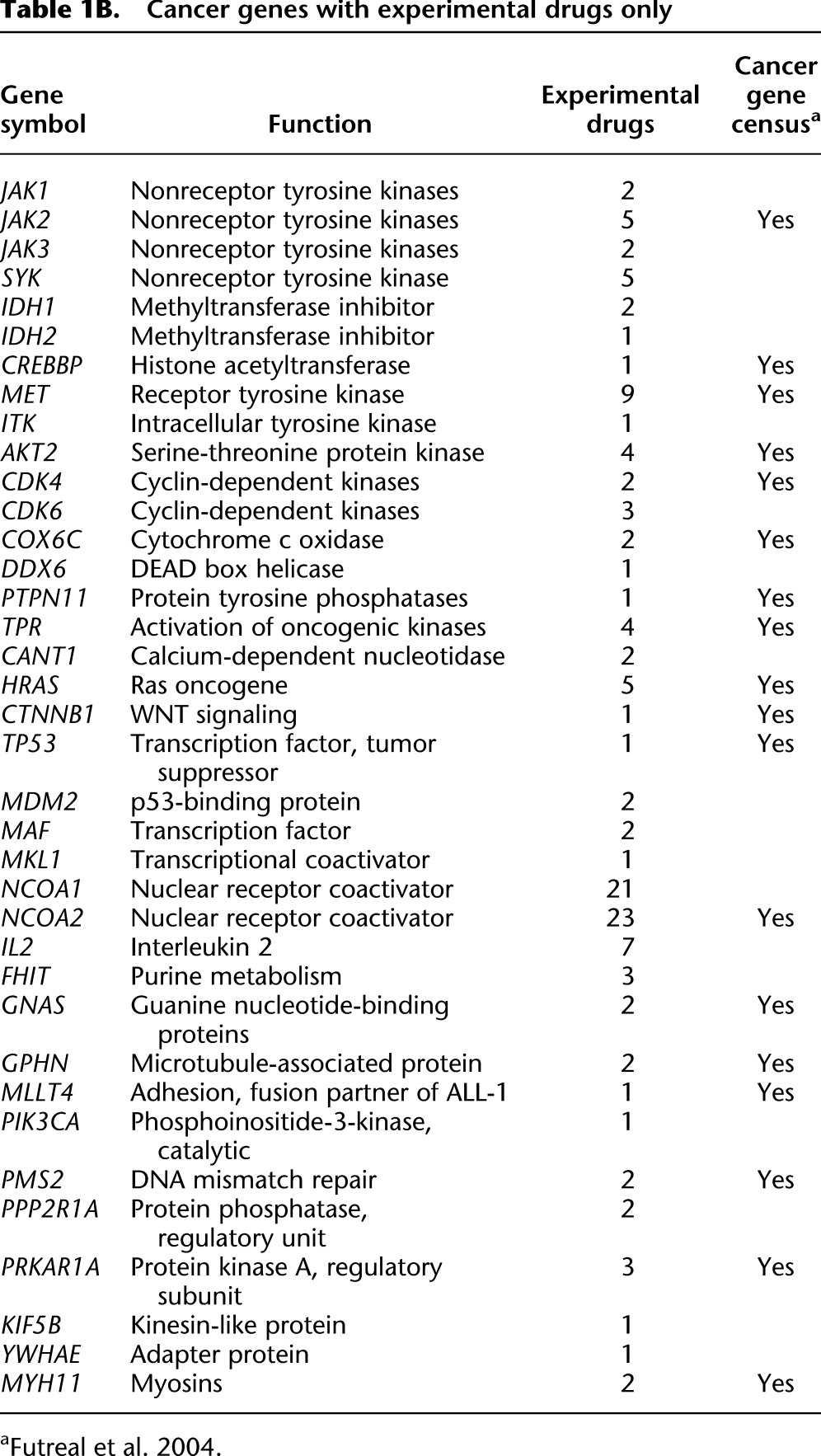
There have been substantial gains on all fronts from the war on cancer. The 10 years since the finishing of the human reference genome has been particularly fruitful, primarily as a consequence of the improved ability to identify and catalog somatic changes in cancer genomes. Looking forward, perhaps the greatest challenge now is translating discoveries into treatments based on strategies mapped out from patients' individual mutation profiles. The number of approved targeted cancer drugs today is still limited (Table 1A). By-and-large, these reflect targets that have been available for a long time: Three-fourths of the targeted genes were in the 2004 census. The list of drugs under development (Table 1B) includes one-half newly discovered targets, suggesting the impact of discovery fostered by the genomic era is starting to make its mark. With the data generation continuing unabated, revealing even more detail of the working of the cancer cell, the list of targets can be expected to accelerate. We are realizing the vision of Dulbecco and benefiting tremendously from the effort to finish the human genome.
Table 1A.
Cancer genes with FDA approved drugs
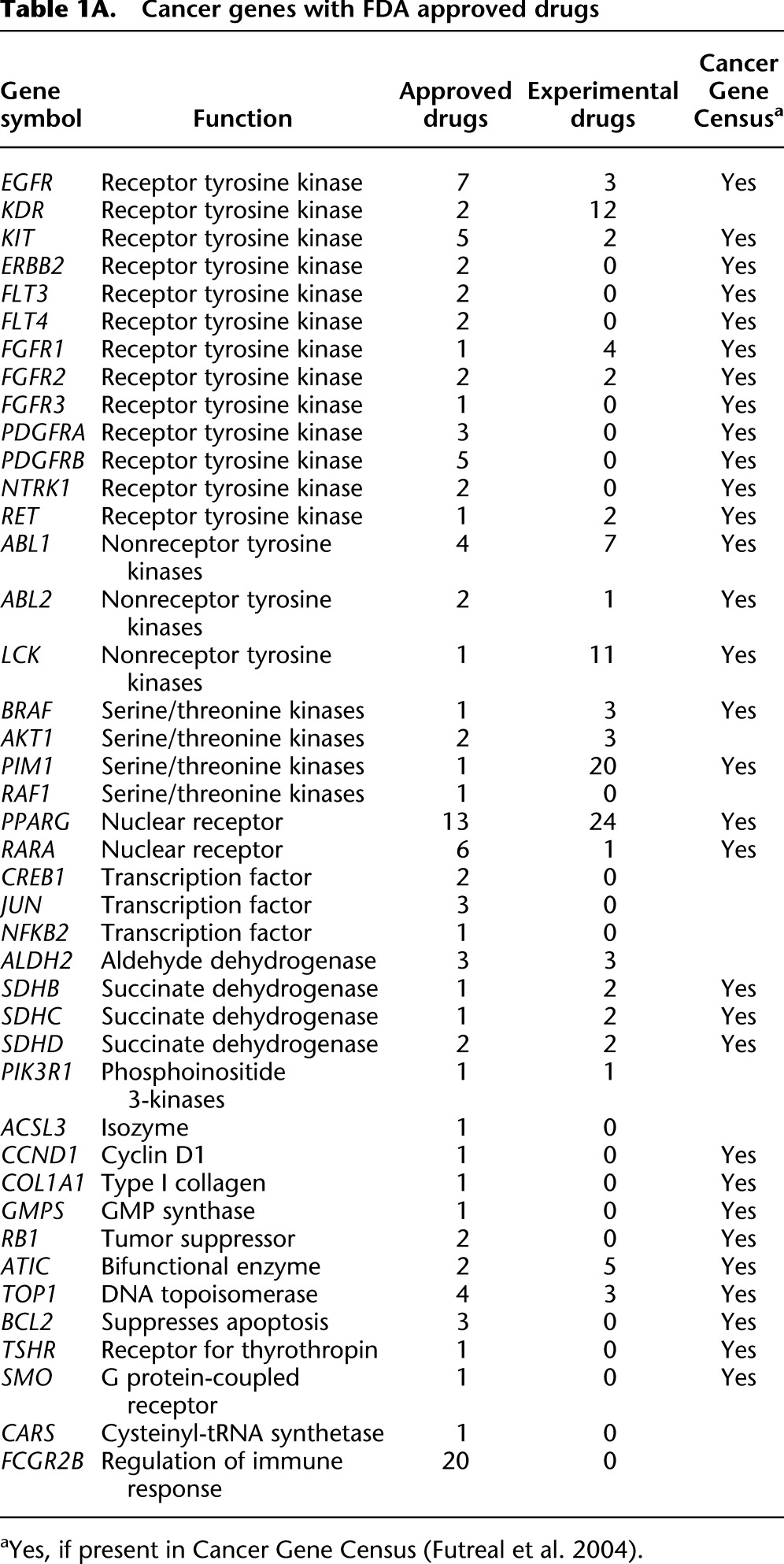
Table 1B.
Cancer genes with experimental drugs only
Footnotes
References
- Albert TJ, Molla MN, Muzny DM, Nazareth L, Wheeler D, Song X, Richmond TA, Middle CM, Rodesch MJ, Packard CJ, et al. 2007. Direct selection of human genomic loci by microarray hybridization. Nat Methods 4: 903–905 [DOI] [PubMed] [Google Scholar]
- Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, Kempski H, Moorman AV, Titley I, Swansbury J, et al. 2011. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 469: 356–361 [DOI] [PubMed] [Google Scholar]
- Bainbridge MN, Warren RL, Hirst M, Romanuik T, Zeng T, Go A, Delaney A, Griffith M, Hickenbotham M, Magrini V, et al. 2006. Analysis of the prostate cancer cell line LNCaP transcriptome using a sequencing-by-synthesis approach. BMC Genomics 7: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Levin JZ, Vijayendran K, Sivachenko A, Adiconis X, Maguire J, Johnson LA, Robinson J, Verhaak RG, Sougnez C, et al. 2010. Integrative analysis of the melanoma transcriptome. Genome Res 20: 413–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott M, Brevet M, Taylor BS, Shimizu S, Ito T, Wang L, Creaney J, Lake RA, Zakowski MF, Reva B, et al. 2011. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet 43: 668–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, et al. 2010. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467: 1109–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network 2012a. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network 2012b. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489: 519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network 2013. Integrated genomic characterization of endometrial carcinoma. Nature 497: 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, et al. 2011. Initial genome sequencing and analysis of multiple myeloma. Nature 471: 467–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Jacobsen JC, Ernst C, Hanscom C, Heilbut A, Blumenthal I, Mills RE, Kirby A, Lindgren AM, Rudiger SR et al. 2012. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nature genetics 44: 390–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Hahn WC, Getz G, Meyerson M 2011. Making sense of cancer genomic data. Genes Dev 25: 534–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L, Jensen RV, De Rienzo A, Gordon GJ, Xu Y, Sugarbaker DJ, Bueno R 2009. Differentially expressed alternatively spliced genes in malignant pleural mesothelioma identified using massively parallel transcriptome sequencing. BMC Med Genet 10: 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing JR, Wilson RK, Zhang J, Mardis ER, Pui CH, Ding L, Ley TJ, Evans WE 2012. The Pediatric Cancer Genome Project. Nat Genet 44: 619–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulbecco R 1986. A turning point in cancer research: Sequencing the human genome. Science 231: 1055–1056 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. 2011. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 39: D945–D950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR 2004. A census of human cancer genes. Nat Rev Cancer 4: 177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Lander ES 2013. Lessons from the cancer genome. Cell 153: 17–37 [DOI] [PubMed] [Google Scholar]
- Gerlinger M, Swanton C 2010. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer 103: 1139–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. 2012. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366: 883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer FC, Weigelt B, Natrajan R, Lambros MB, de Biase D, Vatcheva R, Savage K, Mackay A, Ashworth A, Reis-Filho JS 2010. Molecular analysis reveals a genetic basis for the phenotypic diversity of metaplastic breast carcinomas. J Pathol 220: 562–573 [DOI] [PubMed] [Google Scholar]
- Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, et al. 2009. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 27: 182–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, et al. 2012. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet 44: 53–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greulich H, Kaplan B, Mertins P, Chen TH, Tanaka KE, Yun CH, Zhang X, Lee SH, Cho J, Ambrogio L, et al. 2012. Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc Natl Acad Sci 109: 14476–14481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, Bowcock AM 2010. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330: 1410–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer J, Kwong LN, Lowe SW, Chin L 2010. Non-germline genetically engineered mouse models for translational cancer research. Nat Rev Cancer 10: 470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch D, Kemmerling R, Davis S, Camps J, Meltzer PS, Ried T, Gaiser T 2013. Chromothripsis and focal copy number alterations determine poor outcome in malignant melanoma. Cancer Res 73: 1454–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. 2013. TERT promoter mutations in familial and sporadic melanoma. Science 339: 959–961 [DOI] [PubMed] [Google Scholar]
- Hou Y, Song L, Zhu P, Zhang B, Tao Y, Xu X, Li F, Wu K, Liang J, Shao D, et al. 2012. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell 148: 873–885 [DOI] [PubMed] [Google Scholar]
- Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA 2013. Highly recurrent TERT promoter mutations in human melanoma. Science 339: 957–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, Bhan MK, Calvo F, Eerola I, Gerhard DS, et al. 2010. International network of cancer genome projects. Nature 464: 993–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. 2012. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150: 1107–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium 2004. Finishing the euchromatic sequencing of the human genome. Nature 431: 931–945 [DOI] [PubMed] [Google Scholar]
- Kloosterman WP, Guryev V, van Roosmalen M, Duran KJ, de Bruijn E, Bakker SC, Letteboer T, van Nesselrooij B, Hochstenbach R, Poot M, et al. 2011. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum Mol Genet 20: 1916–1924 [DOI] [PubMed] [Google Scholar]
- Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, et al. 2008. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 456: 66–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, et al. 2010. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363: 2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, Curran JA, Balasubramanian S, Bloom T, Brennan KW, et al. 2012. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med 18: 382–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrangeas F, Avet-Loiseau H, Munshi NC, Minvielle S 2011. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood 118: 675–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N, Chinnaiyan AM 2009. Transcriptome sequencing to detect gene fusions in cancer. Nature 458: 97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, et al. 2012. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 119: 3203–3210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra A, Lindberg M, Faust GG, Leibowitz ML, Clark RA, Layer RM, Quinlan AR, Hall IM 2013. Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome Res 23: 762–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, et al. 2012. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483: 589–593 [DOI] [PubMed] [Google Scholar]
- Navin N, Krasnitz A, Rodgers L, Cook K, Meth J, Kendall J, Riggs M, Eberling Y, Troge J, Grubor V, et al. 2010. Inferring tumor progression from genomic heterogeneity. Genome Res 20: 68–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et al. 2011. Tumour evolution inferred by single-cell sequencing. Nature 472: 90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE 2005. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2: e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. 2008. An integrated genomic analysis of human glioblastoma multiforme. Science 321: 1807–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, et al. 2012. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 366: 1079–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S, Yamasaki T, Zhrebker L, Sivanand S, Spence P, et al. 2012. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet 44: 751–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radivojac P, Clark WT, Oron TR, Schnoes AM, Wittkop T, Sokolov A, Graim K, Funk C, Verspoor K, Ben-Hur A, et al. 2013. A large-scale evaluation of computational protein function prediction. Nat Methods 10: 221–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson M 1986. The proper study of mankind. Nature 322: 11. [DOI] [PubMed] [Google Scholar]
- Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, et al. 2011. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3: 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, et al. 2009. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 461: 809–813 [DOI] [PubMed] [Google Scholar]
- Shain AH, Pollack JR 2013. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE 8: e55119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. 2011. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144: 27–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor TE, Furnari FB, Cavenee WK 2012. Targeting EGFR for treatment of glioblastoma: Molecular basis to overcome resistance. Curr Cancer Drug Targets 12: 197–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuch BB, Laborde RR, Xu X, Gu J, Chung CB, Monighetti CK, Stanley SJ, Olsen KD, Kasperbauer JL, Moore EJ, et al. 2010. Tumor transcriptome sequencing reveals allelic expression imbalances associated with copy number alterations. PLoS ONE 5: e9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin ML, Teague J, et al. 2011. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469: 539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, Larson DE, McLellan MD, Dooling D, Abbott R, et al. 2012. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med 366: 1090–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D 2009. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer 9: 489–499 [DOI] [PubMed] [Google Scholar]
- Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca DS, Zhang L, et al. 2011. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 365: 2497–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Tsutsumi S, Kawaguchi T, Nagasaki K, Tatsuno K, Yamamoto S, Sang F, Sonoda K, Sugawara M, Saiura A, et al. 2012. Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res 22: 208–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, et al. 2008. The complete genome of an individual by massively parallel DNA sequencing. Nature 452: 872–876 [DOI] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. 2007. The genomic landscapes of human breast and colorectal cancers. Science 318: 1108–1113 [DOI] [PubMed] [Google Scholar]
- Wu X, Northcott PA, Dubuc A, Dupuy AJ, Shih DJ, Witt H, Croul S, Bouffet E, Fults DW, Eberhart CG, et al. 2012. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 482: 529–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Hou Y, Yin X, Bao L, Tang A, Song L, Li F, Tsang S, Wu K, Wu H, et al. 2012. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell 148: 886–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al. 2010. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467: 1114–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Ye D, Guan KL, Xiong Y 2012. IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin Cancer Res 18: 5562–5571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap TA, Gerlinger M, Futreal PA, Pusztai L, Swanton C 2012. Intratumor heterogeneity: Seeing the wood for the trees. Sci Transl Med 4: 127ps10. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. 2011. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478: 64–69 [DOI] [PubMed] [Google Scholar]