Protein disulfide isomerase may facilitate the efflux of nitrite derived S-nitrosothiols from red blood cells (original) (raw)
Abstract
Protein disulfide isomerase (PDI) is an abundant protein primarily found in the endoplasmic reticulum and also secreted into the blood by a variety of vascular cells. The evidence obtained here, suggests that PDI could directly participate in the efflux of NO+ from red blood cells (RBC). PDI was detected both in RBC membranes and in the cytosol. PDI was S-nitrosylated when RBCs were exposed to nitrite under ∼50% oxygen saturation but not under ∼100% oxygen saturation. Furthermore, it was observed that hemoglobin (Hb) could promote PDI S-nitrosylation in the presence of ∼600 nM nitrite. In addition, three lines of evidence were obtained for PDI–Hb interactions: (1) Hb co-immunoprecipitated with PDI; (2) Hb quenched the intrinsic PDI fluorescence in a saturable manner; and (3) Hb–Fe(II)–NO absorption spectrum decreased in a [PDI]-dependent manner. Finally, PDI was detected on the surface RBC under ∼100% oxygen saturation and released as soluble under ∼50% oxygen saturation. The soluble PDI detected under ∼50% oxygen saturation was _S_-nitrosylated. Based on these data it is proposed that PDI is taken up by RBC and forms a complex with Hb. Hb–Fe(II)–NO that is formed from nitrite reduction under ∼50% O2, then transfers NO+ to either Hb–Cys β93 or directly to PDI resulting in S-nitroso-PDI which transverses the RBC membrane and attaches to the RBC surface. When RBCs enter tissues the S-nitroso-PDI is released from the RBC-surface into the blood where its NO+ is transferred into the endothelium thereby inducing vasodilation, suggesting local oxygen-dependent dynamic interplays between nitrite, NO and S-nitrosylation.
Abbreviations: BCA, bicinchoninic acid; EDTA, ethylenediaminetetraacetic acid; Hb, hemoglobin; NOx, nitric oxide related species; NP-40, nonyl phenoxypolyethoxylethanol; PDI, protein disulfide isomerase; PMSF, penylmethylsulfenylfluoride; RBC, red blood cells; SNO-Hb, S-nitrosohemoglobin; SNO, _S_-nitrosothiol; SDS-PAGE, sodium dodecyl sulfate, poly acrylamide gel electrophoresis
Keywords: Nitrite reductase, S-nitrosohemoglobin, Hypoxic vasodilation, Protein disulfide isomerase, Red blood cells, S-nitroso-protein disulfide isomerase
Graphical abstract
Highlights
- •
Red blood cells (RBC) contain protein disulfide isomerase (PDI) that can associate with hemoglobin. - •
Formation of S-nitroso-PDI is an oxygen- and Hb-dependent process. - •
S-nitroso-PDI associates with RBC surface in an oxygen dependent manner that facilitates its release under hypoxia.
Introduction
When RBCs enter a hypoxic region of the vasculature they release effector(s) that induce vasodilation thus ensuring that the oxygen they release is effectively distributed. This phenomenon termed hypoxic vasodilation is highly conserved and although first reported some 90 years ago [1–3], the sensing mechanisms as well as the vasodilatory substances released by RBCs remain to be clearly identified. Current research in this area supports either that nitric oxide (NO) and related compounds (NOx) [4–6] or ATP [7–12] as vasodilator-triggers released from RBCs.
The first hypothesis put forward for RBC-mediated hypoxic vasodilatation is through release of ATP upon decrease in HbO2 saturation. The ATP released diffuses to the endothelium and binds to the purinergic receptors leading to increase NO production via eNOS activation [7–13]. Furthermore, recent studies have shown that deoxy-Hb interacts with nitrite and dislodges the membrane bound glycolytic regulatory subunits enhancing intracellular ATP that is released under hypoxic conditions [14–16]. The ATP is released from RBC not only when RBC deoxygenates but also in response to mechanical deformation when RBC travel through narrow vessels [11,12]. Various factors are likely to regulate the role of ATP in vasodilatation such as the activity of transporters that regulate ATP release, enzymes that regulate ATP concentrations and purinergic receptor expression levels [17].
The NO (or NOx) based hypotheses can be further subdivided into those that depend on the scavenging of endothelia-generated or more recently RBC-eNOS-generated NO [18–21] to yield S-nitrosohemoglobin (SNO-Hb) or those that transform nitrite to NO within the RBC by hemoglobin acting as a nitrite reductase. In the SNO-Hb hypothesis, deoxygenated Hb in its T-state scavenges the endothelial NO to yield a mixture of HbFe(II)–NO and HbFe(III)–NO. When the RBCs arrive in the lungs the Hb undergoes a conformational change to the R-state where the O2 displaces the heme-bound NO to Cys β93 of Hb to form SNO-Hb. When the RBC reach the hypoxic tissues, Hb then undergoes conformational changes to the T state which leads to the concomitant release of O2 and transfer the SNO-bound NO bioactivity to the outside of the RBC possibly via transnitrosation reactions to induce endothelial vasodilatation [4,22–24].
The nitrite/nitrite reductase hypothesis involves transport of nitrite to RBC, reaction of nitrite with deoxy-Hb, transport of NO bioactivity from RBC and finally vasodilation [5,15,25,26]. The plasma levels of nitrite are reportedly between ∼120 nM and 290 nM [17,27]. Recent studies suggest that plasma nitrite can accumulate to near μM levels in RBC under hypoxia, via the deoxyHb-mediated inhibition of the anion transporter (AE1) which is responsible for nitrite efflux from the RBC [28]. Within RBC the nitrite could be converted to NO by the previously demonstrated nitrite reductase activities of xanthine oxidoreductase, hemoglobin [28] and eNOS [21,29–31].
The next important question concerns the mechanisms by which intracellular NO or NO-equivalents exit the RBC which contains ∼30 mM Hb. The amount of NO produced by the SNO-Hb or the nitrite routes are expected to be in the submicromolar levels. Under these conditions, any NO that is formed can react with deoxyhemoglobin (Fe2+) and yield heme-nitrosylHb (HbNO) which can either react with oxygen to form nitrate plus methemoglobin (Fe3+) or react with RBC-thiols to yield S-nitrosothiols (SNO). The efflux of SNO-bound NO from RBC could be plausible via a series of transnitrosation reactions where the SNO moiety would be transferred/shuttled from Hb to other intracellular proteins then to membrane spanning proteins eventually ending up as cell surface or secreted SNO-proteins which can deliver their NO into endothelia thus effecting vasodilation. In fact, several studies have implicated the transmembrane anion exchanger 1 (AE1) or band 3, one of the most abundant RBC-proteins, of accepting SNO-Hb-bound NO via transnitrosation [23,24,28,32,33].
Protein disulfide isomerase (PDI) is another enzyme that could potentially play a role in the efflux of NO equivalents from RBCs for the following reasons: PDI accounts for ∼1% of total cellular proteins in mammalian cells. Although it is largely an endoplasmic reticulum—(ER)—resident enzyme, it is secreted or leaks out of cells where it forms weak associations with the cell surfaces of many cell types including pancreatic cells [34,35], B cells [36,37], hepatocytes [38], platelets [39,40], endothelial cells [41], leukocytes [42,43] and platelet derived microparticles [44]. Several studies in RBCs have identified membrane associated PDI. However, the physiological role of PDI in RBCs is unknown [40,45–47].
Previous studies have shown that in endothelial cell surface PDI facilitates the transfer of extracellular SNO to the cytosol [48] and that PDI catalyze the release of NO from SNO-PDI as well as other S-nitrosothiols [49]. In this study, we report the potential involvement of PDI in a nitrite-dependent and oxygen regulated process for the efflux of NO (or NO-equivalents) from RBCs.
Materials and methods
Materials
Buffer salts, diethylenetriaminepentaacetic acid (DTPA), ethylenediaminetetraacetic acid (EDTA), penylmethylsulfenylfluoride (PMSF), nonyl phenoxypolyethoxylethanol (NP-40), sodium dodecyl sulfate (SDS), hemoglobin, sodium dithionite, biotin-maleimide and immunoblotting reagents were obtained from Sigma-Aldrich (St. Loius Mo). All antibodies were purchased from AbCam (Cambridge MA). The bicinchoninic acid assay (BCA assay), Aminolink Plus coupling resin and spin columns were purchased from Thermo Scientific (Rockford, Ill).
RBC preparation
RBCs were prepared for experiments under different oxygen saturations using previously established protocols. Fresh blood was collected from healthy human volunteers by venipunture into BD tubes containing anticoagulant. Blood was centrifuged at 1000×g for 10 min to remove plasma and buffy coat. RBCs were washed with buffer (pH 7.4) of following composition 6.9 g/L NaCl, 2.28 g/L NaHCO3, 0.35 g/L KCl, 0.136 g/L KH2PO4, 0.144 g/L MgSO4, 2.0 g/L D-glucose to prevent hemolysis. Experiments with different oxygen saturations were performed in septa sealed vials. The buffer used in the experiments was also pre-equilibrated for 30 min at respective oxygen saturations. Isolated and washed RBCs in buffer (pH 7.4) were held under 16% O2 or hypoxia 4% O2 for 15 min [28,50]. Nitrite stock solution was prepared in phosphate buffered saline (PBS) with DTPA (100 μM) and added to RBC suspension to a final concentration of 600 nM using a syringe and further incubated for 10 min.
Immunoprecipitation
RBCs membranes were prepared using standard protocols as described previously [17,27,51]. Briefly, to the RBC pellet (1 mL) 40 volumes of ice-cold 5 mM phosphate buffer containing 0.1 mM PMSF, 20 mM NEM and 100 μM DTPA was added. RBCs were then incubated on ice for 20 min to induce hemolysis. After centrifugation at 12,000×g for 10 min at 4 °C, RBC membranes were washed twice with the same buffer.
RBC membranes were dissolved as described earlier [51,52]. RBC membranes were solubilized in lysis buffer containing Hepes (50 mM), NaCl (150 mM), EDTA (5 mM), EGTA (5 mM), sodium pyrophosphate (20 mM), NEM (20 mM), orthovanadate (1 mM), NaF (20 mM), K3Fe (CN)6 (10 mM), NP-40 (1%), PMSF (0.1 mM) and protease inhibitor (1:200). The samples (100 μg) were precleared with protein A/G (40 μL) by incubation and mixing for 1 h at 4 °C. Samples were then incubated with anti-PDI antibody (1:50 dilution) or mouse anti AE1 antibody (1:100 dilution) or rabbit anti-GLUT 1 antibody (1:100 dilution). After incubation for 2 h at 4 °C, protein A/G beads (50 μL) were added to the samples and further incubated for 3 h. The beads were washed three times with lysis buffer. Proteins were eluted from beads using SDS-PAGE sample buffer devoid of β-mercaptoethanol by incubating at 95° for 10 min and analyzed by immunoblotting. For experiments with RBC homogenates, RBC samples (100 μL) were homogenized in PBS with NEM (20 mM), K3Fe(CN)6 (10 mM), DTPA (100 μM), NP-40 (1%), PMSF (0.1 mM) and protease inhibitor (1:200) [53] followed by immunoprecipitation as described above.
Detection of S-nitrosylated PDI by immunoblotting
Nitrite supporting PDI-S-nitrosylation in-vitro in presence of oxy-hemoglobin (oxyHb) was determined as follows: Hb(1 mM) was reduced with dithionite (50 mM) under argon in septa sealed vials and transferred to septa sealed vials containing constant PDI (1 μM), Hb (0.6 mM) and varying amounts of nitrite (78 nM–5 μM) in PBS. The headspace of the vial contained 20 ppm O2. After incubation for 5 min at room temperature the samples were are subjected to biotin switch assay as previously described [50,53,54]. The supernatant was treated with 100 μM DTPA, 20 mM NEM, 10 mM K3Fe(CN)6 and 1% SDS and incubated at 50 °C for 30 min with frequent vortexing. Two volumes of ice-cold acetone were added to precipitate the proteins. The precipitant was further washed with 70% acetone. Protein pellets were resuspended in 100 μL of PBS followed by addition of 1 mM ascorbic acid and 1 mM biotin-maleimide and incubated in dark at room temperature for 1 h. Biotin-labeled proteins were precipitated using pre-chilled acetone and resuspended in PBS. The concentrations of protein in samples were determined by BCA assay. Protein samples (8 μg/well) were resolved on non-reducing SDS-PAGE followed by immunoblotting. The membranes were probed with anti- Hb (1:1000), mouse anti-PDI (1:1000) and streptavidin-HRP (1:100,000). The blots were then incubated with anti-mouse secondary antibodies and visualized using chemiluminescence substrate.
Preparation of Hb samples for UV spectroscopy
All reactions were carried out in PBS pH 7.4 at room temperature. Deoxy-hemoglobin was prepared as previously described [55] Briefly, hemoglobin (Hb) was dissolved in PBS pH 7.4 (8.3 mg/ml) followed by centrifugation at 12,000 rpm for 2 min. The supernatant was used for preparation of deoxy-Hb. Hb (1 mM) was reduced with dithionite (0.5 mM) under argon (Ar) in septa sealed cuvette followed by addition of sodium nitrite (50 μL of 1 mM stock, total _V_=1.05 mL). The concentration of Hb(III), Hb(II) and Hb(II)-NO were determined using the previously reported mM extinction coefficients [56,57]. Small amount of air (500 μL) was introduced by the aid of syringe to displace the NO from the heme to yield SNO-Hb (βCys93) [58]. The change in the [Hb(II)-NO] was monitored at 418 nm (mM extinction coefficient=130 mM−1 cm−1 [55]) with an Agilent 8453 UV spectrophotometer.
NO measurements
The gas phase NO was measured using Sievers® Nitric Oxide Analyzer (NOA 280i). In these experiments, small aliquots (10 μL) of the immunoprecipitated PDI was injected into the NOA chamber containing I3− dissolved in acetic acid. And compared to a NO standard curve generated by injecting NO(aq) solutions into the NOA.
Fluorescence measurements
Fluorescence measurements were performed on Cary Eclipse fluorescence spectrophotometer (Agilent, Canada). The excitation wavelength was set at 280 nm to limit fluorescence measurements mainly to tryptophan. The PDI concentration used was 0.75 μM and all measurements were performed in PBS pH 7.4. The Hb was added from 2 mg/ml stock solution. The measured fluorescence intensity F was corrected for inner filter effects (Fcorr) using the measured absorbance A280 at each Hb concentrations Fcorr=F/(1−10−–A280) (this correction corresponds to the fraction of light absorbed as deduced from the Beer–Lambert law).
Flow cytometry
RBC surface proteins were detected by flow cytometry using previously established protocols with some modifications [59–61]. RBCs were isolated and adjusted to ∼106 cells/ml and held at different oxygen conditions with and without nitrite as described above. RBC samples were immediately fixed with 1% paraformaldehyde for 20 min, followed by washing with PBS (3×1 mL). RBCs were incubated with mouse monoclonal anti-PDI antibody (1:100) for 1 h. After washing with PBS (3×1 mL), RBCs were incubated with sheep anti-mouse IgG-FITC (Stressgen Biotechnologies) for 30 min in dark. RBCs were then washed with PBS (3×1 mL) and analyzed by flow cytometry. RBCs labeled with only sheep anti-mouse IgG-FITC or only anti-PDI antibody were used as controls. Data was collected for 100,000 events of RBC population.
Detection of soluble PDI
The buffer of the RBC suspension under 16% O2 and 4% O2 oxygen with and without nitrite treatment were probed for soluble PDI. The soluble PDI was isolated as described previously [62]. The supernatant of the RBC suspension was subjected to two centrifugation steps first at 1000×g to remove the cells then at 13,000×g to remove cellular debris. The resulting supernatant containing soluble proteins was used for immunoprecipitation of PDI. The supernatant (1 mL) was precleared with protein A/G (40) by incubation and mixing for 1 h at 4 °C. The suspension was centrifuged at 1000×g for 1 min. 10 μg mouse anti-PDI antibodies was immobilized on Aminolink Plus coupling resin, according to the manufacturer′s instructions, followed by addition of 500 μL of supernatant. After incubation for 2 h at 4 °C, the suspension was centrifuged at 1000×g for 1 min. The beads were washed three times with wash buffer and incubated with 100 μL of elution buffer for 5 min. Immunoprecipitated proteins were collected by centrifugation at 1000×g for 2 min. Protein concentrations were determined by BCA assay. Protein samples, ∼20 μg/well, were resolved on non-reducing SDS-PAGE gel followed by immunoblotting. The membranes were then incubated with rabbit anti-PDI antibody (1:1000) antibody for 2 h followed by HRP conjugated anti-rabbit secondary antibodies (1:2500) for 1 h and visualized using chemiluminescence substrate. For determining S-nitrosylation of soluble PDI at various oxygen saturations, 10 μL of imunoprecipitated protein was injected into NO analyzer. The concentration of NO was determined from standard curve.
Results and discussion
PDI is detected in RBC homogenates and membrane fractions
Recent studies indicate that PDI along with several other chaperones are lost in erythroid progenitor cells as they mature to become RBCs [63]. Despite this, PDI was detected on the surface of RBCs [39,40] as well as in proteomic profiles of RBCs [46,47]. Here, the various components of human RBCs were probed with the aid of anti-PDI antibodies (RL-90). Western blots indicated PDI was present in RBC homogenates and in the plasma membrane (Fig. 1).
Fig. 1.

Western immunoblots of: RBC membrane fraction-Lane 1; RBC homogenate-Lane 2; and standard human PDI-Lane 3, all probed with anti-PDI primary antibodies.
RBC-PDI can be S-nitrosated in the presence of Hb plus nitrite under oxygenated conditions
The nitrite reductase hypothesis requires a significant portion of the RBC Hb to be in the deoxyHb form [5,6,26]. In addition, the intra RBC nitrite accumulation proposed by Vitturi et al. [28] also requires large amounts of deoxyHb to inhibit Band3 thereby blocking the efflux of nitrite. In current study, we explored the ability of RBC-PDI to become S-nitrosylated upon incubation of intact RBCs with nitrite under 4% O2 or 16% O2 saturation conditions. These two O2 levels were chosen because 4% O2 corresponds to ∼27 Torr or ∼36 μM oxygen which is the P50 for human Hb, representing oxygenation levels in venous blood. Therefore, at this oxygen tension, Hb is half saturated. Previous studies [6] have demonstrated that the highest rate of nitrite reduction takes place at the P50 of Hb which is 4% O2. The 16% level was chosen as this represents the oxygen tension in the arteries ∼144 μM. At this level Hb is fully saturated with oxygen.
The S-nitrosylation status of PDI was determined by the biotin switch assay. Our data indicate that RBC-PDI is S-nitrosylated under 16% O2 (Fig. 2, Lane 3), whereas it is not S-nitrosylated at 4% O2 (Fig. 2, Lane 1).
Fig. 2.
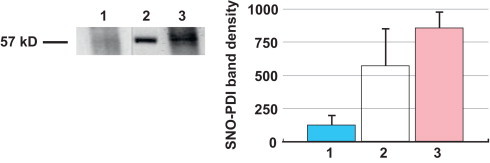
Nitrite promotes RBC-PDI _S_-nitrosylation under normoxia but not under hypoxia: (A) Freshly isolated RBCs were equilibrated with either 4% O2 (Lane 1) or 16% O2 (Lane 3) in septa sealed vials nitrite (600 nM) was introduced and incubated for 10 min. The RBCs were lysed and the S-nitrosylation status of RBC–PDI was determined by the biotin switch assay visualized with streptavidin-HRP. The band corresponding to PDI was identified from the electrophoretic mobility of standard human PDI subjected to SDS-PAGE under identical conditions (Lane 2). (B) Digitized blot densities (ImageJ) of the bands obtained from 3 different experiments with conditions identical to A in each lane. Error bars represent standard deviation (_n_=3).
The next question was to determine whether Hb was responsible for nitrite-mediated PDI-S-nitrosylation under normoxia. To do this, constant amounts of PDI (1 μM), Hb (0.6 mM-dithionite reduced) was incubated with varying amounts of nitrite (78 nM to 5 μM) in PBS, in septa sealed vials equilibrated with 16% O2.
The PDI-S-nitrosylation was detected by the biotin switch assay visualized with streptavidin-HRP. The results indicate that [nitrite] as low as ∼625 nM can support PDI-S-nitrosylation (Fig. 3, Lane 4). Furthermore, these experiments dramatically demonstrate the requirement of Hb in the S-nitrosylation of PDI: ∼50% of PDI is S-nitrosylated with 5 μM nitrite in the presence of Hb (Fig. 3, Lane 7). However, there is no detectable PDI-S-nitrosylation when Hb is excluded (Fig. 3, SNO–PDI: strepatavidin-HRP–Hb or Lane 8).
Fig. 3.
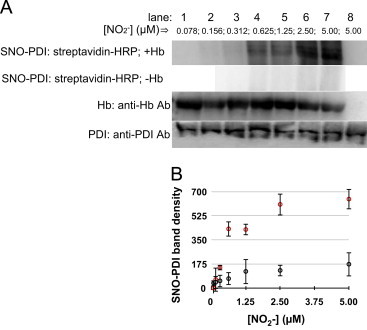
Hb promotes NO2−-dependent nitrosylation of PDI under normoxic conditions (16%–O2): (A) These experiments were performed using constant PDI (1 μM), Hb (0.6 mM) and varying amounts of nitrite (78 nM–5 μM) in PBS-Lanes 1–7. The headspace of the vial contained 16% O2. The mixtures were incubated at 37 °C for 10 min. Aliquots were then removed and added to cold acetone and prepared for either the SNO–PDI determination by the biotin switch assay-visualized by streptavidin-HRP or detecting HB or PDI by Western immunoblots utilizing anti-Hb or anti-PDI, respectively as the primary antibodies. Lane 8-only contained PDI (1 μM) plus nitrite (5 μM) and no Hb. (B) Digitized blot densities (ImageJ) of SNO-PDI as a function of [NO2−] in the presence of Hb (red circles) and absence of Hb (black circles). Error bars represent standard deviation (_n_=3).
PDI co-immunoprecipitates with Hb and Hb quenches the PDI-intrinsic fluorescence
In order for Hb to be able to S-nitrosylate PDI these two proteins must come in close contact so that the nitrosonium ion on moiety on Cys β93-S-NO of Hb can be transferred to the free thiols of PDI. Here we tested the potential interaction of Hb and PDI by two different methods. First RBC homogenates were immunoprecipitated with anti-PDI: protein A/G-sepahrose beads and the immunoprecipitation product was immunoprobed with either anti-PDI or anti-HB primary antibodies (Fig. 4).
Fig. 4.
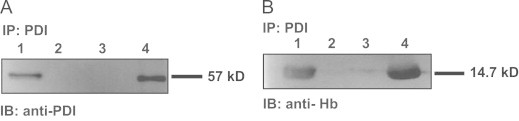
Hb co-immunoprecipitates with PDI: RBC were immunoprecipitated with anti-PDI:ProteinA/G-Agarose beads and immunoprecipited proteins and various controls were immunoblotted with either anti-PDI (A) or anti-Hb (B) primary antibodies: (A) Lane 1: PDI control; Lane 2: anti-PDI:protein A/G agarose; Lane 3: protein A/G agarose; Lane 4: immunoprecipitation product; (B) Lane 1: Hb control; Lane 2: anti-PDI:protein A/G agarose; Lane 3: protein A/G agarose; Lane 4: immunoprecipitation product.
As can be clearly observed (Fig. 4A and B, Lane 4) the immunoprecipitation product contained both PDI and Hb an indication that the two proteins interact in RBC.
The second method used for assessing PDI:Hb interactions was to monitor the intrinsic fluorescence change of PDI as a function of Hb-dose. PDI has five tryptophan residues in its sequence and its Trp fluorescence was assumed to be similarly sensitive to the detection of protein:protein interactions as thioredoxin [64,65]. Here, the intrinsic fluorescence of PDI (0.75 μM) (_λ_ex 278 nm, _λ_em 339 nm) decreased in a [Hb]-dependent manner (Fig. 5) with a half-maximal decrease occurring at ∼1 μM Hb, suggesting a dissociation constant between these two proteins of ∼1 μM or less
Fig. 5.
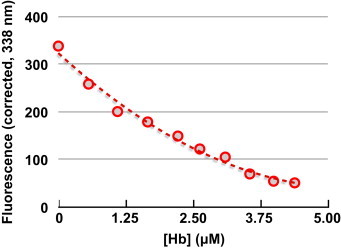
PDI intrinsic fluorescence is quenched by Hb: The intrinsic Trp fluorescence (_λ_ex 278 nm, _λ_em 339 nm) of PDI (0.75 μM) was monitored as a function of [Hb(III)]. The fluorescence was corrected for inner-filter effects by measuring the absorbance of the solution after each addition of Hb and using the equation. Fcorrected=F/1−10A(280nm).
PDI can denitrosate Hb-NO
Having obtained evidence for PDI-Hb interactions the next question was, can PDI denitrosate Hb-NO under normoxic conditions? To test this Hb, under Ar, was reduced with dithionite, exposed to nitrite. The formation of Hb-NO was identified via its characteristic absorption spectrum with a maximum at 418 nm (Fig. 6A).
Fig. 6.
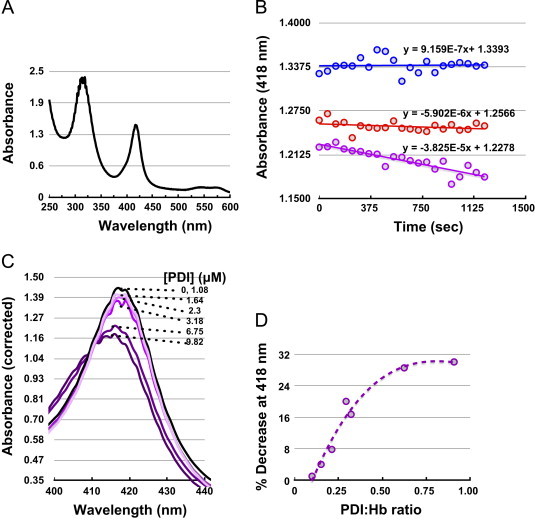
PDI denitrosates Hb-NO: (A) Hb-(II)–NO was formed by incubating dithionite-reduced Hb(II) (10.8 μM) with nitrite resulting in the characteristic Hb-(II)–NO UV/vis spectrum; (B) The Hb(II)–NO was monitored spectrophotometrically, with respect to time, at 418 nm under Ar (blue circles); or in the presence of air (500 μL ) (red circles); or in the presence of PDI (3.4 μM) plus of air (500 μL) (purple circles). The Hb-NO extinction coefficient used to convert Δ_A_(418 nm) to [Hb-NO] was 130,000 M−1 cm−1**;** (C) Hb-(II)–NO (10.8 μM) spectrum in the presence of air (500 μL ) was recorded 10 min after incubation with and varying concentration of PDI to give Hb-NO:PDI ratio between 0 and 10; **(**D) The re-plot of Δ_A_(418 nm) from C, corrected for the absorbance decrease by air alone, as a function of PDI:Hb-(II)-NO ratio.
As previously reported, the conversion Hb-NO to SNO-Hb (Cys β93) requires the presence of oxygen [58,66]. To this end, 500 μL of air was added to septa-sealed (Fig. 6B) samples containing Hb-NO. The Hb-NO was stable in the presence of Ar. Upon introduction of air, the Hb-NO peak decreased at a rate of 2.72 nM/min.
However, the rate of the decrease of the Hb-NO peak increased ∼6.5-fold to 17.7 nM/min in the presence of PDI, suggesting that PDI can denitrosate Hb-NO. Next Hb-NO was titrated with PDI in an attempt to determine the stoichiometry of the interaction (Fig. 6C and D). The Hb-NO peak decreased with increasing amounts of PDI and saturated at a ratio of 1:1 [Hb-NO] to [PDI]. The maximal decrease in the Hb-NO peak was ∼30%. Potential explanations for this include that the amount of O2 introduced into the cuvette was not sufficient to displace all of the Hb-NO. Another possibility is that only some of the Hb-NO sites are accessible to PDI at this oxygen tension.
PDI associates with the RBC-surface in an O2 and nitrite-dependent manner
As outlined in the introduction, PDI is secreted from many cells comprising the vascular system. Fully active PDI has been detected in the blood, in its soluble form as well as in its microparticle-associated and cell surface-associated forms [33–42].
We wanted to determine if PDI associated with the RBC surface and whether the surface association was dependent on O2 and nitrite levels. To do this, RBCs were treated with and without nitrite (50 μM) under 16% O2 and 4% O2 saturation. The PDI on the RBC-surface was detected using flow cytometer (Cytomics FC500, Beckman Coulter, USA) using monoclonal mouse anti-PDI antibody (primary Ab) and sheep-antimouse IgG-FITC (secondary Ab).
There was essentially no PDI detected on the surface of RBCs under 4% O2. However, at 16% O2, in the absence of added nitrite there was a low amount of PDI (corresponding to ∼0.65%±0.2% of total fluorescence). Upon exposure to nitrite, the population distribution increased to ∼8.6%±3.8% (Fig. 7A). These results suggest that PDI associates with the RBC surface at 16% and is released at low (4%) O2. If this hypothesis is correct, then there should be more soluble PDI in the suspension buffer of RBCs exposed to 4% O2 than 16% O2. To test this, we removed the RBCs from the buffer by gentle centrifugation and probed the RBC suspension buffer for PDI by immunoprecipitation. Furthermore, the immunoprecipitation products, isolated from the suspension buffer, were probed for the degree of S-nitrosylation by directly injecting them into an NO-analyzer (acetic acid-iodine).
Fig. 7.
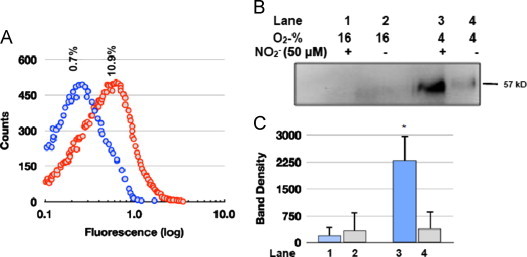
PDI associates with the RBC surface in an O2 and nitrite-dependent manner: (A) Representative flow cytometer RBC population distributions, probed for extracellular PDI. The RBCs (∼106 cells/mL) suspended in PBS were equilibrated in 16% O2 with either no nitrite (blue circles) or 50 μM nitrite (red circles) for 30 min then probed with mouse monoclonal anti-PDI antibody and sheep anti-mouse IgG-FITC and analyzed by flow cytometry; The results obtained for 4% O2 (not graphically displayed) were −nitrite 0.40%±0.10%, +nitrite 0.35%±0.12%; for 16% O2 were −nitrite 0.70%±0.085%, +nitrite 8.6%±3.8% (S.D., _n_=4); (B) Representative immunoblots of soluble PDI detected in the RBC suspension buffer by immunoprecipitation of the RBCs exposed to either 16% or 4% O2±nitrite. The immunoprecipitation product was subjected to SDS-PAGE and immunoblotted with anti-PDI primary antibodies. (D) Digitized blot densities (ImageJ) of the immunoblots (B) error bars represent S.D. (_n_=4).
Interestingly, the amount of soluble PDI found in RBC suspensions was ∼11-fold larger under 4% O2 plus nitrite in comparison to 16% O2 plus or minus nitrite, (Fig. 7B and C) the exact opposite of the RBC surface associated PDI levels determined by flow cytometry (Fig. 7A). This supports our hypothesis that PDI is strongly associated with the RBC surface under normoxia and weakly associated under conditions of hypoxia.
Next, we determined the S-nitrosation status of soluble PDI with the chemiluminescent NO analyzer (NOA). In these experiments, immunoprecipitation product was injected into the NOA containing iodine in acetic acid. Under these conditions protein-SNO would be reduced to NO by I3− and detected by the NOA. There was no detectable NO in the immunoprecipitated samples exposed to 16% O2±nitrite or those exposed to 4% O2 −nitrite. In contrast, RBC exposed to nitrite under 4% O2 yielded ∼274 pmol±74 pmol of NO per mg protein (_n_=4). This corresponds to ∼2% SNO/mol of Hb, not surprizing in view of the lability of SNO functionality in proteins and the ∼2 h workup required for the isolation and analysis of the soluble PDI fraction.
We believe the data presented in this in vitro study makes a compelling case for PDI to have a role in the efflux of NO equivalents from RBCs. First, RBCs reportedly become devoid of PDI as they mature from erythroid progenitor cells [64] yet this protein is detected on the RBC surface [38,39] and in total cellular proteomic profiles [45,46]. In addition, in the present study, we detected PDI in membrane fractions and total cell homogenates (Fig. 1). In addition, the fact that PDI was found to co-immunoprecipitate with Hb (Fig. 4) indicates that PDI is also in the cytosol of RBCs.
We can therefore speculate that RBCs pick up soluble PDI from blood as PDI is readily secreted into the vasculature from many cell types [33–43]. PDI is also known to readily effluxed from cells. The current observation that it is detected intracellularly in RBC suggests that PDI can freely cross plasma membranes in both directions.
Our results also show that RBC-PDI is S-nitrosylated upon exposure of RBCs to nitrite, that nitrite levels as low as 0.6 μM could support significant SNO–PDI formation and there was an absolute requirement for Hb in this process (Fig. 3). In order for PDI to get transnityrosylated from SNO-Hb (Cys β93) or directly denitrosate Hb–Fe(II)–NO these two proteins must interact. Here, we presented multiple lines of evidence for Hb:PDI interactions: (1) these two proteins co-immunoprecipitate (Fig. 4); (2) the intrinsic PDI-fluorescence is perturbed in a saturable manner with Hb (Fig. 5); and (3) we have clear UV/vis spectral evidence that PDI can denitrosate Hb–Fe(II)–NO with a ∼1:1 stoichiometry (Fig. 6).
In order for PDI to “carry” NO-equivalents out of RBCs it must be secreted. Here we were able to use flow cytometry to show that the PDI associates with the RBC surface in the presence of nitrite under normoxia but not under hypoxia. And that under hypoxia most of the PDI is secreted as soluble PDI. Furthermore, the secreted PDI was S-Nitrosated (Fig. 7). These potentially redox regulated mechanisms for PDI–RBC surface associations are currently under investigation.
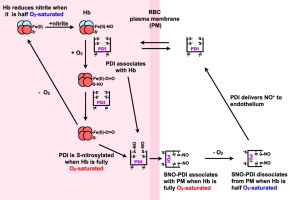
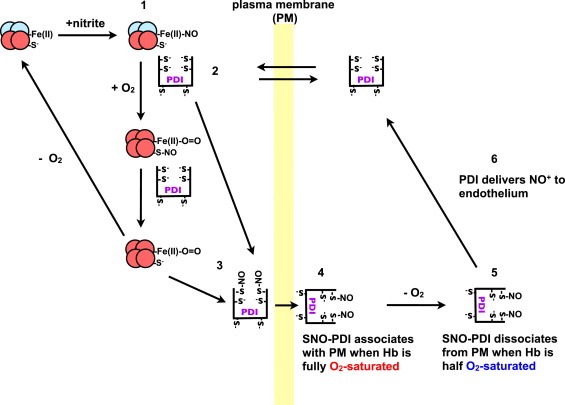
On the basis of these observations, we propose the following mechanism for PDI mediated NO-equivalent efflux from RBC (Scheme 1).
Scheme 1.
.
Under hypoxic conditions nitrite reacts with Hb to form Fe(II)–NO (1). PDI from the blood can equilibrate across the RBC plasma membrane (PM) and interacts with Hb, to form a complex (2). When the RBCs arrive at the lungs, the O2 displaces the NO from the heme to either PDI-thiols or Hb(Cys β93) yielding SNO–PDI (3) or Hb-SNO. We can also speculate that under normoxia, PDI interacts with HbSNO to yield SNO–PDI (3). Under normoxia the SNO–PDI is attached to the RBC extracellular surface (4). Upon entering the tissues the PDI–SNO is released from the RBC surface (5). PDI–SNO then interacts with the endothelia releasing its NO+ or as previously demonstrated [48,49] its NO, triggering hypoxic vasodiation (6) (Scheme 1).
In summary, the role of PDI in the export of NOx from the RBC is supported by: (1) an oxygen-dependent mechanism of PDI binding to the RBC membrane, PDI being bound to the membrane in a PDI–SNO form under normoxia while being in a soluble form readily able to cross the membrane and deliver NOx under hypoxia; (2) this mechanism is coupled with a hypoxic nitrite reductase activity; because we could evidence a direct interaction between PDI and Hb, we propose an Hb-dependent redox process. We cannot rule out the involvement of the nitrite reductase activity of eNOS as this enzyme was reported in RBC [19,29,31]. Nitrite reduction requires nitrite influx within RBC, either in the form of HNO2 or via active transport possibly through AE-1 as suggested [28]. This mechanism amounted in vitro 2% of SNO efflux per mg Hb, a quantity that would be likely to be much smaller in vivo, given the mM concentration of Hb in RBC. Importantly, our work clearly suggests a dynamic equilibrium between “reservoirs of NO” as nitrite, RSNO and NOx species, governed by oxygen levels and mediated by PDI, leading to multiple ways for efficient hypoxic vasodilation to take place when necessary. Given the interactions of PDI with many cell surfaces including platelets, platelet derived-microparticles and endothelial cells [33–43], this efflux mechanism may take place at additional interfaces besides RBC/blood.
Acknowledgments
This work was supported by a NSERC Discovery Grant to B.M. and by a Grant of the French Agence Nationale de la Recherche to A.S. V.M.K. was supported by a University of Windsor Post Graduate Tuition Scholarship.
Footnotes
☆
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Anny Slama-Schwok, Email: anny.slama-schwok@inserm.fr.
Bulent Mutus, Email: bulent.mutus@gmail.com, mutusb@uwindsor.ca.
References
- 1.Hilton R., Eichholtz F. The influence of chemical factors on the coronary circulation. Journal of Physiology. 1925;59:413–425. doi: 10.1113/jphysiol.1925.sp002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gremels H., Starling E.H. On the influence of hydrogen ion concentration and of anoxaemia upon the heart volume. Journal of Physiology. 1926;61:297–304. doi: 10.1113/jphysiol.1926.sp002294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ross J.M., Fairchild H.M., Weldy J., Guyton A.C. Autoregulation of blood flow by oxygen lack. American Journal of Physiology. 1962;202:21–24. doi: 10.1152/ajplegacy.1962.202.1.21. [DOI] [PubMed] [Google Scholar]
- 4.Jia L., Bonaventura C., Bonaventura J., Stamler J.S. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380:221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 5.Cosby K., Partovi K.S., Crawford J.H., Patel R.P., Reiter C.D., Martyr S., Yang B.K., Waclawiw M.A., Zalos G., Xu X., Huang K.T., Shields H., Kim-Shapiro D.B., Schechter A.N., Cannon R.O., 3rd, Gladwin M.T. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nature Medicine. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 6.Huang Z., Shiva S., Kim-Shapiro D.B., Patel R.P., Ringwood L.A., Irby C.E., Huang K.T., Ho C., Hogg N., Schechter A.N., Gladwin M.T. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. Journal of Clinical Investigation. 2005;115:2099–2107. doi: 10.1172/JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellsworth M.L. The red blood cell as an oxygen sensor: what is the evidence? Acta Physiologica Scandinavica. 2000;168:551–559. doi: 10.1046/j.1365-201x.2000.00708.x. [DOI] [PubMed] [Google Scholar]
- 8.Ellsworth M.L. Red blood cell-derived ATP as a regulator of skeletal muscle perfusion. Medicine and Science in Sports and Exercise. 2004;36:35–41. doi: 10.1249/01.MSS.0000106284.80300.B2. [DOI] [PubMed] [Google Scholar]
- 9.Ellsworth M.L., Ellis C.G., Goldman D., Stephenson A.H., Dietrich H.H., Sprague R.S. Erythrocytes: oxygen sensors and modulators of vascular tone. Physiology (Bethesda) 2009;24:107–116. doi: 10.1152/physiol.00038.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sprague R.S., Ellsworth M.L., Stephenson A.H., Lonigro A.J. ATP: the red blood cell link to NO and local control of the pulmonary circulation. American Journal of Physiology. 1996;271:H2717–H2722. doi: 10.1152/ajpheart.1996.271.6.H2717. [DOI] [PubMed] [Google Scholar]
- 11.Sprague R.S., Ellsworth M.L., Stephenson A.H., Lonigro A.J. Participation of cAMP in a signal-transduction pathway relating erythrocyte deformation to ATP release. American Journal of Physiology—Cell Physiology. 2001;281:C1158–C1164. doi: 10.1152/ajpcell.2001.281.4.C1158. [DOI] [PubMed] [Google Scholar]
- 12.Sprague R.S., Stephenson A.H., Ellsworth M.L. Red not dead: signaling in and from erythrocytes. Trends in Endocrinology and Metabolism. 2007;18:350–355. doi: 10.1016/j.tem.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Jagger J.E., Bateman R.M., Ellsworth M.L., Ellis C.G. Role of erythrocyte in regulating local O2 delivery mediated by hemoglobin oxygenation. American Journal of Physiology—Heart and Circulatory Physiology. 2001;280:H2833–H2839. doi: 10.1152/ajpheart.2001.280.6.H2833. [DOI] [PubMed] [Google Scholar]
- 14.Campanella M.E., Chu H., Low P.S. Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2402–2407. doi: 10.1073/pnas.0409741102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao Z., Bell J.B., Mohanty J.G., Nagababu E., Rifkind J.M. Nitrite enhances RBC hypoxic ATP synthesis and the release of ATP into the vasculature: a new mechanism for nitrite-induced vasodilation. American Journal of Physiology—Heart and Circulatory Physiology. 2009;297:H1494–H1503. doi: 10.1152/ajpheart.01233.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia J.I., Seabra A.B., Kennedy R., English A.M. Nitrite and nitroglycerin induce rapid release of the vasodilator ATP from erythrocytes: relevance to the chemical physiology of local vasodilation. Journal of Inorganic Biochemistry. 2010;104:289–296. doi: 10.1016/j.jinorgbio.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 17.Crawford J.H., Isbell T.S., Huang Z., Shiva S., Chacko B.K., Schechter A.N., Darley-Usmar V.M., Kerby J.D., Lang J.D., Jr, Kraus D., Ho C., Gladwin M.T., Patel R.P. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood. 2006;107:566–574. doi: 10.1182/blood-2005-07-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen K.J., Popel A.S. Nitric oxide production pathways in erythrocytes and plasma. Biorheology. 2009;46:107–119. doi: 10.3233/BIR-2009-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kleinbongard P., Schulz R., Rassaf T., Lauer T., Dejam A., Jax T., Kumara I., Gharini P., Kabanova S., Ozuyaman B., Schnurch H.G., Godecke A., Weber A.A., Robenek M., Robenek H., Bloch W., Rosen P., Kelm M. Red blood cells express a functional endothelial nitric oxide synthase. Blood. 2006;107:2943–2951. doi: 10.1182/blood-2005-10-3992. [DOI] [PubMed] [Google Scholar]
- 20.Ozuyaman B., Grau M., Kelm M., Merx M.W., Kleinbongard P. RBC NOS: regulatory mechanisms and therapeutic aspects. Trends in Molecular Medicine. 2008;14:314–322. doi: 10.1016/j.molmed.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 21.Wood K.C., Cortese-Krott M.M., Kovacic J.C., Noguchi A., Liu V.B., Wang X., Raghavachari N., Boehm M., Kato G.J., Kelm M., Gladwin M.T. Circulating blood endothelial nitric oxide synthase contributes to the regulation of systemic blood pressure and nitrite homeostasis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013 doi: 10.1161/ATVBAHA.112.301068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stamler J.S., Jia L., Eu J.P., McMahon T.J., Demchenko I.T., Bonaventura J., Gernert K., Piantadosi C.A. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science. 1997;276:2034–2037. doi: 10.1126/science.276.5321.2034. [DOI] [PubMed] [Google Scholar]
- 23.Pawloski J.R., Hess D.T., Stamler J.S. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409:622–626. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- 24.Pawloski J.R., Stamler J.S. Nitric oxide in RBCs. Transfusion. 2002;42:1603–1609. doi: 10.1046/j.1537-2995.2002.00278.x. [DOI] [PubMed] [Google Scholar]
- 25.Doyle M.P., Hoekstra J.W. Oxidation of nitrogen oxides by bound dioxygen in hemoproteins. Journal of Inorganic Biochemistry. 1981;14:351–358. doi: 10.1016/s0162-0134(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 26.Nagababu E., Ramasamy S., Abernethy D.R., Rifkind J.M. Active nitric oxide produced in the red cell under hypoxic conditions by deoxyhemoglobin-mediated nitrite reduction. Journal of Biological Chemistry. 2003;278:46349–46356. doi: 10.1074/jbc.M307572200. [DOI] [PubMed] [Google Scholar]
- 27.Dejam A., Hunter C.J., Pelletier M.M., Hsu L.L., Machado R.F., Shiva S., Power G.G., Kelm M., Gladwin M.T., Schechter A.N. Erythrocytes are the major intravascular storage sites of nitrite in human blood. Blood. 2005;106:734–739. doi: 10.1182/blood-2005-02-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vitturi D.A., Teng X., Toledo J.C., Matalon S., Lancaster J.R., Jr, Patel R.P. Regulation of nitrite transport in red blood cells by hemoglobin oxygen fractional saturation. American Journal of Physiology—Heart and Circulatory Physiology. 2009;296:H1398–H1407. doi: 10.1152/ajpheart.01303.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mikula I., Durocher S., Martasek P., Mutus B., Slama-Schwok A. Isoform-specific differences in the nitrite reductase activity of nitric oxide synthases under hypoxia. Biochemical Journal. 2009;418:673–682. doi: 10.1042/BJ20080987. [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez F.M., Shiva S., Vincent P.S., Ringwood L.A., Hsu L.Y., Hon Y.Y., Aletras A.H., Cannon R.O., 3rd, Gladwin M.T., Arai A.E. Nitrite anion provides potent cytoprotective and antiapoptotic effects as adjunctive therapy to reperfusion for acute myocardial infarction. Circulation. 2008;117:2986–2994. doi: 10.1161/CIRCULATIONAHA.107.748814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gautier C., van Faassen E., Mikula I., Martasek P., Slama-Schwok A. Endothelial nitric oxide synthase reduces nitrite anions to NO under anoxia. Biochemical and Biophysical Research Communications. 2006;341:816–821. doi: 10.1016/j.bbrc.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 32.Walder J.A., Chatterjee R., Steck T.L., Low P.S., Musso G.F., Kaiser E.T., Rogers P.H., Arnone A. The interaction of hemoglobin with the cytoplasmic domain of band 3 of the human erythrocyte membrane. Journal of Biological Chemistry. 1984;259:10238–10246. [PubMed] [Google Scholar]
- 33.Shingles R., Roh M.H., McCarty R.E. Direct measurement of nitrite transport across erythrocyte membrane vesicles using the fluorescent probe, 6-methoxy-N-(3-sulfopropyl) quinolinium. Journal of Bioenergetics and Biomembranes. 1997;29:611–616. doi: 10.1023/a:1022491220299. [DOI] [PubMed] [Google Scholar]
- 34.Akagi S., Yamamoto A., Yoshimori T., Masaki R., Ogawa R., Tashiro Y. Localization of protein disulfide isomerase on plasma membranes of rat exocrine pancreatic cells. Journal of Histochemistry and Cytochemistry. 1988;36:1069–1074. doi: 10.1177/36.8.3292644. [DOI] [PubMed] [Google Scholar]
- 35.Yoshimori T., Semba T., Takemoto H., Akagi S., Yamamoto A., Tashiro Y. Protein disulfide-isomerase in rat exocrine pancreatic cells is exported from the endoplasmic reticulum despite possessing the retention signal. Journal of Biological Chemistry. 1990;265:15984–15990. [PubMed] [Google Scholar]
- 36.Kroning H., Kahne T., Ittenson A., Franke A., Ansorge S. Thiol-proteindisulfide-oxidoreductase (proteindisulfide isomerase): a new plasma membrane constituent of mature human B lymphocytes. Scandinavian Journal of Immunology. 1994;39:346–350. doi: 10.1111/j.1365-3083.1994.tb03384.x. [DOI] [PubMed] [Google Scholar]
- 37.Tager M., Kroning H., Thiel U., Ansorge S. Membrane-bound proteindisulfide isomerase (PDI) is involved in regulation of surface expression of thiols and drug sensitivity of B-CLL cells. Experimental Hematology. 1997;25:601–607. [PubMed] [Google Scholar]
- 38.Terada K., Manchikalapudi P., Noiva R., Jauregui H.O., Stockert R.J., Schilsky M.L. Secretion, surface localization, turnover, and steady state expression of protein disulfide isomerase in rat hepatocytes. Journal of Biological Chemistry. 1995;270:20410–20416. doi: 10.1074/jbc.270.35.20410. [DOI] [PubMed] [Google Scholar]
- 39.Chen K., Detwiler T.C., Essex D.W. Characterization of protein disulphide isomerase released from activated platelets. British Journal of Haematology. 1995;90:425–431. doi: 10.1111/j.1365-2141.1995.tb05169.x. [DOI] [PubMed] [Google Scholar]
- 40.Essex D.W., Chen K., Swiatkowska M. Localization of protein disulfide isomerase to the external surface of the platelet plasma membrane. Blood. 1995;86:2168–2173. [PubMed] [Google Scholar]
- 41.Hotchkiss K.A., Matthias L.J., Hogg P.J. Exposure of the cryptic Arg–Gly–Asp sequence in thrombospondin-1 by protein disulfide isomerase. Biochimica et Biophysica Acta. 1998;1388:478–488. doi: 10.1016/s0167-4838(98)00211-8. [DOI] [PubMed] [Google Scholar]
- 42.Bennett T.A., Edwards B.S., Sklar L.A., Rogelj S. Sulfhydryl regulation of L-selectin shedding: phenylarsine oxide promotes activation-independent L-selectin shedding from leukocytes. Journal of Immunology. 2000;164:4120–4129. doi: 10.4049/jimmunol.164.8.4120. [DOI] [PubMed] [Google Scholar]
- 43.Hahm E., Li J., Kim K., Huh S., Rogelj S., Cho J. Extracellular protein disulfide isomerase regulates ligand-binding activity of alphaMbeta2 integrin and neutrophil recruitment during vascular inflammation. Blood. 2013;121:3789–3800. doi: 10.1182/blood-2012-11-467985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raturi A., Miersch S., Hudson J.W., Mutus B. Platelet microparticle-associated protein disulfide isomerase promotes platelet aggregation and inactivates insulin. Biochimica et Biophysica Acta. 2008;1778:2790–2796. doi: 10.1016/j.bbamem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 45.Alloisio N., Texier P., Denoroy L., Berger C., Miraglia del Giudice E., Perrotta S., Iolascon A., Gilsanz F., Berger G., Guichard J. The cisternae decorating the red blood cell membrane in congenital dyserythropoietic anemia (type II) originate from the endoplasmic reticulum. Blood. 1996;87:4433–4439. [PubMed] [Google Scholar]
- 46.Low T.Y., Seow T.K., Chung M.C. Separation of human erythrocyte membrane associated proteins with one-dimensional and two-dimensional gel electrophoresis followed by identification with matrix-assisted laser desorption/ionization-time of flight mass spectrometry. Proteomics. 2002;2:1229–1239. doi: 10.1002/1615-9861(200209)2:9<1229::AID-PROT1229>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 47.Goodman S.R., Kurdia A., Ammann L., Kakhniashvili D., Daescu O. The human red blood cell proteome and interactome. Experimental Biology and Medicine (Maywood) 2007;232:1391–1408. doi: 10.3181/0706-MR-156. [DOI] [PubMed] [Google Scholar]
- 48.Ramachandran N., Root P., Jiang X.M., Hogg P.J., Mutus B. Mechanism of transfer of NO from extracellular S-nitrosothiols into the cytosol by cell-surface protein disulfide isomerase. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9539–9544. doi: 10.1073/pnas.171180998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sliskovic I., Raturi A., Mutus B. Characterization of the S-denitrosation activity of protein disulfide isomerase. Journal of Biological Chemistry. 2005;280:8733–8741. doi: 10.1074/jbc.M408080200. [DOI] [PubMed] [Google Scholar]
- 50.Deem S., Berg J.T., Kerr M.E., Swenson E.R. Effects of the RBC membrane and increased perfusate viscosity on hypoxic pulmonary vasoconstriction. Journal of Applied Physiology. 2000;88:1520–1528. doi: 10.1152/jappl.2000.88.5.1520. [DOI] [PubMed] [Google Scholar]
- 51.Zhang J.Z., Ismail-Beigi F. Activation of Glut1 glucose transporter in human erythrocytes. Archives of Biochemistry and Biophysics. 1998;356:86–92. doi: 10.1006/abbi.1998.0760. [DOI] [PubMed] [Google Scholar]
- 52.Zhang J.Z., Hayashi H., Ebina Y., Prohaska R., Ismail-Beigi F. Association of stomatin (band 7.2b) with Glut1 glucose transporter. Archives of Biochemistry and Biophysics. 1999;372:173–178. doi: 10.1006/abbi.1999.1489. [DOI] [PubMed] [Google Scholar]
- 53.Gladwin M.T., Wang X., Reiter C.D., Yang B.K., Vivas E.X., Bonaventura C., Schechter A.N. S-Nitrosohemoglobin is unstable in the reductive erythrocyte environment and lacks O2/NO-linked allosteric function. Journal of Biological Chemistry. 2002;277:27818–27828. doi: 10.1074/jbc.M203236200. [DOI] [PubMed] [Google Scholar]
- 54.Wang X., Kettenhofen N.J., Shiva S., Hogg N., Gladwin M.T. Copper dependence of the assay: modified assay for measuring cellular and blood nitrosated proteins. Free Radical Biology and Medicine. 2008;44:1362–1372. doi: 10.1016/j.freeradbiomed.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Di Iorio E.E. Preparation of derivatives of ferrous and ferric hemoglobin. Methods in Enzymology. 1981;76:57–72. doi: 10.1016/0076-6879(81)76114-7. [DOI] [PubMed] [Google Scholar]
- 56.van Assendelft O.W., Zijlstra W.G. Extinction coefficients for use in equations for the spectrophotometric analysis of haemoglobin mixtures. Analytical Biochemistry. 1975;69:43–48. doi: 10.1016/0003-2697(75)90563-1. [DOI] [PubMed] [Google Scholar]
- 57.Romeo A.A., Capobianco J.A., English A.M. Heme nitrosylation of deoxyhemoglobin by s-nitrosoglutathione requires copper. Journal of Biological Chemistry. 2002;277:24135–24141. doi: 10.1074/jbc.M202221200. [DOI] [PubMed] [Google Scholar]
- 58.Herold S., Rock G. Reactions of deoxy-, oxy-, and methemoglobin with nitrogen monoxide. Mechanistic studies of the S-nitrosothiol formation under different mixing conditions. Journal of Biological Chemistry. 2003;278:6623–6634. doi: 10.1074/jbc.M210275200. [DOI] [PubMed] [Google Scholar]
- 59.Wang Z., Shi J., Zhou Y., Ruan C. Detection of red blood cell-bound immunoglobulin G by flow cytometry and its application in the diagnosis of autoimmune hemolytic anemia. International Journal of Hematology. 2001;73:188–193. doi: 10.1007/BF02981936. [DOI] [PubMed] [Google Scholar]
- 60.de Isla N.G., Riquelme B.D., Rasia R.J., Valverde J.R., Stoltz J.F. Quantification of glycophorin A and glycophorin B on normal human RBCs by flow cytometry. Transfusion. 2003;43:1145–1152. doi: 10.1046/j.1537-2995.2003.00471.x. [DOI] [PubMed] [Google Scholar]
- 61.Beckmann R., Smythe J.S., Anstee D.J., Tanner M.J. Functional cell surface expression of band 3, the human red blood cell anion exchange protein (AE1), in K562 erythroleukemia cells: band 3 enhances the cell surface reactivity of Rh antigens. Blood. 1998;92:4428–4438. [PubMed] [Google Scholar]
- 62.Mezghrani A., Courageot J., Mani J.C., Pugniere M., Bastiani P., Miquelis R. Protein-disulfide isomerase (PDI) in FRTL5 cells. pH-dependent thyroglobulin/PDI interactions determine a novel PDI function in the post-endoplasmic reticulum of thyrocytes. Journal of Biological Chemistry. 2000;275:1920–1929. doi: 10.1074/jbc.275.3.1920. [DOI] [PubMed] [Google Scholar]
- 63.Patterson S.T., Li J., Kang J.A., Wickrema A., Williams D.B., Reithmeier R.A. Loss of specific chaperones involved in membrane glycoprotein biosynthesis during the maturation of human erythroid progenitor cells. Journal of Biological Chemistry. 2009;284:14547–14557. doi: 10.1074/jbc.M809076200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Holmgren A. Tryptophan fluorescence study of conformational transitions of the oxidized and reduced form of thioredoxin. Journal of Biological Chemistry. 1972;247:1992–1998. [PubMed] [Google Scholar]
- 65.Ado K., Takeda N., Kikuchi M., Taniguchi Y. The pressure effect on the structure and functions of protein disulfide isomerase. Biochimica et Biophysica Acta. 2006;1764:586–592. doi: 10.1016/j.bbapap.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 66.Nakamura T., Lipton S.A. Emerging role of protein–protein transnitrosylation in cell signaling pathways. Antioxidants and Redox Signaling. 2013;18:239–249. doi: 10.1089/ars.2012.4703. [DOI] [PMC free article] [PubMed] [Google Scholar]